

Abstract
We showed previously that inactivation of TSC2 induces death in cancer cells lacking the Retinoblastoma (Rb) tumor suppressor under stress conditions, suggesting that inactivation of TSC2 can potentially be used as an approach to specifically kill cancers that have lost WT Rb. As Rb is often inactivated in cancers by overexpression of cyclin D1, loss of p16ink4a cdk inhibitor, or expression of viral oncoproteins, it will be interesting to determine if such functional inactivation of Rb would similarly sensitize cancer cells to TSC2 inactivation induced cell death. In addition, many cancers lack functional Pten, resulting in increased PI3K/Akt signaling that has been shown to modulate E2F-induced cell death. Therefore it will be interesting to test whether loss of Pten will affect TSC2 inactivation induced killing of Rb mutant cancer cells. Here, we show that overexpression of Cyclin D1 or the viral oncogene E1a sensitizes cancer cells to TSC2 knockdown induced cell death and growth inhibition. On the other hand, knockdown of p16ink4a sensitizes cancer cells to TSC2 knockdown induced cell death in a manner that is likely dependant on serum induction of Cyclin D1 to inactivate the Rb function. Additionally, we demonstrate that loss of Pten does not interfere with TSC2 knockdown induced cell death in Rb mutant cancer cells. Together, these results suggest that TSC2 is potentially a useful target for a large spectrum of cancer types with an inactivated Rb pathway.
Keywords: Rb pathway, Cyclin D1, p16ink4a, E1a, Pten, TSC2
1. Introduction
The Retinoblastoma protein pRb functions by binding to the E2F family of transcription factors and regulates the expression of diverse cellular targets including genes involved in cell cycle regulation, DNA replication, DNA repair, cell cycle checkpoint, apoptosis and differentiation [1; 2]. pRb is targeted for inactivation in a large majority of human cancers. Therefore approaches that can specifically target the loss of Rb function can potentially be used in the treatment of a large majority of human cancers.
In an unbiased genetic screen in Drosophila, we found that inactivation of TSC2 and Rb show synthetic lethality in developing Drosophila tissues. TSC2 functions in complex with TSC1, and negatively regulates the Rheb/mTOR pathway and cell growth [3]. Interestingly, we found that knockdown of TSC2 significantly induces cell lethality in human cancer cells when pRb is inactivated by mutation or shRNA-mediated knockdown [4]. These observations raise the possibility that inactivation of TSC2 function can potentially be used to specifically target cancers with loss of Rb function either by mutation or by loss of expression [5].
In addition to mutation or loss of expression, pRb is found to be inactivated in cancers by a variety of mechanisms. Amplification or overexpression of cyclin D1 has been observed in cancers [6], which activates cyclin dependant kinases and causes functional inactivation of Rb. Similarly, loss of the cdk inhibitor p16ink4a leads to Rb inactivation due to deregulated cyclin D/cdk4 activity. Importantly, loss of p16ink4a, and overexpression of cyclin D1 are linked to a poor prognosis in cancer treatment [7; 8]. In addition to alterations in the status of Rb regulators, cellular expression of viral oncogenes such as adenovirus E1a, SV40 large T, and HPV E7 [9; 10] can inactivate Rb through direct binding [11]. Therefore, it is of interest to know if inactivation of TSC2 can also be used to specifically kill cancer cells with these different forms of Rb pathway inactivation.
Rb/E2F-induced cell death is modulated by other regulators and signaling pathways such as the growth factor-stimulated activation of PI3K and Akt survival signaling [12; 13]. Pten, an inhibitor of the PI3K/Akt pathway, is also often mutated in human cancers and Pten loss is implicated in poor prognosis for cancer treatment [14]. It is interesting to note that PI3K survival signaling modulates E2F1-induced cell proliferation or death [13]. In addition, PI3K/Akt also has a major role in the regulation of energy metabolism and the coordination of key metabolic pathways [15]. Consistent with this, activation of PI3K/Akt can inhibit cell death induced by a variety of stimuli but not by oxidative stress. In fact hyperactivity of Akt sensitizes cells to ROS induced cell death [16]. As synergistic cell death induced by inactivation of Rb and TSC2 are mediated, at least in part, by increased ROS levels, it is of interest to determine if Pten loss might interfere with the synergistic cell death associated with Rb and TSC2 loss.
In this report, we use lentiviral mediated overexpression or knockdown to investigate if different ways of Rb pathway inactivation will sensitize cancer cells to TSC2 inactivation induced cell death and if cell death induced by TSC2 inactivation in Rb mutant cancer cells are modulated by Pten loss.
2. Materials and Methods
2.1 Cell Culture
Du145 and PC3 cells were obtained from the American Type Culture Collection. Cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with 8% fetal bovine serum, 50 IU penicillin/streptomycin and 2 mmol/l L-glutamine in a humidified atmosphere with 5% CO2 at 37 °C.
2.2 Lentiviral preparation and infection
Viral E1A and human cyclin D1 were cloned into the lentiviral expression vector pCDH-CMV-EF1-puro. RNAi sequences were cloned into the RNAi expression vector pLKO.1. Sequences for p16ink4a knockdown[17] and Pten knockdown[18] were obtained from literature. The sequences for Pten knockdown were (a) ACGGGAAGACAAGTTCATG, and (b) AAGATCTTGACCAATGGCTAA. Sequence (b) was used in the majority of Pten studies. The sequence for p16ink4a knockdown were: shp16a, GGGAGCAGCATGGAGCCTTCGG; shp16b, GGTCGGGTAGAGGAGGTGCGGG; and shp16c, CTGCCCAACGCACCGAATA. The sequence forming the loop region was CTCGAG. Caffeine was used for viral preparation [19]. The protocol for viral concentration was similar to the one described previously [4], and the viral pellet was resuspended in DMEM with serum, and stored in aliquots at −80 °C.
Each virus preparation was calibrated using the following procedure to ensure equal viral load in our experiments between control and experimental conditions. PC3 cells were infected with lentivirus at different dilutions of virus for 2 days. Cells were then transferred to new wells and incubated in 1 μg/ml puromycin for 2 days. Cell death was assessed using trypan blue stain for each dilution and puromycin killing curves were obtained for each virus preparation. Comparison of these curves was done to ensure equal levels of infection, and therefore viral load, between control and experimental conditions. For experiments examining synergistic cell death between sh-p16, shPten, E1a or CyclinD1 and shTSC2 viral treatment, cells were plated at 1–2 × 104 cells/cm2, and then first infected with sh-p16, shPten, E1a or CyclinD1, while replicate cells were infected at an equal viral load of control virus in parallel. Cells were incubated for 2 days, then transferred to new wells and selected in 1 μg/ml puromycin for 2 days. Cells were then transferred to new wells and infected with a level of shTSC2 that was demonstrated to yield 100% efficiency in puromycin selection, or an equal load of control lentivirus, for 2 days, and then used in experiments. For Rb rescue experiments of E1a treated cells, a similar procedure was followed, except that E1a puromycin-selected cells were plated, and then infected the next day with Rb or a control virus with pCDH vector, and then the next day viral media was removed and cells were infected with shTSC2 virus or an equal load of control virus and then incubated for 2 days before being used in experiments.
