

Abstract
Adenosine-to-inosine (A-to-I) RNA editing is a neurodevelopmentally-regulated epigenetic modification shown to modulate complex behavior in animals. Little is known about human A-to-I editing, but it is thought to constitute one of many molecular mechanisms connecting environmental stimuli and behavioral outputs. Thus, comprehensive exploration of A-to-I RNA editing in human brains may shed light on gene-environment interactions underlying complex behavior in health and disease. Synaptic function is a main target of A-to-I editing, which can selectively recode key amino acids in synaptic genes, directly altering synaptic strength and duration in response to environmental signals. Here we performed a high-resolution survey of synaptic A-to-I RNA editing in a human population, and examined how it varies in autism, a neurodevelopmental disorder in which synaptic abnormalities are a common finding. Using ultra-deep (>1000×) sequencing, we quantified the levels of A-to-I editing of 10 synaptic genes in postmortem cerebella from 14 neurotypical and 11 autistic individuals. A high dynamic range of editing levels was detected across individuals and editing sites, from 99.6% to below detection limits. In most sites, the extreme ends of the population editing distributions were individuals with autism. Editing was correlated with isoform usage, clusters of correlated sites were identified, and differential editing patterns examined. Finally, a dysfunctional form of the editing enzyme ADARB1 was found more commonly in postmortem cerebella from individuals with autism. These results provide a population-level, high-resolution view of A-to-I RNA editing in human cerebella, and suggest that A-to-I editing of synaptic genes may be informative for assessing the epigenetic risk for autism.
Keywords: RNA editing, neurodevelopment, autism, epigenetics, human cerebellum, A-to-I
Introduction
Site-specific adenosine-to-inosine (A-to-I) RNA base conversions, carried out by adenosine deaminase acting on RNA (ADAR) enzymes, exhibit precise regional specificity in the brain and modulate complex behavior in model organisms1–3. A-to-I RNA editing is an efficient means to increase RNA complexity, thereby fine-tuning both gene function and dosage4–6. The cellular machinery recognizes inosine as guanosine, so A-to-I editing of codons and splicing signals directly modifies protein-coding gene function6–11, while editing of microRNAs12–14 and their binding sites15 alters gene expression. This is particularly important in the human brain, the single most complex organ in cellular diversity, connectivity, morphogenesis, and responses to environmental stimuli16.
Synaptic function is a major target of A-to-I editing17, which can fine-tune neurophysiological properties in response to environmental stimuli18, 19. Canonical signaling pathways acting on the editing enzymes link A-to-I RNA editing to environmental cues: ADARB1 function requires inositol hexakisphosphate20, and the expression of ADAR is interferon-inducible21. Several recoding events that directly alter synaptic strength or duration in response to environmental signals have been characterized in rodents7–11. For example, mRNAs of the serotonin receptor HTR2C undergo editing in five sites, which dramatically alters its G-protein coupling activity, and hence the relationship between serotonin levels and postsynaptic signal transduction2, 11. This editing is regulated by exposure to acute stress and chronic treatment with antidepressants22. Another example is the neurodevelopmentally-regulated editing of transcripts encoding the AMPA receptors GRIA2, GRIA3 and GRIA423, where arginine to glycine (R/G) recoding of the ligand binding domains leads to faster desensitization recovery7. Moreover, glutamine to arginine (Q/R) editing of the transmembrane domains in the kainate receptors GRIK1 and GRIK2 reduces their calcium permeability8, with varying degrees of editing throughout mouse neurodevelopment23. Since 0 to 100% of mRNA molecules can be edited at any given point24, Q/R and R/G editing of ionotropic glutamate receptors provides an efficient means for fine-tuning the glutamatergic synapse in response to the changing environment.
Little is known about A-to-I RNA editing in humans, but it has been postulated to be one of the molecular mechanisms connecting environmental inputs and behavioral outputs18, 25. The increased editing in humans26 as compared to other animals27, including nonhuman primates28, has been proposed to generate molecular complexity that might constitute the basis of higher-order cognition18. Therefore, characterizing A-to-I editing in typically and atypically developed individuals may shed light on environment-dependent epigenetic mechanisms central to human neurodevelopment.
Autism Spectrum Disorders (ASD; [MIM 209850]) are highly heritable common neurodevelopmental disorders of complex genetic etiology, characterized by deficits in reciprocal social interaction and repetitive behaviors29. Several studies characterize synaptic abnormalities in ASD29–32. However, the mechanisms for gene-environment interactions and their contribution to the observed synaptic alterations remain unknown. The number of candidate genes is rapidly increasing33–35 and a main challenge is to identify the context in which they confer risk to ASD. Recent twin studies suggest that the contribution of environmental factors to ASD is larger than previously thought, with lower monozygotic concordance estimates (77 to 88%), and a surprisingly high dizygotic concordance of 31% (as compared to a sibling recurrence rate of 19%)36, 37. Hence, the identification of mechanisms governing gene-environment interactions relevant to ASD could be informative for risk assessment38. A-to-I RNA editing is potentially one such mechanism, linking environmental stimuli with synaptic transmission.
Several lines of evidence support an examination of the link between A-to-I editing and ASD. First, model organisms with altered A-to-I editing exhibit maladaptive behaviors characteristic of ASD2, 39, sometimes with seizures1, 40 or Prader-Willi-like symptoms41, both of which are typically detected in 25% of children with ASD42, 43. Second, altered editing of the serotonin receptor HTR2C has been detected in a mouse model of autism44 and in disorders that aggregate in families with ASD45, including schizophrenia46 and major depression47. Third, a fly model of Fragile × syndrome, the most common single gene cause of ASD48, was recently shown to exhibit significant editing alterations, via a direct interaction between the Fragile × gene dFMR1 and the editing enzyme dADAR49. Finally, independent genomic studies have implicated variants in synaptic genes, the most edited type of genes17, as a recurring theme in ASD29–32.
Here we focus on neurodevelopmentally-regulated A-to-I editing that directly alters synaptic function. We precisely quantify and compare the levels of editing across individuals, and characterize the distinct editing landscapes of ten synaptic genes acting in the human cerebellum, contrasting postmortem brains from typically developed individuals with brains isolated from individuals with ASD. We then (i) specifically examine editing patterns in the glutamatergic and serotonergic systems, (ii) correlate editing with isoform usage, and (iii) identify clusters of correlated sites. Importantly, we find that the relative usage of a dysfunctional form of the editing enzyme ADARB1 is significantly higher in ASD.
Materials and Methods
Subjects
Thirty fresh-frozen cerebellar samples of deidentified individuals with nonsyndromic autism and carefully-matched neurotypical individuals were obtained from the National Institute of Child Health and Human Development Brain and Tissue Bank (NICHD BTB) and the Harvard Brain Tissue Resource Center (HBTRC), through the Autism Tissue Program (ATP). To minimize confounding factors, matching was based on age, gender, race, and post-mortem-interval (PMI). Supplementary Table 1 details the 25 samples that passed quality control measures.
