

Abstract
Neuroligins are postsynaptic neural cell adhesion molecules that mediate synaptic maturation and function in vertebrates and invertebrates, but their mechanisms of action and regulation are not well understood. At the Drosophila larval neuromuscular junction (NMJ), previous analysis demonstrated a requirement for Drosophila neuroligin 1 (dnlg1) in synaptic growth and maturation. The goal of the present study was to better understand the effects and mechanisms of loss-of-function and overexpression of dnlg1 on synapse size and function, and to identify signaling pathways that control dnlg1 expression. Consistent with a reduced synapse size, evoked excitatory junctional currents (EJCs) were diminished in dnlg1 mutants but displayed normal Ca2+ sensitivity and short-term plasticity. However, postsynaptic function was also perturbed, in that glutamate receptor staining and the distribution of amplitudes of miniature excitatory junctional currents (mEJCs) were abnormal in mutants. All the above phenotypes were rescued by a genomic transgene. Overexpression of dnlg1 in muscle resulted in synaptic overgrowth, but reduced the amplitudes of EJCs and mEJCs. Overgrowth and reduced EJC amplitude required dnrx1 function, suggesting that increased dnlg1/dnrx1 signaling attenuates synaptic transmission and regulates growth through a retrograde mechanism. In contrast, reduced mEJC amplitude was independent of dnrx1. Synaptic overgrowth, triggered by neuronal hyperactivity, absence of the E3 ubiquitin ligase highwire, and increased phosphoinositide-3-kinase (PI3K) signaling in motor neurons reduced synaptic DNlg1 levels. Likewise, postsynaptic attenuation of PI3K, which increases synaptic strength, was associated with reduced DNlg1 levels. These observations suggest that activity and PI3K signaling pathways modulate growth and synaptic transmission through dnlg1-dependent mechanisms.
Keywords: Drosophila, neuroligin, neurexin, synapse, activity, phosphoinositide-3-kinase
Introduction
During the development of neural circuits, cell adhesion molecules play critical roles in the formation, specification, maturation, and maintenance of synapses. Neuroligins (NLGs) encode a family of postsynaptic transmembrane proteins that regulate synapse formation in vitro and synapse maturation in vivo through trans-synaptic signaling with a presynaptic ligand, neurexin (NRX) (Dean et al., 2003; Graf et al., 2004). NLG/NRX complexes organize synaptic structure by recruiting PDZ domain-containing scaffolding proteins and additional molecules via protein-protein interactions (Irie et al., 1997; Meyer et al., 2004; Poulopoulos et al., 2009). In rodents, three genes encode structurally homologous NLG isoforms (Ichtchenko et al., 1995; Ichtchenko et al., 1996) that are enriched at postsynaptic densities, have overlapping distributions in the brain, and are co-localized at some synapses (Budreck and Scheiffele, 2007; Graf et al., 2004; Song et al., 1999; Varoqueaux et al., 2004), indicating possible redundant function.
Experiments in cultured cells suggested that vertebrate NLGs regulate synapse formation during development (Chih et al., 2005; Fu and Vicini, 2009; Levinson et al., 2005; Prange et al., 2004; Scheiffele et al., 2000), but analysis of triple knockout mice revealed that NLGs function in vivo to regulate synapse maturation (Varoqueaux et al., 2006). Mutations in human nlg genes that decreased NLG levels are associated with rare cases of autism (Jamain et al., 2003; Laumonnier et al., 2004; Zhang et al., 2009) and alterations of NLG levels correlate with perturbations of synaptic function and behavioral phenotypes in mouse models (Dahlhaus et al., 2009; Hines et al., 2008; Kolozsi et al., 2009). Thus, analysis of the mechanisms of NLG signaling and regulation is an essential goal toward understanding the biology of disease and possible modes of therapeutic intervention.
The Drosophila larval neuromuscular junction (NMJ) is an experimental model to study synapse formation and function (Collins and DiAntonio, 2007) and the mechanisms of synaptic plasticity (Griffith and Budnik, 2006). Previous analysis suggested that Drosophila neuroligin1 (dnlg1) (Banovic et al., 2010) and Drosophila neuroligin2 (dnlg2) (Sun et al., 2011), signal in concert with Drosophila neurexin1 (dnrx1) (Chen et al., 2010; Li et al., 2007; Sun et al., 2009) to regulate synapse development. At the larval NMJ, trans-synaptic signaling by dnrx1 in motor neurons and dnlg1 in muscle contributes in part to the regulation of synaptic growth and maturation of glutamatergic synapses (Banovic et al., 2010) but the precise role(s) of dnlg1 in regulating neurotransmitter release and glutamate receptor (GluR) function are unknown. Further, although increasing NLG/NRX signaling affects synapse size (Banovic et al., 2010; Li et al., 2007), the effect of dnlg1 overexpression on synaptic transmission has not been described. Finally, because mechanisms that underlie NLG regulation in any organism are unknown, we sought to identify signaling pathways that regulate dnlg1 expression. In this report, we examined the effects of loss-of-function and overexpression of dnlg1 on synaptic growth and function and identify signaling pathways that alter DNlg1 expression levels. In the absence of dnlg1 function, the number of synaptic boutons was reduced, with a concomitant decrease in evoked release, but the amplitude of postsynaptic GluR responses to spontaneous release was increased. Overexpression of dnlg1 in muscle increased synapse size but attenuated evoked release and decreased the amplitude of postsynaptic GluRs responses to single vesicles. Dnrx1 mutants suppressed synaptic overgrowth and reduced synaptic transmission caused by overexpression of dnlg1, but had no effect on the postsynaptic response to spontaneous release. These data suggest that dnlg1 modulates synaptic transmission by negatively regulating GluR function, and regulates growth and neurotransmitter release via a retrograde mechanism with dnrx1. We found that synapse overgrowth or increased neural activity, and perturbation of PI3K signaling reduced dnlg1 expression, suggesting that attenuation of dnlg1 signaling may contribute to mechanisms of synaptic plasticity.
Materials and Methods
Drosophila stocks and genetics
dnlg1Δ46 deletion mutant was generated following FLP- mediated recombination between the FRT sites of the Exelixis pBAC insertions f00735 and f00713 as described (Thibault et al., 2004) and verified by PCR. Wildtype control strain (w;cs) was a gift of Josh Dubnau (Cold Spring Harbor Laboratories, CSH, NY). The larval muscle drivers, BG487-Gal4 and C57-Gal4 were a gift of Vivian Budnik and are described (Budnik et al., 1996). Overgrowth mutants used in this study: hiwΔN, hiwnd8 and eag1 sh14 were gifts of Aaron DiAntonio (Washington University, St. Louis, MO) and Barry Ganetzky (University of Wisconsin, Madison, WI) respectively. dnrx1273 was a gift of Manzoor Bhat (University of North Carolina, Chapel Hill, NC). The UAS-AP-1 strain (w;UAS-dfos;UAS-djun) was a gift of Subhabrata Sanyal (Emory University, Atlanta, GA). All other strains are described in the Bloomington Stock Center.
Molecular Biology and Gene Rescue
P[acman, dnlg1+] 72A15 was obtained from BACPAC Resources (clone CH321-72A15). DNA was purified following CsCl gradient centrifugation and verified by end sequencing as described (Venken et al., 2009). UAS-dnlg1 transgene was generated by PCR amplification of dnlg1 open reading frame from cDNA clone RE29404 (Stapleton et al., 2002; Tweedie et al., 2009) obtained from the Drosophila Genomics Resource Center, directionally cloned into the Not1 and Asp718 cloning site of pUAST (Brand and Perrimon, 1993) and verified by sequencing. Transgenic flies containing UAS-dnlg1 were generated using standard P-element procedures (Rubin and Spradling, 1982) and two homozygous viable insertions on the first and second chromosomes were used in this study. Transgenic strains targeting P[acman, dnlg1+] 72A15 to PBac{y[+]-attP}VK00037 were generated using phiC31 integrase expressed in the germline as described (Bischof et al., 2007). Two independently derived transgenic strains of P[acman, dnlg1+] 72A15 were used and gave identical results. DNA injections and isolation of transgenic lines were performed by Genetic Services, Inc. (Sudbury, MA).
Antibody production, immunocytochemistry and Western blotting
To generate a polyclonal antibody, DNA corresponding to the complete cytoplasmic domain (AA 855–1354) of dnlg1 was directionally cloned into the BsrG1 and HindIII sites of pET45b and expressed in E.coli using the Novagen pET system (EMD4Biosciences, USA). Soluble His-tagged DNlg1-cyto protein was purified from bacterial extracts using Ni-Agarose chromatography following standard procedures, dialyzed against 1X PBS, and used as antigen to immunize guinea pigs (Openbiosystems/Thermoscientific, USA).
For immunocytochemistry, larvae were dissected in 1X PBS and fixed in 1X PBS, 4% paraformaldehyde (20 min.) or Bouin’s fixative (5 min.), washed in PBS and blocked in 1X PBS, 0.1% Triton X-100, 3% normal goat sera. After antibody incubations and washes, larval fillets were mounted in Vectashield (Vector Laboratories, Burlingame, CA). NMJs of muscle 6/7, segment A3 were imaged with a Leica SP1 or Nikon C1 confocal microscope. For each experiment, image settings were optimized for wildtype specimens and not changed during subsequent imaging of experimental genotypes. Csp puncta were counted from projections of image stacks that were printed and quantified manually. Muscle size was determined using NIH ImageJ software from single confocal sections. Primary antibody dilutions used were guinea pig anti-dnlg1 (1/500), mouse anti-csp (1/50, clone 6D6, Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA), rabbit anti-DPak (1/2000, Nicholas Harden, Simon Frasier University, Vancouver, BC), rabbit anti-GluRIII (1/5000, Aaron DiAntonio, Washington University, St. Louis, MO). Dylight568 Anti-HRP (1/100) and secondary antibodies anti mouse, rabbit and guinea pig IgG conjugated with Dylight488 or Dylight549 (1/200) were obtained from Jackson ImmunoResearch (West Grove, PA).