Control virus used in our experiments was prepared in the same fashion using empty pCDH-CMV-EF1-puro, empty pLKO.1-puro vectors, or a pLKO.1 containing the sequence CCTTATATCACTAAGGGTTT, which is not specific to any human gene as determined by BLAST [4]. No differences were observed between the empty pLKO.1 vector and the pLKO.1 vector containing the non-specific sequence (Fig. S1). TSC2 knockdown constructs were described previously [4] and knockdown of TSC2 was verified using current preparations of the virus.
2.3 Soft Agar Growth Assay
104 cells were seeded into a top layer consisting of 1 part 2x DMEM and FBS and 1 part 0.7 % agarose, over a bottom layer consisting of 1 part 2x DMEM and FBS, and 1 part 1.2 % agarose. Cells were kept in humidified standard cell culture conditions. All soft agar growth assay data are representative of at least 3 independent experiments. Soft agar growth rescue assays were performed in the presence of 12mM NAC or vehicle control added to the top layer mix at the time of plating. All NAC rescue data is representative of 2 independent experiments.
2.4 Cell Death Assays
Cells were treated with viruses and plated at approximately 2×104 cells/cm2. For low serum stress assays, cells were plated directly into DMEM with 0.7% serum. Cells were then incubated for 4–6 days until cell death was apparent. For oxidative stress assays, cells were incubated overnight, and subjected to complete media with 1mM H2O2 the next day, and then incubated for 2 days in Cyclin D1 and sh-p16 experiments, or one day for E1a experiments. Cell death percentage was assessed using trypan blue stain. All data are representative of 3 independent assays.
2.5 DHE staining and quantification
Cells were treated with lentivirus and subjected to low serum media for 3–4 days in the case of PC3 cells, or 1–2 days in the case of Du145 cells. Cells were then trypsinized, and suspended in 20 μM DHE fluorescent stain, and allowed to incubate at 37 °C in standard cell culture conditions for 30 minutes. Cells were then imaged and photographed with identical parameters across all samples and replicates for a given experiment. For quantification, cell number was assessed for each image using ImageJ software, and average luminosity was determined for each image using Adobe Photoshop. The luminosity was then divided by cell number for each image to obtain an average luminosity per cell for the image.
2.6 Western Blotting
After experimental treatments, cells were washed twice with 1×PBS, and lysed in RIPA buffer (50 mm Tris.Cl [pH 7.4], 150 mm NaCl, 2 mm EDTA, 1% NP40, 0.1% SDS, 0.5% sodium deoxycholate, plus protease inhibitors). Equal amounts of protein were loaded. A Li-Cor Odyssey image reader was used for western detection. The following antibodies were used: (Santa Cruz Biotechnology) sc 4689 against p16ink4a, sc 69879 against β-actin, sc 898 against TSC2, and (Cell Signaling Technology) #9559 against Pten, # 9271S against p-Akt Ser 473, and #92G2 against Cyclin D1. The goat anti-mouse immunoglobulin G (IgG) and goat anti-rabbit IgG secondary antibodies were obtained from Li-Cor.
2.7 Transcriptional Reporter Assay
Cells were treated with lentivirus as described above and were plated into a 24-well plate, followed by transfection by lipofetamine 2000 (Invitrogen) according to the manufacturer’s directions. Each tranfection contained 800 ng of E2F4B-Luc, and 5 ng of phRL-Luc. Cell extracts were prepared 48 h post-tranfection, and the luciferase activity was measure using Dual Luciferase Reporter Assay System (Promega) according to the manufacturer’s directions. Luciferase activity was read on a BD Monolight 3010 Luminometer. All data points presented are the average measurement of three independent transfections.
3. Results
3.1 Cyclin D1 overexpression sensitizes cancer cells to TSC2 inactivation induced cell death
Although many forms of cancer demonstrate perturbations in the Rb pathway, this is not always due to mutation or deletion of the Rb gene itself. Overexpression or amplification of Cyclin D1, which can result in the hyperphosphorylation and inactivation of pRb function, is often observed in various cancers [20]. As earlier work done in our lab has demonstrated that loss of Rb sensitizes cells to TSC2-inactivation induced cell death [4], we investigated the effect of inactivating the Rb pathway via overexpression of cyclin D1.
Myc-tagged Cyclin D1 was overexpressed in PC3 cells and a concentration of virus was chosen that caused marked ectopic expression of Cyclin D1 (Fig. 1A). As expected, Cyclin D1 overexpression induced Rb inactivation, leading to the activation of E2F luciferase reporter activity (Fig. 1B, p<0.018). PC3 cancer cells demonstrate aggressive growth in comparison to other prostate cancer cell lines, with both androgen independence and ability to grow in serum free media [21], and it is likely that this ability is due to an autocrine signaling loop from extrusion of TGF alpha into the media, [22] and this growth independence is characteristic of aggressive malignancies. Cyclin D1 overexpressing or control PC3 cells were treated with shTSC2 virus or controls and tested for the ability to survive in 0.7% low serum media. Moderately increased cell death was observed in cells with TSC2 loss, but when Cyclin D1 was overexpressed in conjunction with TSC2 knockdown, we found significantly increased cell death over all other conditions (Fig. 1D, p<0.02). Another feature of PC3 cancer cells is the ability to form anchorage independent colonies in soft agar at high efficiency [21]. To test for alterations to this ability when Cyclin D1 is overexpressed in conjunction with TSC2 knockdown, these cells were also tested for the ability to grow in soft agar. Significantly reduced ability to form colonies was observed in cells with increased Cyclin D1 levels in addition to TSC2 loss in comparison to all other conditions (Fig. 1C, p<0.009). These results mirror earlier findings that knockdown of Rb and TSC2 synergistically increased cell death and inhibited tumor growth [4] and indicate that inactivation of TSC2 can potentially be used to target cancer types with deregulated Cyclin D1 activity.
Fig. 1.