Selection of Target Genes
With the accumulation of multidisciplinary studies highlighting synaptic alterations in ASD29, 31, 32, 34, 50–54, we chose to focus on neurodevelopmentally-regulated A-to-I recoding that alters synaptic function. All synaptic genes shown to undergo developmentally dependent A-to-I recoding by Wahlstedt et al.23 were included in this study (Supplementary Table 2).
Molecular Methods
RNA isolation and quality assurance
Tissue samples were disrupted and RNA isolated in triplicate (mirVana™ miRNA Isolation Kit, Life Technologies, Grand Island, NY, USA), followed by DNase I treatment (DNA-free™, Life Technologies). Samples that seemed intact on 1% agarose gels were quantified on the ND-1000 Spectrophotometer (NanoDrop, Wilmington, DE, USA) and their integrity analyzed using the Agilent 2100 Bioanalyzer Eukaryote Total RNA Nano Series II (Agilent Technologies, Santa Clara, CA, USA). Only samples with RNA Integrity Number (RIN) >7 were included in the study, minimizing post mortem degradation artifacts55. The mean RIN of the samples used was 8.0 (Supplementary Table 1).
454 cDNA library preparation
A stringent PCR-based library preparation protocol was designed to ensure unbiased templates for multiplexed 454 sequencing, as detailed in Supplementary Information and Supplementary Figure 1.
454 sequencing
Bidirectional GS FLX sequencing was performed as described56 by the 454 Life Sciences Sequencing Center (Branford, CT, USA). ASD and neurotypical pools were sequenced on opposite sides of a 2-region PicoTiterPlate. 457,104 reads were obtained, containing 85,709,299 high quality bases (Supplementary Figure 2).
DNA isolation
About 2g of the same cerebellar tissue samples were disrupted with a mortar and pestle on dry ice, and genomic DNA (gDNA) was extracted using the QIAamp DNA Mini Kit (QIAGEN, Valencia, CA, USA).
Genomic DNA genotyping
Sequenom iPLEX genotyping (Sequenom, Inc, San Diego, CA, USA) was done at Boston Children’s Hospital's SNP Genotyping Facility. Twenty-five SNPs were genotyped in four populations. Every sample was genotyped in triplicate, and all triplicates had to agree on the genotype to be considered successful. All passing SNPs had a genotype success rate of >96.7%.
Method validation
cDNA and gDNA from all samples were independently analyzed by parallel capture and Illumina sequencing, as detailed in Supplementary Information. On average, 3420 cDNA reads and 627 gDNA reads were obtained for every sample at each site tested. The results of the two editing detection methods were tightly correlated, with an average Pearson’s r of 0.923 (mean p= 0.002) across six sites. See Supplementary Table 4 for details.
Relative ADARB1 isoform usage analysis
Semiquantitative RT-PCR and Sanger sequencing were used to measure the relative frequencies of the normal and dysfunctional (NR_027672) ADARB1 isoforms, as detailed in Supplementary Information. Custom TaqMan® Gene Expression Assays were used to quantitate the abundance of the dysfunctional ADARB1 isoform relative to GAPDHas detailed in Supplementary Information.
Correlation between editing and splicing of FLNA
When we amplified across the FLNA Q/R site using one primer in exon 43 and one in exon 44, an unexpected 720bp band of varying brightness appeared in all samples. Bidirectional Sanger sequencing revealed that it is a result of intron 43 retention. Its relative abundance was quantitated using QIAGEN’s QIAxcel electrophoresis system, and correlated with Q/R editing levels detected by 454 sequencing.
Gene expression profiling
Affymetrix Exon 1.0 ST arrays (Santa Clara, CA, USA) were used to measure global gene expression of each RNA sample. Following quantile normalization, gene expression levels were calculated using the Probe Log Iterative ER (PLIER) algorithm and differences in Gene Core expression were determined using the Mann-Whitney U test.
RNA-seq
Ten of the surveyed samples were subject to high-throughput RNA sequencing on Applied Biosystem’s SOLiD platform (Life Technologies). Poly(A)+ RNA was captured (Oligotex mRNA Mini Kit, QIAGEN), heat-fragmented (95°C for 20 minutes), and prepared for sequencing (Small RNA Expression Kit, Life Technologies) that yielded 114M reads. Life Technologies’ WT Analysis software package was used to align the reads to the reference genome and expressed sequence tags, identifying 33.4M uniquely mapped reads.
Computational Methods
454 variant calling and quantification
The 454 GS Amplicon Variant Analyzer software was used to deconvolute samples, align reads to reference sequences, and call variants and haplotypes based on bidirectional sequence changes from the reference.
Beta-binomial modeling of editing levels
For each sample in each site, the posterior editing density, f(editing), was a beta distribution with parameters α=1+number of reads with G mapped to the site, and β=1+number of reads with A mapped to the site (Supplementary Figure 3).
Differential editing analyses
Kolmogorov-Smirnov (KS) tests were used to assess the significance of differences in continuous measurements, such as those of editing levels between neurotypical individuals and individuals with ASD. To compare discrete distributions, such as the count of GRIK2 or HTR2C isoforms resulting from combinatorial RNA editing, Pearson’s chi square test was used. The total number of reads belonging to each of the possible editing isoforms were summed across each group and only isoforms supported by > 5 reads in both groups were included in the analysis (following this test’s assumptions).
Correlation between dysfunctional ADARB1 Isoform frequency and overall editing levels
For each sample i, RFi is the relative frequency of the dysfunctional ADARB1 isoform. A sample’s overall editing level, Ei, is the sum of its standardized editing levels across all sites, ∑Zij, where Zij is the standardized editing level of sample i at site j. Pearson’s correlation was calculated between these two metrics, RF and E, across all samples, to measure their linear dependence.
Association between editing and splicing of AMPA Receptors
Two-by-two contingency tables were used to summarize the relationships between isoform selection and editing (Supplementary Table 5), and Fisher’s exact test was used to determine the significance of the association between them.
Correlations among editing sites
The Pearson correlation coefficient was calculated to quantify the linear relationships between editing at different sites, among all individuals. Biclustering was then used to identify modules of tightly correlated sites, with the EXPANDER software57.
Independence of editing at neighboring sites
To summarize the relationships between editing at GRIA2 Q/R and Q/R+4 among all individuals, a two-by-two contingency table is shown in Supplementary Table 5. Fisher’s exact test was used for power and significance calculations.
Multiple testing correction
All p-values were Benjamini-Hochberg corrected for multiple testing58 to ensure that the false discovery rate of this entire study is below 0.05.
