

Abstract
Objectives
Antibiotic resistance in bacterial pathogens is a serious clinical problem. Novel targets are needed to combat increasing drug resistance in Escherichia coli. Our objective is to demonstrate that 2-(3,4-dimethoxyphenyl)-5-[5-(4-methylpiperazin-1-yl)-1H-benzimidazol-2yl]-1H-benzimidazole (DMA) inhibits E. coli DNA topoisomerase I more strongly than human topoisomerase I. In addition, DMA is non-toxic to mammalian cells at antibiotic dosage level.
Methods
In the present study, we have established DMA as an antibacterial compound by determining MICs, post-antibiotic effects (PAEs) and MBCs for different standard as well as clinical strains of E. coli. We have described the differential catalytic inhibitory mechanism of bis-benzimidazole, DMA, for human and E. coli topoisomerase I and topoisomerase II by performing different assays, including relaxation assays, cleavage–religation assays, DNA unwinding assays, ethidium bromide displacement assays, decatenation assays and DNA gyrase supercoiling assays.
Results
DMA significantly inhibited bacterial growth at a very low concentration, but did not affect human cell viability at higher concentrations. Activity assays showed that it preferentially targeted E. coli topoisomerase I over human topoisomerase I, topoisomerase II and gyrase. Cleavage–religation assays confirmed DMA as a poison inhibitor of E. coli topoisomerase I. This study illuminates new properties of DMA, which may be further modified to develop an efficient topoisomerase inhibitor that is selective towards bacterial topoisomerase I.
Conclusions
This is the first report of a bis-benzimidazole acting as an E. coli topoisomerase I inhibitor. DMA is a safe, non-cytotoxic molecule to human cells at concentrations that are needed for antibacterial activity.
Keywords: Hoechst 33342, DMA, MICs, MBCs, PAEs
Introduction
Urinary tract infection (UTI) represents one of the most common diseases encountered in medical practice today, and occurs in all age groups from neonates to the elderly.1,2 The incidence of UTI is higher in women than in men. E. coli is the most frequent urinary tract pathogen, causing around 59% of all UTIs, as it is present in the gastrointestinal tract and provides a pool for initiation of UTI. Frequent use of antibiotics for empirical therapy leads to the development of antibiotic resistance in uropathogens.3 Multidrug-resistant pathogens travel not only locally but globally, and newly introduced pathogens spread rapidly in susceptible hosts.4 Fluoroquinolones are the preferred initial agents for empirical therapy of UTI in areas where antibiotic resistance is of grave concern. The targets of fluoroquinolones and aminocoumarins are DNA gyrase or topoisomerase IV,4 with gyrase the preferred target in E. coli. Owing to an increase in antibiotic resistance, there is a requirement to explore new targets, one of which is DNA topoisomerase I.5
A large number of topoisomerase inhibitors are known to play a major role as anticancer, antiparasitic and antibacterial agents.6 DNA topoisomerase I is a ubiquitous enzyme responsible for the relaxation of DNA supercoils arising as a consequence of processes such as transcription and replication.7 Type I topoisomerases change the DNA linking number by transiently cutting a single DNA strand, whereas type II topoisomerases do so by transiently cutting both strands of duplex DNA. In general, the catalytic activity of type I topoisomerase may be divided into a series of steps, as follows: (i) non-covalent binding to DNA; (ii) cleavage of a single strand with generation of a covalent phosphotyrosyl bond and a free hydroxyl; (iii) passage of the intact strand through the nick; and (iv) religation.8 Type I topoisomerases fall into two categories (type IA and IB) depending on the polarity of their covalent attachment. Human topoisomerase I (HuTOPI) forms a 3′-phosphotyrosyl bond with Tyr-723 (type IB) and can relax both positively and negatively supercoiled DNA, whereas E. coli topoisomerase I (EcTOPI) forms a 5′-phosphotyrosyl bond with Tyr-319 (type IA) and can relax only negatively supercoiled DNA in the presence of magnesium ions.9 Topoisomerase I inhibition can be achieved either by stabilizing the enzyme–DNA complex or by preventing its formation.10–12 Recently, we have evaluated a new bis-benzimidazole, 2-(3,4-dimethoxyphenyl)-5-[5-(4-methylpiperazin-1-yl)-1H-benzimidazol-2yl]-1H-benzimidazole (DMA), a synthetic analogue of Hoechst 33342, which is less cytotoxic and does not affect cell viability in mammalian cells up to a concentration of 100 μM, but inhibits growth of E. coli cultures even at a concentration of 2 μM when grown in co-culture with human cells.13 We have synthesized several analogues of Hoechst 33342 with dichloro {DCA, 2-(2,4-dichlorophenyl)-5-[5-(4-methylpiperazin-1-yl)-1H-benzimidazol-2-yl]-1H-benzimidazole} and difluoro {DFA, 2-(2,4-difluorophenyl)-5-[5-(4-methylpiperazinyl)-1H-benzimidazol-2-yl]-1H-benzimidazole} substitutions on the phenyl ring and evaluated their activity against HuTOPI (Figure 1).14
Figure 1.
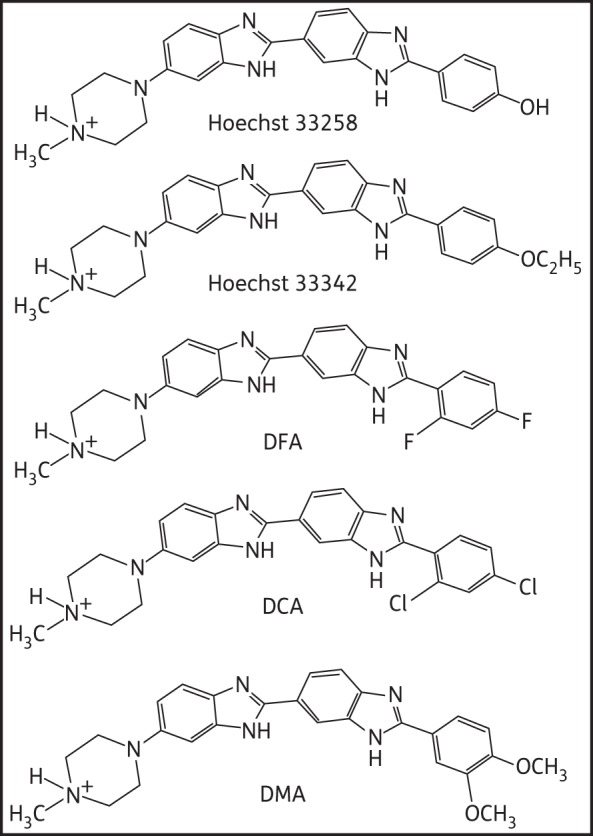
Chemical structure of bis-benzimidazole derivatives.
