

Abstract
BCR-ABL+ K562 cells hold clinical promise as a component of cancer vaccines, either as bystander cells genetically modified to express immunostimulatory molecules, or as a source of leukemia antigens. To develop a method for detecting T-cell reactivity against K562 cell-derived antigens in patients, we exploited the dendritic cell (DC)-mediated cross-presentation of proteins generated from apoptotic cells. We used UVB irradiation to consistently induce apoptosis of K562 cells, which were then fed to autologous DCs. These DCs were used to both stimulate and detect antigen-specific CD8+ T-cell reactivity. As proof-of-concept, we used cross-presented apoptotic influenza matrix protein-expressing K562 cells to elicit reactivity from matrix protein-reactive T cells. Likewise, we used this assay to detect increased anti-CML antigen T-cell reactivity in CML patients that attained long-lasting clinical remissions following immunotherapy (donor lymphocyte infusion), as well as in 2 of 3 CML patients vaccinated with lethally irradiated K562 cells that were modified to secrete high levels of granulocyte macrophage colony-stimulating factor (GM-CSF). This methodology can be readily adapted to examine the effects of other whole tumor cell-based vaccines, a scenario in which the precise tumor antigens that stimulate immune responses are unknown.
Keywords: GM-CSF, K562, chronic myeloid leukemia, cross-presentation, immune monitoring, whole cell vaccine
Introduction
Chronic myeloid leukemia (CML) is characterized by the presence of the BCR-ABL translocation,1 and is known to be particularly immunosensitive. The high susceptibility of CML to immune destruction has been evidenced by the relatively high rates of durable remission achieved following hematopoietic stem cell transplantation (HSCT) and donor lymphocyte infusion (DLI).2 However, the utility of these treatments is often undermined by the risks of severe toxicity, most commonly in the form of acute or chronic graft-versus-host disease (GVHD). To overcome these limitations, therapeutic cancer vaccines for CML have been developed for the specific activation of the immune system against CML cells.3-7 Whole tumor cell-based vaccination has emerged as a promising strategy to treat a variety of malignancies,8-12 and has the advantage - over peptide-based approaches - of providing a range of antigens for the stimulation of polyclonal immune responses. In particular, the BCR-ABL+ K562 cell line has been shown to be amenable to large-scale manufacture, and has been developed as a clinical grade vaccine reagent.13,14
To determine the efficacy of cancer vaccines, methods are needed to monitor the development of antitumor immunity. This can readily be accomplished with peptide-based vaccines, as the immunizing epitopes are known. In contrast, whole tumor cell-based vaccines by definition comprise a complex mixture of antigens, and those specically targeted by therapy-induced immunity are unknown. An attractive approach to this challenge is to employ whole tumor cells themselves as as an antigen source, enabling the screening of all tumor antigens to which a patient was exposed. On the other hand, whole tumor cells in their natural state are often poorly immunogenic, and may only weakly stimulate an immune response.
One strategy for displaying antigens derived from whole tumor cells is through their cross-presentation by antigen presenting cells (APCs), such as dendritic cells (DCs). Apoptotic bodies can be efficiently ingested, processed and their antigens cross-presented on MHC class I molecules by immature dendritic cells (iDCs), leading to T cell activation-15,16 This cross- strategy has previously been developed for DC-based vaccines.17-19 Here, we describe the optimization and application of a K562 cell-based assay to detect CML-associated immunity following the administration of an effective immunotherapy for CML, DLI, as well as following K562 cell-based vaccination. Our studies demonstrate that developing a whole cell-based disease-specific assay that can be readily applied to the monitoring of cell-based cancer vaccines is feasible.
Results
Immature DCs effectively phagocytose apoptotic K562 cells
Several studies have identified that apoptosis results in efficient antigen cross-presentation to T cells.15,16,18 We sought to adapt this concept for developing an assay to monitor the immunologic activity of K562 cell-based tumor vaccines. As schematically represented in Figure 1, our workflow was to first optimize conditions for K562 cell death and for phagocytosis by DCs. We then wanted to ensure that cellular antigens derived from apoptotic K562 cells could be cross-presented to stimulate antigen-specific T-cell responses. To this aim, we tested our assay first for the detection of anti-viral responses against a known immunogenic viral antigen introduced into K562 cells, and second, for the detection of CML-specific immunity in CML patients successfully treated with DLI. Finally, we applied our assay to peripheral blood mononuclear cells (PBMCs) collected from CML patients who had been vaccinated with an experimental anti-CML immunotherapy, namely lethally irradiated K562 cells modified to secrete granulocyte macrophage colony-stimulating factor (GM-CSF) adjuvant.

Figure 1. Schema for developing a cross-presentation assay for measuring immunoreactivity to K562 cells.
Early apoptosis, defined as the stage at which cells are AnnexinV+ (have exposed phosphatidylserine on the cell surface) and PI- (have intact plasma membranes), has been identified as an optimal stage the uptake of apoptotic cells by DCs.20,21 We selected UVB as the method of apoptosis, as this is an established, inexpensive, and simple approach.17,18,21,22 We investigated a range of intensities (50–2000 mJ) and followed cells over a period of 2 to 48 h (Figure 2A). Intensities ranging from 300 to 500mJ consistently produced a high rate of early K562 apoptosis, which was not seen at lower intensities. Intensities higher than 500 mJ caused rapid necrosis. Irradiation with 400 mJ consistently resulted in approximately 35–45% early apoptosis, and 65–75% total apoptosis (AnnexinV+PI+) at 15 h post exposure (Fig. 2B). We thus selected these energies and time points for inducing apoptosis of K562 cells in subsequent experiments.

Figure 2. High stable rates of apoptosis of K562 cells occur upon UVB irradiation (400 mJ). (A) K562 cells were cultured for 15 h after irradiation with UVB and then stained with AnnexinV and PI. Early apoptotic cells were defined as AnnexinV+ and PI-, and late apoptotic cells were described as double positive. Dot plots are representative of a minimum of 5 experiments. (B) Average results of AnnexinV and PI staining of K562 cells over 24 h following UVB irradiation at 0–500 mJ. All results shown are based on the average of a minimum of three experiments.