Protein extracts of larval body wall were prepared by homogenizing tissue in RIPA buffer supplemented with protease inhibitors (Roche, USA). Western blotting was performed using standard procedures. To detect dnlg1 protein, primary antibody was diluted 1/1000 and visualized with HRP-conjugated secondary antibody (Jackson ImmunoResearch) and Pierce ECL reagent (Thermoscientific, Rockford, IL). To verify loading amounts, blots were stripped and re-probed with anti-α tubulin (1/2000, clone 12G10, Developmental Studies Hybridoma Bank).
Quantitative PCR
Total RNA was prepared from larval body walls using the RNAeasy Fibrous Tissue Mini Kit (Qiagen, USA) and used as a template for cDNA synthesis with oligo dT or random primers. Dnlg1 levels were determined using the StepOnePlus Real-Time PCR system (ABI, Carlsbad, CA) and normalized to dRas1 levels. The sequence of the dnlg1 primers used is 5′-GGAATTGGCGTTTATCTCG-3′ forward and 5′-CCAAGGCATGTGAATTGATG-3′ reverse. DRas primers were 5′-CTGCTGTCGAGGAAGGAAAA-3′ forward and 5′-AGTCTTCAATGGTGGGATCG- 3′ reverse.
Electrophysiology
Neuromuscular recordings were performed on muscle 6 from abdominal segment 3 using two-electrode voltage clamp (Sandstrom, 2004). Recordings were performed using with thin-walled glass electrodes in HL3.1 Saline (Feng et al., 2004) with 0.3 mM Ca2+ except as indicated in the text. Synaptic currents were amplified using an AxoClamp 2B and data were recorded with pClamp 9 software. EJCs were evoked via shock to the cut end of the nerve while muscle 6 was held at −70 mV. To avoid potential confusion from short-term synaptic plasticity, only the first EJC in a series was used for analysis.
To analyze mEJCs, 100 sec. of data were recorded from preparations that had not been subjected to nerve shock. mEJCs were measured using MiniAnalysis software (Synaptosoft, Ft. Lee, NJ), and amplitude data for each preparation were plotted and fitted with a Gaussian distribution to determine the mean (Origin, OriginLab, Northhampton MA; Sandstrom, 2004). To examine temporal parameters, mEJCs in each preparation were binned according to size, averaged, and fitted to measure the stated parameter using MiniAnalysis. All other data were analyzed and plotted with Sigmaplot 11.0 (Systat Software, San Jose, CA).
Statistical Analysis
Numbers in text are given as mean ± standard error of the mean (SEM). Differences between means were compared using one-way ANOVA, followed by appropriate post-hoc tests. In cases of multiple comparisons, the α level was adjusted appropriately to avoid type I errors. The Kruskal-Wallis test was used for datasets that did not satisfy the criteria for parametric statistics, as measured by the Levene test. Ca2+ vs. EJC amplitude data were fitted with the Hill equation in Origin, with the data weighted instrumentally by standard errors; all parameters were allowed to vary. Cumulative probability histograms were fitted with 3-parameter logistic equations in SigmaPlot. Because the data were normalized, the maximum value was fixed at 1.0, while other parameters were allowed to vary. Distributions were compared using the two-sample Kolmogorov-Smirnov (K-S) test (Sokal and Rohlf, 1995).
Results
dnlg1 mutants have fewer, but normal release sitess
The synaptic terminal at the Drosophila NMJ comprises dozens of varicosities, or boutons, each of which contains multiple synaptic sites where the neurotransmitter glutamate is released presynaptically and GluRs are clustered postsynaptically. Dnlg1 function is required for synaptic growth and maturation at the larval NMJ because bouton number and evoked neurotransmitter release are reduced in mutants (Banovic et al., 2010), but it is not known whether the synaptic sites that remain function normally.
Using an independently generated deletion mutant, dnlg1Δ46, that removes the transcription start site and most of the extracellular domain of the protein (Fig. 1A; see Methods for details), we confirmed that dnlg1 mutants have smaller, weaker synapses (Figs 1B –1F). The molecular analysis (Fig. 1A), absence of DNlg1 staining at synapses and on Western blots (see below), and the fact that dnlg1 synaptic phenotype was not changed when combined with a deficiency for the region (Df(3R)ED5223, hereafter referred to as Df) indicates that the deletion is a null allele. In dnlg1Δ46/Df mutants, the number of synaptic boutons stained with Cysteine String Protein (Csp) was reduced 50% (Figure 1C, E) as compared to w; cs control (Fig. 1B, E). The amplitudes of EJCs evoked by nerve shock were significantly reduced in dnlg1Δ46 homozygotes and dnlg1Δ46/Df compared to wildtype (Fig. 1F; p < 0.05; Kruskal-Wallis). Importantly, a single copy of a genomic transgene (P[acman, dnlg1+] 72A15, hereafter called P[acman dnlg1+]), rescued the number of boutons (Figure 1D, E) and size of evoked synaptic currents (Fig. 1F) in P[acman, dnlg1+]/+; dnlg1Δ46/Df larvae.
Figure 1.

Reduced but normal synaptic transmission in dnlg1 mutants is due to fewer release sites. A) Molecular map of dnlg1 locus and genetic reagents used in this study. The genes encoding dnlg1 (CG31146) and dnlg3 (CG34127) are adjacent on Chromosome 3, and are transcribed in opposite directions. The intron/exon structure of the dnlg1 gene has been described (FLYBASE; see Methods for reference). Deletion of the region between PBac{wh}f00735 and PBac{wh}f00713 removes ~29 KB of DNA that includes the transcription start site and most of the extracellular domain of the protein. The extent of the genomic rescue transgene, P[acman, dnlg1+]72A15 is indicated by the grey bar at the bottom. (B–D) Confocal projections of NMJs of larval muscle 6 and 7 stained with anti-cysteine string protein (CSP) to visualize synaptic boutons. B) Wild-type (w;cs) terminal. C) dnlg1Δ46/ Df (“dnlg1−“) mutant terminal, with fewer boutons. D) P[acman, dnlg1+]: dnlg1Δ46/ Df terminal, showing rescue of synapse size by a single copy of a genomic transgene. E) Quantification of bouton number. Bouton number is reduced significantly in dnlg1Δ46/Df, and is returned to normal by P[acman, dnlg1+]. Number indicates individual NMJs analyzed. Asterisk indicates significant difference from the wildtype. F) Average EJC amplitude is significantly reduced in dnlg1Δ46 homozygotes and dnlg1Δ46/Df, and rescued by genomic transgene. Insets show representative traces of wild-type, dnlg1Δ46 homozygote and dnlg1Δ46/ Df EJCs. G) mEJC distribution is altered in dnlg1 mutants, with more events above and below the modal (peak) value of approximately 0.7 nA. Inset shows examples of mEJCs in wildtype and mutant larvae. Scale bar, 1 nA, 500 ms. H) Average quantal content, calculated by dividing average EJC amplitude by average mEJC amplitude. Standard errors were calculated from the sums of squares of the errors of EJC and mEJC amplitudes (Meyer, 1975). Quantal content is reduced drastically in the mutants and rescued by a single copy of P[acman dnlg1+]. I) Effect of external Ca2+ concentration on EJC amplitude in wildtype and dnlg1 mutants. Data were fitted with the Hill equation (see Methods). The slope and midpoint for the relationship between Ca2+ and EJC amplitude were not different between dnlg1Δ46/ Df and the wildtype. The maximum value of synaptic current (Imax) is significantly lower for the mutant, suggesting fewer available release sites. J) Paired-pulse facilitation, an indicator of vesicle release probability. dnlg1Δ46/ Df (grey bars) does not differ significantly from the wildtype (w1118, black bars), at either 0.2 mM or 0.3 mM Ca2+. The insets above the bars at 0.3 mM Ca2+ show representative traces at 0.3 mM Ca2+, illustrating the size disparity between the genotypes.
GluR clusters in dnlg1 mutants are larger and more intense than those of the wildtype (supplementary Fig. 1; Banovic et al., 2010), suggesting that postsynaptic GluR function may be compromised. Indeed, amplitude distributions of miniature EJCs (mEJCs), which reflect the responses of GluRs to single vesicles, were abnormal in dnlg1 mutants (Fig. 1G). Wild-type mEJC amplitudes followed a roughly Gaussian distribution with a peak at approximately 0.7 nA and a slight rightward tail (Fig. 1G, filled region). mEJC amplitude distributions in dnlg1 mutants were significantly broader, with a higher proportion of events that were both larger and smaller than the mean (Fig. 1G, unfilled region; P < 0.001, Kolmogorov-Smirnov). The additional scatter is also seen as increased coefficients of variation for the mutants (w = 0.43 ± 0.011, dnlg1Δ46/Df = 0.50 ± 0.014, p < 0.01 one-way ANOVA). Therefore, altered GluR staining in dnlg1 mutants was paralleled by changes in GluR responsiveness.
Despite changing their distribution, loss of dnlg1 did not affect the mean amplitude of mEJCs (w1118 = 0.78 ± 0.03, dnlg1Δ46 = 0.87 ± 0.07, dnlg1Δ46/Df = 0.75 ± 0.05; Kruskal-Wallis p > 0.1; see also Banovic et al, 2010). Therefore, the primary effect of dnlg1 mutants was to reduce quantal content, the number of vesicles released per action potential, by more than 60% (Fig. 1H).
The reduction of quantal content could result from inhibiting the vesicle fusion machinery, diminishing Ca2+ sensitivity of fusion, or by lowering the number of functional release sites. To distinguish between these possibilities, we examined the effects of external Ca2+ concentration and repetitive activity on glutamate release (Fig. 1I). The slope of the relationship between external Ca2+ and EJC amplitude was close to the expected value of 4 (Dodge and Rahamimoff, 1967) in both genotypes (w1118 = 4.61 ± 0.98, dnlg1Δ46/Df = 3.77 ± 1.05), and release saturated at approximately 1 mM (Fig. 1I). The concentration of Ca2+ at half-maximal release (EC50) did not differ between mutants and wildtype (w1118 = 0.424 ± 0.089 mM, dnlg1Δ46/Df = 0.516 ± 0.140 mM). The similar slopes and EC50s for mutant and wildtype suggest that the Ca2+ sensitivity of vesicle fusion is unaltered in dnlg1 mutants. The primary difference between mutant and wildtype was the size of the synaptic current at maximal release (Imax; w1118, 86.1 ± 28.4 nA/nF; dnlg1Δ46/Df, 35.4 ± 15.1 nA/nF). Because average mEJC size, and therefore quantal size, is not altered in the mutants, the reduction of Imax is best explained by a reduction in the maximum number of functional release sites.