In the present study, we have evaluated DCA, DFA and DMA for their antibacterial activity against standard E. coli and resistant, clinical strains of E. coli isolated from UTI patients. The MIC and MBC study showed that DMA is more active against most of the resistant E. coli strains. Our study demonstrated that DMA can inhibit EcTOPI efficiently. In addition, DMA was found to increase the DNA cleavage product formed by EcTOPI, identifying DMA as a poison inhibitor of type IA topoisomerase. Our study confirmed that DMA has no significant inhibitory effect on HuTOPI and E. coli gyrase. Although DMA inhibited human topoisomerase II at higher concentrations, it did not act as a human topoisomerase II poison inhibitor. Hoechst 33342 was found to be mutagenic, clastogenic and cytotoxic to human cells, but DMA appeared to be a safer molecule, as it is non-mutagenic and non-cytotoxic to mammalian cells up to a concentration of 100 μM.15 These results establish DMA as a good antibacterial molecule selectively targeting EcTOPI over HuTOPI.
Materials and methods
Materials and enzymes
Camptothecin, etoposide and novobiocin were obtained from Sigma (St Louis, MO, USA). Camptothecin and etoposide were dissolved in DMSO, and novobiocin was dissolved in sterile water. Synthesis of the bis-benzimidazole derivatives DCA, DFA and DMA has been described previously.14 These derivatives were dissolved in methanol. Catenated kinetoplast DNA (kDNA) and pBAD/Thio and pHOT1 plasmid DNA were purchased from TopoGen Inc. (Port Orange, FL, USA). E. coli DNA gyrase and its relaxed substrate were purchased from New England Biolabs (GmBH, Germany).
Bacterial strains and growth of bacteria
ATCC 25922 strain and clinical strains of E. coli were used for the study. These strains were chosen as they are resistant to the commercial antibiotics. Bacteria were grown in cation-adjusted Mueller–Hinton (MH) broth. MICs (MIC50s) were determined in the same medium by the microdilution method according to the CLSI. All the MIC, post-antibiotic effect (PAE) and MBC experiments were done in triplicate and values shown are averages of three experiments (±SD).
Determination of MICs, MBCs and PAEs
The MIC50s of bis-benzimidazoles (DCA, DFA and DMA) were determined by standard microdilution procedures in 96-well plates with low evaporation lids (Falcon, Becton Dickinson) and bis-benzimidazoles were present at concentrations of 0.125, 0.25, 0.5, 1, 2, 4, 8, 16, 32, 64 and 128 μM. The MIC50 value was defined as the minimal concentration that inhibited growth to 50% at 24 h.
Cultures of E. coli were grown to early exponential phase (109 cells/mL) in MH broth in a Versa Max spectrophotometer with shaking for PAE determination. OD600 measurements were taken at 20 min intervals. The bacterial cultures were diluted to 2 × 105 cells/mL in MH broth, and 80 μL aliquots from this dilution were added to the wells of a low-attachment 96-well plate. Aliquots of 20 μL of each ligand (having five times the MIC value of the ligand) were added to each well of a 96-well plate. The 96-well plate was incubated for 60 min at 37°C as described above. Bacteria were harvested by centrifugation at 2300 g for 5 min, and cell pellets were resuspended in fresh pre-warmed MH broth (100 μL). The rinse cycle was repeated twice and plates were incubated for 24 h at 37°C. PAE duration was calculated by subtracting the time taken for the control culture to reach 50% of the OD600 from the time taken for antimicrobial-treated cultures to reach the same OD.16,17
MBCs were calculated using the literature method.18 The MBC for each strain was determined by pelleting the culture of the MIC50 value and three values beyond the MIC50 value. The pellet was resuspended in ligand-free MH broth and washed three times to remove the ligand. Then the ligand-free pellet was again resuspended in 100 μL of fresh MHB and plated on MH agar plates without ligand. MBC endpoints were read as the lowest dilution of ligand with no growth (>99.9% killing) after overnight incubation at 37°C using culture without ligand as a control.
Cloning, expression and purification of HuTOPI and EcTOPI
The recombinant HuTOPI gene was amplified from cDNA synthesized from the RNA of U87 cells and the EcTOPI gene was amplified from the genomic DNA of E. coli K-12 by PCR, as described previously.13 The amplified HuTOPI gene was inserted in-frame into the multiple cloning site of pGEX-4T-I between the BamHI and EcoRI sites. Similarly, the amplified EcTOPI gene was inserted in-frame into the multiple cloning site of pGEX-4T-I between the XhoI and NotI sites. This plasmid encodes the entire topoisomerase I protein with the addition of a glutathione-S-transferase at the N-terminal. The accuracy of cloning was confirmed by DNA sequencing. Recombinant proteins were expressed in E. coli DE3 and purified using glutathione Sepharose.13
Relaxation assay of EcTOPI
EcTOPI was diluted with a buffer of 10 mM Tris-HCl, pH 8.0, 50 mM NaCl, 0.1 mg/mL gelatin and 0.5 mM MgCl2. DMA was added to 10 ng of the diluted enzyme present in a volume of 10 μL, before addition of 10 μL of the same buffer containing 250 ng of supercoiled pBAD/Thio plasmid DNA purified by caesium chloride gradient centrifugation. The mixture was incubated at 37°C for 30 min before termination of the reaction and analysis by agarose gel electrophoresis as described previously.16 The ethidium bromide-stained gel was photographed over UV light for densitometry analysis. The percentage relaxation was determined by dividing the distance between the negatively supercoiled band (SC) and the weighted centre of the partially relaxed band (PR) by the distance between the supercoiled band (SC) and the fully relaxed band (FR) [i.e. (SC − PR)/(SC − FR)].17 Percentage inhibition by DMA was then calculated by subtracting percentage relaxation in the presence of DMA from 100% relaxation obtained with enzyme only.
Assay of DNA cleavage–religation equilibrium of EcTOPI
The effect of DMA on the DNA cleavage–religation equilibrium of EcTOPI was assayed using a 5′-32P-labelled 216 bp single-stranded DNA substrate generated by denaturation of a DNA PCR product prepared as described previously.18 The labelled DNA (50 ng) was incubated with 100 ng of EcTOPI in 5 μL of 10 mM Tris, pH 8.0, at 37°C for 30 min to allow DNA cleavage product to form. DMA was then added to each reaction and incubated for 2 min followed by addition of 2 mM MgCl2 to determine whether DMA can inhibit the shifting of the cleavage–religation equilibrium towards religation upon the addition of Mg2+. After further incubation at 37°C for 5 min, the reaction was terminated by the addition of 6.5 μL of sequencing gel loading buffer. The level of DNA cleavage products was analysed by electrophoresis in a 7% sequencing gel followed by Phosphor-Imager analysis of the dried gel.