To ensure that iDCs are capable of taking up apoptotic K562 cells, iDCs from normal donors were stained with the green fluorescent dye PKH2. These labeled iDCs were then co-cultured with K562 cells that had been labeled with a red fluorescent dye (PKH26) for 24 h at ratios of 1:1, 1:2, 1:5 and 1:9. A higher ratio of K562 cells to iDCs generally resulted in higher rates of phagocytosis. High levels of phagocytosis of approximately 80–85% (visualized as double positive cells on flow cytometry) were observed at physiological temperature (Fig. 3). On the other hand, minimal phagocytosis was observed when the same process was performed at 4°C.
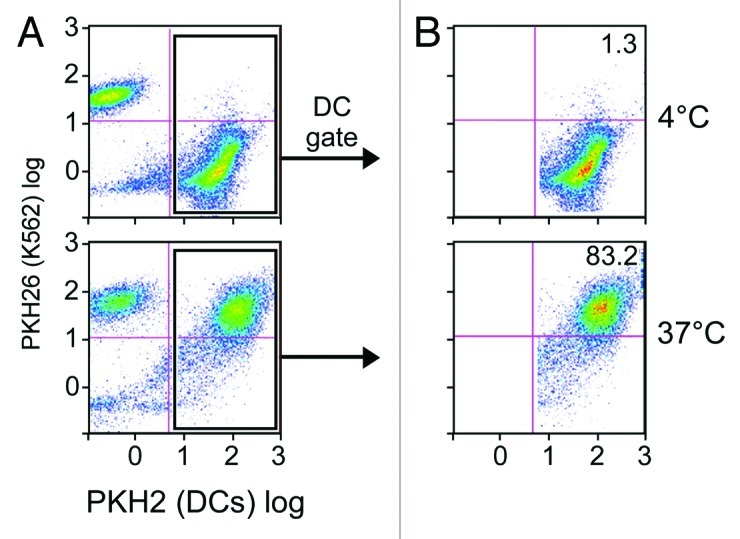
Figure 3. DCs effectively phagocytose apoptotic K562 cells. Immature DCs labeled in green (with PKH2) and apoptotic K562 cells labeled in red (with PKH26) were co-cultured at either 4°C (top) or 37°C (bottom) for 24 h at a 1:9 ratio. Shown are FACS results for the total population (A), and cells gated on the DC population (B). Dot plots are representative of 3 experiments.
Cross-presenting DCs effectively stimulate antigen-specific T-cells.
We next examined whether apoptotic K562 cells can be effectively processed and cross-presented by DCs to stimulate antigen-specific T cells. For these experiments, we used the well-characterized HLA-A*0201 restricted and immunodominant influenza M1 epitope as a model antigen.18,23,24 We introduced the influenza matrix protein into K562 cells through DNA plasmid nucleofection. We reasoned that if we could induce expression of this protein (and consequently of the M1 epitope), detection of M1-specific CD8+ T-cell immunity from normal HLA-A2+ volunteers by ELISpot assay would confirm that cross-presentation of K562 antigens had taken place.
As shown in Figure 4A (inset), we could achieve expression of influenza matrix protein in K562 cells (M1-K562) by nucleofection. We then induced early apoptosis of M1-expressing K562 cells using the conditions described above. These apoptotic cells were fed to HLA-A2+ DCs isolated from normal adult volunteers, which were then used to stimulate autologous CD8+ T cells. As expected, we found that DCs fed with apoptotic M1-K562 cells could induce M1-specific CD8+ T-cell responses (average 5.25 spots/100,000 CD8+ T cells; p = 0.04) (Fig. 4A). In contrast, apoptotic K562 cells or non-cross-presented M1-K562 cells alone (average 1.5 spots and 0.5 spots/100,000 cells, respectively), or DCs fed with apoptotic K562 cells lacking expression of the influenza matrix protein (0.75 spots/100,000 cells) generated only minimal reactivity. These results demonstrate that the cross-presentation of antigens by DCs can effectively be used to detect T-cell reactivity.

Figure 4. Apoptotic K562 cells when presented on DCs can stimulate antigen-specific immunity against either introduced or native antigen. (A) K562 cells were transfected to express M1 (see inset) and then irradiated with 400 mJ UVB to induce apoptosis. Following irradiation, these cells were co-cultured with iDCs for 24 h and matured using a cytokine cocktail for 24 h. On ELISpot, we used CD8+ T cells alone as a negative control, M1-pulsed DCs and PHA as positive controls, and apoptotic M1-K562 and DCs alone on CD8+ T-cells to establish background reactivity. There was a significant increase in reactivity of apoptotic M1-K562 when cross-presented by DCs compared with apoptotic M1-K562 cells alone (*p = 0.04) or DC cross-presented apoptotic K562 cells (p = 0.04). Significance was determined using the two-tailed Student’s t test. All experiments were performed in duplicate wells, and mean spot-forming cells per 100,000 CD8+ T cells are reported. Error bars represent two standard deviations. The ELISpot image is representative of more than 3 experiments, while the bar graph displays average results from four experiments, with standard deviation as shown. (B) T cells from pre- and post-DLI patients were tested on ELISpot against CML antigens in the form of apoptotic K562 cross-presented on DCs, and results were reported as spots per 100,000 CD8+ T cells (minus background, defined as reactivity of apoptotic K562 cells alone or autologous DCs alone on CD8+ T cells); p = 0.08 (Student’s t test). Three patients treated with imatinib were tested as comparison, and showed no reactivity. Results are shown as means with error bars representing two standard deviations.
CML-specific reactivity can be detected following anti-CML immunotherapy
DLI is a highly effective immunotherapy for the treatment of transplanted patients with relapsed CML. In this procedure, donor lymphocytes are infused into patients, typically in the absence of further chemo- or radio-therapy, leading to durable remission in 75–80% patients.25 Numerous reports have described the generation of CML antigen-specific antibodies and CD8+ T-cell responses following DLI.26-28 We therefore examined this group of patients for evidence of increased anti-K562 T-cell immunity developing upon DLI. We selected PBMC samples from 4 DLI-responsive patients (Patients DLI1–4), collected before and after DLI (at the time of clinical response) (Table 1). This patient group was clinically homogeneous: all patients relapsed 6–15 months following myeloablative allogeneic HSCT, received CD8-depleted donor lymphocytes for the treatment of relapsed stable-phase CML, and promptly developed cytogenetic and molecular responses (median 2.75 and 8 months post-DLI, respectively).