Paired-pulse facilitation (PPF) provides another measure of release probability, in that the amplitude ratio of two closely spaced EJCs (EJC2/EJC1) increases as release probability decreases (Zucker and Regehr, 2002). EJCs of wild-type and mutant synapses facilitated by approximately 1.5 fold in 0.2 mM Ca2+, while showing no facilitation in 0.3 mM Ca2+ (Fig. 1J). The paired-pulse ratio did not differ significantly between mutant and wildtype at either Ca2+ concentration (p> 0.10, one-way ANOVA), despite the differences in initial amplitudes (inset; see also Fig. 1F), suggesting that dnlg1 mutants have normal probability of release. Therefore, the anatomical and physiological data converge on the conclusion that dnlg1 function is required to determine the correct number of synapses at the NMJ, rather than regulating the efficacy or Ca2+ sensitivity of exocytosis.
Postsynaptic overexpression of dnlg1 causes synaptic overgrowth but reduces synaptic efficacy
We isolated a DNlg1-specific antiserum raised against the cytoplasmic domain of the protein and localized DNlg1 to the postsynaptic compartment of the NMJ (Fig. 2A). DNlg1 exhibited a punctate pattern beneath all synaptic boutons and was co-localized with the postsynaptic marker DPAK (Fig. 2A). Immunoreactivity was not detected in the mutant (Fig. 2B), demonstrating the specificity of the antiserum. Furthermore, inducing expression of dnlg1 in larval muscle 6 and 7 with the BG487-gal4 driver produced a dramatic increase in synaptic staining in both mutant (Fig. 2D) and wild-type backgrounds (data not shown)
Figure 2.

DNlg1 is postsynaptic and regulates growth and synaptic function. (AD) Confocal projections of NMJs from wildtype and dnlg1− larval muscle 6 and 7 segment A3, imaged at 63X. Insets show single confocal sections at 100X. A) DNlg1 (green) co-localized with postsynaptic DPAK (red). B) Absence of DNlg1 (green) in dnlg1Δ46/ Df NMJ co-stained with anti-DPAK (red). Scale bar, 25 μm main images, 5 μm insets. C, D) DNlg1 levels are dramatically increased in BG487>dnlg1.2; dnlg1− NMJs compared to control (wt). Because of the high signal intensity in BG487>dnlg1.2; dnlg1− NMJs (D), the gain of the detector was decreased to prevent saturation. As a consequence, DNlg1 expression is not apparent in the wild-type sample (C). Neuronal membranes are marked with anti-HRP to show the presynaptic terminal. E) Overexpression of dnlg1 in larval muscle significantly increased bouton number in mutant and wildtype (asterisks). F) overexpression of dnlg1 in larval muscle attenuates synaptic transmission by a dominant mechanism. EJCs remained small in BG487-Gal4/UAS-dnlg1-2; dnlg1− mutant larva despite restoration of dnlg1 expression in muscle and increased synapse size. Evoked EJCs were significantly reduced in wildtype NMJs overexpressing dnlg1 in larval muscle (BG487-Gal4/UAS-dnlg1-2;+) despite the increase in synapse bouton number. Asterisks indicate significant difference from the wild-type control, UAS-dnlg1-2; +. G. Effects of dnlg1 transgenes on mEJC distribution in dnlg1Δ46 homozygotes. A genomic transgene, P[acman, dnlg1+] (blue) rescues amplitude distribution to the level of wild-type control (UAS-nlg1-1, black) compared to dnlgD46 mutants (UAS-nlg1-1; dnlgD46, red). Muscle-specific overexpression has the opposite effect of the mutant, shifting the distributions to the left in both mutant (BG487-Gal4/UAS- dnlg1-1;dnlg1Δ46, green) and wild-type backgrounds (BG487/UAS-dnlg1;+, cyan). H. Amplitude distribution is narrowed and shifted to the left in BG487-Gal4/UAS- dnlg1-1;dnlg1Δ46 (black line) compared to the wildtype (grey region), although the modal value remains the same at 0.65 nA. I. Overexpression of dnlg1 in muscle reduces paired-pulse facilitation significantly (asterisk).
Driving expression of wild-type dnlg1 in the muscles of dnlg1 mutants using BG487-GAL4 increased bouton number above wildtype levels by greater than 40% (Fig. 2E; P < 0.05, Kruskal-Wallis). Likewise, BG487-gal4 driven overexpression of dnlg1 in a wild-type background, increased bouton number by a similar extent (Fig. 2E). Thus, postsynaptic expression of dnlg1 is sufficient to rescue the synaptic growth defects of dnlg1 mutants while overexpression had a dominant effect on synapse size, increasing bouton numbers above wildtype. This contrasts with the results of a previous study (Banovic et al., 2010), in which expression of a dnlg1-GFP fusion transgene in muscle using a different Gal4 driver (mef2-Gal4) inhibited synapse growth in a wild-type background.
Unlike bouton number, EJC amplitude in mutants was not restored by postsynaptic expression of dnlg1, remaining unchanged in BG487-Gal4/UAS-dnlg1-1; dnlg1Δ46/Df larva compared to negative controls (dnlg1-1; dnlg1Δ46/Df and BG487-Gal4; dnlg1Δ46/Df; Figure 2F). Neither of two UAS-dnlg1 transgenes driven in muscle rescued EJC amplitude in dnlg1Δ46or dnlg1Δ46/Df mutants, with amplitude remaining significantly smaller than the positive control (UAS-dnlg1-1, which behaved identically to wildtype; Fig. 2F). Importantly, overexpression of dnlg1 in the muscles of wild-type larvae reduced EJC amplitude to mutant levels (Fig. 2F), indicating a dominant inhibition of synaptic transmission. In all cases, when stimulus strength was varied, two units were clearly visible (data not shown), indicating that reduced EJC amplitude was not due to elevated threshold or conduction block in one of the motor neurons.
The decrease in EJC amplitude caused by dnlg1 overexpression was partly explained by reduced mEJC amplitude (Figs. 2G and H). Although the genomic transgene completely rescued mEJC amplitude (Fig. 2G; P > 0.05, K-S; P[acman dnlg1+]; dnlgΔ46, n = 1013 events), driving dnlg1 in either wildtype or mutant backgrounds with BG487-Gal4 shifted the distribution significantly to the left of the wildtype (Figure 2G; p < 0.001 Kolmogorov-Smirnov, UAS-dnlg1-1;+, n = 1611 events, BG487-Gal4/UAS-dnlg1-1; dnlg1Δ46, n = 548 events; BG487-Gal4/UAS-dnlg1-1; +, n = 964 events). This resulted in a narrow, Gaussian distribution with a single peak overlapping that of the wildtype (Fig. 2H), essentially the opposite of loss of function (Fig. 1G). Because of the reduced number of large events, average mEJC amplitude was reduced by ~25% (UAS-dnlg1-1 = 0.82 ± 0.03 nA vs UAS-dnlg1-1; BG487-Gal4; dnlg1Δ46/Df = 0.62 ± 0.02 nA; p < 0.05). The mEJC data therefore suggest that overexpression of dnlg1 alters postsynaptic sensitivity by reducing glutamate receptors cluster size and/or density, consistent with models of NLGs organizing postsynaptic proteins (Irie et al., 1997).
Whereas dnlg1 overexpression reduced mEJC amplitude by 25%, EJC amplitude was inhibited by >60% (Fig. 2F). Therefore, glutamate release must also be inhibited, either via a reduction in the number of presynaptic release sites or their efficacy. The number of anatomical release sites was not reduced, in that there were more synaptic boutons (Fig. 2E), each of which had normal numbers of puncta stained with the active zone marker Bruchpilot (Supplementary Fig. 1; w1118 = 7.9 ± 1.1 vs BG487/UAS-dnlg1-1 = 7.5 ± 0.96 puncta/bouton; p > 0.25). However, paired-pulse facilitation was significantly reduced (Fig. 2I; P < 0.05, one-way ANOVA), indicating that a greater than normal proportion of available release sites were active during the first pulse. Therefore, rather than reducing the efficacy of all synapses in the terminal, dnlg1 overexpression rendered a subpopulation of active zones unavailable for release under our recording conditions.
In summary, both GluR sensitivity and vesicle release are reduced when dnlg1 is overexpressed in the muscle. The mEJC phenotype is essentially the opposite of that caused by loss of dnlg1 function, and, combined with the effects of dnlg1 mutations on GluR staining, indicates that DNlg1 contributes to postsynaptic function by regulating GluR clustering and organization. Inhibition of release by dnlg1 overexpression further suggests that the muscle signals to the presynaptic terminal to reduce the number of active release sites.
Dnrx1 is required for some, but not all dnlg1 functions
Dnlg1 overexpression in muscle causes synaptic overgrowth and inhibition of glutamate release (Fig. 2), both of which require coordination of pre- and postsynaptic molecular pathways. Because Drosophila Neurexin1 (DNrx1) has been proposed to mediate trans-synaptic signaling via DNlgs (Li et al., 2007; Sun et al., 2009), we examined how DNrx1 signaling contributes to the synaptic phenotypes associated with DNlg1 overexpression.
We found that synaptic overgrowth caused by dnlg1 overexpression was suppressed by dnrx loss-of-function (Fig. 3A). Bouton number was significantly reduced in dnrx mutants overexpressing dnlg1 in larval muscle (BG487-Gal4/UAS-dnlg1.1; dnrx1273/Df; p < 0.05, one-way ANOVA) as compared to heterozygous dnrx1 controls (BG487-Gal4/UAS-dnlg1.1; dnrx1273/+). In fact, overexpression of dnlg1 in the dnrx1 mutant background did not increase bouton number above that of the negative control (BG487-gal4/+; dnrx1273/Df), indicating that dnrx1 is epistatic to, and therefore downstream of, dnlg1 for this phenotype.
Figure 3.