Assay of DNA cleavage by HuTOPI
To observe the DNA cleavage product of HuTOPI, 5 U of enzyme was incubated with 300 ng of supercoiled pBAD/Thio plasmid DNA in 20 μL of the reaction buffer (10 mM Tris, pH 8.0, 150 mM NaCl, 0.1% BSA, 0.1 mM spermidine and 5% glycerol) at 37°C for 30 min before the addition of 2 μL of 10% SDS/20 mg/mL proteinase K. Incubation was continued at 37°C for 30 min before analysis by agarose gel electrophoresis in the presence of 0.5 μg/mL ethidium bromide.
DNA unwinding assay
The ability of ligand to bind to the minor groove of DNA was studied with the topoisomerase I unwinding assay. The unwinding assay was performed with 1 μg of pHOT1 plasmid DNA in the presence or absence of DMA, etoposide and adriamycin in 25 μL of reaction mixture. Relaxed DNA was prepared by treating the supercoiled plasmid DNA with an excess of HuTOPI, and purified by proteinase K digestion at 37°C, phenol/chloroform extraction and ethanol precipitation. After incubation at 37°C for 15 min, reactions were terminated by the addition of pre-warmed stop solution (5% SDS/0.25% bromophenol blue) and electrophoresed on a 1% agarose gel. The DNA was stained with 0.5 μg/mL ethidium bromide and visualized by UV light.
Ethidium bromide displacement assay
DNA intercalation by DMA, N-4-9-acridinylamino-2-methoxyphenyl-methanesulphonamide (o-AMSA) and N-4-9-acridinylamino-3-methoxyphenyl-methanesulphonamide (m-AMSA) was determined with an ethidium bromide displacement assay modified from published procedures.19 A 90 μL mixture of 28 mM MOPS, pH 6.5, 2.2 μM ethidium bromide and 130.5 ng of fish sperm DNA was added to each well of a 96-well microplate. Dilutions of DMA, m-AMSA or o-AMSA in water were then added in 10 μL aliquots. Fluorescence was measured on a Cary Eclipse Varian fluoresce spectrophotometer using excitation and emission wavelengths of 545 and 595 nm, respectively.
Assay for inhibition of DNA relaxation by human topoisomerase II α
DMA was added to 0.25 μg of supercoiled pBAD/Thio plasmid DNA before addition of 2 U of human topoisomerase II α enzyme (from Affymetrix) in a final volume of 20 μL of reaction buffer supplied by the manufacturer. The mixture was incubated at 37°C for 30 min before termination of the reaction and analysis by gel electrophoresis as described for EcTOPI relaxation activity.
Topoisomerase II α decatenation assay
Decatenation of kDNA was assayed by monitoring the appearance of decatenated DNA in the presence of ligand of different concentrations. One unit of human topoisomerase II α is defined as the amount of enzyme required to fully decatenate 0.1 μg of kDNA in 15 min at 37°C. Reactions were carried out in buffer of 30 mM Tris-HCl, pH 7.6, 3 mM ATP, 15 mM 2-mercaptoethanol, 8 mM MgCl2 and 60 mM NaCl in a final volume of 20 μL. Reactions were incubated with 1 U of enzyme in the presence or absence of the indicated inhibitor for 30 min at 37°C. The reactions were terminated with 2 μL of 10% SDS, followed by proteinase K treatment for 15 min at 37°C. After addition of 0.1 volume of loading dye (50% glycerol/0.025% bromophenol blue), samples were extracted once with an equal volume of chloroform/isoamyl alcohol (24 : 1). Following a brief centrifugation in a microfuge, the blue upper layer was loaded directly onto an agarose gel. The decatenated products were analysed by electrophoresis in 1% agarose gels with ethidium bromide (0.5 μg/mL) at 100 V.4,18 All the assays were conducted three times and IC50 values are shown as means ± SD.
Assay of DNA cleavage by human topoisomerase II
Five units of human topoisomerase II α enzyme was incubated with 0.3 μg of supercoiled pBAD/Thio plasmid DNA in 20 μL of the relaxation reaction buffer supplied by Affymetrix at 37°C for 20 min before the addition of 2 μL of 10% SDS/20 mg/mL proteinase K. The samples were incubated further at 37°C for 30 min before analysis by agarose gel electrophoresis in the presence of 0.5 μg/mL ethidium bromide.
E. coli DNA gyrase supercoiling assay
In a DNA supercoiling assay, 0.5 μg of relaxed plasmid DNA was incubated with 1 U of E. coli DNA gyrase at 37°C for 30 min in 25 μL reactions containing ligands at 1, 5, 10, 25, 50, 75 and 100 μM. Reactions were terminated by adding 10 mM EDTA, 0.5% SDS, 0.25 μg/mL bromophenol blue and 15% glycerol. Methanol concentrations in each reaction were maintained at 1% by the addition of serially diluted drug stocks so as to avoid solvent-mediated inhibition of gyrase.20 The gels were stained with 5 μg/mL ethidium bromide, destained in water and photographed under UV illumination using an AlphaImager 2200.
Statistical analysis
Results are given as means ± SD. An independent two-tailed Student's t-test was performed. Differences were considered statistically significant at P < 0.05.
Results
Effect of bis-benzimidazoles on growth of E. coli
We have previously reported that DMA, a novel bis-benzimidazole, shows inhibition of EcTOPI and a low MIC50 for E. coli K-12.12 The MIC50 value of DMA was 8 μM and a PAE of >24 h was obtained for E. coli K-12.12 These results have driven us to undertake a detailed systematic study of bis-benzimidazole ligands as antibacterial agents (Table 1). The lowest MIC50 value of DMA determined for clinical strains of E. coli was 3.74 mg/L (8 μM). Susceptibility of bacteria to different classes of antibiotics was observed in the range 0.125–128.0 μM, with varying MIC50s for different strains. Interestingly, the MIC50 values of DMA decreased from 3.74 to 3.06 mg/L and from 7.49 to 5.37 mg/L, respectively, when DH5α and BL21 E. coli strains were transformed with the EcTOPI expression clone, whereas transformation with the vector without a gene insert did not affect the MIC values for these two E. coli strains.
Table 1.