Table 1. Clinical characteristics of DLI-treated CML patients samples whose PBMC were tested by ELISpot against the K562 cross-presentation assay.
| Patient | Age/Sex | Months from HSCT to relapse | Months from HSCT to DLI | Months from DLI to cytogenetic remission | Months from DLI to molecular remission |
|---|---|---|---|---|---|
| DLI1 |
40/F |
6 |
24 |
3 |
7 |
| DLI2 |
50/M |
14 |
25 |
3 |
8 |
| DLI3 |
59/F |
15 |
14 |
2.5 |
8 |
| DLI4 | 25/F | 11 | 54 | 2 | 12 |
As shown in Figure 4B, we observed increased CD8+ T-cell reactivity to cross-presented apoptotic K562 cells in a DLI-responsive patient via ELIspot assays (average 16.5 spots/100,000 cells), compared with samples from the same patient prior to DLI (average 0 spots/100,000 cells, (p = 0.08). Furthermore, we detected only minimal reactivity in three CML patients who had been treated with non-immunologic therapy (imatinib, 800 mg daily for 1–7 y; Table 2). Our results are consistent with the detection of CML-specific T-cells following successful immunotherapy for CML, and suggest the potential utility of this assay for detecting CML specific T-cell responses following other forms of immunotherapy for CML.
Table 2. Clinical characteristics of Imatinib treated CML patient samples whose PBMC were tested by ELISpot against the K562 cross-presentation assay.
| Patient | Age/Sex | Previous therapy | Imatinib maximum dose; length of treatment |
|---|---|---|---|
| IM1 |
65/M |
Hydroxyures, interferon, busulfan |
800mg/daily; 3 y |
| IM2 |
67/M |
Hydroxyurea, interferon |
800mg/daily; 7 y |
| IM3 | 64/M | Hydroxyurea, interferon | 800mg/daily; 1 y |
GM-K562 vaccination elicits a T-cell response against CML antigens
At Dana-Farber Cancer Institute (DFCI, Boston), we launched a clinical trial to test the effects of whole tumor cell-based vaccines in patients with evidence of persistent minimal residual disease following treatment with imatinib. The vaccine consisted of lethally irradiated K562 cells (as a source of CML antigen) genetically modified to express GM-CSF (as adjuvant), and was administered as a series of 9 subcutaneous/intradermal injections. These vaccines were well-tolerated, although transient skin reactions at the site of injection were common (Fig. 5). To identify possible responses to all commonly expressed CML antigens, we screened patients for CD8+ T-cell immune responses against K562 cells.
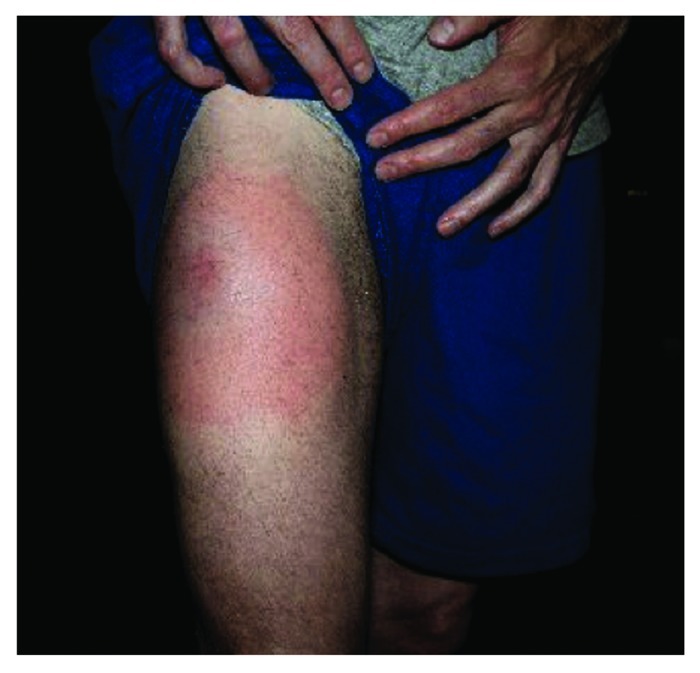
Figure 5. Skin reactions characterized by erythema, swelling and warmth occurred commonly at the immunization site, following sc/id injection of irradiated GM-CSF secreting K562 cells. These reactions were transient, lasting 1–5 d following vaccination.
We analyzed PBMC samples collected from three patients (Patients A-C) who were vaccinated with the GM-CSF expressing K562 vaccine (Table 3). We focused our analysis of CD8+ T-cell reactivity to apoptotic/cross-presented K562 cells at three time points: approximately 30 days prior to vaccination; at the middle of the vaccination cycle (vaccine #5, or day 50); and 30 days after the final vaccine (~day 170). We tested CD8+ T cells against autologous DCs that were fed apoptotic K562 cells on ELISpot assays, compared with apoptotic K562 cells or autologous DCs alone. Results of the ELIspot assays were integrated with counts of absolute white blood cell (WBC) numbers, molecular detection of tumor burden, and immunophenotype of peripheral blood T cells throughout the vaccination period (Fig. 6).
Table 3. Clinical characteristics of GM-CSF secreting irradiated K562 cell treated CML patient samples whose PBMC were tested by ELISpot against the K562 cross-presentation assay .
| Patient | Age/Sex | Previous therapy | Imatinib maximum dose; length of treatment |
|---|---|---|---|
| A |
42/M |
- |
800mg/daily; 2 y |
| B |
45/M |
Hydroxyurea |
800mg/daily; 1 y |
| C | 55/M | Hydroxyurea | 800mg/daiy; 5 y |

Figure 6. Immunologic monitoring of CML patients subjected to therapeutic vaccination with irradiated GM-CSF-secreting K562 cells. Reactivity to K562 antigens by cross-presentation assay (measured by ELISpot and shown on bottom row) in relation to: timing of vaccination (indicated by arrows, top row); to absolute WBC count (top row); molecular response, measured by %BCR-ABL/GUS transcript in PBMC (mid row); and ratio of CD8+ cells to regulatory T cells (Tregs) (defined as FOXP3+/CD25+), measured by flow cytometry (bottom row).
