

Abstract
Painful bladder syndrome is a debilitating condition that affects 3–6% of women in the United States. Multiple lines of evidence suggest that changes in CNS processing are key to the development of chronic bladder pain conditions but little is known regarding the underlying cellular, molecular, and neuronal mechanisms. Using a mouse model of distention-induced bladder pain, we found that the central nucleus of the amygdala (CeA) is a critical site of neuromodulation for processing of bladder nociception. Furthermore, we demonstrate that metabotropic glutamate receptor 5 (mGluR5) activation in the CeA induces bladder pain sensitization by increasing CeA output. Thus, pharmacological activation of mGluR5 in the CeA is sufficient to increase the response to bladder distention. Additionally, pharmacological blockade or virally mediated conditional deletion of mGluR5 in the CeA reduced responses to bladder distention suggesting that mGluR5 in the CeA is also necessary for these responses. Finally, we used optogenetic activation of the CeA and demonstrated that this caused a robust increase in the visceral pain response. The CeA-localized effects on responses to bladder distention are associated with changes in extracellular signal-regulated kinases 1/2 (ERK1/2) phosphorylation in the spinal cord. Overall, these data demonstrate that mGluR5 activation leads to increased CeA output that drives bladder pain sensitization.
Introduction
Visceral pain is the most common reason that patients seek medical attention and the most common form of pain produced by disease (Cervero and Laird, 1999). Visceral pain associated with interstitial cystitis or painful bladder syndrome (PBS/IC) affects 3–8 million women in the United States (Berry et al., 2011), yet PBS/IC is poorly understood and treated (Dimitrakov et al., 2007). Up to 91% of these patients carry a diagnosis of another chronic disorder such as chronic fatigue, migraine, fibromyalgia, anxiety, and/or depression (Warren et al., 2009). Factors such as stress and depression (Macaulay et al., 1987; Baldoni et al., 1995) increase PBS/IC pain, and chronic pain is associated with increases in both stress and depression. Activity in the amygdala, a primary limbic structure, is positively correlated with stress, anxiety, and pain behavior (Neugebauer et al., 2004; Carrasquillo and Gereau, 2007; Ikeda et al., 2007; Ji et al., 2007; Neugebauer, 2007). The fact that emotion and stress modulate visceral pain and that the amygdala processes stress and nociceptive signals suggests that the amygdala is involved in the pathogenesis of chronic visceral pain.
The central nucleus of the amygdala (CeA) receives both indirect and direct nociceptive information (Bernard and Besson, 1990; Burstein and Potrebic, 1993; Bernard et al., 1996; Bourgeais et al., 2001). Noxious colorectal distention increases c-fos expression in the CeA (Traub et al., 1996), and the excitability of CeA neurons increases after induction of colitis in rats (Han and Neugebauer, 2004). Afferent outputs from the CeA to the hypothalamus and brainstem regions such as the periaqueductal gray (PAG) also make the amygdala well positioned to modulate responses to painful stimuli. Activation of the CeA with chronic corticosterone implants increases visceromotor responses to distention in rats (Greenwood-Van Meerveld et al., 2001; Myers and Greenwood-Van Meerveld, 2010). However, it is undetermined whether acute changes in excitability of CeA neurons modulate the response to noxious bladder stimulation.
The excitability of neurons in the CeA during visceral stimulation is modulated in part by metabotropic glutamate receptor 5 (mGluR5) (Li and Neugebauer, 2004). mGluR5 activation of extracellular signal regulated kinases 1/2 (ERK1/2) has been hypothesized to play a role in the modulation of pain responses (Ji, 2004; Kolber et al., 2010). Pharmacological activation of mGluR5 in the CeA increases rectal distention-induced neuronal responses (Ji and Neugebauer, 2010) and behavioral vocalizations (Li et al., 2011). However, it is unknown whether mGluR5 in the CeA plays a key role in bladder pain.