Susceptibilities of E. coli strains to bis-benzimidazoles
| DCA |
DFA |
DMA |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Strain description | MIC (μM)/mg/L) | PAE (h) | MBC (μM) | MIC (μM)/mg/L) | PAE (h) | MBC (μM) | MIC (μM)/mg/L) | PAE (h) | MBC (μM) |
| E. coli DH5α | 8.0 ± 0.017/3.8 | 22.2 ± 1.8 | 16.0 | 4.0 ± 0.007/1.78 | 23.6 ± 0.4 | 8.0 | 8.0 ± 0.012/3.74 | 20.8 ± 1.1 | 16.0 |
| E. coli BL21 | 16.0 ± 0.072/7.6 | 21.9 ± 2.1 | 32.0 | 8.0 ± 0.013/3.55 | 22.0 ± 2.0 | 16.0 | 16.0 ± 0.032/7.49 | 23.8 ± 0.2 | 32.0 |
| ATCC 25922 | 32.0 ± 0.066/15.2 | 24 ± 0.1 | 64.0 | 32.0 ± 0.031/14.21 | 21.2 ± 2.8 | 64.0 | 32 ± 0.054/14.98 | 22.3 ± 1.3 | 64.0 |
| E. coli 72R | ND | ND | ND | 32.0 ± 0.057/14.21 | ND | 64.0 | 8.0 ± 0.011/3.74 | ND | 16.0 |
| E. coli 118R | 32.0 ± 0.057/15.2 | >24 ± 0.2 | 64.0 | 32.0 ± 0.044/14.21 | >24 ± 0.1 | 64.0 | 8.0 ± 0.042/3.74 | 24 ± 0.9 | 16.0 |
| E. coli 85R | 64.0 ± 0.055/30.4 | ND | 128.0 | 32.0 ± 0.039/14.21 | 24 ± 0.2 | 64.0 | 8.0 ± 0.015/3.74 | 24 ± 0.7 | 16.0 |
| E. coli 360R | 8.0 ± 0.028/3.8 | 20.8 ± 1.3 | 16.0 | 8.0 ± 0.029/3.55 | 2.3 ± 0.6 | 16.0 | 8.0 ± 0.023/3.74 | 24 ± 0.1 | 16.0 |
| E. coli 451R | 128 ± 0.090/60.8 | ND | ND | 64.0 ± 0.077/28.43 | ND | 128.0 | 8.0 ± 0.003/3.74 | 22 ± 0.5 | 16.0 |
| E. coli 59R | 64.0 ± 0.064/30.4 | ND | 128.0 | 16.0 ± 0.054/7.1 | 22.2 ± 1.6 | 32.0 | 16.0 ± 0.072/7.49 | 1.6 ± 0.1 | 32.0 |
| E. coli 81R | 64.0 ± 0.069/30.4 | ND | 128.0 | 32.0 ± 0.030/14.21 | 20.3 ± 1.1 | 64.0 | 16.0 ± 0.031/7.49 | 23.6 ± 0.6 | 32.0 |
| E. coli 132R | 32.0 ± 0.042/15.2 | 10 ± 1.3 | 64.0 | 32.0 ± 0.047/14.21 | 0.9 ± 0.1 | 64.0 | 16.0 ± 0.046/7.49 | >24 ± 0.1 | 32.0 |
| E. coli 151R | 32.0 ± 0.037/15.2 | >24 ± 0.1 | 64.0 | 32.0 ± 0.041/14.21 | 15.6 ± 1.4 | 64.0 | 16.0 ± 0.033/7.49 | 21.6 ± 0.4 | 32.0 |
| E. coli 401R | 32.0 ± 0.029/15.2 | >24 ± 0.5 | 64.0 | 128.0 ± 0.063/56.86 | ND | ND | 32.0 ± 0.023/14.98 | 16 ± 1.5 | 64.0 |
| E. coli 555R | ND | ND | ND | ND | ND | ND | 32.0 ± 0.071/14.98 | 24 ± 2.2 | 64.0 |
AMP, ampicillin; CHL, chloramphenicol; CIP, ciprofloxacin; GEN, gentamicin; KAN, kanamycin; NAL, nalidixic acid; TMP, trimethoprim; TET, tetracycline; ND, not determined.
All the MIC, PAE and MBC experiments were done in triplicate and values shown are averages of three experiments (±SD).
E. coli DH5α, E. coli BL21 and E. coli ATCC 25922 were reference strains for E. coli susceptibility testing.
E. coli 72R, resistant to CIP; E. coli 118R, resistant to AMP, TMP and NAL; E. coli 85R, resistant to CHL, AMP, KAN, TMP, CIP and NAL; E. coli 360R, resistant to GEN, KAN, TET and NAL; E. coli 451R, resistant to AMP, TET and GEN; E. coli 59R, resistant to CHL, AMP, TMP, CIP and NAL; E. coli 81R, resistant to AMP, KAN, TMP, TET, CIP and NAL; E. coli 132R, resistant to CHL, AMP, KAN, TMP, CIP and NAL; E. coli 151R, resistant to CHL, AMP, KAN, TMP, CIP and NAL; E. coli 401R, resistant to AMP, CHL, CIP, GEN and TET; E. coli 555R, resistant to AMP, CIP, GEN and TET.
E. coli strains 72, 118, 151, 360, 451, 59, 81, 85, 132, 401 and 555, obtained from patients suffering from UTIs, were tested in the study and showed varying resistance patterns to antibiotics. All were found to be susceptible to benzimidazoles with longer durations of PAE (Table 1). It was observed that growth of the E. coli reference and clinical isolates was inhibited after a brief exposure of cultures to DMA at five times the MIC. All the E. coli strains tested were found to be susceptible to benzimidazoles with longer durations of PAE. The PAE of bisubstituted ligands was observed to be longer than the PAE of other known antibiotics (result not shown).
DMA showed the lowest MIC50/MBC and highest PAE for most of the E. coli strains among the three ligands; thus, further experiments were carried out on DMA only to elucidate its mechanism of growth inhibition in E. coli. We had already established the low cytotoxicity of DMA towards several cancer as well as transformed cell lines.14 Interestingly, in normal cell lines, the IC50 of DMA for HDF and MCF10A viability could not be determined up to 72 h post-DMA treatment.21 These results suggest that DMA is not cytotoxic towards normal cells at concentrations much higher than the antibacterial concentrations.
Effect of DMA on the DNA relaxation activity of EcTOPI
The effect of DMA on the relaxation activity of EcTOPI was investigated using negatively supercoiled plasmid DNA as a substrate. The amount of enzyme present in each assay reaction (10 ng of EcTOPI) was required for complete relaxation of 250 ng of plasmid DNA after incubation at 37°C for 30 min. Inhibition of EcTOPI relaxation activity by DMA was readily observed (Figure 2). The IC50 of DMA was found to be 3.8 μM from the results of three independent experiments (Figure 2). These results showed that DMA inhibits EcTOPI efficiently.
Figure 2.