Results
Precise Multiplex Quantitation of A-to-I RNA Editing in Human Cerebella
For a high-resolution view of A-to-I RNA editing in a human brain population, ultra-deep (>1000×) 454 cDNA sequencing and genomic DNA (gDNA) genotyping were used to detect A/G mixtures on cDNA mapping to homozygous genomic A/A (Supplementary Figure 1). This study focused on all ten synaptic genes shown in mouse to undergo neurodevelopmentally-regulated A-to-I recoding23 that results in well-characterized neurophysiological alterations7–9, 59–61 (Supplementary Table 2). Editing was measured in postmortem cerebellum, one of several brain regions implicated in ASD62 by both imaging63 and autopsy64 studies. Cerebellar tissue samples were obtained from neurotypical individuals and individuals with non-syndromic ASD, matched by gender, age, race, and postmortem interval (Supplementary Table 1). Pooled, barcoded, bidirectional 454 cDNA sequencing yielded on average 1344 reads of 212bp per individual per amplicon (Supplementary Figure 2). gDNA was genotyped at 18 well-characterized editing sites with known functional consequences and 7 positions aligned to a mixture of guanosines and adenosines on cDNA. A-to-I edited sites were identified by the presence of a bidirectional cDNA A/G mixture at homozygous A/A gDNA positions. Editing levels were modeled by a beta-binomial distribution, resulting in a posterior editing density for each individual at each site. This model considers both the fraction of edited reads and the total number of reads covering a site, to produce a distribution describing the level of editing and our confidence in that measurement (Supplementary Figure 3).
This approach provided a high confidence dataset of synaptic A-to-I RNA editing from individuals with ASD and matched neurotypical individuals, with an average 95% confidence interval length of 0.038. For independent validation, the same cDNA and gDNA were analyzed by parallel capture and >3000× Illumina sequencing in six sites among all individuals (Supplementary Information). A tight correlation between the two editing detection methods is shown in Supplementary Figure 4 and Supplementary Table 4, with an average correlation coefficient of 0.923.
High Dynamic Range of Editing across Sites and Individuals
The editing levels of 25 sites were robustly measured and found to range from 2.5% to 99.6% (Figure 1A and Supplementary Figure 5). Unexpectedly broad distributions of editing levels were found across neurotypical individuals and carefully-matched individuals with ASD in those sites previously shown to be neurodevelopmentally regulated in mice23 (average coefficient of variation 54.8%). Only one site, GRIA2 Q/R, showed similar levels of editing among all individuals (CV=0.5%), consistent with reports that editing of this site is essential and unchanged throughout development23, 65. No relationships between editing levels and age, gender, race, postmortem interval, or RNA integrity were detected (Supplementary Figure 6).
Figure 1.

Individuals with ASD at the extremes of the population editing distributions in 20 of 25 sites. (A) High dynamic range of mean editing across sites, from 99.6% to below detection limit. Standard deviation bars, shown in purple, indicate the large variability in neurodevelopmentally-regulated editing among carefully matched individuals. (B) The individual standardized deviations from the mean editing level across all sites show that the extreme of the population editing distributions tend to be individuals with ASD (orange triangles), and they are the major contributor to the large variability shown in (A). Horizontal lines denote the standardized 95% confidence interval of an individual’s posterior editing distribution. Synaptic editing levels at least two standard deviations away from the mean are highly informative of ASD, with a positive predictive value of 78%.
By inspection of the individual posterior editing densities across all sites (Supplementary Figure 5), we noticed that the extremes of most editing distributions are individuals with ASD. To quantify this observation, the point estimates of the editing levels of each individual at each site were transformed to Z scores and those ≥2 or ≤−2 were considered extreme, representing editing levels that are at least two standard deviations away from the mean. In 20 of 25 sites, individuals with ASD were at the extreme of the spectrum of editing seen for that site (Figure 1B). Having extreme synaptic editing levels is highly informative of ASD, with a positive predictive value p(ASD | |Zediting | > 2) of 0.78. Outliers at different sites are different individuals with ASD (Supplementary Figures 5 and 7), and editing of more than three standard deviations away from the mean was specific to individuals with ASD (Supplementary Figure 7). The overall editing variance in ASD was more than two-fold that of neurotypical individuals (ASD variance=0.58, median neurotypical variance of equally sized subsamples=0.24, Brown-Forsythe p=5.6e-3). The major contributors to this increased variance are 30% of the individuals with ASD (Supplementary Figure 8).
Patterns of Editing in the Glutamatergic and Serotonergic Systems
Next, editing patterns were specifically examined in the glutamatergic and serotonergic systems, which have been shown to be implicated in ASD29, 66. GRIK2, whose genomic locus has been repeatedly linked and associated with ASD66, is edited in three sites, leading to the formation of eight protein isoforms that differ in their calcium permeability23. Differential GRIK2 editing between individuals with ASD and neurotypical individuals is depicted in Figure 2, both on a single site and isoform-wide levels. Kolmogorov-Smirnov (KS) tests were used to assess the significance of differences in continuous measurements, such as those of editing levels between neurotypical individuals and individuals with ASD. To compare discrete distributions, such as the count of GRIK2 or HTR2C isoforms resulting from combinatorial RNA editing, Pearson’s chi square test was used. The relative frequencies of GRIK2 isoforms were significantly different between individuals with ASD and neurotypical individuals (p<1e-4, chi-square test). Differences were also detected in the editing of GRIA4 R/G (p<3.6e-2, KS testSupplementary Figure 9). Other comparisons of glutamate receptors did not reach statistical significance, as this study is not powered to detect small effect sizes (Supplementary Figure 10).
Figure 2.

Differences in editing-mediated GRIK2 isoforms between neurotypical individuals (blue) and individuals with ASD (orange). GRIK2 is edited at three sites: I/V, Y/C (both part of TM1) and Q/R (TM2), altering the receptor’s calcium permeability. Editing at these sites was found to be tightly correlated (Figure 5B). (A) Large variability in GRIK2 editing at each site among all individuals. The extremes of the observed editing spectra are individuals with ASD. Consult Supplementary Figure 5 for more details. (B) Combinatorial editing of the three sites results in eight isoforms, with distinct molecular properties. Significant group differences in GRIK2 isoform distributions between individuals with ASD and neurotypical individuals were identified (p<1e-4, chi-square test). (C) The strongest differences are in IYQ and VCQ isoforms, which are ~1.5-fold over-represented in ASD. Shown are differences in the cumulative density functions (CDFs) of the VCR, ICR, IYQ, and VCQ isoforms.
Editing was also examined in the serotonin receptor HTR2C, which is targeted by the only FDA-approved drugs to treat autistic symptoms, risperidone and aripiprazole. This receptor undergoes RNA editing at five sites, which dramatically alters its G-protein coupling activity. HTR2C editing may create up to 24 different protein isoforms, characterized by distinct molecular and behavioral phenotypes2, 6. Comparing the relative isoform frequencies of individuals with ASD and neurotypical individuals revealed significant differences (p<1e-4, chi square test), the strongest in IDV, MNV and ISI isoforms - all under-represented in ASD (Figure 3). However, the variance within ASD is much larger than that between neurotypical and ASD individuals, suggesting that this finding should be considered with caution and revisited with a larger sample size.