For both Patients A and B, transiently increased anti-CML T-cell reactivity was detected at mid-vaccination cycle, while Patient C demonstrated an absence of reactivity at all time points. Patient A exhibited increased anti-K562 antigen reactivity from 101 spots/100,000 cells prior to vaccination, to 154.5 spots/100,000 cells following the 5th vaccine, that then declined to 82.5 spots/100,000 cells at day 170. Likewise, T cells in Patient B revealed increased reactivity from 12.5 spots/100,000 cells prior to vaccination to 25 spots/100,000 cells following the 5th vaccine, which then declined to 13.5 spots/100,000 cells at day 170. None of the patients demonstrated alterations in their circulating WBCs. For Patients A and B, however, concomitant decrements of approximately one log were observed in molecular tumor burden (%BCR-ABL/GUS transcript) At the same time, an increased ratio of effector CD8+ to regulatory T cells (Tregs) was observed in Patient B. As for Patient C, the absence of T-cell reactivity was associated with a gradual one log increase in molecular tumor burden over the 170 days. Of note, both Patients A and B subsequently demonstrated prolonged stable disease on drug therapy despite persistent minimal residual disease, while Patient C, with rising tumor burden, subsequently underwent HSCT.
Discussion
Among multiple approaches for the design of anticancer vaccines, whole cell-based strategies have so far proved to be the most promising.8,29 Whole cell-based vaccines can be autologous or allogeneic, and may be modified (e.g., to express immunomodulatory molecules),30-33 fused with DCs,34-36 or unmodified (e.g., in the form of lysates or irradiated whole cells).37,38 The promise of this vaccine formulation has been primarily attributed to the multi-epitope nature of the immunogen. However, this same feature also creates challenges for monitoring, since the stimulating antigen(s) are unknown. One commonly employed monitoring approach uses candidate tumor-associated antigens as representative targets for immune responses. For example, in the case of CML, BCR-ABL, proteinase 3 and Wilms tumor 1 (WT1) all are highly expressed and the latter two are known to be immunogenic.39-42 However, these individual antigens may not be consistently immunogenic across patients, and only a subset of patients may bear the restricting HLA-allele associated with known antigen-derived epitopes. This was suggested by Smith et al.,13 since they observed decrement in tumor burden following vaccination of CML patients with irradiated, modified K562 cells and development of humoral immunity against whole tumor-cell lysates, but absence of detectable T-cell immunity against a panel of known CML associated antigens.43 Presumably, T-cell reactivity developed against unknown CML antigens.
Our study demonstrates that developing a robust assay to measure T cell reactivity to leukemia antigens from whole tumor cells is feasible. While other sources of whole-cell tumor antigens for this purpose are available, such as total RNA or tumor lysates, we favored using apoptotic bodies since they have been demonstrated to efficiently provoke T-cell responses.20,22,44 We focused on developing a K562 cell-based assay, since clinical studies at our center and elsewhere have used irradiated K562 cells as a vaccine reagent.45,46 These studies include approaches using K562 cells either directly as a therapeutic vaccine for myeloid leukemia or as bystander cells, for cell-based delivery of cytokine adjuvants or other immunologically active molecules.47-51 Thus, the development of a whole cell-based assay to detect anti-K562 T-cell responses might enable the screening of T-cell responses to leukemia antigens, for determining the immunogenicity of bystander cells. Finally, by detecting anti-leukemia T-cell reactivity, our assay can provide a stepping-stone for the discovery of novel leukemia antigens, if used in combination with approaches such as T-cell based cDNA expression library cloning.
We applied our assay to monitor three CML patients who were exposed to a series of 9 vaccinations with GM-CSF-secreting K562 cells over 20 weeks, and integrated these results with information on molecular tumor burden and T-cell immunophenotype. Despite the limitations of a small sample size, the results of our monitoring studies stimulate further investigation. In two of three patients, we observed a 50% increase in T-cell reactivity to CML antigens during the course of vaccination. This mid-cycle reactivity coincided with 1-log decreases in molecular tumor burden. Patient B further demonstrated a dramatic decrease in ratio of Tregs to effector T cells at this time. While the results for each assay measurement were modest, taken together they suggest that the vaccine has a biologic activity against CML.
Both Patients A and B were further observed to lose anti-K562 reactivity later in the vaccine course. On the one hand, this pattern could reflect the effects of the vaccine administration schedule. All vaccines were initially administered weekly (× 3) and then gradually tapered off to bi-monthly (× 3), then finally as monthly vaccines (× 3). Possibly, a high frequency of vaccination is vital to inducing antitumor responses, and for overcoming endogenous tumor-associated immunosuppression.52 Alternatively, induction of immunity could also activate counter regulatory mechanisms that function to suppress excessive immune activity, i.e., generation of exhausted and inefficient T cells with continuous antigenic stimulation.53 Furthermore, GM-CSF itself can function as both an immunostimulatory and immunosuppressive molecule.54
In summary, our studies using a K562 cross-presentation assay are suggestive of the development of anti-CML T-cell reactivity following whole leukemia-cell vaccination. Moreover, our results suggest that alternative adjuvants and/or administration schedules might be required to generate more consistent responses. The availability of our K562 cross-presentation assay renders benchmark antitumor immunity measurements possible, from which the effects of further modifications to this promising immunogen formulation can be assessed.
Materials and Methods
Patient samples
Heparinized peripheral blood from normal volunteers and from CML patients were collected during their course of treatment while participating on clinical research protocols at the Dana-Farber Cancer Institute (DFCI), Boston MA. All clinical protocols were approved by the DFCI Human Subjects Protection Committee. The K562-GM-CSF trial also received approval from the Food and Drug Administration and the NIH Recombinant Advisory Committee on Gene Transfer. Peripheral blood mononuclear cells (PBMCs) from normal donors and patients were isolated by Ficoll/Hypaque density-gradient centrifugation (GE Healthcare Bio-Sciences AB, Piscataway), cryopreserved with 10% DMSO, and stored in vapor-phase liquid nitrogen until the time of analysis.
Induction of apoptotic death of K562 cells
The BCR-ABL+ erythroleukemia cell line K562 (ATCC) was cultured in 162 cm2 tissue culture flasks (Corning Inc.) from an initial concentration of 1x106 cells/mL, using media consisting of RPMI 1640 (Cellgro Mediatech Inc.) with 10% fetal bovine serum (FBS) (Lonza). K562 cells were harvested on day 3 of culture, washed and suspended in PBS (Mediatech Inc.) at a concentration of 5 × 106 cells/mL. 5x106 of these cells were plated in PBS in each well of 6-well plates (Corning Inc.) and irradiated with UVB (0 to 2000 mJ; Stratalinker UV Crosslinker 1800, Stratagene, Cedar Creek TX). Following irradiation, the cells were incubated at 37°C until harvest. Two to 24 h following irradiation, K562 cells were labeled with AnnexinV and propidium iodide (PI) stains, according to the manufacturer’s instructions (BD PharMingen), and analyzed by flow cytometry (Cytomics FC 500, Beckman Coulter Inc.). The extent of apoptosis was defined as the percentage of AnnexinV positive and PI negative cells based on 50,000 events.