Cyclin D overexpression in PC3 cancer cells synergizes with TSC2 knockdown to produce increased ROS and cell death in stressed conditions. (A) PC3 cells were infected with control or Cyclin D1 lentivirus, selected with puromycin, and western blotting was performed on cell lysates to assess Cyclin D1 and Actin protein levels. (B) PC3 cells were treated with Cyclin D1 lentivirus or control and puromycin-selected, and transfected with an E2F-Luciferase reporter and assayed for luciferase activity. (C) PC3 cells were treated with Cyclin D1 lentivirus and puromycin-selected, and treated with shTSC2 and control viruses, seeded into soft agar, and colonies counted. (D) PC3 cells overexpressing Cyclin D1 and lacking TSC2 were incubated in low serum media and assayed for cell death using trypan blue staining. (E–H) PC3 cells overexpressing Cyclin D1 were treated with control or shTSC2 virus, incubated in low serum media, trypsinized, stained with DHE and imaged. (I) Images from DHE staining were quantified for average luminescence per cell. (J) Cyclin D1 overexpressing PC3 cells were incubated in 1 mM H2O2 for 2 days and cell death was assayed by trypan blue staining. (K) PC3 cells overexpressing cyclin D1 were treated with shTSC2 or control were seeded into soft agar with or without 12 mM NAC and colonies were counted.
3.2 Cyclin D1 overexpression and TSC2 inactivation synergistically increase intracellular ROS levels and mediate anchorage-independent cell growth
Earlier findings indicated that the increased cell death observed in cells lacking Rb and TSC2 correlated with increased ROS levels [4]. We therefore assayed ROS levels in PC3 cells with overexpressed Cyclin D1 and TSC2 knockdown using DHE staining and fluorescent imaging. PC3 cells were treated with Cyclin D1, shTSC2 or control lentivirus, and stressed in low serum media, and stained with DHE (Fig. 1E–H). Quantification of these results showed a marked increase in DHE staining of cells lacking TSC2 and overexpressing Cyclin D1 (Fig. 1I, p<0.04), which is correlated to the significantly increased cell death and reduced colony formation in soft agar that was observed (Fig. 1C and 1D). These results indicated a potential vulnerability in Cyclin D1/shTSC2 treated cells to oxidative stress, which is also a form of stress encountered by cancer cells in tumors, which in turn makes cancer cells more dependent on cellular antioxidant mechanisms [23; 24]. We therefore tested cells treated with Cyclin D1 and shTSC2 lentivirus for sensitization to cell death at low levels of H2O2. The pattern of cell death observed in low H2O2 (Fig. 1J) was similar to that in low serum conditions (Fig. 1D), with some increased death in shTSC2 cells, and further elevated cell death when Cyclin D1 was overexpressed in conjunction with and shTSC2 (p<0.045 in comparison to all controls). To test if increased ROS might play a key role in the observed inhibition of anchorage independent cell growth, soft agar assays were performed on cells overexpressing Cyclin D1 and treated with shTSC2 lentivirus or controls, in the presence or absence of the antioxidant NAC. As shown in Fig. 1K, TSC2 knockdown in combination with Cyclin D1 overexpression inhibited colony growth in soft agar in the absence of NAC (p<0.003) but not in the presence of NAC (p>0.1). Together these results suggest that cancer cell types with inactivation of the Rb pathway via overexpression of Cyclin D1 will be susceptible to TSC2 inactivation induced cell death due to increased cellular stress such as ROS.
3.3 Inactivation of the cdk inhibitor p16ink4a and TSC2 results in serum-dependant sensitization to stress induced cell death and growth inhibition
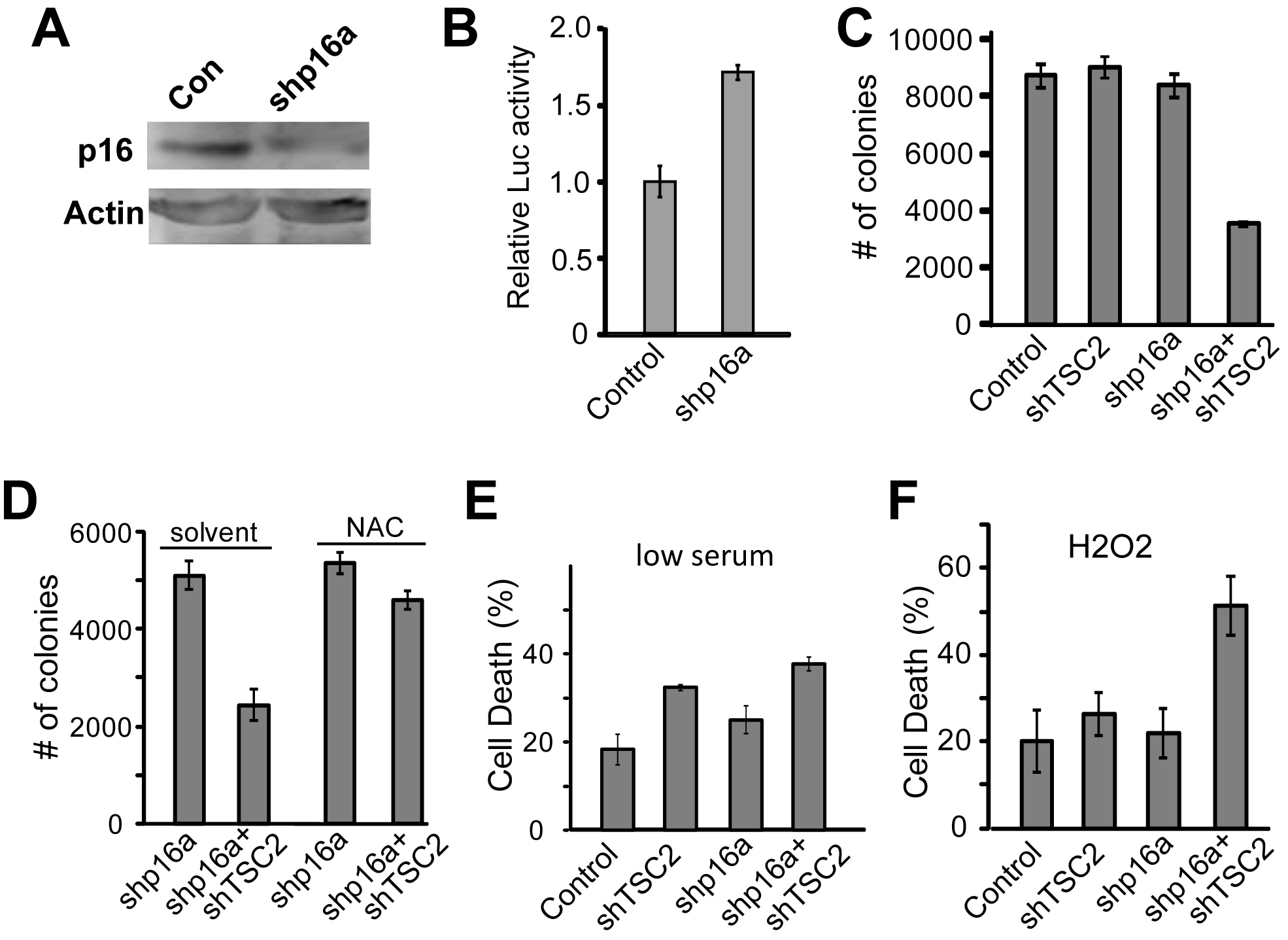
In addition to Cyclin D1 overexpression or amplification, loss of Cyclin D1/cdk4 inhibitor p16ink4a can also lead to Rb pathway inactivation. In fact, loss of p16ink4a is common in cancers, and often associated with poor prognosis [7]. We tested if inactivation of p16ink4a will sensitize cancer cells to TSC2 inactivation-induced cell death. Lentivirus with shRNA targeted against p16ink4a was generated and knockdown of p16ink4a was verified (Fig. 2A). We further tested the effect of p16ink4a knockdown on the Rb pathway, and found that p16ink4a loss in PC3 cells also significantly increased E2F activity (p<0.01) as measured by a transfected E2F-luciferase reporter (Fig. 2B). To determine if knockdown of p16ink4a also sensitizes cancer cells to TSC2 inactivation-induced anchorage independent cell growth inhibition, cell growth assays in soft-agar were performed. Reduced colony formation in comparison to all controls was apparent in cells treated with sh-p16a and shTSC2 lentivirus (Fig. 2C, p<0.004). The observed synergistic death effect of shp16 with shTSC2 are likely mediated by deregulated E2F activity since the two shp16 constructs (shp16a and shp16c) that deregulated E2F activity (Fig. S3A–B, p<0.002) induced synergistic death and inhibited colony in soft agar in conjunction with shTSC2 (Fig. 2C and 2F; S3D and S3F, p<0.009) while shp16b, which did not deregulate E2F, failed to induce synergistic death or inhibit colony growth (Fig. S3C and S3E p>0.5 between shTSC2 and shp16+shTSC2).
