

Abstract
Autophagy is generally considered to be antipathogenic. The autophagy gene ATG16L1 has a commonly occurring mutation associated with Crohn disease (CD) and intestinal cell abnormalities. Mice hypomorphic for ATG16L1 (ATG16L1HM) recreate specific features of CD. Our recent study shows that the same ATG16L1HM mice that are susceptible to intestinal inflammatory disease are protected from urinary tract infections (UTI), a common and important human disease primarily caused by uropathogenic E. coli (UPEC). UPEC colonize the bladder and exhibit both luminal and intra-epithelial stages. The host responds by recruiting innate immune cells and shedding infected epithelial cells to clear infection. Despite these countermeasures, UPEC can persist within the bladder epithelium as membrane-enclosed quiescent intracellular reservoirs (QIRs) that can seed recurrent UTI. The mechanisms of persistence remain unknown. In this study, we show that ATG16L1 deficiency protects the host against acute UTI and UPEC latency. ATG16L1HM mice clear urinary bacterial loads more rapidly and thoroughly due to ATG16L1-deficient innate immune components. Furthermore, ATG16L1HM mice exhibit superficial urothelial cell-autonomous architectural aberrations that also result in significantly reduced QIR numbers. Our findings reveal a host-protective effect of ATG16L1 deficiency in vivo against a common pathogen.
Keywords: Atg16L1, Atg5, Crohn disease, autophagy, bladder, urinary tract infections, uropathogenic E. coli
Recent reviews highlight the complex interplay between autophagy and microbial adaptations governing host-pathogen interaction outcomes. Autophagic degradation of intracellular pathogens is a significant host defense. However, pathogens employ many strategies to evade or subvert the autophagy machinery for survival including retarding the maturation of autophagosomes, impairing fusion with lysosomes, escaping to the cytosol, and adapting to survive and replicate within the autophagosomal or lysosomal compartment. Recently, for example, Kim and coworkers demonstrated the importance of host autophagy in modulating effective antimicrobial responses to Mycobacterium tuberculosis, and the Celli group showed that Brucella subverts autophagy complexes to facilitate its intracellular cycle. However, little is known about how UPEC interact with autophagy. We previously showed that UPEC are enclosed in LAMP1-positive vesicles (QIRs) that resemble LAMP1-positive spacious Listeria-containing phagosomes (SLAPs) described by Brumell’s group. In this study, we show that UPEC are also targeted by the ATG16L1, LC3, and SQSTM1/p62 proteins. Our findings for UPEC, thus, resemble other studies on intracellular recognition of Salmonella enterica serovar Typhimurium, Shigella flexneri, and Listeria monocytogenes. We also find UPEC in double-membranous structures in transmission electron micrographs of infected bladders (Fig. 1).
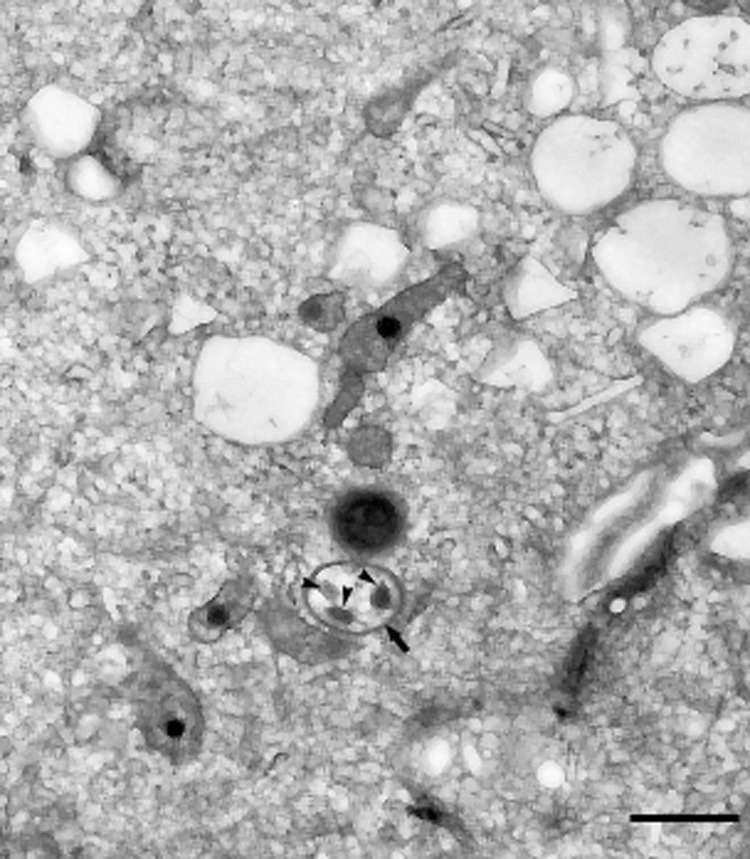
Figure 1. UPEC enclosed within a double-membraned autophagosomal structure. TEM of bladder tissue from ATG16L1HM mice 14 d post-infection, depicting UPEC (arrowheads) enclosed in a double-membraned autophagosomal structure (arrow). Bar: 1 µm.