Here, we used right amygdala-specific pharmacological activation and inhibition of mGluR5 as well as conditional deletion of mGluR5 to determine the role of CeA-specific mGluR5 signaling in bladder pain. Next, we used optogenetic approaches to stimulate CeA neurons to determine whether increased activation of the CeA neurons increases the visceromotor response to noxious bladder distention. Overall, we demonstrate that either mGluR5 activation in the CeA or optogenetic activation of the CeA is sufficient to sensitize responses to painful bladder distention; we have identified a novel role for mGluR5 in the ongoing control of acute visceral pain.
Materials and Methods
Animals
All mouse protocols were in accordance with National Institutes of Health guidelines and were approved by the Animal Care and Use Committees of Washington University (St. Louis, MO) and Duquesne University (Pittsburgh, PA). Female mice (all C57BL/6J background), aged 10–13 weeks, were housed on a 12 h light/dark schedule with ad libitum access to rodent chow and water. Unless otherwise noted, wild-type (WT) mice were used for all experiments.
Visceromotor response to urinary bladder distention
The visceromotor response (VMR) is a spinobulbospinal reflex to bladder distention that has been validated as a measure of pain, as the response is suppressed by analgesics and potentiated by bladder inflammation. The VMR is observed in decerebrate rodents and not in rodents with a transected spinal cord (Castroman and Ness, 2001; Ness et al., 2001; Ness and Elhefni, 2004) but can also be modulated by higher brain centers (Qin et al., 2003). Bladder distention reliably produces pain and/or discomfort in humans (Ness et al., 1998) and is frequently used in rodents as a model of visceral pain. Procedures for bladder distention and VMR recording were similar to those previously described (Lai et al., 2011) (for illustration of setup, see Fig. 1A). Experimenter was blinded to treatment and/or genotype in all experiments. Briefly, on the day of VMR recording, the mice were anesthetized with 2% isoflurane. Chlorinated silver wire electrodes were placed on the external oblique abdominal muscle and subcutaneously across the abdominal wall (as a ground) to allow differential amplification of the abdominal VMR signals. A lubricated angiocatheter was inserted into the bladder via the urethra for bladder distention. After surgical preparation, isoflurane was reduced to ∼1.0% until a flexion reflex response was present (evoked by pinching the paw), but spontaneous escape behavior and righting reflex were absent. The use of low levels of isoflurane during urinary bladder distention (UBD) has been validated previously (Ness and Elhefni, 2004). After the desired level of anesthesia was achieved, the level of isoflurane was not changed for the duration of the experiment. The animals were not restrained in any fashion and body temperature was maintained using an overhead radiant light and monitored throughout the experiment. Phasic bladder distention with compressed air was then used to evoke bladder nociception. The air pressure was controlled by an automated distention control device custom-made in the Washington University School of Medicine Electronic Shop (St. Louis, MO). The distention stimulus applied was 20–80 mmHg pressure for 20 s with 2 min intertrial interval (ITI). The precise pressure gradient used varied depending on the specific experiment (see Results). The VMR signal was relayed in real time using a Grass CP511 preamplifier (Grass Technologies) to a PC via a WinDaq DI-720 module (Dataq Instruments). Data were exported to Igor Pro 6.05 software (Wavemetrics). Using a custom script, the VMR signals were subtracted from the baseline, rectified, and integrated over 20 s to quantify the evoked response.
Figure 1.

Intra-amygdala DHPG results in bladder hyperalgesia. A, Illustration of in vivo urinary bladder distention VMR setup. Compressed air guided into the bladder with a catheter is used to distend the bladder. Distention induces a visceromotor response that is recorded as an EMG from the abdominal muscles. Representative EMG traces show that the VMR response increases with increasing bladder distention pressure. B, Mice are cannulated in the right CeA, and drug is delivered via the cannula. C, Infusion of an mGluR1/5 agonist, DHPG (0.1 nmol), into the right CeA (n = 7) induces a hyperalgesic response to graded bladder distention compared with baseline (pretreatment) values. EMG inset shows representative EMG trace to 50 mmHg distention at baseline and following DHPG treatment (step represents 20 s distention stimulus). Brain atlas insets show cannula targeting with black stars indicative of cannula tip. D, Treatment with intra-amygdala vehicle (aCSF) (n = 7) had no effect on the evoked response to graded bladder distention. Brain atlas insets show cannula targeting with white stars indicative of cannula tip (***p < 0.001, two-way ANOVA main effect of treatment; *p < 0.05, Bonferroni's post test compared with baseline value). Error bars indicate SEM. BLA, Basolateral amygdala; CeL, lateral division of CeA; CeLC, laterocapsular division of CeA; CeM, medial division of CeA.