Figure 3.

Differences in the relative abundance of HTR2C isoforms between individuals with ASD and neurotypical individuals (p<1e-4, chi-square test). 21 of the possible 24 isoforms created by combinatorial editing of five HTR2C sites were detected. The greatest differences were in the IDV, MNV and ISI isoforms, all under-represented in ASD by >2-fold.
Differential Relative ADARB1 Isoform Usage in Individuals with ASD
The sites examined here are predominantly edited by ADARB1 (Supplementary Table 2). To explore the regulatory basis of the observed editing differences, ADARB1 expression and relative isoform usage were examined. In rodents, the function of ADARB1 has been shown to be developmentally regulated via alternative splicing that creates an inactive protein, while its overall expression remains fairly constant throughout development23, 67. In humans, a dysfunctional ADARB1 isoform (ADAR2g, NR_027672), resulting from alternative skipping of the exon harboring two double stranded RNA binding domains (dsRBDs), has been found to constitute about 20% of adult cerebellar ADARB1 mRNA68. As such, the presence of this dsRBDs-encoding exon was assayed by semi-quantitative RT-PCR in all samples. While individuals with ASD and neurotypical individuals showed similar levels of overall ADARB1 expression (Supplementary Figure 11), the relative usage of the inactive ADARB1 isoform was significantly more common in individuals with ASD (Figure 4 and Supplementary Figure 12, p=3.0e-3, Mann-Whitney U test). Exon skipping was confirmed by bidirectional sequencing in all samples. Ten of the surveyed samples were also subject to RNA-seq, and the relative ADARB1 exon usage determined by RNA-seq correlated with the semi-quantitative RT-PCR results (Supplementary Information). Additionally, an increased relative usage of the dysfunctional ADARB1 isoform in ASD was observed in an independent cohort of nine cerebellar tissue samples (Supplementary Information). The relative frequency of the dysfunctional isoform is correlated with overall editing levels (Pearson’s r= 0.48, p=3.2e-2), as measured by the sum of the standardized editing scores across all sites. Finally, the abundance of the inactive ADARB1 isoform was quantitated in all samples using TaqMan® Gene Expression Assays (Supplementary Information). The dysfunctional form was over-expressed in individuals with ASD (p=7.3e-3, Mann-Whitney U test), with a significant correlation between semi-quantitative and quantitative PCR results (Pearson’s p= 2.0e-5).
Figure 4.
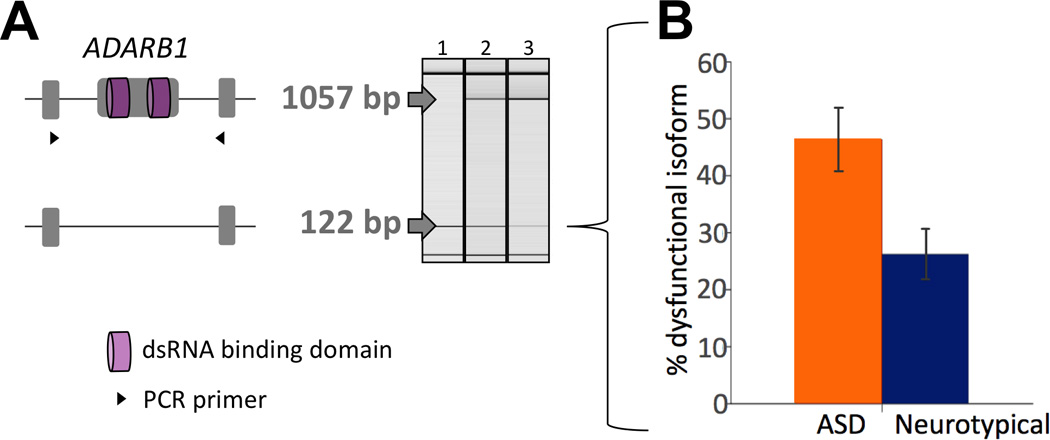
Differences in the relative usage of ADARB1 inactive form between individuals with ASD and neurotypical individuals (p=3.0e-3, Mann-Whitney U test). This isoform lacks the enzyme’s dsRBDs and has been shown to be untranslated68. ADARB1 is the predominant editing enzyme of the sites analyzed in this study. (A) Semiquantitative RT-PCR was used to measure the relative frequencies of functional (1057 bp product) and dysfunctional (NR_027672, 122 bp product) ADARB1 isoforms, using primers (black triangles) which detect both. (B) The mean relative frequency of the dysfunctional ADARB1 isoform was 46.3% ± 5.6% in ASD and 26% ± 4.5% in neurotypical individuals, similar to that previously reported in neurotypical human cerebellum68. All 25 samples included in the RNA editing survey were also part of this analysis. The relative frequency of the dysfunctional isoform is correlated with overall editing levels (Pearson’s p=3.2e-2). Additionally, the abundance of the dysfunctional form as quantitated by TaqMan® Gene Expression Assays was higher in individuals with ASD (p=7.3e-3, Mann-Whitney U test, Supplementary Figure 12), with a significant correlation between quantitative and semi-quantitative PCR results (Pearson’s p= 2.0e-5).
Tight Relationships between Recoding and Splicing in Three AMPA Receptors and Filamin-A
The long 454 reads (Supplementary Figure 2) were used to study regulatory characteristics of human RNA editing, including the previously hypothesized6; 69 relationships between recoding and alternative splicing selection in three AMPA receptors (GRIA2, GRIA3, and GRIA4) and filamin-A (FLNA). GRIA2, GRIA3, and GRIA4 each contain two mutually exclusive exons that modulate desensitization kinetics, termed flip and flop7. Editing at the 3’ end of the exon immediately preceding the flip/flop module was strongly associated with the flop isoform in GRIA4 (OR=101.9 [95% CI 87.8–118.2]), GRIA3 (OR= 30.6 [26.8–34.8]), and to a lesser extent GRIA2 (OR= 2.6 [2.4–2.8]) (All p-values <1e-300, Fisher’s exact test, Figure 5A and Supplementary Table 5). Consistent with the detection of differential editing at GRIA4 R/G between individuals with ASD and neurotypical individuals, differential GRIA4 isoform usage was also identified (p<3.6e-2, KS test, Supplementary Figure 13).
Figure 5.