The Virgin group had previously demonstrated that ATG16L1HM mice develop intestinal abnormalities in Paneth cells. Our work shows that ATG16L1 deficiency induces multiple, baseline abnormalities in cellular components that UPEC encounter during infection. ATG16L1-deficient cells dramatically accumulate multivesicular bodies, lysosomes and the UPEC receptor (UPK1A/UP1a). The aberrations are intrinsic to ATG16L1-deficient epithelial cells, because transferring ATG16L1-deficient hematopoietic cells does not induce them in wild-type epithelium, and even newly regenerating ATG16L1-deficient superficial cells show the same accumulations. Our study shows that UPEC are less able to occupy their intracellular niches to persist as QIRs in the ATG16L1HM epithelium. We propose, thus, that UPEC may normally utilize ATG16L1 and possibly other autophagy proteins to establish latency, thus ATG16L1 deficiency can be protective in this regard. In other words, UPEC may need the normal autophagic machinery to persist. The mechanisms underlying how UPEC avoid degradation or survive in the autophagosomal niches remain to be elucidated.
Autophagy plays multiple roles in both innate and adaptive immunity. Recent studies have suggested that autophagy governs the balance between defending against pathogens and modulating innate immunity to prevent excessive inflammatory responses and inflammasome signaling. Lee et al., demonstrated that ATG16L1 deficiency leads to a hyperinflammatory response by removing the restriction on IL1B/IL-1β signaling cascades and IL-6 production. Similarly, Saitoh et al., showed that ATG16L1-deficient macrophages produce high amounts of the inflammatory cytokines IL1B and IL18. In our study, we observed significantly increased levels of IL-6 and IL1A/IL-1α in infected ATG16L1HM mice relative to controls. Furthermore, our findings demonstrated that ATG16L1-deficient hematopoietic cells, especially neutrophils and macrophages, contribute critically to mounting a hypervigilant innate immune response, which likely promotes the rapid clearance of extracellular UPEC. Cadwell and Virgin previously demonstrated enhanced transcription of pro-inflammatory cytokines in aberrant Paneth cells of ATG16L1HM mice. In the setting of CD and the presence of commensal bacteria, elevated proinflammatory cytokine levels induced by ATG16L1 deficiency lead to intestinal pathology, which is detrimental to the host. However, in the ATG16L1-deficient urinary tract, the elevated proinflammatory cytokine levels may have a beneficial effect, as UPEC has been shown previously to inhibit proinflammatory cytokine production. Thus, an ATG16L1 deficiency-induced hyperinflammatory response may help clear the infection by countering UPEC’s ability to dampen innate immune responses.
It is important to determine if the protection we observed is ATG16L1-specific or represents a general effect of the autophagy pathway on UTI pathogenesis. The autophagy machinery comprises many essential proteins including ATG5, ATG7 and ATG12. We demonstrated that innate immune cell-specific knockdown of Atg5 induces a similar protective phenotype as that induced by ATG16L1 deficiency, suggesting that UPEC may hijack multiple autophagy components to colonize and persist in the urinary tract.
A polymorphism in ATG16L1 associated with CD can be found in up to 50% of individuals in certain populations. It is unclear why a seemingly unfavorable allelic variant of ATG16L1 would occur at such high frequency. One explanation is that the alleles were originally selected for a beneficial property such as protection against chronic or recurrent infectious diseases. Our results demonstrating that ATG16L1 mutation can confer protection against UTIs could explain why mutant alleles are unexpectedly frequent: protection against UTIs could be a mechanism to counter the negative selection imposed by the inflammation-promoting effects of the ATG16L1 mutation. A better understanding of the link between autophagy, latency, and inflammation could lead to therapeutic approaches, for example by targeting ATG16L1 or autophagy in general to treat recurrent UTI. Thus, the use of inhibitors of autophagy or ATG16L1 to eliminate latent bacterial reservoirs may have substantial clinical benefits to combat refractory and recurrent UTIs, because conventional antibiotics are unable to penetrate urothelial barriers to clear bacteria sequestered as QIRs.
Acknowledgments
I.U.M. holds a K99/R00 Pathway to Independence award, DK080643.
Glossary
Abbreviations:
- UTI
urinary tract infections
- UPEC
uropathogenic E. coli
- ATG16L1HM mice
ATG16L1 hypomorphic mice
- CD
Crohn disease
- QIRs
quiescent intracellular reservoirs
Footnotes
Previously published online: www.landesbioscience.com/journals/autophagy/article/21600