Fig. 2.

p16ink4a knockdown synergizes with TSC2 knockdown resulting in Increased cell death in response to stress in complete media, but not in response to low serum stress. (A) PC3 cells were treated with control or sh-p16a lentivirus and selected in puromycin, and lysates were subjected to western blotting, and probed with p16ink4a and Actin antibodies. (B) PC3 cells were selected for control or p16ink4a knockdown, and transfected with an E2F-Luciferase reporter, and Luciferase activity was measured. (C) PC3 cells were selected for p16ink4a knockdown and treated with shTSC2 and control viruses, seeded into soft agar, and colonies counted. (D) PC3 cells selected for p16 knockdown and treated with shTSC2 or control were seeded into soft agar with or without 12 mM NAC and colonies were counted. (E) PC3 cells treated with sh-p16 and shTSC2 lentivirus were incubated in low serum media, and assayed for cell death using trypan blue staining. (F) PC3 cells lacking p16ink4a were incubated in 1 mM H2O2 for 2 days and cell death was assayed by trypan blue staining.
Consistent with our previous finding that Rb and TSC2 inactivation-induced cell death and growth inhibition are mediated by increased ROS [4], we found that addition of antioxidant NAC restored colony growth of p16ink4a and TSC2 double knockdown cells and no significant difference was observed between sh-p16 and sh-p16+shTSC2 samples after NAC addition (Fig. 2D, p=0.65). This suggests an important role for ROS in the inhibition of colony growth of the p16ink4a and TSC2 knockdown cells. Interestingly, while p16ink4a and TSC2 double knockdown cells were much more sensitized to undergo cell death than the control or each of the single knockdown cells when cells were grown in the presence of low levels of H2O2 in normal serum (Fig. 2F, p<0.0005), no synergistic cell death was observed when cells were grown in low serum conditions (Fig. 2E). These observations were distinct from the synergistic cell death induction of the Rb and TSC2 double knockdown cells [4] or the CyclinD1 overexpressing TSC2 knockdown cells (Fig. 1D). A likely explanation for the decreased ability of sh-p16 and shTSC2 to synergistically induce cell death in serum free conditions is that the regulation of pRb by p16ink4a is mediated by Cyclin D kinase activities. In serum free conditions, the level of Cyclin D and therefore the associated kinase would be low and therefore not be able to inactivate the function of pRb. To test this, PC3 cells were treated with control or Cyclin D1 lentivirus, and incubated for 3 days in medium with or without serum. As predicted, endogenous Cyclin D1 levels were significantly decreased in PC3 cells grown in serum free media. In contrast, high levels of myc tagged Cyclin D1 were still detected in cells in serum free conditions (Fig. S2). These results suggest that TSC2 could serve as a useful target in some p16ink4a-negative cancer cells, however the efficacy in those cases could be dependent on mitogenic signaling and the downstream activity of cyclin/cdk complexes.
3.4 Inactivation of the Rb pathway via the viral oncogene E1a sensitizes cancer cells to TSC2 inactivation-induced cell death and growth inhibition
The Rb pathway can also be inactivated by the expression of viral oncoproteins such as adenovirus E1a and Human Papiloma Virus protein E7 [9; 10]. Due to the link of E1a-like proteins to cancer, it is of interest to investigate if cancer cells with inactivated Rb pathway by the expression of viral proteins are sensitive to TSC2 inactivation-induced cell death. We therefore tested if ectopic E1a expression sensitizes cancer cells to TSC2 inactivation-induced death.
As expected, E1a expression inactivated Rb and significantly upregulated E2F activity (Fig. 3A, p<0.001). To test if TSC2 knockdown synergized with E1a to cause sensitization to cell death, PC3 cells with ectopic E1a were treated with shTSC2 or control virus, and the ability to survive in low serum was tested (Fig. 3C). We found that shTSC2 treatment caused some increase in cell death, while cells treated with E1a and shTSC2 showed markedly increased cell death in comparison to all other samples (p<0.002). Furthermore, cells similarly treated were tested for the ability to grow in anchorage independent conditions in soft agar (Fig. 3B). We found that E1a alone caused some reduction in anchorage independent growth, but E1a and shTSC2 treatment together caused a marked reduction in colony formation compared to all controls (p<0.005).
Fig. 3.

Ectopic E1A expression in PC3 cancer cells synergizes with TSC2 knockdown to produce increased ROS and cell death in stressed conditions. (A) PC3 cells were treated with E1A lentivirus or control and puromycin-selected, and transfected with an E2F-Luciferase reporter and assayed for luciferase activity. (B) PC3 cells were selected for ectopic E1A expression and treated with shTSC2 and control viruses, seeded into soft agar, and colonies counted. (C) PC3 cells with ectopic E1A and lacking TSC2 were incubated in low serum media and assayed for cell death using trypan blue staining. (D) PC3 with ectopic E1A were incubated in 1 mM H2O2 for 1 day and cell death was assayed by trypan blue staining. (E–H) PC3 cells with ectopic E1A expression were treated with control or shTSC2 virus, and incubated in low serum media. Cells were then trypsinized and stained with DHE before imaging. (I) Images from DHE staining were quantified for average luminescence per cell. (J) PC3 cells selected for ectopic E1A expression and treated with shTSC2 or control were seeded into soft agar with or without 12 mM NAC and colonies were counted. (K) E1A-selected PC3 cells were treated with control or Rb lentivirus, and then control or shTSC2, and incubated in 1mM H2O2 for 1 day and cell death was assayed by trypan blue staining.