Inhibition of relaxation activity of EcTOPI by DMA. Lane 1, negatively supercoiled pBAD/Thio plasmid DNA; lane 2, relaxed plasmid DNA by treatment with EcTOPI; lanes 3–10, plasmid DNA + EcTOPI in the presence of 1, 5, 7.5, 10, 12.5, 15, 20 and 30 μM DMA, respectively. FR, fully relaxed; PR, partially relaxed; OC, open circle; SC, supercoiled. The percentage inhibition values were averages obtained from three independent experiments. Error bars denote standard deviations.
DMA affects the cleavage–religation equilibrium of EcTOPI and increases the accumulation of DNA cleavage product
Many clinically important inhibitors of topoisomerase are known to be topoisomerase poisons because they shift the DNA cleavage–religation equilibrium of topoisomerase towards DNA cleavage, resulting in increased accumulation of cleaved DNA and cell death.20,21 We therefore determined whether DMA can act with the mechanism of a poison inhibitor towards EcTOPI. In the case of type IA topoisomerase, Mg2+ is not required for DNA cleavage, but is required for DNA religation and the complete catalytic cycle of removal of the negative supercoil form DNA.22,23 The addition of Mg2+ after initial DNA cleavage by topoisomerase allows a DNA cleavage–religation equilibrium to be established and only a low-level DNA cleavage product was observed. The presence of DMA led to an increase in the level of single-stranded DNA cleavage products (Figure 3). This increased level of DNA cleavage product could be observed at inhibitor concentrations significantly below the IC50 value for inhibition of the complete catalytic cycle, similar to previous results for other topoisomerase poison inhibitors.24,25 In separate experiments, it was determined that the presence of up to 16 μM DMA had no effect on the formation of DNA cleavage product in the absence of Mg2+ (Figure S1, available as Supplementary data at JAC Online). Mg2+ is required for the religation step of the EcTOP1 catalytic cycle. Thus, the result suggests that DMA inhibits the relaxation activity of EcTOPI by inhibiting DNA religation. The interaction between DMA and the EcTOP1 covalent complex may stabilize the covalent complex and shift the cleavage religation equilibrium, leading to an increase in the accumulation of DNA cleavage product in the presence of Mg2+.
Figure 3.
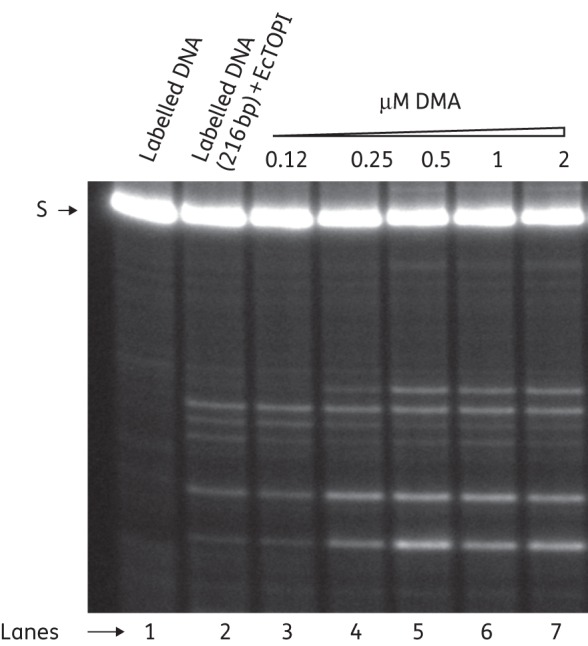
Phosphor-Imager analysis of cleavage products formed from 5′-32P-labelled single-stranded DNA by electrophoresis in a sequencing gel. Lane 1, 5′-32P-labelled single-stranded DNA; lane 2, 5′-32P-labelled single-stranded DNA with enzyme; lanes 3–7, 5′-32P-labelled single-stranded DNA with enzyme in the presence of 0.12, 0.25, 0.5, 1 and 2 mM DMA, respectively. S, single-stranded DNA.
DMA does not act as poison inhibitor of HuTOPI
To determine whether DMA can act as a poison inhibitor of HuTOPI, the reaction mixture of HuTOPI and DNA was treated with SDS and proteinase K to detect the cleaved DNA intermediate (Figure S2, available as Supplementary data at JAC Online). The HuTOPI poison inhibitor camptothecin24 was used as a positive control to show the increase in the amount of nicked DNA in the presence of a poison inhibitor. An increase in nicked DNA was not observed in the presence of up to 50 μM DMA, thus demonstrating that DMA does not act as a poison inhibitor of HuTOPI.
Analysis of DMA–DNA binding
The ability of bis-benzimidazoles to bind to the minor groove of DNA was determined by a topoisomerase I-catalysed unwinding assay, which is based on the ability of intercalating compounds to unwind the DNA duplex and thereby change the DNA twist. Supercoiled pHOT1 plasmid DNA was treated with an excess of topoisomerase IB (HuTOPI), such that no supercoiled DNA was left in the reaction mixture. The relaxed DNA substrate was purified and used as a substrate for the unwinding assay. In the presence of a strong intercalative drug, such as adriamycin, supercoiling of the relaxed substrate DNA was induced at concentrations of 50, 100 and 300 μM. Conversely, no unwinding of DNA was observed with the non-intercalative drug etoposide and DMA at concentrations of 50, 100 and 300 μM (Figure 4). Thus, DMA does not intercalate into DNA even at very high concentrations well above the inhibitory concentration for EcTOPI.
Figure 4.

Analysis of binding mode of DMA with DNA. (a) Lane 1, pHOT1 plasmid DNA (1 μg); lanes 2 and 3, relaxed pHOT1 plasmid DNA generated by treatment of pHOT1 plasmid DNA (1 μg) with HuTOPI (4 U), followed by phenol/chloroform extraction and ethanol precipitation; lanes 4–6; pHOT1 plasmid DNA + HuTOPI in the presence of 50, 100 and 300 μM etoposide, respectively. (b) Lane 1, pHOT1 plasmid DNA (1 μg); lanes 2 and 3, relaxed pHOT1 plasmid DNA generated by treatment of pHOT1 plasmid DNA (1 μg) with HuTOPI (4 U), followed by phenol/chloroform extraction and ethanol precipitation; lanes 4–6, pHOT1 plasmid DNA + HuTOPI in the presence of 50, 100 and 300 μM adriamycin, respectively. (c) Lane 1, pHOT1 plasmid DNA (1 μg); lanes 2 and 3, relaxed pHOT1 plasmid DNA generated by treatment of pHOT1 plasmid DNA (1 μg) with HuTOPI (4 U), followed by phenol/chloroform extraction and ethanol precipitation; lanes 4–6; pHOT1 plasmid DNA + HuTOPI in the presence of 50, 100 and 300 μM DMA, respectively. OC, open circle; FR, fully relaxed; SC, supercoiled.