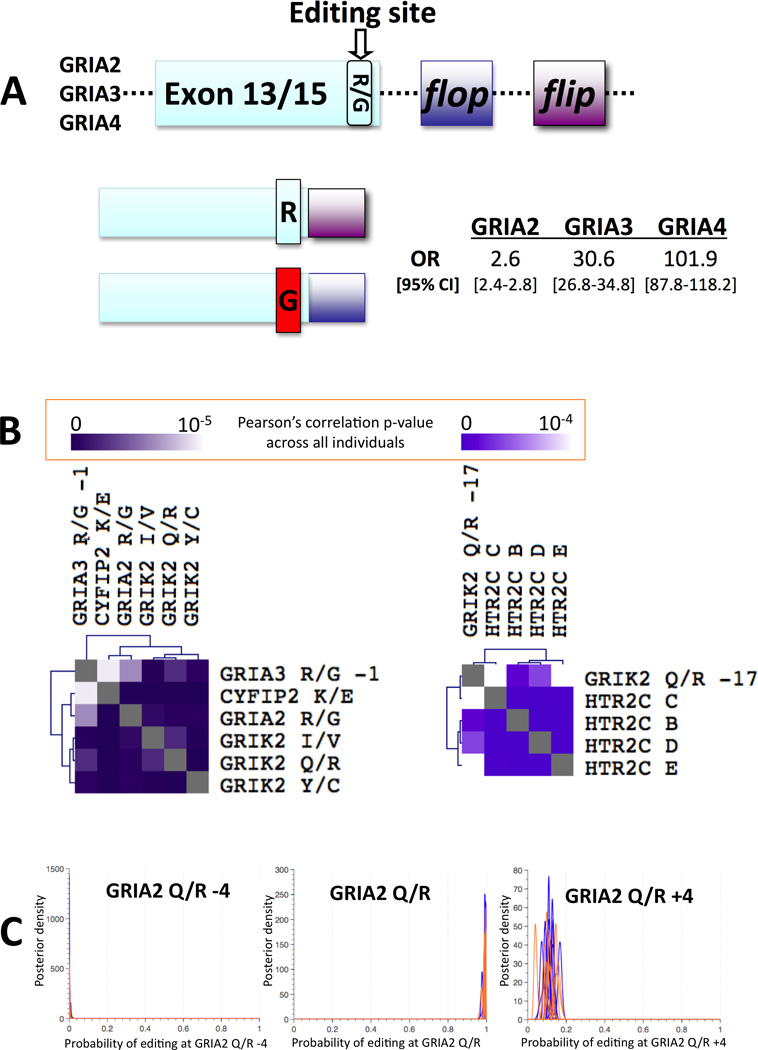
Relationships between editing and splicing, and among sites. (A) Editing at the 3’ end of the exon immediately preceding the flip/flop module is strongly associated with the flop isoform in GRIA4 (OR=101.9 [95% CI 87.8–118.2]), GRIA3 (OR= 30.6 [26.8–34.8]), and to a lesser extent GRIA2 (OR= 2.6 [2.4–2.8]) (All p-values <1e-300, Fisher’s exact test). (B) The linear correlation of editing levels across sites was measured among all individuals and biclustered to reveal modules of tightly correlated sites, suggesting coregulation by the same factors. The tightest module contains 6 sites in 4 genes, demonstrating highly correlated editing across these sites among all individuals. Consult Supplementary Figure 5 for broader perspective. (C) Three neighboring sites at a 4 bp distance from one another undergo independent editing (p=1, power>0.97, Fisher’s exact test), with GRIA2 Q/R −4 fully unedited, GRIA2 Q/R fully edited, and GRIA2 Q/R+4 variably edited across individuals, between 4–17%.
Another relationship between recoding and splicing was examined in FLNA, where editing at the 3’ end of exon 43 was found to be significantly correlated with intron 43 retention (Pearson’s p<6e-3, Supplementary Figure 14). This intron retention introduces a frameshift and is not included in any human Refseq FLNA isoform. Alignment of the retained intron sequence to expressed sequence tags and short transcriptome reads revealed its expression in several tissues from multiple organisms. Thus it likely represents a conserved splicing event that possibly regulates FLNA dosage. All retained introns were found to contain an unknown A-to-I editing site at their center, verified by gDNA genotyping. Whether this editing is a cause or effect of the intron retention remains to be elucidated.
cis and trans Relationships across Editing Sites
To learn more about the regulation of human RNA editing, clusters of tightly correlated editing sites across all individuals were identified. First, Pearson’s correlation was calculated to quantify the linear dependencies between editing at different sites, among all individuals. Biclustering was then used to identify modules of tightly correlated sites with an average correlation coefficient > 0.7 between all pairs of sites in the cluster, across all samples (Supplementary Table 6). The cluster with the strongest correlation contained 6 sites in 4 genes (GRIA2 R/G, CYFIP2 K/E, GRIA3 R/G-1, and GRIK2 I/V, Q/R and Y/C) (Pearson’s r=0.8, p<1e-4), suggesting that these sites may be co-regulated (Figure 5B). Some neighboring sites, including GRIA2 Q/R and Q/R+4, showed independent editing (Fisher’s Exact p=1, Power>0.97, Figure 5C and Supplementary Table 5), suggesting that their editing is either spatiotemporally distinct and/or carried out by different complexes.
Discussion
This study represents an initial examination in ASD of A-to-I RNA editing, a form of gene-environment interaction that fine-tunes synaptic function in response to environmental stimuli. Human genes that undergo editing are more often involved in processes that are affected in neurodevelopmental disorders17, 18, and A-to-I editing is overall increased in the human lineage26–28. While little is known about the impact of RNA editing in humans, there is compelling evidence supporting a key role for A-to-I editing in modulating complex behavior in animals1–3, 41, 44, 70. Mice and flies with altered editing levels recapitulate several behavioral homologues to human ASD1, 41, 44. Gene-specific studies in animals demonstrate the causal relationship between editing levels and specific maladaptive behaviors1, 10, and between specific environmental exposures and editing levels71. Therefore, the results presented here shed light on the role of an epigenetic mechanism that connects environmental signals and downstream behavioral outputs, integrating genetic and environmental information.
DNA sequence variation in a number of different loci is strongly associated with ASD, but no individual locus is altered in more than 2% of cases66. In contrast, 30% of the individuals with ASD examined here showed extreme levels of synaptic RNA editing, suggesting that A-to-I editing may be both a marker for and mechanism of ASD. It has been recently shown that typical synaptic protein synthesis occurs within an optimal range and deviations in either direction can lead to cognitive impairments72. This study suggests that a similar inverse-U relationship might exist between synaptic A-to-I editing levels and the ASD phenotype. In ASD, it now appears that increased variance can be found in different levels of function, ranging from synaptic A-I editing and protein synthesis72, through mitochondrial73 and immune74 function, to cellular and systems neuroanatomy75. A plausible generalization of these findings is that many biological processes exhibit increased variance in ASD as a result of many etiologies disrupting ASD function and homeostasis. We speculate that altered A-to-I editing could act as a common compensatory mechanism for the wide range of synaptic abnormalities in ASD, as affected neurons try to maintain synaptic homeostasis. Alternatively, altered editing could be a direct consequence of ASD-causing mutations. In either case, larger sets of genes, brain regions, and individuals should be examined to understand the potential contribution of A-to-I editing to ASD.