We next tested whether the death effects we observed in E1a and shTSC2 treated cells correlated to increased ROS levels as it had in other forms of Rb pathway inactivation. Cells were treated with E1a, shTSC2 and control viruses, and subjected to low serum stress and stained with DHE to assess ROS levels (Fig. 3E–H). A mild and variable increase in DHE staining was observed in shTSC2 treated cells, while a clear increase in DHE staining was observed in cells that expressed ectopic E1a in conjunction with TSC2 knockdown (Fig. 3I, p<0.03). These data indicated a correlation between increased ROS levels and the cell death effects observed in E1a and shTSC2 treated cells (Fig. 3B–C). Therefore, we tested for a direct ability of oxidative stress to cause death in E1a and shTSC2 treated cells by incubating cells in a low level of H2O2. Indeed a clear increase in cell death in E1a and shTSC2 treated cells was present over controls under this condition (Fig. 3D, p<0.0005). To test for a potential necessary role of ROS-induced stress in these cell death effects, PC3 cells with ectopic E1a expression were treated with shTSC2 or control lentivirus and subjected to soft-agar assay in the presence or absence of the antioxidant NAC (Fig. 3J). We found that in the presence of vehicle control, the ability of E1a/shTSC2 cells ability to grow in soft agar was significantly inhibited (p<2×10−6) while no significant inhibition of E1a/shTSC2 cells was seen with the addition of NAC (p=0.33).
To verify that the sensitization to death of E1a and shTSC2 treated cells was due to E1a inactivation of the Rb pathway, as opposed to other effects of ectopic viral E1a, we tested the ability of overexpressed WT Rb to rescue the synergistic cell death phenotype. As shown in Fig. 3K, while cells expressing E1a and shTSC2 exhibited significantly increased cell death (Fig. 1K, control, p<0.03), this effect was rescued by overexpressed Rb (Fig. 1K, +Rb, p=0.11). Together, these data suggest that cancers induced by the expression of viral oncoproteins that inactivate Rb can also be targeted by approaches involving inactivation of TSC2.
3.5 Pten does not affect TSC2 inactivation-induced ROS levels or cell death in Rb mutant cancer cells
Many forms of cancer exhibit loss of functional Pten [25]. Loss of Pten results in increased PI3K/Akt survival signaling, which might have the potential to interfere with cell death induced by inactivation of Rb and TSC2 [13]. Conversely, deregulated Akt signaling can also upregulate energy metabolism and sensitize cells to ROS-related death [16]. Therefore it is of interest to test if Pten loss might modulate the synergistic cell death induced by inactivation of both Rb and TSC2.
Lentiviral constructs containing shRNA against Pten were generated. Both constructs were able to significantly reduce the endogenous levels of Pten protein and lead to a corresponding increase in the level of phosphorylated active Akt (p-Akt) (Fig. 4A). However, Pten knockdown did not significantly affect TSC2 inactivation-induced cell death in Rb mutant DU145 cancer cells grown in low-serum (Fig. 4B). While both control cells and shPten treated cells showed significant increases in cell death when treated with shTSC2 (p<0.002 and p<0.0009), no significant difference in cell death levels were seen with Pten knockdown. Similar results were observed with the second shPten construct (Fig. S4). Furthermore, Pten knockdown did not significantly affect shTSC2-induced inhibition of anchorage-independent growth in soft agar (Fig. 4C, p=0.6).
Fig. 4.

Pten knockdown in Du145 cells does not rescue synergistic cell death between shTSC2 and lack of functional Rb, and does not alter ROS levels in these cells. (A) Du145 cells were treated with lentivirus encoding two shRNA sequences against Pten, selected with puromycin, then subjected to western blotting and probed with antibodies against Pten, p-Akt and Actin. (B) Du145 cells lacking Pten and infected with shTSC2 were incubated in low serum, and cell death was assayed with trypan blue staining. (C) Du145 cells were selected for Pten knockdown and treated with shTSC2 and control virus, seeded into soft agar, and colonies counted. (D–G) Du145 cells were selected for Pten knockdown, and treated with shTSC2 and incubated in low serum media. Cells were then trypsinized and stained with DHE before imaging. (H) Images from DHE staining were quantified for average luminescence per cell.
While Pten loss activates survival signaling, activation of the Akt pathway has been shown to have the potential to increase ROS levels in cells [16]. Earlier work showed that increased ROS levels in cells lacking Rb and TSC2 was observed to correlate with synergistically increased death effects [4]. We therefore further tested the effect of Pten knockdown on ROS levels in Du145 cells both with or without TSC2 knockdown. TSC2 knockdown markedly increased intracellular ROS levels assessed by DHE stain both in control and shPten treated cells (Fig. 4E and 4G), and quantification of this data (Fig. 4H) showed significant increases (p<0.04, p<0.003) in DHE staining. In contrast, knockdown of Pten did not significantly change the ROS levels either with or without TSC2 knockdown (Fig. 4D–G and Fig. 4H). Therefore knockdown of Pten did not significantly affect the shTSC2-induced ROS levels or cell death in Rb mutant DU145 cancer cells. These observations suggest that inactivation of TSC2 can potentially be used to kill Rb mutant cancer cells irrespective of their Pten status.
4. Discussion
Targeted cancer therapies are of clear interest. Rb is inactivated in many cancers, and often loss of p16ink4a and overexpression of cyclin D1 are associated with poor prognosis [7; 8]. The results here further build on previous results by our laboratory, where loss of Rb was shown to synergize with loss of TSC2, resulting in cell death in Drosophila developing tissues and in human cancer cell lines [4]. Our findings suggest that any approach developed for targeting TSC2 in Rb deficient cancers could potentially be extended to include some instances of cancers with Cyclin D1 overexpression, loss of p16ink4a, or cancer associated with the presence of viral oncoproteins that inactivate Rb such as human papiloma virus-associated cervical cancers (Fig. 5).
Fig. 5.

A model for synergistic cancer cell death in response to stress when TSC2, but not Pten is inactivated in the context of a deregulated Rb pathway.
A key role for ROS was previously demonstrated in stress induced cell death of Rb and TSC2 negative cells [4]. In our assays, significant increases in cell death were similarly correlated with significant increases in ROS levels visualized by DHE stain. Previous work in our lab additionally showed that SOD2 induction in shTSC2 treated cells was Rb dependant, with reduced SOD2 induction in response to stress and TSC2 loss in the absence of Rb. Similarly, knockdown of p16 reduced shTSC2-induced SOD2 levels (Fig. S5). In addition, loss of functional TSC2 is also associated with increased ROS production [4]. These findings suggest that a synergistic effect between increased ROS due to TSC2 loss and reduced SOD2 induction due to Rb inactivation may play a role in our observed cell death. Consistent with this idea, significantly increased ROS levels were observed when shTSC2 treatment was combined with Rb pathway inactivation (Fig. 1I and 3I), indicating that a potential ROS-linked sensitivity to shTSC2-induced cell death may be unmasked by Rb pathway inactivation in our experiments (Fig. 5). Indeed, the addition of the antioxidant NAC was able to significantly rescue colony formation (Fig. 1K, 2D and 3J).