Isolation and preparation of monocyte-derived DCs
To generate DCs, frozen PBMCs were thawed and monocytes were selected through immunomagnetic CD14+ bead selection (Miltenyi Biotech). The CD14+ cells were cultured at 2 million cells in 3 mL of RPMI supplemented with 2% non-HI FCS (Cellgro), 2 mM glutamine, 50 µg/mL human transferrin (Roche), 5 µg/mL human insulin (Sigma-Aldrich), and 15 µg/mL gentamycin (Invitrogen) in the presence of 120 ng/mL GM-CSF (R&D Systems) and 70 ng/mL IL-4 (R&D Systems) in 6-well plates. Additional quantities of GM-CSF (120 ng/mL) and IL-4 (70 ng/mL) were added on days 3 and 5 to generate iDCs. DCs were matured on day 7 by addition of a cocktail consisting of GM-CSF (50 ng/mL), IL-4 (20 ng/mL), tumor necrosis factor α (TNFα; 10 ng/mL) and prostaglandin E-2 (PGE-2; 1 μM) (all from R&D Systems). We confirmed that DCs were consistently immature at day 6 and mature at day 8 using flow cytometric analysis with the markers CD80, CD86, HLA-DR and CD83.
Uptake of apoptotic bodies by antigen-presenting dendritic cells
For most experiments, apoptotic K562 cells served as source of antigenic ‘food’ for dendritic cells. Day 6 iDCs were co-cultured together with irradiated K562 cells at a 1:9 ratio at 37°C in DC media. After 24 h, fresh DC media containing the DC maturation cytokine cocktail was added to the co-culture, and incubated for a further 24 h. For some experiments, we introduced the influenza matrix protein into K562 cells by DNA nucleofection (Nucleofector II, Amaxa Biosystems) of a plasmid encoding the influenza matrix protein (gift of Matthew Albert, Rockefeller University, NY) on day 2 of culture to create M1-expressing K562 cells (M1-K562). M1-K562 cells were cultured overnight before apoptosis induction by UVB irradiation on day 3.
To confirm phagocytosis of apoptotic K562 cells, we stained K562 cells red with PKH26 (Sigma-Aldrich), according to the manufacturer’s instructions, prior to induction of apoptosis. iDCs were stained green with PKH2 (Sigma-Aldrich), according to the manufacturer’s instructions, following harvest on day 6 and immediately prior to co-culture of the two cell populations. The two stained cell populations were combined immediately following K562 irradiation in a 1:9 ratio and co-cultured in DC media for 24 h at 37°C (to encourage phagocytosis) or 4°C (to inhibit phagocytosis). Phagocytosis of apoptotic bodies by DCs was defined by the percentage of cells that were double positive for both PHK2 and PKH26 stains on flow cytometry.
In vitro expansion of antigen-specific T-cells
To expand autologous K562 reactive T cells, we thawed autologous PBMCs and stimulated them for 7 days by co-culture with mature DCs that had been fed apoptotic K562 cells in T-cell media in 24-well plates (Becton Dickinson). 2–3 × 106 PBMCs were placed in each well and maintained in Iscove’s modified Dulbecco’s media (IMDM) (Cellgro Mediatech Inc.) that was supplemented with 20% FBS, 1% glutamine (2 mM), penicillin (50 IU/mL; Mediatech Inc.) and streptomycin (50 µg/mL; Mediatech Inc.). IL-7 (10 ng/mL) was added on day 0 and 4; IL-2 (50 U/mL) (both R&D Systems) was also added on day 4. For some experiments, T cells were expanded against peptide-pulsed controls. DCs were generated as described and pulsed with M1 peptide (10 µg/mL) for 2 h prior to co-culture with autologous PBMCs for 7 d.
Detection of antigen specific T-cell responses
We used the ELISpot assay to measure the frequency of reactive IFNγ secreting CD8+ T-cells at 7 d after initiation of in vitro stimulation. Nitrocellulose membrane 96-well plates (Multiscreen, Millipore) were coated with anti-human IFNγ monoclonal antibody (1-D1K; Mabtech). One day prior to plating the antigen-primed T-cells on the ELISpot plate together with different test antigens, we selected cytotoxic T-cells by immunomagnetic CD8 selection (Miltenyi Biotech). The CD8+ T-cells were starved overnight in serum-free AIM-V medium (Gibco) and 1% glutamine (2 mM) to reduce ELISpot background. IFNγ secretion was detected using capture and detection antibodies as directed (Mabtech AB) and imaged using an ImmunoSpot Series Analyzer (Cellular Technology Ltd.).
K562 vaccination clinical protocol
Patients received a series of 9 vaccinations on a clinical trial conducted at DFCI from 2005 to 2008 (registered as NCT00301093 on www.clinicaltrials.gov). Each vaccine contained 5x106 GM-K562 cells that had been lethally irradiated with 10,000 cGy γ rays. Vaccines were administered once a week (+/− 1 d) for three weeks, then every other week (+/− 1 d) for three doses and then every 28 d (+/− 2 d) for three doses. Peripheral blood samples were collected monthly from study subjects for molecular BCR-ABL monitoring (Department of Molecular Diagnostics, Brigham and Women’s Hospital, Boston) and immunophenotyping for the first nine months after initiation of vaccines. The monoclonal antibodies used for immunophenotyping were directed against CD8 (clone B9.11; Beckman-Coulter); CD4 (clone 13B8.2; Beckman-Coulter); FOXP3 (clone PCH101; eBioscience), and CD25 (clone B1.49.9; Beckman-Coulter).
Statistical Analyses
Two-tailed Student’s t-test with pooled variance were applied to determine the difference between groups. p values < 0.05 were considered significant.
Disclosure of Potential Conflicts of Interest
The authors declare no competing financial interests.