Analysis of ethidium bromide displacement assay
The intercalation of DNA by DMA was further evaluated using a fluorescence intercalator displacement assay.19 The results (Figure 5) showed that DMA exhibited negligible DNA intercalation, as demonstrated by displacement of the intercalator ethidium bromide from DNA at concentrations well above the IC50 value for inhibition of EcTOPI activity. Less than 4% displacement of ethidium bromide was observed at 100 μM DMA, while addition of 25 μM of the well-known DNA intercalators m-AMSA and o-AMSA resulted in >50% displacement of ethidium bromide. It was unlikely that the effect of DMA on EcTOPI activity observed here was due to DNA intercalation. The weak DNA intercalation activity of DMA was also consistent with its limited side effects on HuTOPI and human cultured cell viability.
Figure 5.
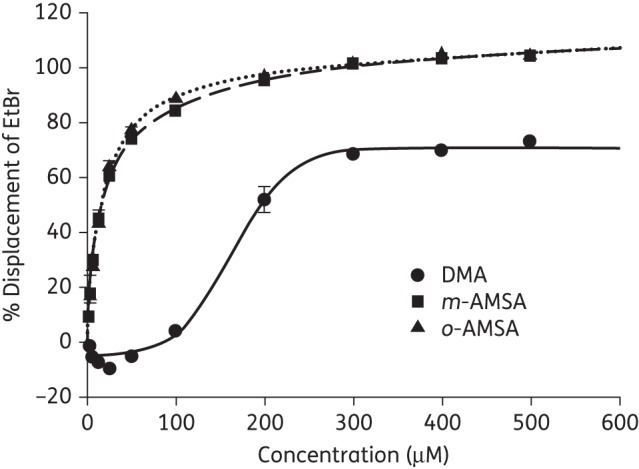
Analysis of ethidium bromide displacement by fluorometric titration assay. Averages of triplicate fluorometric titration results are shown for DMA, m-AMSA and o-AMSA. Error bars denote standard deviations. EtBr, ethidium bromide.
Effect of bis-benzimidazoles on type II topoisomerase activities
DMA was found to inhibit the relaxation of negatively supercoiled DNA by human topoisomerase II α, but to a lesser extent than EcTOPI (Figure 6a). Based on quantified results from three independent experiments, the IC50 concentration of 13 μM of DMA corresponded to half of the maximal inhibition observed (Figure 6a). Etoposide was used as positive control for inhibition of the decatenation reaction. At a concentration of 100 μM, 30.25 ± 1% decatenation inhibition of kDNA by etoposide was observed (data not shown), whereas 53.88 ± 2% decatenation inhibition of kDNA was obtained in the presence of DMA at the same concentration, suggesting less inhibitory potential of DMA towards human topoisomerase II (Figure 6b).
Figure 6.

Relaxation of pBAD/Thio by human topoisomerase II α in the presence of DMA. (a) Lane 1, negatively supercoiled pBAD/Thio plasmid DNA; lane 2, relaxation of pBAD/Thio by human topoisomerase II α; lanes 3–10; pBAD/Thio + human topoisomerase II α in the presence of 1, 5, 7.5, 10, 12.5, 15, 20 and 30 μM DMA, respectively. HuTOPIIα, human topoisomerase II α; FR, fully relaxed; PR, partially relaxed; OC, open circle; SC, supercoiled. The percentage inhibition values were averages obtained from three independent experiments. Error bars denote standard deviations. (b) Decatenation of kDNA with human topoisomerase II α in the presence of DMA. Lane 1, kDNA as control; lane 2; decatenation of kDNA by human topoisomerase II α, lane 3; kDNA with 100 μM DMA, lanes 4–10; kDNA + human topoisomerase II α in the presence of 1, 5, 10, 25, 50, 75 and 100 μM DMA, respectively. HuTOPIIα, human topoisomerase II α; N, nicked monomer; C, closed monomer.
Treatment of the reaction mixture of human topoisomerase II and DNA with SDS and proteinase K allowed detection of the cleaved DNA intermediate. The human topoisomerase II poison inhibitor m-AMSA was used in control reactions to demonstrate the action of a topoisomerase II poison inhibitor for increasing the amount of linear cleaved DNA.25 No increase in linear DNA was observed in the presence of up to 50 μM DMA (data not shown), indicating that DMA does not act as a poison inhibitor of human topoisomerase II.
DMA also did not show any inhibition of DNA gyrase supercoiling activity at up to a concentration of 100 μM DMA (Figure 7), while novobiocin, as a positive control, showed complete inhibition of DNA gyrase at 100 μM (Figure S3, available as Supplementary data at JAC Online).
Figure 7.

Analysis of supercoiling of relaxed pHOT1 by E. coli gyrase in the presence of DMA. Lane 1, relaxed DNA as control; lane 2, supercoiling of relaxed DNA by E. coli DNA gyrase; lane 3; relaxed DNA in the presence of 100 μM DMA; lanes 4–10, supercoiled DNA in the presence of 1, 5, 10, 25, 50, 75 and 100 μM DMA, respectively. FR, fully relaxed; SC, supercoiled.
Discussion
Modifying known bioactive molecules and identifying novel cellular targets are the strategies to be followed to discover new antibiotics. On the basis of the knowledge of medicinal chemistry, novel moieties can be developed to target either old cellular targets or new targets.26–30 We followed the first approach and modified the old bioactive molecule (bis-benzimidazole) to develop a non-cytotoxic, non-mutagenic and non-clastogenic potent antibacterial agent targeting topoisomerase I in E. coli. The type IA topoisomerase found in bacteria represents a novel target that remained to be utilized for development of much needed antibacterial therapy for drug-resistant pathogens.5 Owing to the essential function of bacterial topoisomerase I in removal of negative supercoils generated during transcription,7 poison inhibitors of bacterial topoisomerase I should be highly effective during response to host defence and other antibiotics.31 For the first time phenanthrene and fluorene derivatives have shown good E. coli topoisomerase inhibition and have been demonstrated to be poison inhibitors of type IA topoisomerase.32 These compounds have also been shown to be poison inhibitors of the mitochondrial type II topoisomerase in African trypanosomes.33 Recently, phenanthrene analogues were reported to target Streptococcus pneumoniae topoisomerase I without affecting human cell viability.34 These inhibitors of S. pneumoniae topoisomerase I were found to be DNA intercalators and did not affect the growth of E. coli significantly.34 In this study, a novel non-cytotoxic, non-mutagenic and non-clastogenic potent antibacterial agent has been identified and shown to act as a poison inhibitor of topoisomerase I in E. coli.