Cancer metastasis, growth, and the conditions of the tumor environment all induce stresses on cancer cells (Fig. 5). Advanced prostate cancers can escape androgen ablation treatment and exhibit autonomous growth as well as tumorigenicity. PC3 cells are a model for this type of cancer, as they have been shown to be able to form colonies in soft agar efficiently, which is considered to be correlated with tumorigenicity [21], as well as grow independently of androgens in the absence of serum. A likely major cause of this mitogen independent growth has been demonstrated to be an autocrine signal loop consisting of PC3 production of TGF alpha signaling back to the cell through the EGF receptor to promote cell growth [22]. The assays chosen in this work assess some key aspects of the cancer cell’s ability to overcome some of these obstacles and stresses outlined above. In assays for anchorage independent growth, cells with overexpressed Cyclin D1, p16ink4a knockdown, or ectopic E1a all demonstrated a significant loss of ability to form colonies in soft agar when TSC2 was knocked down (Fig. 1C, 2C and 3B). Furthermore, TSC2 knockdown cells that were treated with Cyclin D1, sh-p16ink4a or E1a lentivirus demonstrated significantly increased death over both shTSC2 and other controls in conditions of oxidative stress (Fig. 1J, 2F and 3D). Alternately, under conditions of low serum, Cyclin D1 and E1a lentivirus-treated cells, but not sh-p16 cells, synergized with TSC2 knockdown to show increased death over shTSC2 treated cells and other controls. Since these experiments model some of the stresses and challenges faced by cancer cells during growth and metastasis, these data suggest that in cancers with elevated Cyclin D1 activity, as well as those demonstrating ectopic viral oncogenes such as E7, TSC2 may be a potential target for therapy. On the other hand, the data suggest that in cancers characterized by p16ink4a loss, the usefulness of TSC2 as a target may be conditional (Fig. 5).
Although Rb is often inactivated in cancer cells, this does not necessarily mean Rb is completely inactivated in all phases of the cell cycle. It is possible that different mechanisms of Rb inactivation in different cancer cells will have different levels and/or durations of Rb inactivation. For example, inactivation of pRb by direct regulators such as the viral oncoprotein E1a can potentially lead to pRb inactivation in all the phases of the cell cycle as long as there is sufficient E1a level, while inactivation of pRb by p16 ink4a loss will depend on the level of Cyclin D/Cdk4 activity in the cell. In soft agar and H2O2 assays, which are performed in complete media, sh-p16 cells synergized with TSC2 loss resulting in elevated death effects over controls. On the other hand, sh-p16 and shTSC2 treated cells did not show significant increase in cell death in low serum media. Previous work has indicated that Cyclin D1 and its binding partner cdk4/6, as well as their ability to associate, are serum dependant [20], and we have verified in our system that while levels of ectopic Cyclin D1 remain high in serum free media, endogenous Cyclin D1 does not (Fig. S2). While autocrine signaling through TGF alpha production, as well as other possible factors, may confer growth independence on PC3 cells, levels of unrestrained Cyclin D1 signaling in sh-p16 treated PC3 cells in low serum media may be insufficient to deregulate the Rb pathway past the threshold needed for synergistic effects with shTSC2. Therefore, our results indicate that an argument can be made for the potential usefulness of TSC2 as a target in p16ink4a negative cells, but any usefulness of this target might be highly dependent on the context of Cyclin D/cdk4/6 signaling in the given cell type.
The tumor suppressor Pten is often mutated in cancer, and its loss is also tied with poor prognosis [14], and resistance to targeted therapies. Therefore it was of interest to us to address whether Pten loss has an effect on survival of Rb-mutant cancer cells when challenged with TSC2 inactivation. No rescue effect was observed in DU145 cells subjected to TSC2 knockdown in soft agar or low serum stress, which is consistent with the observation that Akt activation is unable to protect death induced by ROS [16] and the fact that PC3 cells, which lack functional Pten, are sensitized to synergistic death from Rb/TSC2 loss. On the other hand, although one might expect knockdown of Pten and activation of Akt will increases ROS levels [16], we found Pten knockdown does not significantly affect ROS levels either alone or in conjunction with shTSC2. As Akt activation is regulated by insulin/IRS signaling and TORC2, which are under feedback negative regulation by TORC1 activity [26; 27], it is possible that such negative feedback regulations prevent strong enough Akt activation that can lead to detectable changes in ROS levels or cell death in our experimental conditions.
The importance of targeting cancers with inactivation of the Rb pathway is of clear importance, as these cancers can be resistant to therapy. Although the loss of functional Rb pathway results in certain advantages for a cancer cell in terms of unregulated entry into S-phase and proliferation, other apoptotic programs and vulnerabilities can be unmasked by the deregulated Rb pathway [28; 29], which can potentially be exploited in developing novel cancer therapies. Indeed, ER negative breast cancers with high Rb loss signature were associated with better outcome when treated with chemotherapy [30]. Our data presented here along with that published before [4], suggest that the metabolic stresses of activated mTOR feeds into and synergizes with these vulnerabilities opened up by an inactivated Rb pathway (Fig. 5). Further research into targeting the disadvantageous effects to a cancer cell of a deregulated Rb pathway could yield new targeted therapies for cancers with disrupted Rb signaling.
Supplementary Material
Comparison of empty and non-specific shRNA sequence control lentivirus. (A) PC3 cells were infected with pCDH-CyclinD1 or empty pCDH lentivirus and selected with puromycin. Cells were then infected with pLKO.1-shTSC2, empty pLKO.1 or pLKO.1 with a shRNA sequence not specific for any human gene, and the cells were incubated in 1mM H2O2 and assayed for death with trypan blue. (B) PC3 cells were infected with pLKO.1-shp16a, empty pLKO.1 or pLKO.1 lentivirus containing a nonspecific shRNA sequence and selected with puromycin. Cells were then infected with pLKO.1-shTSC2, empty pLKO.1 or pLKO.1 with nonspecific sequence. The cells were then incubated in 1mM H2O2 and assayed for death with trypan blue. (C) PC3 cells were infected with pCDH-E1A or empty pCDH lentivirus and selected with puromycin. Cells were then infected with pLKO.1-shTSC2, empty pLKO.1 or pLKO.1 with a nonspecific shRNA sequence, and the cells were incubated in 1mM H2O2 and assayed for death with trypan blue. * indicate significant difference compared to both the empty vector and the nonspecific controls.