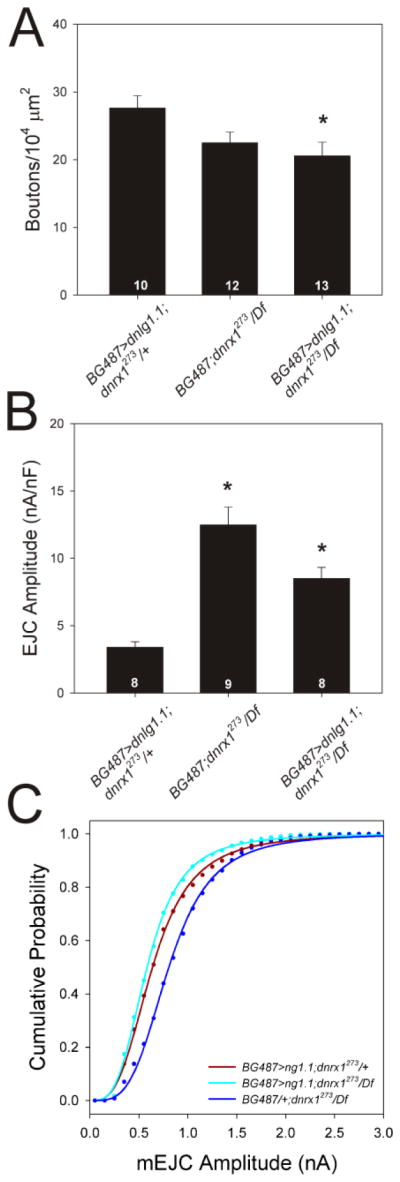
dnrx1 mediates some effects of dnlg1 overexpression. A. dnlg1-dependent overgrowth is blocked in dnrx1 mutants. Asterisk indicates that bouton number is significantly reduced when dnlg1 is overexpressed in dnrx1 mutants (BG487>ng1.1; dnrx1273/Df) compared to dnrx1 heterozygotes (BG487>ng1.1; dnrx1273/+). There is no significant difference between dnrx1 mutants overexpressing dnlg1 and those carrying only the gal4 driver. B. Inhibition of evoked release by dnlg1 depends on dnrx1. Overexpression of dnlg1 in muscles strongly reduces EJC size in dnrx1 heterozygotes, an effect that is significantly reduce in dnrx1 homozygotes, which do not differ from dnrx1 mutants carrying only the gal4 transgene. Asterisks indicate significant difference from BG487>ng1.1; dnrx1273/+. C. Muscle overexpression of dnlg1 reduces mEJC amplitude equally in dnrx1 mutants and heterozygotes (cyan and dark red lines, respectively), compared to dnrx1 mutant controls (dark blue line), indicating that dnrx1 function is not required for this phenotype.
Likewise, the inhibitory effects of dnlg1 overexpression on evoked synaptic transmission were suppressed in dnrx1 mutants (Fig. 3B). EJC amplitude was significantly restored in dnrx1 mutants overexpressing dnlg1 in larval muscle (BG487-Gal4/UAS-dnlg1.1; dnrx1273/Df; p < 0.05, Kruskal-Wallis) compared to positive control drnx1 heterozygotes (BG487-Gal4/UAS-dnlg1.1; dnrx1273/+). As with bouton number, EJC amplitude did not differ significantly between the experimental genotype and the negative control (BG487-gal4/+; dnrx1273/Df; p > 0.05; Kruskal-Wallis). Therefore, the effects of dnlg1 overexpression on growth and synaptic transmission require inter-cellular communication through dnrx1.
GluR cluster size and mean mEJC amplitudes are increased in dnrx1 loss-of-function mutants (Li et al., 2007), resembling in part the dnlg1 mutant phenotype (Fig. 1), suggesting that trans-synaptic signaling mediates these postsynaptic defects. However, unlike the effects on synaptic growth and EJC amplitude, alterations of mEJC amplitude distributions resulting from dnlg1 overexpression did not depend on dnrx1 function (Fig. 3C). BG487-driven overexpression of dnlg1 in dnrx1 heterozygous and dnrx1 homozygous mutant backgrounds exhibited a similar leftward shift of mEJC amplitudes (Fig. 3C; p < 0.001, Kolmogorov-Smirnov; BG487-gal4/UAS-dnlg1.1; dnrx1273/+, n = 1876 events, BG487-gal4/UAS-dnlg1.1; dnrx1273/Df(3R)Exel6191, n = 1411 events) compared to negative controls (BG487-gal4/+; dnrx1273/Df(3R)Exel6191, n = 3122 events). Our data suggest that retrograde non-cell autonomous signaling by DNlg1 stimulate growth and inhibit glutamate release in the presynaptic neuron via DNrx1, and that additional cell autonomous signaling by DNlg1 in the muscle regulates GluR responsiveness.
Alteration of PI3K signaling in neurons or muscle reduces dnlg1 expression
Activation of tyrosine kinase receptors by growth factors such as insulin regulates cell growth and organism size by signaling through the PI3K/AKT pathway and the activation of mTOR kinase (Hay and Sonenberg, 2004). Signaling through this pathway in vertebrate neurons plays an important role in multiple forms of synaptic plasticity through a mechanism that increases translation (Wang et al., 2010), but the molecular targets that mediate changes in synaptic strength and function have not been identified. In Drosophila, increased PI3K signaling is sufficient to drive the formation of additional functional synapses in larval and adult neurons (Martin-Pena et al., 2006) while decreased PI3K signaling reduces synapse number and suppresses activity dependent synaptic overgrowth (Howlett et al., 2008). In addition, PI3K negatively regulates synaptic activity (Howlett et al., 2008) as constitutive activation of PI3K signaling in motor neurons increased synapse size but reduced EJC amplitude, a phenocopy of dnlg1 overexpression in larval muscle. To address the potential linkage between PI3K and dnlg1, we examined DNlg1 expression in NMJs in which PI3K signaling was altered in motor neurons or larval muscle. Synapse overgrowth triggered by expression of a constitutively active form of the catalytic subunit, PI3KCAAX in motor neurons correlated with reduced DNlg1 levels in D42>PI3KCAAX larvae (Figure 4B, 4G) compared to wildtype controls (Fig. 4A, 4F) implying that synaptic overgrowth or decreased synaptic transmission mediated by elevated PI3K signaling down-regulates dnlg1 expression.
Figure 4.

Phospho-Inositol-3 Kinase (PI3K) signaling in motor neurons and muscle regulates dnlg1 expression. (A–E) Confocal projections of synapses, labeled with anti-DNlg1 (green) and anti-HRP (red). (F – J) DNLg1 (single channel) only. (A–C, F–H) Increased or decreased presynaptic PI3K signaling down regulates dnlg1 expression. Synaptic overgrowth triggered by increased PI3K signaling reduced DNlg1 levels in NMJs of D42>PI3KCAAX (B, G) compared to control (A, F). Attenuation of PI3K signaling in motor neurons also reduced DNlg1 levels in NMJs of D42>PI3KDN (C, H) compared to controls (A, F). PI3K signaling in muscle is required for normal dnlg1 expression. Postsynaptic attenuation of PI3K signaling reduced DNlg1 levels in NMJs of C57> PI3KDN (E,J) compared to controls (D, I). (K) Western blot of DNlg1 protein in larval body wall extracts from experimental and control genotypes used for imaging. Dnlg1 antibody detects a polypeptide of ~150 KD (arrow) in the wildtype that is absent in mutants and reduced by genetic alterations of PI3K signaling in motor neurons and muscle (K). (L, M) Attenuation of PI3K signaling in muscle reduced the number of synaptic boutons. Confocal projections of larval NMJs labeled with anti-CSP. L) Wild-type control. M) C57> PI3KDN. (N) C57>PI3KDN reduces bouton number significantly (asterisk) (O) Average EJC amplitude is significantly increased in C57>PI3KDN compared to control.
Surprisingly, attenuation of presynaptic PI3K signaling also caused down-regulation of dnlg1 expression. Decreased synaptic growth and increased neuronal excitability triggered by expression of the dominant negative PI3KDN in motor neurons correlated with reduced DNlg1 levels in D42>PI3KDN larvae (Fig. 4C, 4H) suggesting that reduced PI3K signaling negatively regulates dnlg1 expression either directly or via activity-dependent mechanisms. The same genetic alterations of PI3K signaling in motor neurons reduced total levels of DNlg1 relative to controls on Western blots (Fig. 4K) confirming the results obtained by imaging studies. Thus, presynaptic PI3K functions in motor neurons to modulate DNlg1 levels in postsynaptic muscle through a trans-synaptic signaling pathway that regulates synapse size and strength in response to changes in activity or growth.
To determine if cell autonomous signaling by PI3K plays a role in dnlg1 regulation, we altered PI3K signaling in larval muscle using the C57-Gal4 driver. Increased postsynaptic PI3K signaling had no effect on DNlg1 levels in C57>PI3K or C57> PI3KCAAX larvae (data not shown). In contrast, DNlg1 levels were reduced in NMJs of C57>PI3KDN larvae (Fig. 4E, J) in separate experiments using two independent UAS-PI3KDN transgenic lines, as were total DNlg1 levels compared to control on Western blots (Fig. 4K). Thus, PI3K signals to positively regulate dnlg1 expression in muscle and is required for wildtype DNlg1 levels at the synapse.
Attenuation of PI3K signaling in larval muscle and reduced levels of DNlg1 correlated with decreased bouton number and muscle size. Adjusting for muscle size, bouton number was significantly reduced by ~20% in C57> PI3KDN larvae (Figs. 4M, N) compared to wildtype (Figs. 4L, N). In C57>PI3KDN larvae, muscle capacitance was reduced compared to wildtype, consistent with reduced muscle size (wt = 2.9 ± 0.35 nF, C57>PI3KDN = 1.6 + 0.27 nF, one-way ANOVA, p < 0.05). Curiously, the amplitudes of EJCs from C57>PI3KDN terminals were significantly increased (Fig. 4O, one-way ANOVA, p < 0.05) although the frequency (wt = 1.91 + 0.34, C57>PI3KDN = 2.29 + 0.32; p > 0.05, one-way ANOVA), mean amplitude, and distribution of mEJCs were normal (P > 0.05 Kolmogorov-Smirnov). Thus, reduced postsynaptic PI3K signaling and lower levels of DNlg1 correlated with reduced synapse size and increased quantal content without affecting quantal size. These observations suggest that PI3K signaling in muscle regulates synapse size and strength and contributes to dnlg1 regulation.