Acknowledgments
The authors would like to acknowledge Nathalie Blachere and Nir Hacohen for their insightful discussions in the development of this research. We thank Mr. Quinlan L. Sievers for excellent technical assistance. We would like to acknowledge the generous support from the Pasquarello Tissue Bank, and from the clinical transplant teams at the DFCI, Boston. We also acknowledge support from the Department of Defense for this project. C. J. W. acknowledges support from the Department of Defense (W81XWH-07–1-0080), the Miles and Eleanor Shore Award, NCI (5R21CA115043–2), the Early Career Physician-Scientist Award of the Howard Hughes Medical Institute, and is a Damon-Runyon Cancer Research Foundation Clinical Investigator (CI-38–07).
Authors Contributions
AB and CJW designed the study, performed research, analyzed data, and wrote the manuscript. UH, MS, and TS performed research. JSL and RM generated the clinical grade GM-K562 vaccination reagent. DN provided statistical analysis for the vaccine trial and this study. MW, RS, CC, JR, GD, and DD conducted the clinical trial and provided critical patient samples. All authors edited the paper.
Footnotes
Previously published online: www.landesbioscience.com/journals/oncoimmunology/article/20954
References
- 1.Cortes J. Natural history and staging of chronic myelogenous leukemia. Hematol Oncol Clin North Am. 2004;18:569–84, viii. doi: 10.1016/j.hoc.2004.03.011. [viii.] [DOI] [PubMed] [Google Scholar]
- 2.Wu CJ. Immunological targeting of the cancer stem cell. In: The Stem Cell Research Community, ed. Stembook: Stembook, 2008. [Google Scholar]
- 3.Cathcart K, Pinilla-Ibarz J, Korontsvit T, Schwartz J, Zakhaleva V, Papadopoulos EB, et al. A multivalent bcr-abl fusion peptide vaccination trial in patients with chronic myeloid leukemia. Blood. 2004;103:1037–42. doi: 10.1182/blood-2003-03-0954. [DOI] [PubMed] [Google Scholar]
- 4.Bocchia M, Gentili S, Abruzzese E, Fanelli A, Iuliano F, Tabilio A, et al. Effect of a p210 multipeptide vaccine associated with imatinib or interferon in patients with chronic myeloid leukaemia and persistent residual disease: a multicentre observational trial. Lancet. 2005;365:657–62. doi: 10.1016/S0140-6736(05)17945-8. [DOI] [PubMed] [Google Scholar]
- 5.Jain N, Kantarjian HM, Garcia-Manero G, Borthakur G, Ebarb T, Cortes JE. Synthetic tumor-specific breakpoint peptide vaccine in patients (pts) with chronic myeloid leukemia (CML) and minimal residual disease: A phase II trial. Journal of Clinical Oncology, 2008 ASCo Annual meeting Proceedings (Post-Meeting Edition) 2008; 26:7056. [Google Scholar]
- 6.el-Shami K, Smith BD. Immunotherapy for myeloid leukemias: current status and future directions. Leukemia. 2008;22:1658–64. doi: 10.1038/leu.2008.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mittal S, Marshall NA, Barker RN, Vickers MA. Immunomodulation against leukemias and lymphomas: a realistic future treatment? Crit Rev Oncol Hematol. 2008;65:101–8. doi: 10.1016/j.critrevonc.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 8.Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10:909–15. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Gruijl TD, van den Eertwegh AJ, Pinedo HM, Scheper RJ. Whole-cell cancer vaccination: from autologous to allogeneic tumor- and dendritic cell-based vaccines. Cancer Immunol Immunother. 2008;57:1569–77. doi: 10.1007/s00262-008-0536-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ward S, Copier J, Dalgleish A. Technical challenges facing therapeutic cancer vaccines. Curr Opin Drug Discov Devel. 2008;11:168–77. [PubMed] [Google Scholar]
- 11.Di Nicola M, Zappasodi R, Carlo-Stella C, Mortarini R, Pupa SM, Magni M, et al. Vaccination with autologous tumor-loaded dendritic cells induces clinical and immunologic responses in indolent B-cell lymphoma patients with relapsed and measurable disease: a pilot study. Blood. 2009;113:18–27. doi: 10.1182/blood-2008-06-165654. [DOI] [PubMed] [Google Scholar]
- 12.Soiffer R, Hodi FS, Haluska F, Jung K, Gillessen S, Singer S, et al. Vaccination with irradiated, autologous melanoma cells engineered to secrete granulocyte-macrophage colony-stimulating factor by adenoviral-mediated gene transfer augments antitumor immunity in patients with metastatic melanoma. J Clin Oncol. 2003;21:3343–50. doi: 10.1200/JCO.2003.07.005. [DOI] [PubMed] [Google Scholar]
- 13.Smith BD, Kasamon YL, Kowalski J, Gocke C, Murphy K, Miller CB, et al. K562/GM-CSF immunotherapy reduces tumor burden in chronic myeloid leukemia patients with residual disease on imatinib mesylate. Clin Cancer Res. 2010;16:338–47. doi: 10.1158/1078-0432.CCR-09-2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dessureault S, Alsarraj M, McCarthy S, Hunter T, Noyes D, Lee D, et al. A GM-CSF/CD40L producing cell augments anti-tumor T cell responses. J Surg Res. 2005;125:173–81. doi: 10.1016/j.jss.2004.11.036. [DOI] [PubMed] [Google Scholar]
- 15.Albert ML, Sauter B, Bhardwaj N. Dendritic cells acquire antigen from apoptotic cells and induce class I-restricted CTLs. Nature. 1998;392:86–9. doi: 10.1038/32183. [DOI] [PubMed] [Google Scholar]