In continuation of an earlier study, DMA showed significant E. coli growth inhibition even for multidrug-resistant (MDR) strains of E. coli isolated from the patients with UTI, reported in Safdurjung Hospital of New Delhi. The PAE results suggested that the effect of the molecule lasted up to 24 h after treatment, even in MDR strains of E. coli. The decrease in the MIC50 value of transformed E. coli cells in both DH5α and BL21 E. coli strains with the EcTOPI expression clone was consistent with the accumulation of the topoisomerase I cleavage complex being responsible for cell killing. Assays with topoisomerases established DMA as a topoisomerase IA poison inhibitor that could inhibit human topoisomerase II α at a higher concentration, but did not act as a poison inhibitor of this class. It was previously reported that Hoechst 33258 and 3334235,36 specifically interrupt the breakage–reunion reaction of mammalian DNA topoisomerase I by trapping the reversible topoisomerase I-cleavable complexes and stabilizing them. However, interaction of DMA with HuTOPI did not result in poisoning of HuTOPI.
The 50% inhibition of EcTOPI relaxation activity was achieved at a DMA concentration of around 3.8 μM. Our results suggest that DMA inhibits the relaxation activity of EcTOPI by shifting the cleavage–religation equilibrium and increasing the accumulation of DNA cleavage product. DMA might act with the mechanism of an interfacial inhibitor to account for the trapping of the DNA–enzyme cleavage complex. Camptothecin has been shown to act at the protein–DNA interface as an interfacial poison inhibitor of HuTOPI.11 Our experiments showed that DMA does not act as a HuTOPI poison inhibitor even though the related bis-benzimidazoles Hoechst 33258 and 33342 are known to be poison inhibitors of mammalian topoisomerase I.35,36
The selective action of DMA as a poison inhibitor of EcTOPI, but not HuTOPI, and its low toxicity towards human topoisomerase II could be important for the low cytotoxicity observed against human culture cells. The increased level of EcTOPI–DNA cleavage product was observed at DMA concentrations that were below the IC50 value for inhibition of relaxation activity. Other topoisomerase poison inhibitors, including quinolones, have also been shown to increase the target topoisomerase cleavage product at concentrations below the concentrations required for inhibition of the complete catalytic cycle.24,25,37 Attempts will be made to determine whether resistance to DMA can be mapped to the topA gene encoding topoisomerase I.
DMA did not show a significant effect on E. coli DNA gyrase activity even at a concentration of 100 μM, suggesting that DNA gyrase is not a target of the antibacterial activity of DMA.
Conclusions
This is the first report of a bis-benzimidazole acting as an EcTOPI poison inhibitor. DMA is safe and non-cytotoxic for human cells up to the concentration required for use as an antibiotic. The finding that EcTOPI is more susceptible to this class of compound than HuTOPI may be exploited in developing rational approaches to treat E. coli infections. Bis-benzimidazoles can be developed into more specific antibiotics targeting this pivotal enzyme.
Funding
This work was partly supported by the Department of Science and Technology (DST), the Department of Biotechnology (DBT) and the Council for Scientific and Industrial Research (CSIR). The work at New York Medical College was supported by grant number R01AI069313 (to Y.-C. T.-D.) from the National Institute of Allergy and Infectious Diseases.
Transparency declarations
None to declare.
Supplementary data
Acknowledgements
We acknowledge Maria V. Abrenica for technical assistance. S. B. and M. S. are thankful to the CSIR for senior research fellowships and D. S. is thankful to the CSIR for a junior research fellowship.
References
- 1.Kaper JB, Nataro JP, Mobley HL. Pathogenic Escherichia coli. Nat Rev Microbiol. 2004;2:123–40. doi: 10.1038/nrmicro818. [DOI] [PubMed] [Google Scholar]
- 2.Schaeffer AJ, Rajan N, Cao Q, et al. Host pathogenesis in urinary tract infections. Int J Antimicrob Agents. 2001;17:245–51. doi: 10.1016/s0924-8579(01)00302-8. [DOI] [PubMed] [Google Scholar]
- 3.Biswas D, Gupta P, Prasad R, et al. Choice of antibiotic for empirical therapy of acute cystitis in a setting of high antimicrobial resistance. Indian J Med Sci. 2006;60:53–8. [PubMed] [Google Scholar]
- 4.Alt S, Mitchenall LA, Maxwell A, et al. Inhibition of DNA gyrase and DNA topoisomerase IV of Staphylococcus aureus and Escherichia coli by aminocoumarin antibiotics. J Antimicrob Chemother. 2011;66:2061–9. doi: 10.1093/jac/dkr247. [DOI] [PubMed] [Google Scholar]
- 5.Tse-Dinh YC. Bacterial topoisomerase I as a target for discovery of antibacterial compounds. Nucleic Acids Res. 2009;37:731–7. doi: 10.1093/nar/gkn936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pommier Y, Leo E, Zhang H, et al. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem Biol. 2010;17:421–33. doi: 10.1016/j.chembiol.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang JC. Cellular roles of DNA topoisomerases: a molecular perspective. Nat Rev Mol Cell Biol. 2002;3:430–40. doi: 10.1038/nrm831. [DOI] [PubMed] [Google Scholar]
- 8.Schoeffler AJ, Berger JM. DNA topoisomerases: harnessing and constraining energy to govern chromosome topology. Q Rev Biophys. 2008;41:41–101. doi: 10.1017/S003358350800468X. [DOI] [PubMed] [Google Scholar]
- 9.Boege F, Straub T, Kehr A, et al. Selected novel flavones inhibit the DNA binding or the DNA religation step of eukaryotic topoisomerase I. J Biol Chem. 1996;271:2262–70. doi: 10.1074/jbc.271.4.2262. [DOI] [PubMed] [Google Scholar]
- 10.Bridewell DJ, Finlay GJ, Baguley BC. Differential actions of aclarubicin and doxorubicin: the role of topoisomerase I. Oncol Res. 1997;9:535–42. [PubMed] [Google Scholar]
- 11.Pommier Y. DNA topoisomerase I inhibitors: chemistry, biology, and interfacial inhibition. Chem Rev. 2009;109:2894–902. doi: 10.1021/cr900097c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bansal S, Tawar U, Singh M, et al. Old class but new dimethoxy analogue of benzimidazole: a bacterial topoisomerase I inhibitor. Int J Antimicrob Agents. 2010;35:186–90. doi: 10.1016/j.ijantimicag.2009.07.018. [DOI] [PubMed] [Google Scholar]