Additional presynaptic signaling pathways down-regulate dnlg1 expression during overgrowth
Neuronal activity and ubiquitin dependent proteolysis contribute to presynaptic signaling mechanisms that regulate synaptic growth. To address possible linkage of these pathways to dnlg1, we measured DNlg1 levels in synaptic overgrowth mutants caused by increased neuronal excitability or the absence of the E3 ubiquitin ligase, highwire (hiw).
The ether-a-go-go, and Shaker (eag Sh) double mutant is a genetic model for the study of activity dependent synaptic plasticity. In these mutants, chronic neuronal hyperexcitability results in synaptic overgrowth (Budnik et al., 1990) due to changes in gene expression mediated by the AP-1 transcription factor (Sanyal et al., 2002). We examined the effects of increased signaling through this pathway on dnlg1 expression in NMJs from eag sh mutants or following neuronal overexpression of AP-1. DNlg1 levels were significantly decreased in NMJs from eag1 Sh14 (Fig. 5B, G) and in elav>AP1 larvae (Fig. 5E, J), suggesting that synaptic overgrowth or increased activity signal to down-regulate dnlg1 expression. Decreased DNlg1 expression in eag Sh mutants was confirmed in Western blots of body wall extracts (Fig. 5K). In wildtype extracts, DNlg1 antiserum detects a polypeptide with the expected molecular weight of 150 KD that was absent in dnlg1 mutants and reduced in eag1 Sh14 mutants. The reduction of DNlg1 levels in eag Sh mutants was likely due to decreased dnlg1 transcription or RNA stability, because QPCR measurements of dnlg1 mRNA revealed a two-fold reduction in mutant as compared to wildtype (Fig. 5L).
Figure 5.

Presynaptic signaling acts non-cell autonomously to down-regulate DNlg1 levels during overgrowth. A–E) Confocal projections of NMJs from synapse overgrowth genotypes and controls labeled with anti-DNlg1 (green) and anti-HRP (red). DNlg1 staining (single channel) is shown in panels F – J. Activity-dependent signaling down regulates dnlg1 expression. Synaptic DNlg1 levels are reduced in eag1Sh14 (G) compared to wild-type controls (F), and following neural overexpression of AP-1 in elav-Gal4; UAS-AP1 NMJs (J) compared to elav-Gal4 controls (I). hiw signaling is required for normal levels of dnlg1 expression. DNlg1 levels are reduced at hiwΔN mutant synapses (H) compared to controls (F). K) Western blot of larval body wall extracts from wildtype, dnlg1− and two overgrowth mutants probed with anti-DNlg1 antisera. A polypeptide of molecular weight 150kD (arrow) is detected in wildtype that is absent in dnlg1Δ46/ Df and reduced in eag1 Sh14 and hiwΔN mutants. L) dnlg1 RNA levels measured by Q-PCR are reduced in body walls from eag1 Sh14 as compared to wild-type controls.
Mutants lacking the E3 ubiquitin ligase, hiw exhibit dramatic synaptic overgrowth, but surprisingly have reduced quantal content (Wan et al., 2000), a phenocopy of BG487-gal4 driven overexpression of dnlg1. We found that Dnlg1 levels were reduced at synapses of hiwΔN (Fig. 5C, H) and hiwnd8 mutants (data not shown), and on Western blots of hiwΔN larval body wall extracts (Figure 5K). These observations implicate hiw as a component of a presynaptic signaling pathway that regulates dnlg1 expression by an anterograde mechanism. Thus, synaptic overgrowth triggered by three different signals in motor neurons (activity, the absence of hiw, and constitutive PI3K) results in a dramatic reduction of dnlg1 expression.
Our data demonstrate that signals that engage the pathways of synapse growth and plasticity always cause the down-regulation of dnlg1 expression. Thus, a reduction of DNlg1 levels appears to be a molecular signature of pathway output and an indication that attenuation of dnlg1 signaling has a functional role. Alternatively, dnlg1 down-regulation during overgrowth may represent a homeostatic response to dampen retrograde signaling that promotes synaptic growth.
Discussion
To gain insight into the role of NLGs in synapse maturation and function, we determined how loss-of-function and overexpression of dnlg1 affected growth and synaptic transmission (Fig. 6A), and identified signaling pathways that regulate dnlg1 expression (Fig. 6B). Based on loss-of-function and overexpression experiments, DNlg1 signals via a retrograde mechanism to regulate synapse size and function in a DNrx1-dependent manner (Fig. 6A). Loss-of-function reduced the number of boutons with a concomitant reduction in the number of functional release sites. Conversely, overexpression drove synaptic growth, but attenuated evoked glutamate release, signaling to the presynaptic neuron via DNrx1. DNlg1 also regulates GluR responsiveness release through a DNrx1-independent pathway. Therefore, in the proposed model, DNlg1 mediates bi-directional signaling across the synapse with DNrx1 to regulate synapse growth and function (Fig 6B).
Figure 6.

A) Model of the proposed function of dnlg1 in the regulation of synaptic growth and activity in the neuromuscular junction based on the analysis of loss of function and gain of function mutants. Overexpression of dnlg1 causes increased growth of the synapse (indicated by arrow), and attenuation of neurotransmitter release (indicated by bar) via retrograde signaling that requires dnrx1 function. Dnlg1 overexpression attenuates GluR function and the response to spontaneous release in muscle (indicated by bar), through a pathway that is independent of dnrx1 function. Loss of dnlg1 function had the opposite effect on synapse size, but did not facilitate synaptic transmission. B. Regulation of dnlg1 expression by signaling pathways in motor neurons and muscle. Presynaptic signals that increase synaptic growth reduce DNlg1 levels (indicated by bar) and implicate activity, ubiquitin dependent proteolysis and PI3K signaling in the regulation of dnlg1 expression. At present, it is unclear whether dnlg1 down-regulation is required for synaptic growth, or is part of a more complex homeostatic pathway. Postsynaptic PI3K signaling modulates synaptic growth and function and is also implicated in dnlg1 regulation. In this model, multiple signaling pathways converge on dnlg1 to contribute to the regulation of synaptic growth and function through non-cell autonomous and cell autonomous mechanisms.
dnlg1 expression is modulated by multiple anterograde signals from presynaptic motor neurons and cell autonomous signaling in muscle (Figure 6B). Synaptic overgrowth triggered by constitutive PI3K signaling in motor neurons, neuronal hyperactivity or the absence of hiw correlated with reduced DNlg1 levels at the NMJ. Likewise, increased synaptic strength and reduced synapse size associated with attenuation of PI3K signaling in the motor neurons or in muscle also correlated with lowered DNlg1 levels. Thus, downregulation of dnlg1 expression appears to be a common output of signaling pathways implicated in synaptic plasticity and suggests that attenuation of dnlg1 signaling contributes to mechanisms that coordinate growth with synaptic activity. While it is difficult to reconcile these observations in a simple model, these data suggest that reduced signaling by dnlg1 contributes to plasticity, but is unlikely the sole determinant of the polarity of the response. Reduced levels of DNlg1 may be a prerequisite for assembly or disassembly of synapses, but other factors (perhaps additional cell adhesion molecules) likely determine whether these changes result in an increase or a decrease in synaptic strength.
Dnlg1 regulates synaptic function but is not essential for synaptic transmission
Previous analysis (Banovic et al., 2010) suggested that dnlg1 did not directly regulate synaptic function, because the reduction of synaptic strength in mutants was a consequence of a defect in synaptic growth. We confirmed that evoked release in dnlg1 mutants generated smaller synaptic currents and find, in addition, that mutant synapses respond normally to changes in extracellular Ca2+ concentration, and have normal release probability. However, our findings that dnlg1 overexpression attenuates release and that DNlg1 levels determine the postsynaptic response to quantal release suggest a more nuanced view.
In dnlg1 loss-of-function mutants, distribution of mEJCs in the mutant was shifted significantly to the right, indicating an increased number of larger responses. Postsynaptic overexpression of dnlg1 had a reciprocal effect, increasing the number of smaller mEJCs. Although changes in vesicle size or alteration in glutamate transport by vesicular ATPases can affect mEJC distributions (Daniels et al., 2004; Karunanithi et al., 2002), ultrastructural analysis of dnlg1 mutant synapses did not reveal defects in synapse vesicle size (Banovic et al., 2010). Instead, the altered GluRIII staining in the mutants and the apparent muscle autonomy of the overexpression phenotype indicate that dnlg1 negatively regulates the clustering or turnover, and possibly the sensitivity of GluRs.
Banovic et al, 2010 demonstrated that dnlg1 is necessary for synaptic growth, and the effects of dnlg1 overexpression presented here show that dnlg1 signaling is sufficient for the formation of nascent synapses in the NMJ. Thus, dnlg1 has a unique function in the regulation of synapse growth and differs from the mammalian neuroligins, which are not required for synapse formation in vivo. However, our data also show that dnlg1 signaling modulates postsynaptic function and can attenuate neurotransmitter release, suggesting a determinative regulatory role in synapse maturation. Vertebrate neuroligins have been proposed to drive synaptic maturation through the recruitment of scaffolding molecules or other PDZ domain binding proteins to the postsynaptic complex (Sudhof, 2008), and in the case of nlg1, promote synaptic GluR localization (Heine et al., 2008; Mondin et al., 2011). The accumulation of synaptic GluRs in dnlg1 mutants and the inhibitory effects of dnlg1 overexpression on synaptic transmission suggest that dnlg1 may inhibit GluR function by antagonizing signals the promote GluR synaptic localization or organization.
Overexpression of dnlg1 in muscle results in overgrowth but attenuates synaptic function
The effect of muscle overexpression of dnlg1 on synaptic growth and neurotransmitter release suggests a retrograde signaling mechanism. Overexpression of wild-type dnlg1 in larval muscle using the BG487-Gal4 driver increased synaptic bouton number but reduced quantal content, and both effects were suppressed by mutation in dnrx1. Thus, increased synaptic growth and inhibition of neurotransmitter release resulting from retrograde DNlg1 signaling is mediated by DNrx1. Consistent with this hypothesis, overexpression of dnrx1 in motor neurons increased synapse bouton number but reduced quantal content (Li et al., 2007), a phenocopy of dnlg1 overexpression in muscle. Furthermore, dnrx1 mutants suppressed the synaptic growth defects of dnlg1-gfp overexpression driven by mef2-gal4 (Banovic et al., 2010).