Surgical procedure for cannula implantation
WT mice were cannulated as previously described (Kolber et al., 2010). Briefly, mice were deeply anesthetized with a combination ketamine/xylazine anesthetic and mounted in a stereotaxic frame. An 8.00 mm stainless-steel guide cannula was implanted above the right CeA (coordinates: 1.25 mm anterior to bregma; 2.70 mm lateral to midline; 4.2 mm ventral to skull). The guide cannula was affixed to the skull with two bone screws and dental cement. An 8.00 mm stylet was inserted in the guide cannula to prevent clogging. Mice recovered for 3 d [for herpes simplex virus (HSV) optogenetic experiments] or 6 d (for pharmacological experiments) before further testing. At the end of experiments, brains were sectioned to verify cannula position and injection site. Briefly, for all experiments, the cannula end was verified by histological or visual analysis (under 4× magnification). This included first using a mouse stereotaxic atlas (Paxinos and Franklin, 2001) and the hippocampus as a rostral–caudal guide to identify the brain section corresponding to the tip of the cannula. Next, a determination of correct CeA targeting was done by using the internal branch of the external capsule and the optic tract as markers for the lateral and medial aspects of the CeA. The center of the cannula target was verified by looking at serial sections across the extent of the cannulated area. For Western experiments in which only 1 mm sections were analyzed, the section was cut directly over the cannula tract and punches were made on either side of the cannula tip after visual verification of CeA targeting. For experiments described in this manuscript, we have included insets in figures that show cannula tip placements. In all experiments, the right amygdala only was targeted for manipulation because previous results from both mice (Carrasquillo and Gereau, 2008; Kolber et al., 2010) and rats (Ji and Neugebauer, 2009; Li et al., 2011) have shown a specific lateralization of the right amygdala in somatic and visceral pain.
Intra-amygdala drug infusion
Mice were cannulated targeting the right amygdala as described above. Six days after cannulation surgery, drugs were infused. Microinjections were performed as previously described (Carrasquillo and Gereau, 2007). Briefly, injections were performed using a 32 gauge injection cannula that extended 0.5 mm beyond the tip of the guide cannula. The injection cannula was attached to flexible plastic tubing, and a microliter syringe (Hamilton) was used to deliver drug or vehicle. A total volume of 0.3 μl was infused over a period of 3 min and the injection cannula was kept in place for an additional 1 min to allow for drug diffusion.
Drugs injected included (RS)-3,5-dihydroxyphenylglycine (DHPG) and 2-methyl-6-(phenylethynyl)pyridine hydrochloride (MPEP). DHPG (Tocris) is an agonist that activates both mGluR5 and mGluR1. On the day of the experiment, DHPG was dissolved in artificial CSF (aCSF) (25 mm NaHCO3, 122 mm NaCl, 1.3 mm CaCl2, 1.2 mm MgSO4, 3 mm KCl, and 0.4 mm KH2PO4, pH 7.35) as a 5 mm stock solution, which was diluted in aCSF to 0.1 nmol (final injected volume, 0.3 μl). Mice were treated with DHPG 30 min following baseline distention and then distended again 30 min after treatment. MPEP (Tocris) is a selective noncompetitive antagonist of mGluR5. On the day of the experiment, MPEP was dissolved in aCSF as a stock 5 mm solution, which was diluted in aCSF to a final concentration of 0.01 nmol (final injected volume, 0.3 μl). For bladder distention experiments with baseline distention, mice were treated with MPEP (or vehicle) immediately following baseline distention and then distended again 60 min after treatment. For bladder distention experiments with no baseline distention, mice were treated with MPEP (or vehicle) 60 min before bladder distention at 80 mmHg. Dosing and timing for DHPG and MPEP was derived from our previous studies examining amygdala-induced somatic hypersensitivity (Kolber et al., 2010).