- 13.Singh M, Tandon V. Synthesis and biological activity of novel inhibitors of topoisomerase I: 2-aryl-substituted 2-bis-1H-benzimidazoles. Eur J Med Chem. 2011;46:659–69. doi: 10.1016/j.ejmech.2010.11.046. [DOI] [PubMed] [Google Scholar]
- 14.Tawar U, Jain AK, Dwarakanath BS, et al. Influence of phenyl ring disubstitution on bisbenzimidazole and terbenzimidazole cytotoxicity: synthesis and biological evaluation as radioprotectors. J Med Chem. 2003;46:3785–92. doi: 10.1021/jm030114w. [DOI] [PubMed] [Google Scholar]
- 15.Reimer LG, Stratton CW, Reller LB. Minimum inhibitory and bactericidal concentrations of 44 antimicrobial agents against three standard control strains in broth with and without human serum. Antimicrob Agents Chemother. 1981;19:1050–5. doi: 10.1128/aac.19.6.1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhu CX, Tse-Dinh YC. The acidic triad conserved in type IA DNA topoisomerases is required for binding of Mg(II) and subsequent conformational change. J Biol Chem. 2000;275:5318–22. doi: 10.1074/jbc.275.8.5318. [DOI] [PubMed] [Google Scholar]
- 17.Domanico PL, Tse-Dinh YC. Mechanistic studies on E. coli DNA topoisomerase I: divalent ion effects. J Inorg Biochem. 1991;42:87–96. doi: 10.1016/0162-0134(91)80035-g. [DOI] [PubMed] [Google Scholar]
- 18.Annamalai T, Dani N, Cheng B, et al. Analysis of DNA relaxation and cleavage activities of recombinant Mycobacterium tuberculosis DNA topoisomerase I from a new expression and purification protocol. BMC Biochem. 2009;10:18. doi: 10.1186/1471-2091-10-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boger DL, Fink BE, Brunette SR, et al. A simple, high-resolution method for establishing DNA binding affinity and sequence selectivity. J Am Chem Soc. 2001;123:5878–91. doi: 10.1021/ja010041a. [DOI] [PubMed] [Google Scholar]
- 20.Maxwell A, Howells AJ. Overexpression and purification of bacterial DNA gyrase. Methods Mol Biol. 1999;94:135–44. doi: 10.1385/1-59259-259-7:135. [DOI] [PubMed] [Google Scholar]
- 21.Deweese JE, Osheroff N. The DNA cleavage reaction of topoisomerase II: wolf in sheep's clothing. Nucleic Acids Res. 2009;37:738–48. doi: 10.1093/nar/gkn937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tse-Dinh YC. Uncoupling of the DNA breaking and rejoining steps of Escherichia coli type I DNA topoisomerase. Demonstration of an active covalent protein-DNA complex. J Biol Chem. 1986;261:10931–5. [PubMed] [Google Scholar]
- 23.Bhat AG, Leelaram MN, Hegde SM, et al. Deciphering the distinct role for the metal coordination motif in the catalytic activity of Mycobacterium smegmatis topoisomerase I. J Mol Biol. 2009;393:788–802. doi: 10.1016/j.jmb.2009.08.064. [DOI] [PubMed] [Google Scholar]
- 24.Hsiang YH, Hertzberg R, Hecht S, et al. Camptothecin induces protein-linked DNA breaks via mammalian DNA topoisomerase I. J Biol Chem. 1985;260:14873–8. [PubMed] [Google Scholar]
- 25.Nelson EM, Tewey KM, Liu LF. Mechanism of antitumor drug action: poisoning of mammalian DNA topoisomerase II on DNA by 4′-(9-acridinylamino)-methanesulfon-m-anisidide. Proc Natl Acad Sci USA. 1984;81:1361–5. doi: 10.1073/pnas.81.5.1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat Rev Cancer. 2006;6:789–802. doi: 10.1038/nrc1977. [DOI] [PubMed] [Google Scholar]
- 27.Beerman TA, McHugh MM, Sigmund R, et al. Effects of analogs of the DNA minor groove binder Hoechst 33258 on topoisomerase II and I mediated activities. Biochim Biophys Acta. 1992;1131:53–61. doi: 10.1016/0167-4781(92)90098-k. [DOI] [PubMed] [Google Scholar]
- 28.Kim JS, Sun Q, Gatto B, et al. Structure–activity relationships of benzimidazoles and related heterocycles as topoisomerase I poisons. Bioorg Med Chem. 1996;4:621–30. doi: 10.1016/0968-0896(96)00047-8. [DOI] [PubMed] [Google Scholar]
- 29.Singh MP, Joseph T, Kumar S, et al. Synthesis and sequence-specific DNA binding of a topoisomerase inhibitory analog of Hoechst 33258 designed for altered base and sequence recognition. Chem Res Toxicol. 1992;5:597–607. doi: 10.1021/tx00029a003. [DOI] [PubMed] [Google Scholar]
- 30.Sun Q, Gatto B, Yu C, et al. Synthesis and evaluation of terbenzimidazoles as topoisomerase I inhibitors. J Med Chem. 1995;38:3638–44. doi: 10.1021/jm00018a024. [DOI] [PubMed] [Google Scholar]
- 31.Liu IF, Sutherland JH, Cheng B, et al. Topoisomerase I function during Escherichia coli response to antibiotics and stress enhances cell killing from stabilization of its cleavage complex. J Antimicrob Chemother. 2011;66:1518–24. doi: 10.1093/jac/dkr150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cheng B, Liu IF, Tse-Dinh YC. Compounds with antibacterial activity that enhance DNA cleavage by bacterial DNA topoisomerase I. J Antimicrob Chemother. 2007;59:640–5. doi: 10.1093/jac/dkl556. [DOI] [PubMed] [Google Scholar]
- 33.Tang SC, Shapiro TA. Newly identified antibacterial compounds are topoisomerase poisons in African trypanosomes. Antimicrob Agents Chemother. 2010;54:620–6. doi: 10.1128/AAC.01025-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Garcia MT, Blazquez MA, Ferrandiz MJ, et al. New alkaloid antibiotics that target the DNA topoisomerase I of Streptococcus pneumoniae. J Biol Chem. 2011;286:6402–13. doi: 10.1074/jbc.M110.148148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McHugh MM, Woynarowski JM, Sigmund RD, et al. Effect of minor groove binding drugs on mammalian topoisomerase I activity. Biochem Pharmacol. 1989;38:2323–8. doi: 10.1016/0006-2952(89)90472-3. [DOI] [PubMed] [Google Scholar]
- 36.Chen AY, Yu C, Bodley A, et al. A new mammalian DNA topoisomerase I poison Hoechst 33342: cytotoxicity and drug resistance in human cell cultures. Cancer Res. 1993;53:1332–7. [PubMed] [Google Scholar]
- 37.Domagala JM, Hanna LD, Heifetz CL, et al. New structure–activity relationships of the quinolone antibacterials using the target enzyme. The development and application of a DNA gyrase assay. J Med Chem. 1986;29:394–404. doi: 10.1021/jm00153a015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.