- 16.Blachère NE, Darnell RB, Albert ML. Apoptotic cells deliver processed antigen to dendritic cells for cross-presentation. PLoS Biol. 2005;3:e185. doi: 10.1371/journal.pbio.0030185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Benencia F, Courrèges MC, Coukos G. Whole tumor antigen vaccination using dendritic cells: comparison of RNA electroporation and pulsing with UV-irradiated tumor cells. J Transl Med. 2008;6:21. doi: 10.1186/1479-5876-6-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Orange DE, Jegathesan M, Blachère NE, Frank MO, Scher HI, Albert ML, et al. Effective antigen cross-presentation by prostate cancer patients’ dendritic cells: implications for prostate cancer immunotherapy. Prostate Cancer Prostatic Dis. 2004;7:63–72. doi: 10.1038/sj.pcan.4500694. [DOI] [PubMed] [Google Scholar]
- 19.Brusa D, Garetto S, Chiorino G, Scatolini M, Migliore E, Camussi G, et al. Post-apoptotic tumors are more palatable to dendritic cells and enhance their antigen cross-presentation activity. Vaccine. 2008;26:6422–32. doi: 10.1016/j.vaccine.2008.08.063. [DOI] [PubMed] [Google Scholar]
- 20.Kokhaei P, Choudhury A, Mahdian R, Lundin J, Moshfegh A, Osterborg A, et al. Apoptotic tumor cells are superior to tumor cell lysate, and tumor cell RNA in induction of autologous T cell response in B-CLL. Leukemia. 2004;18:1810–5. doi: 10.1038/sj.leu.2403517. [DOI] [PubMed] [Google Scholar]
- 21.Kotera Y, Shimizu K, Mulé JJ. Comparative analysis of necrotic and apoptotic tumor cells as a source of antigen(s) in dendritic cell-based immunization. Cancer Res. 2001;61:8105–9. [PubMed] [Google Scholar]
- 22.Schnurr M, Scholz C, Rothenfusser S, Galambos P, Dauer M, Röbe J, et al. Apoptotic pancreatic tumor cells are superior to cell lysates in promoting cross-priming of cytotoxic T cells and activate NK and gammadelta T cells. Cancer Res. 2002;62:2347–52. [PubMed] [Google Scholar]
- 23.Gianfrani C, Oseroff C, Sidney J, Chesnut RW, Sette A. Human memory CTL response specific for influenza A virus is broad and multispecific. Hum Immunol. 2000;61:438–52. doi: 10.1016/S0198-8859(00)00105-1. [DOI] [PubMed] [Google Scholar]
- 24.Mirmonsef P, Tan G, Zhou G, Morino T, Noonan K, Borrello I, et al. Escape from suppression: tumor-specific effector cells outcompete regulatory T cells following stem-cell transplantation. Blood. 2008;111:2112–21. doi: 10.1182/blood-2007-06-096586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tomblyn M, Lazarus HM. Donor lymphocyte infusions: the long and winding road: how should it be traveled? Bone Marrow Transplant. 2008;42:569–79. doi: 10.1038/bmt.2008.259. [DOI] [PubMed] [Google Scholar]
- 26.Nijmeijer BA, van Schie MLJ, Verzaal P, Willemze R, Falkenburg JHF. Responses to donor lymphocyte infusion for acute lymphoblastic leukemia may be determined by both qualitative and quantitative limitations of antileukemic T-cell responses as observed in an animal model for human leukemia. Exp Hematol. 2005;33:1172–81. doi: 10.1016/j.exphem.2005.06.034. [DOI] [PubMed] [Google Scholar]
- 27.Zhang W, Choi J, Zeng W, Rogers SA, Alyea EP, Rheinwald JG, et al. Graft-versus-leukemia antigen CML66 elicits coordinated B-cell and T-cell immunity after donor lymphocyte infusion. Clin Cancer Res. 2010;16:2729–39. doi: 10.1158/1078-0432.CCR-10-0415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Norde WJ, Overes IM, Maas F, Fredrix H, Vos JCM, Kester MGD, et al. Myeloid leukemic progenitor cells can be specifically targeted by minor histocompatibility antigen LRH-1-reactive cytotoxic T cells. Blood. 2009;113:2312–23. doi: 10.1182/blood-2008-04-153825. [DOI] [PubMed] [Google Scholar]
- 29.Johnson RS, Walker AI, Ward SJ. Cancer vaccines: will we ever learn? Expert Rev Anticancer Ther. 2009;9:67–74. doi: 10.1586/14737140.9.1.67. [DOI] [PubMed] [Google Scholar]
- 30.Wysocki PJ, Kazimierczak U, Suchorska W, Kotlarski M, Malicki J, Mackiewicz A. Gene-modified tumor vaccine secreting a designer cytokine Hyper-Interleukin-6 is an effective therapy in mice bearing orthotopic renal cell cancer. Cancer Gene Ther. 2010;17:465–75. doi: 10.1038/cgt.2010.2. [DOI] [PubMed] [Google Scholar]
- 31.Olivares J, Kumar P, Yu Y, Maples PB, Senzer N, Bedell C, et al. Phase I trial of TGF-β 2 antisense GM-CSF gene-modified autologous tumor cell (TAG) vaccine. Clin Cancer Res. 2011;17:183–92. doi: 10.1158/1078-0432.CCR-10-2195. [DOI] [PubMed] [Google Scholar]
- 32.Nemunaitis J, Nemunaitis M, Senzer N, Snitz P, Bedell C, Kumar P, et al. Phase II trial of Belagenpumatucel-L, a TGF-beta2 antisense gene modified allogeneic tumor vaccine in advanced non small cell lung cancer (NSCLC) patients. Cancer Gene Ther. 2009;16:620–4. doi: 10.1038/cgt.2009.15. [DOI] [PubMed] [Google Scholar]
- 33.Hardwick N, Chan L, Ingram W, Mufti G, Farzaneh F. Lytic activity against primary AML cells is stimulated in vitro by an autologous whole cell vaccine expressing IL-2 and CD80. Cancer Immunol Immunother. 2010;59:379–88. doi: 10.1007/s00262-009-0756-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qi C-J, Ning Y-L, Han Y-S, Min H-Y, Ye H, Zhu Y-L, et al. Autologous dendritic cell vaccine for estrogen receptor (ER)/progestin receptor (PR) double-negative breast cancer. Cancer Immunol Immunother. 2012;•••:1–10. doi: 10.1007/s00262-011-1192-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schwaab T, Schwarzer A, Wolf B, Crocenzi TS, Seigne JD, Crosby NA, et al. Clinical and immunologic effects of intranodal autologous tumor lysate-dendritic cell vaccine with Aldesleukin (Interleukin 2) and IFN-α2a therapy in metastatic renal cell carcinoma patients. Clin Cancer Res. 2009;15:4986–92. doi: 10.1158/1078-0432.CCR-08-3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.De Vleeschouwer S, Fieuws S, Rutkowski S, Van Calenbergh F, Van Loon J, Goffin J, et al. Postoperative adjuvant dendritic cell-based immunotherapy in patients with relapsed glioblastoma multiforme. Clin Cancer Res. 2008;14:3098–104. doi: 10.1158/1078-0432.CCR-07-4875. [DOI] [PubMed] [Google Scholar]