Increased retrograde signaling by DNlg1/DNrx1 promotes growth, but the nascent synapses have reduced function not accounted for by defects in apposition of pre and postsynaptic components or by a reduction in the number of active zones. Overexpression of dnlg1 likely attenuates presynaptic function by increasing synaptic DNrx1 levels in the terminal. Banovic et al., 2010 showed that dnrx1 is required to maintain DNlg1 levels at the synapse, suggesting that trans-synaptic complex formation stabilizes both binding partners. Because NRXs function to recruit molecules implicated in exocytosis (Atasoy et al., 2007; Biederer and Sudhof, 2000; Hata et al., 1996; Ho et al., 2003), increased signaling may have deleterious effects on presynaptic function, by altering subunit stoichiometry of components of the release machinery. Alternatively, overexpression of DNlg1 may preclude binding of additional DNrx1 ligands important for presynaptic function. Three additional neuroligins encoded by the fly genome are potential DNrx1 binding partners including DNlg2 which binds DNrx1 in vitro and is expressed both in muscle and motor neurons (Sun et al., 2011), DNlg3 which is expressed in all neurons (B. Mozer, unpublished observations), and DNlg4.
Presynaptic regulation of dnlg1 expression during overgrowth
Constitutive activation of PI3K signaling in motor neurons (Howlett et al., 2008), mutations in hiw, a neuronal E3 ubiquitin ligase (Collins et al., 2006; Wan et al., 2000; Wu et al., 2005), and shaggy, which encodes the Drosophila homolog of GSK3-B (Franciscovich et al., 2008; Franco et al., 2004) cause an increase in synapse bouton numbers and reduce quantal content. Because of the shared phenotype and site of action in motor neurons PI3K, hiw and shaggy may signal through a common pathway and epistasis experiments suggest that hiw and shaggy act upstream of AP1 to regulate synaptic growth (Franciscovich et al., 2008). We show that dnlg1 overexpression in muscle increased synapse bouton numbers and reduced quantal content. This observation suggests that retrograde signaling by dnlg1 drives growth and attenuates neurotransmitter release by inhibition of hiw or shaggy or the activation of PI3K signaling in motor neurons.
At the Drosophila NMJ, neuronal hyperexcitability increases synaptic growth and function through alterations of cAMP levels and changes of gene expression mediated by transcription factors including AP1 and CREB, the cyclic AMP responsive element binding protein (Budnik et al., 1990; Davis et al., 1996; Sanyal et al., 2002; Zhong et al., 1992). We find that DNlg1 expression is reduced during synapse overgrowth in eag Sh mutants or following neural overexpression of AP1 suggesting that dnlg1 is negatively regulated by activity. Similarly, (Schuster et al., 1996) showed that reduced expression of the homophilic cell adhesion molecular fasciclinII (fasII) is necessary for synaptic overgrowth in eag Sh mutants. Whether down-regulation of dnlg1 is required for synaptic overgrowth or represents a homeostatic response to pro-growth signaling remain unanswered questions. In eag Sh mutants, dnlg1 RNA is substantially reduced, suggesting that the postsynaptic response to overgrowth involves changes in gene expression and not only protein turnover. Future experiments will be needed to determine if transcriptional repression or alterations of mRNA splicing or turnover account for the decreased dnlg1 RNA levels. Furthermore, the fact that dnlg1 expression induces growth under some conditions (the present paper) and inhibits growth in others (Banovic et al., 2010) suggests that spatial and/or temporal regulation of dnlg1 is likely to be complex.
Regulation of dnlg1 expression by PI3K signaling
In vertebrates, PI3K signaling has been implicated in mechanisms that regulate synaptic function and plasticity (Arendt et al., 2009; Cuesto et al., 2011; Kim et al., 2011; Man et al., 2003). PI3K signaling activates mTOR kinase and has been postulated to increase translation of synaptic proteins, but the down stream effectors that mediate synaptic changes have not been identified. In the fly NMJ, presynaptic PI3K signaling regulates synaptic growth (Martin-Pena et al., 2006) and dampens synaptic activity mediated by presynaptic metabotropic glutamate receptors (Howlett et al., 2008). Alterations of synapse size and function mediated by reduced PI3K signaling in neurons results in changes in adult Drosophila behavior affecting odor perception (Acebes et al., 2011) and sensitivity to alcohol (Eddison et al., 2011). We found that increasing PI3K signaling in the presynaptic neuron, or inhibiting it in either the neuron or muscle, caused dramatic reduction of dnlg1 expression at the Drosophila NMJ. Whether reduced levels of DNlg1 are required to mediate PI3K dependent alterations of synaptic size and strength, and the nature of the anterograde signal to the muscle, remain to be determined. These observations suggest that neuroligins are candidate downstream effectors of PI3K signaling pathways and that reduced dnlg1 signaling contributes to PI3K dependent synaptic changes and altered behavior.
A current model of autism proposes that increased translation of synaptic proteins due to elevated PI3K/TOR signaling underlies synaptic dysfunction and the behavioral phenotypes associated with the disease (Stern, 2011). Elevated PI3K signaling has been implicated in autism associated with certain neurodevelopmental syndromes (Bourgeron, 2009; Kwon et al., 2006) including fragile X (Gross et al., 2010; Sharma et al., 2010). In this context, constitutive activation of presynaptic PI3K signaling in the Drosophila NMJ, is a simple model of synaptic function of the autistic brain. Proteins whose levels increase in response to elevated PI3K signaling likely contribute to pathogenic mechanisms of synaptic dysfunction, so the downregulation of dnlg1 we observed may not be relevant to disease. However, expression of Nlg1 was reduced in the hippocampus of fmr1 mutant mouse brain (Dahlhaus and El-Husseini, 2009) suggesting that downregulation of nlg expression in response to increased PI3K signaling occurs in vertebrate synapses in a mouse model of autism. Our study provides evidence of linkage between PI3K/mTOR and nlg signaling at the synapse and warrants further investigation of the role of neuroligins in PI3K-dependent signaling processes relevant to neurodevelopmental diseases.
Supplementary Material
Supplementary Figure 1. Localization of active zones and glutamate receptors in dnlg1 loss- and gain-of-function synapses. A–C) Terminals stained with anti-Bruchpilot (BRP, red) to visualize active zones, and GluRIII (green) to show postsynaptic glutamate receptor clusters. D–F) Show only the GluRIII staining. Scale bar = 5 μ.
Acknowledgments
The work was supported by the intramural research program of the National Institute of Heart, Lung and Blood (B.A.M.) and the National Institute of Mental Health (D.J.S.). We thank Marshall Nirenberg for his early support of this work, Shu-Hwa Yu for assistance with immuno-staining experiments, Devera Schoenberg (DIR, NINDS) for proofreading of the manuscript, the NHLBI Intramural Light Microscope Core Facility, and Jun Zhu and Ting Ni for help with qPCR. We thank Howard Nash, NIMH, for support and critical reading of the manuscript, Benjamin White, NIMH, for critical reading of the manuscript and the use of equipment, and our colleagues at the NIH and elsewhere that provided equipment, reagents and fly strains.