Preparation of CeA-specific mGluR5 knock-out mice (mGluR5CeAKO)
Mice homozygous for the mGluR5loxP allele (Xu et al., 2009) or WT littermates were used. All animals were injected in the right amygdala with virus containing recombinant lentiviral vector LV-Ef1α-Cre (LV-Cre) as previously described (Kolber et al., 2008, 2010). Briefly, mice were anesthetized with a combination ketamine/xylazine anesthetic and mounted in a stereotaxic frame. A small hole was drilled over the right CeA (coordinates: 1.25 mm anterior to bregma; 2.7 mm lateral to midline; 4.7 mm ventral to skull). A 32 gauge needle was lowered into the hole, and 4 × 105 infectious viral particles were injected over 5 min. The injection needle was kept in place after injection for 4 min to allow for stable diffusion. All testing in these mice occurred 2 weeks after recovery from surgery. After testing, brains were sectioned to determine needle targeting and 1 mm diameter × 1 mm high punches were made of the amygdala in two adjacent sections to determine amount of mGluR5 protein disrupted in mGluR5CeAKO mice compared with WT mice injected with LV-Cre. In addition, tissue from the lumbosacral spinal cord was harvested for analysis of ERK phosphorylation. See below for a description of processing of tissue samples.
Optogenetic vectors, infusion, and stimulation parameters
Vectors.
HSV vectors were obtained from McGovern Institute (Massachusetts Institute of Technology, Boston, MA). Briefly, HSV vectors driven by the CMV promoter [channelrhodopsin hChR2(H134R)-EYFP or EYFP only] were prepared as previously described (Neve et al., 2005; Covington et al., 2010). All experiments were performed on day 4 after infection because maximal expression of HSV vectors occurs on days 3–4 (Covington et al., 2010).
Vector infusion.
Mice were cannulated in the right amygdala as described above. Three days after cannulation surgery, HSV vectors were infused. Microinjections were performed similarly to that described above for drugs. A total volume of 1 μl was infused over a period of 5 min, and the injection cannula was kept in place for an additional 1 min to allow for viral infusion.
Optogenetic CeA stimulation during bladder distention.
On the day of the experiment, an 8.00 mm fiber optic fiber (0.2 mm diameter; Thor Labs) was placed in the animal's cannula. Optic stimulation was performed similar to previously published protocols (Covington et al., 2010; Zhang et al., 2010). Briefly, the fiber optic was connected via a FC/PC adaptor to a 473 nm blue laser diode (OEM) connected to a power supply. Laser pulses were controlled using a Techtronix pulse generator. During all stimulations, a 5 ms, 20 Hz (18 ms spike width) pulse train was delivered for 30 min. The stimulation protocol was modeled to mimic the firing pattern of CeA neurons that occurs after painful injury in rodents (Ji and Neugebauer, 2009). The intensity of the fiber optic light was verified before and after each experiment use using a luxometer (Thor Labs). For the distention experiment, we applied a bladder stimulus of 30 mmHg (20 s, three times, with 2 min ITI), and then 60 mmHg (20 s, three times, with 2 min ITI) during baseline testing, 20 min after the laser was turned on, and 15 min after the laser was turned off. For Western blot and immunohistochemistry experiments, mice were stimulated with the laser as above for 30 min and then killed 15 min after laser off with no distention. Mice were killed after the experiment and analyzed by immunohistochemistry and Western blotting for targeting, ERK phosphorylation, and/or eYFP expression (see below).