- 37.Jocham D, Richter A, Hoffmann L, Iwig K, Fahlenkamp D, Zakrzewski G, et al. Adjuvant autologous renal tumour cell vaccine and risk of tumour progression in patients with renal-cell carcinoma after radical nephrectomy: phase III, randomised controlled trial. Lancet. 2004;363:594–9. doi: 10.1016/S0140-6736(04)15590-6. [DOI] [PubMed] [Google Scholar]
- 38.Li G, Andreansky S, Helguera G, Sepassi M, Janikashvili N, Cantrell J, et al. A chaperone protein-enriched tumor cell lysate vaccine generates protective humoral immunity in a mouse breast cancer model. Mol Cancer Ther. 2008;7:721–9. doi: 10.1158/1535-7163.MCT-07-2067. [DOI] [PubMed] [Google Scholar]
- 39.Molldrem JJ, Komanduri K, Wieder E. Overexpressed differentiation antigens as targets of graft-versus-leukemia reactions. Curr Opin Hematol. 2002;9:503–8. doi: 10.1097/00062752-200211000-00006. [DOI] [PubMed] [Google Scholar]
- 40.Inoue K, Ogawa H, Sonoda Y, Kimura T, Sakabe H, Oka Y, et al. Aberrant overexpression of the Wilms tumor gene (WT1) in human leukemia. Blood. 1997;89:1405–12. [PubMed] [Google Scholar]
- 41.Lozzio BB, Lozzio CB. Properties and usefulness of the original K-562 human myelogenous leukemia cell line. Leuk Res. 1979;3:363–70. doi: 10.1016/0145-2126(79)90033-X. [DOI] [PubMed] [Google Scholar]
- 42.Gannagé M, Abel M, Michallet AS, Delluc S, Lambert M, Giraudier S, et al. Ex vivo characterization of multiepitopic tumor-specific CD8 T cells in patients with chronic myeloid leukemia: implications for vaccine development and adoptive cellular immunotherapy. J Immunol. 2005;174:8210–8. doi: 10.4049/jimmunol.174.12.8210. [DOI] [PubMed] [Google Scholar]
- 43.Smith B, Kasamon Y, Miller C, Chia C, Murphy K, Kowalski J, et al. K562/GM-CSF vaccination reduces tumor burden, including achieving molecular remissions, in chronic myeloid leukemia (CML) oatients with residual disease on imatinib mesylate (IM). Blood (ASH Annual Meeting Abstracts) 2005; 106. [Google Scholar]
- 44.Hayashi T, Hideshima T, Akiyama M, Raje N, Richardson P, Chauhan D, et al. Ex vivo induction of multiple myeloma-specific cytotoxic T lymphocytes. Blood. 2003;102:1435–42. doi: 10.1182/blood-2002-09-2828. [DOI] [PubMed] [Google Scholar]
- 45.Ho VT, Vanneman M, Kim H, Sasada T, Kang YJ, Pasek M, et al. Biologic activity of irradiated, autologous, GM-CSF-secreting leukemia cell vaccines early after allogeneic stem cell transplantation. Proc Natl Acad Sci U S A. 2009;106:15825–30. doi: 10.1073/pnas.0908358106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Butler MO, Friedlander P, Milstein MI, Mooney MM, Metzler G, Murray AP, et al. Establishment of antitumor memory in humans using in vitro-educated CD8+ T cells. Sci Transl Med. 2011;3:80ra34. doi: 10.1126/scitranslmed.3002207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ruffini PA, Di Nicola M, Carlo-Stella C, Siena S, Gianni AM. Genetic idiotypic and tumor cell-based vaccine strategies for indolent non Hodgkin’s lymphoma. Curr Gene Ther. 2005;5:511–21. doi: 10.2174/156652305774329221. [DOI] [PubMed] [Google Scholar]
- 48.Nagarajan S, Selvaraj P. Human tumor membrane vesicles modified to express glycolipid-anchored IL-12 by protein transfer induce T cell proliferation in vitro: a potential approach for local delivery of cytokines during vaccination. Vaccine. 2006;24:2264–74. doi: 10.1016/j.vaccine.2005.11.045. [DOI] [PubMed] [Google Scholar]
- 49.Suhoski MM, Golovina TN, Aqui NA, Tai VC, Varela-Rohena A, Milone MC, et al. Engineering artificial antigen-presenting cells to express a diverse array of co-stimulatory molecules. Mol Ther. 2007;15:981–8. doi: 10.1038/mt.sj.6300134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Borrello IM, Levitsky HI, Stock W, Sher D, Qin L, DeAngelo DJ, et al. Granulocyte-macrophage colony-stimulating factor (GM-CSF)-secreting cellular immunotherapy in combination with autologous stem cell transplantation (ASCT) as postremission therapy for acute myeloid leukemia (AML) Blood. 2009;114:1736–45. doi: 10.1182/blood-2009-02-205278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nemunaitis J, Jahan T, Ross H, Sterman D, Richards D, Fox B, et al. Phase 1/2 trial of autologous tumor mixed with an allogeneic GVAX vaccine in advanced-stage non-small-cell lung cancer. Cancer Gene Ther. 2006;13:555–62. doi: 10.1038/sj.cgt.7700922. [DOI] [PubMed] [Google Scholar]
- 52.Wherry EJ, Ahmed R. Memory CD8 T-cell differentiation during viral infection. J Virol. 2004;78:5535–45. doi: 10.1128/JVI.78.11.5535-5545.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Parmiani G, Castelli C, Pilla L, Santinami M, Colombo MP, Rivoltini L. Opposite immune functions of GM-CSF administered as vaccine adjuvant in cancer patients. Ann Oncol. 2007;18:226–32. doi: 10.1093/annonc/mdl158. [DOI] [PubMed] [Google Scholar]
- 54.Jinushi M, Nakazaki Y, Dougan M, Carrasco DR, Mihm M, Dranoff G. MFG-E8-mediated uptake of apoptotic cells by APCs links the pro- and antiinflammatory activities of GM-CSF. J Clin Invest. 2007;117:1902–13. doi: 10.1172/JCI30966. [DOI] [PMC free article] [PubMed] [Google Scholar]