Footnotes
Conflict of interest: none
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Brian A. Mozer, Laboratory of Biochemical Genetics, National Institutes of Heart, Lung and Blood, NIH, Bethesda MD 20892
David J. Sandstrom, Laboratory of Molecular Biology, National Institutes of Mental Health, NIH, Bethesda MD 20892
References
- Acebes A, Martin-Pena A, Chevalier V, Ferrus A. Synapse loss in olfactory local interneurons modifies perception. J Neurosci. 2011;31:2734–2745. doi: 10.1523/JNEUROSCI.5046-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendt KL, Royo M, Fernandez-Monreal M, Knafo S, Petrok CN, Martens JR, Esteban JA. PIP3 controls synaptic function by maintaining AMPA receptor clustering at the postsynaptic membrane. Nat Neurosci. 2009;13:36–44. doi: 10.1038/nn.2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atasoy D, Schoch S, Ho A, Nadasy KA, Liu X, Zhang W, Mukherjee K, Nosyreva ED, Fernandez-Chacon R, Missler M, Kavalali ET, Sudhof TC. Deletion of CASK in mice is lethal and impairs synaptic function. Proc Natl Acad Sci U S A. 2007;104:2525–2530. doi: 10.1073/pnas.0611003104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banovic D, Khorramshahi O, Owald D, Wichmann C, Riedt T, Fouquet W, Tian R, Sigrist SJ, Aberle H. Drosophila neuroligin 1 promotes growth and postsynaptic differentiation at glutamatergic neuromuscular junctions. Neuron. 2010;66:724–738. doi: 10.1016/j.neuron.2010.05.020. [DOI] [PubMed] [Google Scholar]
- Biederer T, Sudhof TC. Mints as adaptors. Direct binding to neurexins and recruitment of munc18. J Biol Chem. 2000;275:39803–39806. doi: 10.1074/jbc.C000656200. [DOI] [PubMed] [Google Scholar]
- Bischof J, Maeda RK, Hediger M, Karch F, Basler K. An optimized transgenesis system for Drosophila using germ-line-specific phiC31 integrases. Proc Natl Acad Sci U S A. 2007;104:3312–3317. doi: 10.1073/pnas.0611511104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgeron T. A synaptic trek to autism. Curr Opin Neurobiol. 2009;19:231–234. doi: 10.1016/j.conb.2009.06.003. [DOI] [PubMed] [Google Scholar]
- Budnik V, Koh YH, Guan B, Hartmann B, Hough C, Woods D, Gorczyca M. Regulation of synapse structure and function by the Drosophila tumor suppressor gene dlg. Neuron. 1996;17:627–640. doi: 10.1016/s0896-6273(00)80196-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budnik V, Zhong Y, Wu CF. Morphological plasticity of motor axons in Drosophila mutants with altered excitability. J Neurosci. 1990;10:3754–3768. doi: 10.1523/JNEUROSCI.10-11-03754.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budreck EC, Scheiffele P. Neuroligin-3 is a neuronal adhesion protein at GABAergic and glutamatergic synapses. Eur J Neurosci. 2007;26:1738–1748. doi: 10.1111/j.1460-9568.2007.05842.x. [DOI] [PubMed] [Google Scholar]
- Chen K, Gracheva EO, Yu SC, Sheng Q, Richmond J, Featherstone DE. Neurexin in embryonic Drosophila neuromuscular junctions. PLoS One. 2010;5:e11115. doi: 10.1371/journal.pone.0011115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chih B, Engelman H, Scheiffele P. Control of excitatory and inhibitory synapse formation by neuroligins. Science. 2005;307:1324–1328. doi: 10.1126/science.1107470. [DOI] [PubMed] [Google Scholar]
- Collins CA, DiAntonio A. Synaptic development: insights from Drosophila. Curr Opin Neurobiol. 2007;17:35–42. doi: 10.1016/j.conb.2007.01.001. [DOI] [PubMed] [Google Scholar]
- Collins CA, Wairkar YP, Johnson SL, DiAntonio A. Highwire restrains synaptic growth by attenuating a MAP kinase signal. Neuron. 2006;51:57–69. doi: 10.1016/j.neuron.2006.05.026. [DOI] [PubMed] [Google Scholar]
- Cuesto G, Enriquez-Barreto L, Carames C, Cantarero M, Gasull X, Sandi C, Ferrus A, Acebes A, Morales M. Phosphoinositide-3-kinase activation controls synaptogenesis and spinogenesis in hippocampal neurons. J Neurosci. 2011;31:2721–2733. doi: 10.1523/JNEUROSCI.4477-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlhaus R, El-Husseini A. Altered neuroligin expression is involved in social deficits in a mouse model of the fragile X syndrome. Behav Brain Res. 2009;208:96–105. doi: 10.1016/j.bbr.2009.11.019. [DOI] [PubMed] [Google Scholar]
- Dahlhaus R, Hines RM, Eadie BD, Kannangara TS, Hines DJ, Brown CE, Christie BR, El-Husseini A. Overexpression of the cell adhesion protein neuroligin-1 induces learning deficits and impairs synaptic plasticity by altering the ratio of excitation to inhibition in the hippocampus. Hippocampus. 2009;20:305–322. doi: 10.1002/hipo.20630. [DOI] [PubMed] [Google Scholar]
- Daniels RW, Collins CA, Gelfand MV, Dant J, Brooks ES, Krantz DE, DiAntonio A. Increased expression of the Drosophila vesicular glutamate transporter leads to excess glutamate release and a compensatory decrease in quantal content. J Neurosci. 2004;24:10466–10474. doi: 10.1523/JNEUROSCI.3001-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis GW, Schuster CM, Goodman CS. Genetic dissection of structural and functional components of synaptic plasticity. III. CREB is necessary for presynaptic functional plasticity. Neuron. 1996;17:669–679. doi: 10.1016/s0896-6273(00)80199-3. [DOI] [PubMed] [Google Scholar]
- Dean C, Scholl FG, Choih J, DeMaria S, Berger J, Isacoff E, Scheiffele P. Neurexin mediates the assembly of presynaptic terminals. Nat Neurosci. 2003;6:708–716. doi: 10.1038/nn1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodge FA, Jr, Rahamimoff R. Co-operative action a calcium ions in transmitter release at the neuromuscular junction. J Physiol. 1967;193:419–432. doi: 10.1113/jphysiol.1967.sp008367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddison M, Guarnieri DJ, Cheng L, Liu CH, Moffat KG, Davis G, Heberlein U. arouser reveals a role for synapse number in the regulation of ethanol sensitivity. Neuron. 2011;70:979–990. doi: 10.1016/j.neuron.2011.03.030. [DOI] [PubMed] [Google Scholar]
- Feng Y, Ueda A, Wu CF. A modified minimal hemolymph-like solution, HL3.1, for physiological recordings at the neuromuscular junctions of normal and mutant Drosophila larvae. J Neurogenet. 2004;18:377–402. doi: 10.1080/01677060490894522. [DOI] [PubMed] [Google Scholar]
- Franciscovich AL, Mortimer AD, Freeman AA, Gu J, Sanyal S. Overexpression screen in Drosophila identifies neuronal roles of GSK-3 beta/shaggy as a regulator of AP-1-dependent developmental plasticity. Genetics. 2008;180:2057–2071. doi: 10.1534/genetics.107.085555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco B, Bogdanik L, Bobinnec Y, Debec A, Bockaert J, Parmentier ML, Grau Y. Shaggy, the homolog of glycogen synthase kinase 3, controls neuromuscular junction growth in Drosophila. J Neurosci. 2004;24:6573–6577. doi: 10.1523/JNEUROSCI.1580-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Z, Vicini S. Neuroligin-2 accelerates GABAergic synapse maturation in cerebellar granule cells. Mol Cell Neurosci. 2009;42:45–55. doi: 10.1016/j.mcn.2009.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graf ER, Zhang X, Jin SX, Linhoff MW, Craig AM. Neurexins induce differentiation of GABA and glutamate postsynaptic specializations via neuroligins. Cell. 2004;119:1013–1026. doi: 10.1016/j.cell.2004.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith LC, Budnik V. Plasticity and second messengers during synapse development. Int Rev Neurobiol. 2006;75:237–265. doi: 10.1016/S0074-7742(06)75011-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross C, Nakamoto M, Yao X, Chan CB, Yim SY, Ye K, Warren ST, Bassell GJ. Excess phosphoinositide 3-kinase subunit synthesis and activity as a novel therapeutic target in fragile X syndrome. J Neurosci. 2010;30:10624–10638. doi: 10.1523/JNEUROSCI.0402-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hata Y, Butz S, Sudhof TC. CASK: a novel dlg/PSD95 homolog with an N-terminal calmodulin-dependent protein kinase domain identified by interaction with neurexins. J Neurosci. 1996;16:2488–2494. doi: 10.1523/JNEUROSCI.16-08-02488.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–1945. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- Heine M, Thoumine O, Mondin M, Tessier B, Giannone G, Choquet D. Activity-independent and subunit-specific recruitment of functional AMPA receptors at neurexin/neuroligin contacts. Proc Natl Acad Sci U S A. 2008;105:20947–20952. doi: 10.1073/pnas.0804007106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hines RM, Wu L, Hines DJ, Steenland H, Mansour S, Dahlhaus R, Singaraja RR, Cao X, Sammler E, Hormuzdi SG, Zhuo M, El-Husseini A. Synaptic imbalance, stereotypies, and impaired social interactions in mice with altered neuroligin 2 expression. J Neurosci. 2008;28:6055–6067. doi: 10.1523/JNEUROSCI.0032-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho A, Morishita W, Hammer RE, Malenka RC, Sudhof TC. A role for Mints in transmitter release: Mint 1 knockout mice exhibit impaired GABAergic synaptic transmission. Proc Natl Acad Sci U S A. 2003;100:1409–1414. doi: 10.1073/pnas.252774899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howlett E, Lin CC, Lavery W, Stern M. A PI3-kinase-mediated negative feedback regulates neuronal excitability. PLoS Genet. 2008;4:e1000277. doi: 10.1371/journal.pgen.1000277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichtchenko K, Hata Y, Nguyen T, Ullrich B, Missler M, Moomaw C, Sudhof TC. Neuroligin 1: a splice site-specific ligand for beta-neurexins. Cell. 1995;81:435–443. doi: 10.1016/0092-8674(95)90396-8. [DOI] [PubMed] [Google Scholar]
- Ichtchenko K, Nguyen T, Sudhof TC. Structures, alternative splicing, and neurexin binding of multiple neuroligins. J Biol Chem. 1996;271:2676–2682. doi: 10.1074/jbc.271.5.2676. [DOI] [PubMed] [Google Scholar]
- Irie M, Hata Y, Takeuchi M, Ichtchenko K, Toyoda A, Hirao K, Takai Y, Rosahl TW, Sudhof TC. Binding of neuroligins to PSD-95. Science. 1997;277:1511–1515. doi: 10.1126/science.277.5331.1511. [DOI] [PubMed] [Google Scholar]