Supplementary Material
Acknowledgments
We thank Oliver St. Lawrence, Jamie Jett, Benjamin Boese and Tim Harkins at 454, our wonderful lab mates, Prof. David Bartel, Thutrang Nguyen, Eran Mick, and Elena Helman for their tremendous help. Human tissue was obtained from the National Institute of Child Health and Human Development Brain and Tissue Bank for Developmental Disorders at the University of Maryland, Baltimore, MD, contract HHSN275200900011C, Re. No. N01-HD-9-0011, and from The Harvard Brain Tissue Resource Center (HBTRC), through the Autism Tissue Program. HBTRC is supported by Grant MH068855. The Molecular Genetics Core Facility at Children’s Hospital Boston Intellectual and Developmental Disabilities Research Center (IDDRC) is supported by Grant NIH-P30-HD18655. This study was generously supported by the Nancy Lurie Marks Family Foundation, The Roche Applied Science Sequencing Grant Program, Autism Speaks, Simons Foundation, and NIH Grant 1R01MH085143-01.
Footnotes
Data Availability
Access to the raw sequence reads can be found at the National Database for Autism Research under accession number NDARCOL0001951.
Conflict of Interest
The authors declare no conflict of interest.
Supplementary information is available at Molecular Psychiatry's website
References
- 1.Jepson JE, Savva YA, Yokose C, Sugden AU, Sahin A, Reenan RA. Engineered alterations in RNA editing modulate complex behavior in Drosophila: regulatory diversity of adenosine deaminase acting on RNA (ADAR) targets. J Biol Chem. 2011;286(10):8325–8337. doi: 10.1074/jbc.M110.186817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mombereau C, Kawahara Y, Gundersen BB, Nishikura K, Blendy JA. Functional relevance of serotonin 2C receptor mRNA editing in antidepressant- and anxiety-like behaviors. Neuropharmacology. 2010;59(6):468–473. doi: 10.1016/j.neuropharm.2010.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tonkin LA, Saccomanno L, Morse DP, Brodigan T, Krause M, Bass BL. RNA editing by ADARs is important for normal behavior in Caenorhabditis elegans. EMBO J. 2002;21(22):6025–6035. doi: 10.1093/emboj/cdf607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maas S. Gene regulation through RNA editing. Discov Med. 2010;10(54):379–386. [PubMed] [Google Scholar]
- 5.Mattick JS. RNA as the substrate for epigenome-environment interactions: rNA guidance of epigenetic processes and the expansion of RNA editing in animals underpins development, phenotypic plasticity, learning, and cognition. Bioessays. 2010;32(7):548–552. doi: 10.1002/bies.201000028. [DOI] [PubMed] [Google Scholar]
- 6.Nishikura K. Functions and regulation of RNA editing by ADAR deaminases. Annu Rev Biochem. 2010;79:321–349. doi: 10.1146/annurev-biochem-060208-105251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lomeli H, Mosbacher J, Melcher T, Hoger T, Geiger JR, Kuner T, et al. Control of kinetic properties of AMPA receptor channels by nuclear RNA editing. Science. 1994;266(5191):1709–1713. doi: 10.1126/science.7992055. [DOI] [PubMed] [Google Scholar]
- 8.Kohler M, Burnashev N, Sakmann B, Seeburg PH. Determinants of Ca2+ permeability in both TM1 and TM2 of high affinity kainate receptor channels: diversity by RNA editing. Neuron. 1993;10(3):491–500. doi: 10.1016/0896-6273(93)90336-p. [DOI] [PubMed] [Google Scholar]
- 9.Rula EY, Lagrange AH, Jacobs MM, Hu N, Macdonald RL, Emeson RB. Developmental modulation of GABA(A) receptor function by RNA editing. J Neurosci. 2008;28(24):6196–6201. doi: 10.1523/JNEUROSCI.0443-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feldmeyer D, Kask K, Brusa R, Kornau HC, Kolhekar R, Rozov A, et al. Neurological dysfunctions in mice expressing different levels of the Q/R site-unedited AMPAR subunit GluR-B. Nat Neurosci. 1999;2(1):57–64. doi: 10.1038/4561. [DOI] [PubMed] [Google Scholar]
- 11.Burns CM, Chu H, Rueter SM, Hutchinson LK, Canton H, Sanders-Bush E, et al. Regulation of serotonin-2C receptor G-protein coupling by RNA editing. Nature. 1997;387(6630):303–308. doi: 10.1038/387303a0. [DOI] [PubMed] [Google Scholar]
- 12.Kawahara Y, Zinshteyn B, Sethupathy P, Iizasa H, Hatzigeorgiou AG, Nishikura K. Redirection of silencing targets by adenosine-to-inosine editing of miRNAs. Science. 2007;315(5815):1137–1140. doi: 10.1126/science.1138050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kawahara Y, Zinshteyn B, Chendrimada TP, Shiekhattar R, Nishikura K. RNA editing of the microRNA-151 precursor blocks cleavage by the Dicer-TRBP complex. EMBO Rep. 2007;8(8):763–769. doi: 10.1038/sj.embor.7401011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang W, Chendrimada TP, Wang Q, Higuchi M, Seeburg PH, Shiekhattar R, et al. Modulation of microRNA processing and expression through RNA editing by ADAR deaminases. Nat Struct Mol Biol. 2006;13(1):13–21. doi: 10.1038/nsmb1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Borchert GM, Gilmore BL, Spengler RM, Xing Y, Lanier W, Bhattacharya D, et al. Adenosine deamination in human transcripts generates novel microRNA binding sites. Hum Mol Genet. 2009;18(24):4801–4807. doi: 10.1093/hmg/ddp443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mehler MF, Mattick JS. Noncoding RNAs and RNA editing in brain development, functional diversification, and neurological disease. Physiol Rev. 2007;87(3):799–823. doi: 10.1152/physrev.00036.2006. [DOI] [PubMed] [Google Scholar]
- 17.Li JB, Levanon EY, Yoon JK, Aach J, Xie B, Leproust E, et al. Genome-wide identification of human RNA editing sites by parallel DNA capturing and sequencing. Science. 2009;324(5931):1210–1213. doi: 10.1126/science.1170995. [DOI] [PubMed] [Google Scholar]