Western blotting
Protein expression in mGluR5CeAKO mice.
After bladder distention (see above), spinal cords were removed by hydraulic extrusion from mGluR5CeAKO mice (compared with LV-Cre-injected WT mice). The lumbosacral portion of the spinal cord that is innervated by bladder afferent input (L6–S1) was isolated by (1) identifying the thickest portion of the lumbar enlargement as L4/L5, (2) measuring 2 mm caudal to this portion as the start of L6, and then (3) isolating a section of cord that started at this L6 point and continuing 7 mm caudal. This method of identification has been validated by our immunohistochemical analysis of pERK1/2 in the dorsal horn after bladder distention (Lai et al., 2011). That is, the Western “lumbosacral” isolates used in this experiment correspond to the immunohistochemical sections that stain positive for phosphorylated ERK1/2 after bladder distention. After the lumbosacral portion of the spinal cord was isolated, the tissue was frozen on dry ice. Brains were removed, sectioned into 1-mm-thick coronal sections using an acrylic brain matrix (Stoelting), and amygdala punches (two per side per animal) were obtained using a custom-made 1 mm punch tool and frozen on dry ice. All tissue samples were homogenized with ice-cold homogenization buffer (20 mm Tris-HCl, pH 7.4, 1 mm EDTA, 1 mm sodium pyrophosphate, 25 mg/ml aprotinin, 25 mg/ml leupeptin, and 100 mm phenylmethylsulfonyl fluoride). The levels of mGluR5 in the left or right amygdala were measured by Western blotting using antibodies specific for mGluR5 and the loading control protein β-Tubulin. Total protein (7.5 μg) for amygdala homogenates was separated using 4–12% SDS-PAGE and transferred to nitrocellulose membranes. For mGluR5 Western blotting, membranes were blocked in Odyssey blocking buffer for 1 h and then coincubated in mouse anti-β-Tubulin (1:10,000; Sigma-Aldrich) and rabbit anti-mGluR5 (1:1000; Millipore) primary antibodies in Odyssey buffer with 0.1% Tween 20 for 1 h at room temperature (RT). After washing in TBS with 0.1% Tween 20, blots were incubated in goat anti-mouse IR800 (1:20,000; Sigma-Aldrich) and goat anti-rabbit Alexa Fluor 680 (1:20,000; Sigma-Aldrich) for 1 h at RT. Blots were washed and scanned using an Odyssey infrared scanner. Densitometry of bands corresponding to mGluR5 and β-Tubulin was performed using Odyssey software (version 3.0). Each mGluR5 band was divided by the pixel value for the β-Tubulin value of that sample and then normalized to the left amygdala because our previous results showed that mGluR5 protein is increased in the right amygdala compared with the left amygdala (Kolber et al., 2010). The levels of ERK1/2 phosphorylation in the spinal cord were measured by Western blotting using antibodies specific for phosphorylated ERK and for total ERK. Total protein (5 μg) for spinal cord homogenates was run as described above. Blots were blocked as above and then coincubated in mouse anti-p44/42 ERK (1:1000; Cell Signaling) and rabbit anti-phospho-p44/42 ERK (1:1000; Cell Signaling) primary antibodies in Odyssey buffer with 0.1% Tween 20 (Sigma-Aldrich) for 1 h at RT. Final processing was done as above. Densitometry of bands corresponding to ERK1 (p44) and ERK2 (p42) was performed using Odyssey, version 3.0, software (LI-COR).
Bladder distention induced ERK1/2 activation.
All mice (sham undistended and distended) were anesthetized and catheterized as described above for urinary bladder distention testing. Distended animals received 20 s, 80 mmHg bladder distention stimulation (five times with 2 min ITI). Mice were killed 5 min after last bladder distention (or equivalent amount of time for sham mice). Spinal cords and brains were processed as above. Westerns were done as above on spinal cord and amygdala tissue for phosphorylated ERK and for total ERK.
Optogenetically induced spinal cord ERK1/2 phosphorylation.