{kind=link}
Endogenous levels of Cyclin D1 are serum-dependant while ectopic myc-Cyclin D1, which is expressed from the pCDH vector, remains highly expressed in the absence of serum. PC3 cells were treated with control or Cyclin D1 lentivirus and selected, and then incubated for 3 days in the absence or presence of 8% FBS, and western blotting was performed for Cyclin D1 and actin.
{kind=link}
sh-p16 lentiviral constructs which elevate E2F activity also show synergistic cell death effects with TSC2 knockdown. (A) PC3 cells were treated with control, sh-p16a or sh-p16b lentivirus, and were then transfected with an E2F-luciferase reporter, and luciferase activity was measured. (B) PC3 cells were treated with control and sh-p16c lentivirus, trasnfected with an E2F-luciferase construct and tested for luciferase activity. (C) Cells treated with sh-p16 b or control virus were then treated with shTSC2 or control lentivirus and subjected to 1mM H2O2 and tested for cell death with trypan blue. (D) PC3 cells were infected with control or sh-p16c lentivirus, and then shTSC2 or control virus, and incubated in 1mM H2O2 and tested for cell death with trypan blue assays. (E) PC3 cells were infected with sh-p16b, shTSC2 and controls, seeded into soft agar, and colony formation was assessed. (F) PC3 cells were infected with sh-p16c, shTSC2 and control lentivirus, and seeded into soft agar, and colony formation was assessed.
{kind=link}
Loss of Pten does not affect synergistic cell death between mutant Rb and TSC2 knockdown in Du145 cancer cells. A second shPten construct (shPten a) was used to knock down Pten levels in Du145 cells. Cells were then treated with shTSC2 or an equal viral load of control lentivirus, and incubated in low serum media, and cell death was assessed using trypan blue staining.
{kind=link}
Knockdown of p16 decreased levels of SOD2 induced by shTSC2. The levels of SOD2 and Actin were detected by Western blots on extracts from cells infected with lentivirus containing control, shTSC2, shp16 or both shTSC2 and shp16. The normalized SOD2 levels were indicated.
{kind=link}
Acknowledgments
We would like to thank Drs. Binghui Li, R. Hiipakka, J. Kokontis, and members of the Du lab, particularly Drs. Jiong Zhao and Gabe Gordon for many discussions and supply of reagents.
Role of the Funding Source
This work was supported in part by the following grants: DOD W81XWH-10-1-0077, NIH/NCCAM AT004418, NIH GM074197, NIH CA149275.
Footnotes
Conflict of Interest
None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bracken AP, Ciro M, Cocito A, Helin K. E2F target genes: unraveling the biology. Trends Biochem Sci. 2004;29:409–417. doi: 10.1016/j.tibs.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 2.Gordon GM, Du W. Conserved RB functions in development and tumor suppression. Protein Cell. 2011;2:864–878. doi: 10.1007/s13238-011-1117-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kwiatkowski DJ. Tuberous sclerosis: from tubers to mTOR. Ann Hum Genet. 2003;67:87–96. doi: 10.1046/j.1469-1809.2003.00012.x. [DOI] [PubMed] [Google Scholar]
- 4.Li B, Gordon GM, Du CH, Xu J, Du W. Specific killing of Rb mutant cancer cells by inactivating TSC2. Cancer Cell. 2010;17:469–480. doi: 10.1016/j.ccr.2010.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gordon GM, Du W. Targeting Rb inactivation in cancers by synthetic lethality. Am J Cancer Res. 2011;1:773–786. [PMC free article] [PubMed] [Google Scholar]
- 6.Hosokawa Y, Arnold A. Cyclin D1/PRAD1 as a central target in oncogenesis. J Lab Clin Med. 1996;127:246–252. doi: 10.1016/s0022-2143(96)90092-x. [DOI] [PubMed] [Google Scholar]
- 7.Gunia S, Erbersdobler A, Hakenberg OW, Koch S, May M. p16(INK4a) is a Marker of Good Prognosis for Primary Invasive Penile Squamous Cell Carcinoma: A Multi-Institutional Study. J Urol. 2012;187:899–907. doi: 10.1016/j.juro.2011.10.149. [DOI] [PubMed] [Google Scholar]
- 8.Nakashima T, Kuratomi Y, Yasumatsu R, Masuda M, Koike K, Umezaki T, Clayman GL, Nakagawa T, Komune S. The effect of cyclin D1 overexpression in human head and neck cancer cells. Eur Arch Otorhinolaryngol. 2005;262:379–383. doi: 10.1007/s00405-004-0831-z. [DOI] [PubMed] [Google Scholar]
- 9.McLaughlin-Drubin ME, Munger K. The human papillomavirus E7 oncoprotein. Virology. 2009;384:335–344. doi: 10.1016/j.virol.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Howley PM, Livingston DM. Small DNA tumor viruses: large contributors to biomedical sciences. Virology. 2009;384:256–259. doi: 10.1016/j.virol.2008.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Whyte P, Buchkovich KJ, Horowitz JM, Friend SH, Raybuck M, Weinberg RA, Harlow E. Association between an oncogene and an anti-oncogene: the adenovirus E1A proteins bind to the retinoblastoma gene product. Nature. 1988;334:124–129. doi: 10.1038/334124a0. [DOI] [PubMed] [Google Scholar]
- 12.Hallstrom TC, Nevins JR. Specificity in the activation and control of transcription factor E2F-dependent apoptosis. Proc Natl Acad Sci U S A. 2003;100:10848–10853. doi: 10.1073/pnas.1831408100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hallstrom TC, Mori S, Nevins JR. An E2F1-dependent gene expression program that determines the balance between proliferation and cell death. Cancer Cell. 2008;13:11–22. doi: 10.1016/j.ccr.2007.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ettl T, Baader K, Stiegler C, Muller M, Agaimy A, Zenk J, Kuhnel T, Gosau M, Zeitler K, Schwarz S, Brockhoff G. Loss of PTEN is associated with elevated EGFR and HER2 expression and worse prognosis in salivary gland cancer. Br J Cancer. 2012;106:719–726. doi: 10.1038/bjc.2011.605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robey RB, Hay N. Is Akt the “Warburg kinase”?-Akt-energy metabolism interactions and oncogenesis. Semin Cancer Biol. 2009;19:25–31. doi: 10.1016/j.semcancer.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nogueira V, Park Y, Chen CC, Xu PZ, Chen ML, Tonic I, Unterman T, Hay N. Akt determines replicative senescence and oxidative or oncogenic premature senescence and sensitizes cells to oxidative apoptosis. Cancer Cell. 2008;14:458–470. doi: 10.1016/j.ccr.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shin JJ, Katayama T, Michaud WA, Rocco JW. Short hairpin RNA system to inhibit human p16 in squamous cell carcinoma. Arch Otolaryngol Head Neck Surg. 2004;130:68–73. doi: 10.1001/archotol.130.1.68. [DOI] [PubMed] [Google Scholar]