- Jamain S, Quach H, Betancur C, Rastam M, Colineaux C, Gillberg IC, Soderstrom H, Giros B, Leboyer M, Gillberg C, Bourgeron T. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat Genet. 2003;34:27–29. doi: 10.1038/ng1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karunanithi S, Marin L, Wong K, Atwood HL. Quantal size and variation determined by vesicle size in normal and mutant Drosophila glutamatergic synapses. J Neurosci. 2002;22:10267–10276. doi: 10.1523/JNEUROSCI.22-23-10267.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JI, Lee HR, Sim SE, Baek J, Yu NK, Choi JH, Ko HG, Lee YS, Park SW, Kwak C, Ahn SJ, Choi SY, Kim H, Kim KH, Backx PH, Bradley CA, Kim E, Jang DJ, Lee K, Kim SJ, Zhuo M, Collingridge GL, Kaang BK. PI3Kgamma is required for NMDA receptor-dependent long-term depression and behavioral flexibility. Nat Neurosci. 2011;14:1447–1454. doi: 10.1038/nn.2937. [DOI] [PubMed] [Google Scholar]
- Kolozsi E, Mackenzie RN, Roullet FI, deCatanzaro D, Foster JA. Prenatal exposure to valproic acid leads to reduced expression of synaptic adhesion molecule neuroligin 3 in mice. Neuroscience. 2009;163:1201–1210. doi: 10.1016/j.neuroscience.2009.07.021. [DOI] [PubMed] [Google Scholar]
- Kwon CH, Luikart BW, Powell CM, Zhou J, Matheny SA, Zhang W, Li Y, Baker SJ, Parada LF. Pten regulates neuronal arborization and social interaction in mice. Neuron. 2006;50:377–388. doi: 10.1016/j.neuron.2006.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laumonnier F, Bonnet-Brilhault F, Gomot M, Blanc R, David A, Moizard MP, Raynaud M, Ronce N, Lemonnier E, Calvas P, Laudier B, Chelly J, Fryns JP, Ropers HH, Hamel BC, Andres C, Barthelemy C, Moraine C, Briault S. X-linked mental retardation and autism are associated with a mutation in the NLGN4 gene, a member of the neuroligin family. Am J Hum Genet. 2004;74:552–557. doi: 10.1086/382137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levinson JN, Chery N, Huang K, Wong TP, Gerrow K, Kang R, Prange O, Wang YT, El-Husseini A. Neuroligins mediate excitatory and inhibitory synapse formation: involvement of PSD-95 and neurexin-1beta in neuroligin-induced synaptic specificity. J Biol Chem. 2005;280:17312–17319. doi: 10.1074/jbc.M413812200. [DOI] [PubMed] [Google Scholar]
- Li J, Ashley J, Budnik V, Bhat MA. Crucial role of Drosophila neurexin in proper active zone apposition to postsynaptic densities, synaptic growth, and synaptic transmission. Neuron. 2007;55:741–755. doi: 10.1016/j.neuron.2007.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man HY, Wang Q, Lu WY, Ju W, Ahmadian G, Liu L, D’Souza S, Wong TP, Taghibiglou C, Lu J, Becker LE, Pei L, Liu F, Wymann MP, MacDonald JF, Wang YT. Activation of PI3-kinase is required for AMPA receptor insertion during LTP of mEPSCs in cultured hippocampal neurons. Neuron. 2003;38:611–624. doi: 10.1016/s0896-6273(03)00228-9. [DOI] [PubMed] [Google Scholar]
- Martin-Pena A, Acebes A, Rodriguez JR, Sorribes A, de Polavieja GG, Fernandez-Funez P, Ferrus A. Age-independent synaptogenesis by phosphoinositide 3 kinase. J Neurosci. 2006;26:10199–10208. doi: 10.1523/JNEUROSCI.1223-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer G, Varoqueaux F, Neeb A, Oschlies M, Brose N. The complexity of PDZ domain-mediated interactions at glutamatergic synapses: a case study on neuroligin. Neuropharmacology. 2004;47:724–733. doi: 10.1016/j.neuropharm.2004.06.023. [DOI] [PubMed] [Google Scholar]
- Meyer S. Data Analysis for Scientists and Engineers. Wiley; New York: 1975. [Google Scholar]
- Mondin M, Labrousse V, Hosy E, Heine M, Tessier B, Levet F, Poujol C, Blanchet C, Choquet D, Thoumine O. Neurexin-neuroligin adhesions capture surface-diffusing AMPA receptors through PSD-95 scaffolds. J Neurosci. 2011;31:13500–13515. doi: 10.1523/JNEUROSCI.6439-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulopoulos A, Aramuni G, Meyer G, Soykan T, Hoon M, Papadopoulos T, Zhang M, Paarmann I, Fuchs C, Harvey K, Jedlicka P, Schwarzacher SW, Betz H, Harvey RJ, Brose N, Zhang W, Varoqueaux F. Neuroligin 2 drives postsynaptic assembly at perisomatic inhibitory synapses through gephyrin and collybistin. Neuron. 2009;63:628–642. doi: 10.1016/j.neuron.2009.08.023. [DOI] [PubMed] [Google Scholar]
- Prange O, Wong TP, Gerrow K, Wang YT, El-Husseini A. A balance between excitatory and inhibitory synapses is controlled by PSD-95 and neuroligin. Proc Natl Acad Sci U S A. 2004;101:13915–13920. doi: 10.1073/pnas.0405939101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokal RR, Rohlf FJ. Biometry: the principles and practice of statistics in biological research. 3. W. H. Freeman and Co; New York: 1995. [Google Scholar]
- Rubin GM, Spradling AC. Genetic transformation of Drosophila with transposable element vectors. Science. 1982;218:348–353. doi: 10.1126/science.6289436. [DOI] [PubMed] [Google Scholar]
- Sandstrom DJ. Isoflurane depresses glutamate release by reducing neuronal excitability at the Drosophila neuromuscular junction. J Physiol. 2004;558:489–502. doi: 10.1113/jphysiol.2004.065748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanyal S, Sandstrom DJ, Hoeffer CA, Ramaswami M. AP-1 functions upstream of CREB to control synaptic plasticity in Drosophila. Nature. 2002;416:870–874. doi: 10.1038/416870a. [DOI] [PubMed] [Google Scholar]
- Scheiffele P, Fan J, Choih J, Fetter R, Serafini T. Neuroligin expressed in nonneuronal cells triggers presynaptic development in contacting axons. Cell. 2000;101:657–669. doi: 10.1016/s0092-8674(00)80877-6. [DOI] [PubMed] [Google Scholar]
- Schuster CM, Davis GW, Fetter RD, Goodman CS. Genetic dissection of structural and functional components of synaptic plasticity. II. Fasciclin II controls presynaptic structural plasticity. Neuron. 1996;17:655–667. doi: 10.1016/s0896-6273(00)80198-1. [DOI] [PubMed] [Google Scholar]
- Sharma A, Hoeffer CA, Takayasu Y, Miyawaki T, McBride SM, Klann E, Zukin RS. Dysregulation of mTOR signaling in fragile X syndrome. J Neurosci. 2010;30:694–702. doi: 10.1523/JNEUROSCI.3696-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song JY, Ichtchenko K, Sudhof TC, Brose N. Neuroligin 1 is a postsynaptic cell-adhesion molecule of excitatory synapses. Proc Natl Acad Sci U S A. 1999;96:1100–1105. doi: 10.1073/pnas.96.3.1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stapleton M, Liao G, Brokstein P, Hong L, Carninci P, Shiraki T, Hayashizaki Y, Champe M, Pacleb J, Wan K, Yu C, Carlson J, George R, Celniker S, Rubin GM. The Drosophila gene collection: identification of putative full-length cDNAs for 70% of D. melanogaster genes. Genome Res. 2002;12:1294–1300. doi: 10.1101/gr.269102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern M. Insulin signaling and autism. Front Endocrinol (Lausanne) 2011;2:54. doi: 10.3389/fendo.2011.00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof TC. Neuroligins and neurexins link synaptic function to cognitive disease. Nature. 2008;455:903–911. doi: 10.1038/nature07456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun M, Liu L, Zeng X, Xu M, Fang M, Xie W. Genetic interaction between Neurexin and CAKI/CMG is important for synaptic function in Drosophila neuromuscular junction. Neurosci Res. 2009;64:362–371. doi: 10.1016/j.neures.2009.04.009. [DOI] [PubMed] [Google Scholar]
- Sun M, Xing G, Yuan L, Gan G, Knight D, With SI, He C, Han J, Zeng X, Fang M, Boulianne GL, Xie W. Neuroligin 2 is required for synapse development and function at the Drosophila neuromuscular junction. J Neurosci. 2011;31:687–699. doi: 10.1523/JNEUROSCI.3854-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibault ST, Singer MA, Miyazaki WY, Milash B, Dompe NA, Singh CM, Buchholz R, Demsky M, Fawcett R, Francis-Lang HL, Ryner L, Cheung LM, Chong A, Erickson C, Fisher WW, Greer K, Hartouni SR, Howie E, Jakkula L, Joo D, Killpack K, Laufer A, Mazzotta J, Smith RD, Stevens LM, Stuber C, Tan LR, Ventura R, Woo A, Zakrajsek I, Zhao L, Chen F, Swimmer C, Kopczynski C, Duyk G, Winberg ML, Margolis J. A complementary transposon tool kit for Drosophila melanogaster using P and piggyBac. Nat Genet. 2004;36:283–287. doi: 10.1038/ng1314. [DOI] [PubMed] [Google Scholar]
- Tweedie S, Ashburner M, Falls K, Leyland P, McQuilton P, Marygold S, Millburn G, Osumi-Sutherland D, Schroeder A, Seal R, Zhang H. FlyBase: enhancing Drosophila Gene Ontology annotations. Nucleic Acids Res. 2009;37:D555–559. doi: 10.1093/nar/gkn788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varoqueaux F, Aramuni G, Rawson RL, Mohrmann R, Missler M, Gottmann K, Zhang W, Sudhof TC, Brose N. Neuroligins determine synapse maturation and function. Neuron. 2006;51:741–754. doi: 10.1016/j.neuron.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Varoqueaux F, Jamain S, Brose N. Neuroligin 2 is exclusively localized to inhibitory synapses. Eur J Cell Biol. 2004;83:449–456. doi: 10.1078/0171-9335-00410. [DOI] [PubMed] [Google Scholar]
- Venken KJ, Carlson JW, Schulze KL, Pan H, He Y, Spokony R, Wan KH, Koriabine M, de Jong PJ, White KP, Bellen HJ, Hoskins RA. Versatile P[acman] BAC libraries for transgenesis studies in Drosophila melanogaster. Nat Methods. 2009;6:431–434. doi: 10.1038/nmeth.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan HI, DiAntonio A, Fetter RD, Bergstrom K, Strauss R, Goodman CS. Highwire regulates synaptic growth in Drosophila. Neuron. 2000;26:313–329. doi: 10.1016/s0896-6273(00)81166-6. [DOI] [PubMed] [Google Scholar]
- Wang DO, Martin KC, Zukin RS. Spatially restricting gene expression by local translation at synapses. Trends Neurosci. 2010;33:173–182. doi: 10.1016/j.tins.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Wairkar YP, Collins CA, DiAntonio A. Highwire function at the Drosophila neuromuscular junction: spatial, structural, and temporal requirements. J Neurosci. 2005;25:9557–9566. doi: 10.1523/JNEUROSCI.2532-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Milunsky JM, Newton S, Ko J, Zhao G, Maher TA, Tager-Flusberg H, Bolliger MF, Carter AS, Boucard AA, Powell CM, Sudhof TC. A neuroligin-4 missense mutation associated with autism impairs neuroligin-4 folding and endoplasmic reticulum export. J Neurosci. 2009;29:10843–10854. doi: 10.1523/JNEUROSCI.1248-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Y, Budnik V, Wu CF. Synaptic plasticity in Drosophila memory and hyperexcitable mutants: role of cAMP cascade. J Neurosci. 1992;12:644–651. doi: 10.1523/JNEUROSCI.12-02-00644.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Localization of active zones and glutamate receptors in dnlg1 loss- and gain-of-function synapses. A–C) Terminals stained with anti-Bruchpilot (BRP, red) to visualize active zones, and GluRIII (green) to show postsynaptic glutamate receptor clusters. D–F) Show only the GluRIII staining. Scale bar = 5 μ.