- 18.Mattick JS, Mehler MF. RNA editing, DNA recoding and the evolution of human cognition. Trends Neurosci. 2008;31(5):227–233. doi: 10.1016/j.tins.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 19.Garrett S, Rosenthal JJ. RNA Editing Underlies Temperature Adaptation in K+ Channels from Polar Octopuses. Science. 2012 doi: 10.1126/science.1212795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Macbeth MR, Schubert HL, Vandemark AP, Lingam AT, Hill CP, Bass BL. Inositol hexakisphosphate is bound in the ADAR2 core and required for RNA editing. Science. 2005;309(5740):1534–1539. doi: 10.1126/science.1113150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Patterson JB, Samuel CE. Expression and regulation by interferon of a double-stranded-RNA-specific adenosine deaminase from human cells: evidence for two forms of the deaminase. Mol Cell Biol. 1995;15(10):5376–5388. doi: 10.1128/mcb.15.10.5376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Englander MT, Dulawa SC, Bhansali P, Schmauss C. How stress and fluoxetine modulate serotonin 2C receptor pre-mRNA editing. J Neurosci. 2005;25(3):648–651. doi: 10.1523/JNEUROSCI.3895-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wahlstedt H, Daniel C, Enstero M, Ohman M. Large-scale mRNA sequencing determines global regulation of RNA editing during brain development. Genome Res. 2009;19(6):978–986. doi: 10.1101/gr.089409.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wulff BE, Nishikura K. Substitutional A-to-I RNA editing. WIREs RNA. 2010;1(1):90–101. doi: 10.1002/wrna.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jepson JE, Reenan RA. RNA editing in regulating gene expression in the brain. Biochim Biophys Acta. 2008;1779(8):459–470. doi: 10.1016/j.bbagrm.2007.11.009. [DOI] [PubMed] [Google Scholar]
- 26.Peng Z, Cheng Y, Tan BC, Kang L, Tian Z, Zhu Y, et al. Comprehensive analysis of RNA-Seq data reveals extensive RNA editing in a human transcriptome. Nat Biotechnol. 2012 doi: 10.1038/nbt.2122. [DOI] [PubMed] [Google Scholar]
- 27.Athanasiadis A, Rich A, Maas S. Widespread A-to-I RNA editing of Alu-containing mRNAs in the human transcriptome. PLoS Biol. 2004;2(12):e391. doi: 10.1371/journal.pbio.0020391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Paz-Yaacov N, Levanon EY, Nevo E, Kinar Y, Harmelin A, Jacob-Hirsch J, et al. Adenosine-to-inosine RNA editing shapes transcriptome diversity in primates. Proc Natl Acad Sci U S A. 2010;107(27):12174–12179. doi: 10.1073/pnas.1006183107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Toro R, Konyukh M, Delorme R, Leblond C, Chaste P, Fauchereau F, et al. Key role for gene dosage and synaptic homeostasis in autism spectrum disorders. Trends Genet. 2010;26(8):363–372. doi: 10.1016/j.tig.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 30.Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S, et al. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. 2011;474(7351):380–384. doi: 10.1038/nature10110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bourgeron T. A synaptic trek to autism. Curr Opin Neurobiol. 2009;19(2):231–234. doi: 10.1016/j.conb.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 32.Gilman SR, Iossifov I, Levy D, Ronemus M, Wigler M, Vitkup D. Rare de novo variants associated with autism implicate a large functional network of genes involved in formation and function of synapses. Neuron. 2011;70(5):898–907. doi: 10.1016/j.neuron.2011.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Anney RJ, Kenny EM, O'Dushlaine C, Yaspan BL, Parkhomenka E, Buxbaum JD, et al. Gene-ontology enrichment analysis in two independent family-based samples highlights biologically plausible processes for autism spectrum disorders. Eur J Hum Genet. 2011 doi: 10.1038/ejhg.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, Regan R, et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466(7304):368–372. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sakai Y, Shaw CA, Dawson BC, Dugas DV, Al-Mohtaseb Z, Hill DE, et al. Protein interactome reveals converging molecular pathways among autism disorders. Sci Transl Med. 2011;3(86):86ra49. doi: 10.1126/scitranslmed.3002166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hallmayer J, Cleveland S, Torres A, Phillips J, Cohen B, Torigoe T, et al. Genetic Heritability and Shared Environmental Factors Among Twin Pairs With Autism. Arch Gen Psychiatry. 2011 doi: 10.1001/archgenpsychiatry.2011.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ronald A, Hoekstra RA. Autism spectrum disorders and autistic traits: A decade of new twin studies. Am J Med Genet B Neuropsychiatr Genet. 2011 doi: 10.1002/ajmg.b.31159. [DOI] [PubMed] [Google Scholar]
- 38.Grafodatskaya D, Chung B, Szatmari P, Weksberg R. Autism spectrum disorders and epigenetics. J Am Acad Child Adolesc Psychiatry. 2010;49(8):794–809. doi: 10.1016/j.jaac.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 39.Singh M, Zimmerman MB, Beltz TG, Johnson AK. Affect-related behaviors in mice misexpressing the RNA editing enzyme ADAR2. Physiol Behav. 2009;97(3–4):446–454. doi: 10.1016/j.physbeh.2009.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jepson JE, Savva YA, Yokose C, Sugden AU, Sahin A, Reenan RA. Engineered alterations in RNA editing modulate complex behavior in Drosophila: regulatory diversity of adenosine deaminase acting on RNA (ADAR) targets. J Biol Chem. 2010 doi: 10.1074/jbc.M110.186817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morabito MV, Abbas AI, Hood JL, Kesterson RA, Jacobs MM, Kump DS, et al. Mice with altered serotonin 2C receptor RNA editing display characteristics of Prader-Willi syndrome. Neurobiol Dis. 2010;39(2):169–180. doi: 10.1016/j.nbd.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Canitano R. Epilepsy in autism spectrum disorders. Eur Child Adolesc Psychiatry. 2007;16(1):61–66. doi: 10.1007/s00787-006-0563-2. [DOI] [PubMed] [Google Scholar]
- 43.Veltman MW, Craig EE, Bolton PF. Autism spectrum disorders in Prader-Willi and Angelman syndromes: a systematic review. Psychiatr Genet. 2005;15(4):243–254. doi: 10.1097/00041444-200512000-00006. [DOI] [PubMed] [Google Scholar]
- 44.Nakatani J, Tamada K, Hatanaka F, Ise S, Ohta H, Inoue K, et al. Abnormal behavior in a chromosome-engineered mouse model for human 15q11-13 duplication seen in autism. Cell. 2009;137(7):1235–1246. doi: 10.1016/j.cell.2009.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Daniels JL, Forssen U, Hultman CM, Cnattingius S, Savitz DA, Feychting M, et al. Parental psychiatric disorders associated with autism spectrum disorders in the offspring. Pediatrics. 2008;121(5):e1357–e1362. doi: 10.1542/peds.2007-2296. [DOI] [PubMed] [Google Scholar]
- 46.Sodhi MS, Burnet PW, Makoff AJ, Kerwin RW, Harrison PJ. RNA editing of the 5-HT(2C) receptor is reduced in schizophrenia. Mol Psychiatry. 2001;6(4):373–379. doi: 10.1038/sj.mp.4000920. [DOI] [PubMed] [Google Scholar]
- 47.Gurevich I, Tamir H, Arango V, Dwork AJ, Mann JJ, Schmauss C. Altered editing of serotonin 2C receptor pre-mRNA in the prefrontal cortex of depressed suicide victims. Neuron. 2002;34(3):349–356. doi: 10.1016/s0896-6273(02)00660-8. [DOI] [PubMed] [Google Scholar]