- 18.Miller TW, Perez-Torres M, Narasanna A, Guix M, Stal O, Perez-Tenorio G, Gonzalez-Angulo AM, Hennessy BT, Mills GB, Kennedy JP, Lindsley CW, Arteaga CL. Loss of Phosphatase and Tensin homologue deleted on chromosome 10 engages ErbB3 and insulin-like growth factor-I receptor signaling to promote antiestrogen resistance in breast cancer. Cancer Res. 2009;69:4192–4201. doi: 10.1158/0008-5472.CAN-09-0042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ellis BL, Potts PR, Porteus MH. Creating higher titer lentivirus with caffeine. Hum Gene Ther. 22:93–100. doi: 10.1089/hum.2010.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Diehl JA. Cycling to cancer with cyclin D1. Cancer Biol Ther. 2002;1:226–231. doi: 10.4161/cbt.72. [DOI] [PubMed] [Google Scholar]
- 21.Kaighn ME, Narayan KS, Ohnuki Y, Lechner JF, Jones LW. Establishment and characterization of a human prostatic carcinoma cell line (PC-3) Invest Urol. 1979;17:16–23. [PubMed] [Google Scholar]
- 22.Hofer DR, Sherwood ER, Bromberg WD, Mendelsohn J, Lee C, Kozlowski JM. Autonomous growth of androgen-independent human prostatic carcinoma cells: role of transforming growth factor alpha. Cancer Res. 1991;51:2780–2785. [PubMed] [Google Scholar]
- 23.Hileman EO, Liu J, Albitar M, Keating MJ, Huang P. Intrinsic oxidative stress in cancer cells: a biochemical basis for therapeutic selectivity. Cancer Chemother Pharmacol. 2004;53:209–219. doi: 10.1007/s00280-003-0726-5. [DOI] [PubMed] [Google Scholar]
- 24.Pelicano H, Carney D, Huang P. ROS stress in cancer cells and therapeutic implications. Drug Resist Updat. 2004;7:97–110. doi: 10.1016/j.drup.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 25.Hollander MC, Blumenthal GM, Dennis PA. PTEN loss in the continuum of common cancers, rare syndromes and mouse models. Nat Rev Cancer. 2011;11:289–301. doi: 10.1038/nrc3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang Q, Inoki K, Kim E, Guan KL. TSC1/TSC2 and Rheb have different effects on TORC1 and TORC2 activity. Proc Natl Acad Sci U S A. 2006;103:6811–6816. doi: 10.1073/pnas.0602282103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shah OJ, Wang Z, Hunter T. Inappropriate Activation of the TSC/Rheb/mTOR/S6K Cassette Induces IRS1/2 Depletion, Insulin Resistance, and Cell Survival Deficiencies. Curr Biol. 2004;14:1650–1656. doi: 10.1016/j.cub.2004.08.026. [DOI] [PubMed] [Google Scholar]
- 28.Polager S, Ginsberg D. E2F - at the crossroads of life and death. Trends Cell Biol. 2008;18:528–535. doi: 10.1016/j.tcb.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 29.Tanaka-Matakatsu M, Xu J, Cheng L, Du W. Regulation of apoptosis of rbf mutant cells during Drosophila development. Dev Biol. 2009;326:347–356. doi: 10.1016/j.ydbio.2008.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ertel A, Dean JL, Rui H, Liu C, Witkiewicz AK, Knudsen KE, Knudsen ES. RB-pathway disruption in breast cancer: differential association with disease subtypes, disease-specific prognosis and therapeutic response. Cell Cycle. 2010;9:4153–4163. doi: 10.4161/cc.9.20.13454. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Comparison of empty and non-specific shRNA sequence control lentivirus. (A) PC3 cells were infected with pCDH-CyclinD1 or empty pCDH lentivirus and selected with puromycin. Cells were then infected with pLKO.1-shTSC2, empty pLKO.1 or pLKO.1 with a shRNA sequence not specific for any human gene, and the cells were incubated in 1mM H2O2 and assayed for death with trypan blue. (B) PC3 cells were infected with pLKO.1-shp16a, empty pLKO.1 or pLKO.1 lentivirus containing a nonspecific shRNA sequence and selected with puromycin. Cells were then infected with pLKO.1-shTSC2, empty pLKO.1 or pLKO.1 with nonspecific sequence. The cells were then incubated in 1mM H2O2 and assayed for death with trypan blue. (C) PC3 cells were infected with pCDH-E1A or empty pCDH lentivirus and selected with puromycin. Cells were then infected with pLKO.1-shTSC2, empty pLKO.1 or pLKO.1 with a nonspecific shRNA sequence, and the cells were incubated in 1mM H2O2 and assayed for death with trypan blue. * indicate significant difference compared to both the empty vector and the nonspecific controls.
Endogenous levels of Cyclin D1 are serum-dependant while ectopic myc-Cyclin D1, which is expressed from the pCDH vector, remains highly expressed in the absence of serum. PC3 cells were treated with control or Cyclin D1 lentivirus and selected, and then incubated for 3 days in the absence or presence of 8% FBS, and western blotting was performed for Cyclin D1 and actin.
sh-p16 lentiviral constructs which elevate E2F activity also show synergistic cell death effects with TSC2 knockdown. (A) PC3 cells were treated with control, sh-p16a or sh-p16b lentivirus, and were then transfected with an E2F-luciferase reporter, and luciferase activity was measured. (B) PC3 cells were treated with control and sh-p16c lentivirus, trasnfected with an E2F-luciferase construct and tested for luciferase activity. (C) Cells treated with sh-p16 b or control virus were then treated with shTSC2 or control lentivirus and subjected to 1mM H2O2 and tested for cell death with trypan blue. (D) PC3 cells were infected with control or sh-p16c lentivirus, and then shTSC2 or control virus, and incubated in 1mM H2O2 and tested for cell death with trypan blue assays. (E) PC3 cells were infected with sh-p16b, shTSC2 and controls, seeded into soft agar, and colony formation was assessed. (F) PC3 cells were infected with sh-p16c, shTSC2 and control lentivirus, and seeded into soft agar, and colony formation was assessed.
Loss of Pten does not affect synergistic cell death between mutant Rb and TSC2 knockdown in Du145 cancer cells. A second shPten construct (shPten a) was used to knock down Pten levels in Du145 cells. Cells were then treated with shTSC2 or an equal viral load of control lentivirus, and incubated in low serum media, and cell death was assessed using trypan blue staining.
Knockdown of p16 decreased levels of SOD2 induced by shTSC2. The levels of SOD2 and Actin were detected by Western blots on extracts from cells infected with lentivirus containing control, shTSC2, shp16 or both shTSC2 and shp16. The normalized SOD2 levels were indicated.