- 48.Hagerman R, Hoem G, Hagerman P. Fragile × and autism: Intertwined at the molecular level leading to targeted treatments. Mol Autism. 2010;1(1):12. doi: 10.1186/2040-2392-1-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bhogal B, Jepson JE, Savva YA, Pepper AS, Reenan RA, Jongens TA. Modulation of dADAR-dependent RNA editing by the Drosophila fragile × mental retardation protein. Nat Neurosci. 2011;14(12):1517–1524. doi: 10.1038/nn.2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Peca J, Ting J, Feng G. SnapShot: Autism and the synapse. Cell. 2011;147(3):706. doi: 10.1016/j.cell.2011.10.015. 706 e701. [DOI] [PubMed] [Google Scholar]
- 51.Stephenson DT, O'Neill SM, Narayan S, Tiwari A, Arnold E, Samaroo HD, et al. Histopathologic characterization of the BTBR mouse model of autistic-like behavior reveals selective changes in neurodevelopmental proteins and adult hippocampal neurogenesis. Mol Autism. 2011;2(1):7. doi: 10.1186/2040-2392-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang X, McCoy PA, Rodriguiz RM, Pan Y, Je HS, Roberts AC, et al. Synaptic dysfunction and abnormal behaviors in mice lacking major isoforms of Shank3. Hum Mol Genet. 2011;20(15):3093–3108. doi: 10.1093/hmg/ddr212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gai X, Xie HM, Perin JC, Takahashi N, Murphy K, Wenocur AS, et al. Rare structural variation of synapse and neurotransmission genes in autism. Mol Psychiatry. 2011 doi: 10.1038/mp.2011.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Farra N, Zhang WB, Pasceri P, Eubanks JH, Salter MW, Ellis J. Rett syndrome induced pluripotent stem cell-derived neurons reveal novel neurophysiological alterations. Mol Psychiatry. 2012 doi: 10.1038/mp.2011.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Durrenberger PF, Fernando S, Kashefi SN, Ferrer I, Hauw JJ, Seilhean D, et al. Effects of antemortem and postmortem variables on human brain mRNA quality: a BrainNet Europe study. J Neuropathol Exp Neurol. 2010;69(1):70–81. doi: 10.1097/NEN.0b013e3181c7e32f. [DOI] [PubMed] [Google Scholar]
- 56.Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437(7057):376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shamir R, Maron-Katz A, Tanay A, Linhart C, Steinfeld I, Sharan R, et al. EXPANDER--an integrative program suite for microarray data analysis. BMC Bioinformatics. 2005;6:232. doi: 10.1186/1471-2105-6-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society. 1995;57(1):125–133. [Google Scholar]
- 59.Belcher SM, Howe JR. Characterization of RNA editing of the glutamate-receptor subunits GluR5 and GluR6 in granule cells during cerebellar development. Brain Res Mol Brain Res. 1997;52(1):130–138. doi: 10.1016/s0169-328x(97)00252-0. [DOI] [PubMed] [Google Scholar]
- 60.Bhalla T, Rosenthal JJ, Holmgren M, Reenan R. Control of human potassium channel inactivation by editing of a small mRNA hairpin. Nat Struct Mol Biol. 2004;11(10):950–956. doi: 10.1038/nsmb825. [DOI] [PubMed] [Google Scholar]
- 61.Nishikura K. Editor meets silencer: crosstalk between RNA editing and RNA interference. Nat Rev Mol Cell Biol. 2006;7(12):919–931. doi: 10.1038/nrm2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Abrahams BS, Geschwind DH. Connecting genes to brain in the autism spectrum disorders. Arch Neurol. 2010;67(4):395–399. doi: 10.1001/archneurol.2010.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mostofsky SH, Powell SK, Simmonds DJ, Goldberg MC, Caffo B, Pekar JJ. Decreased connectivity and cerebellar activity in autism during motor task performance. Brain. 2009;132(Pt 9):2413–2425. doi: 10.1093/brain/awp088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Palmen SJ, van Engeland H, Hof PR, Schmitz C. Neuropathological findings in autism. Brain. 2004;127(Pt 12):2572–2583. doi: 10.1093/brain/awh287. [DOI] [PubMed] [Google Scholar]
- 65.Higuchi M, Maas S, Single FN, Hartner J, Rozov A, Burnashev N, et al. Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature. 2000;406(6791):78–81. doi: 10.1038/35017558. [DOI] [PubMed] [Google Scholar]
- 66.Abrahams BS, Geschwind DH. Advances in autism genetics: on the threshold of a new neurobiology. Nat Rev Genet. 2008;9(5):341–355. doi: 10.1038/nrg2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rueter SM, Dawson TR, Emeson RB. Regulation of alternative splicing by RNA editing. Nature. 1999;399(6731):75–80. doi: 10.1038/19992. [DOI] [PubMed] [Google Scholar]
- 68.Kawahara Y, Ito K, Ito M, Tsuji S, Kwak S. Novel splice variants of human ADAR2 mRNA: skipping of the exon encoding the dsRNA-binding domains, and multiple C-terminal splice sites. Gene. 2005;363:193–201. doi: 10.1016/j.gene.2005.07.028. [DOI] [PubMed] [Google Scholar]
- 69.Levanon EY, Hallegger M, Kinar Y, Shemesh R, Djinovic-Carugo K, Rechavi G, et al. Evolutionarily conserved human targets of adenosine to inosine RNA editing. Nucleic Acids Res. 2005;33(4):1162–1168. doi: 10.1093/nar/gki239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dracheva S, Lyddon R, Barley K, Marcus SM, Hurd YL, Byne WM. Editing of serotonin 2C receptor mRNA in the prefrontal cortex characterizes high-novelty locomotor response behavioral trait. Neuropsychopharmacology. 2009;34(10):2237–2251. doi: 10.1038/npp.2009.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Du Y, Stasko M, Costa AC, Davisson MT, Gardiner KJ. Editing of the serotonin 2C receptor pre-mRNA: Effects of the Morris Water Maze. Gene. 2007;391(1–2):186–197. doi: 10.1016/j.gene.2006.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Auerbach BD, Osterweil EK, Bear MF. Mutations causing syndromic autism define an axis of synaptic pathophysiology. Nature. 2011;480(7375):63–68. doi: 10.1038/nature10658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rossignol DA, Frye RE. Mitochondrial dysfunction in autism spectrum disorders: a systematic review and meta-analysis. Mol Psychiatry. 2012;17(3):290–314. doi: 10.1038/mp.2010.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Garbett K, Ebert PJ, Mitchell A, Lintas C, Manzi B, Mirnics K, et al. Immune transcriptome alterations in the temporal cortex of subjects with autism. Neurobiol Dis. 2008;30(3):303–311. doi: 10.1016/j.nbd.2008.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lainhart JE, Lange N. Increased neuron number and head size in autism. JAMA. 2011;306(18):2031–2032. doi: 10.1001/jama.2011.1633. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.