Mice were anesthetized and catheterized as described above for urinary bladder distention testing. All mice [HSV-EYFP and HSV-hChR2(H134R)-EYFP] received 30 min of stimulation using the optical stimulation protocol described above. Fifteen minutes following termination of optical stimulation, mice were killed, and spinal cord was processed as above. Western blotting was completed using spinal cord tissue and assessing levels of phosphorylated ERK1/2 and total ERK1/2.
Immunohistochemistry
YFP CeA expression.
Following the optogenetic bladder stimulation experiment (described above), mice were deeply anesthetized with pentobarbital, perfused transcardially with PBS (37°C), followed by 25 ml of ice-cold 4% paraformaldehyde (PFA) solution in PBS. The brain was dissected and postfixed in PFA overnight at 4°C. After cryoprotection in 30% sucrose, coronal sections (30 μm) were obtained on a cryostat and stored in PBS at 4°C until immunostaining. Sections were rinsed in TBS and blocked in 1% BSA/0.2% milk/0.1% Triton X-100 for 1 h at RT. Sections were incubated overnight at 4°C in rabbit anti-GFP antibody (1:2000; Invitrogen) in blocking solution. Sections were rinsed in TBS with 0.1% Triton X-100 and incubated in donkey anti-rabbit Alexa Fluor 555 secondary antibody (1:250; Invitrogen) in blocking solution for 1 h at RT. Sections were rinsed in TBS with 0.2% Triton X-100, TBS, and then wet mounted and visualized using a Nikon Eclipse80i fluorescent microscope.
Optogenetically induced spinal cord ERK1/2 phosphorylation.
Following optogenetic CeA stimulation (described above), mice were deeply anesthetized with pentobarbital then perfused with PBS (37°C) followed by ice-cold 4% PFA. The spinal cord was removed and the L4–S2 region was isolated. Samples were fixed in 4% PFA for 4 h, followed by 30% sucrose for 48 h, both at 4°C. Thirty-micrometer transverse sections were cut on a cryostat, and then immediately placed in PBS. Representative sections from L6–S1 of the spinal cord were rinsed in PBS, and then placed in 1.0% sodium borohydride for 10 min. Following additional rinses, sections were washed in PBS with 0.1% Triton X-100, and then rinsed again. Sections were blocked in 5% normal goat serum with PBS containing 0.1% Triton X-100, 1% BSA, and 0.1% fish gelatin at RT for 1 h. Exposure to rabbit anti-pERK (1:200; Cell Signaling) occurred overnight at 4°C in 1.5% NGS blocking solution. Sections were rinsed in PBS then incubated with goat anti-rabbit Alexa Fluor 488 antibody (1:200; Invitrogen) in 1.5% NGS blocking solution at RT for 1 h. After a final rinse, sections were wet mounted and visualized using a Nikon Microphot-SA microscope. pERK was quantified in four L6–S1 sections for each animal. For cell counts, each spinal cord section was divided into four main regions as previously established (Lai et al., 2011): deep dorsal commissure, spinal parasympathetic nuclei, lateral dorsal horn, and medial dorsal horn. The total number of cells in each region was counted for each section. For each animal, an average cell count was determined using the four sections so that presented data represent the average number of positive cells per section per mouse. All spinal cords were removed and processed on the same day. Sections from all animals were stained at the same time. The researcher who performed the cell counts was blinded to the treatment groups.
Data analysis
Results are expressed as means ± SEM. Student's t tests were used to compare pairs of means. In cases of two independent variables (e.g., genotype and pressure), two-way ANOVA with repeated measures was used followed by Bonferroni's post hoc tests when significant main effects were found. A value of p < 0.05 was considered statistically significant for all statistical comparisons. All statistical comparisons were performed with Prism 5 software (GraphPad).
Results
Amygdala mGluR activation induces bladder hypersensitivity
To study pain-like behavior originating from the bladder as directly as possible, we chose to study the response to urinary bladder distention (Fig. 1A). The behavioral response to bladder distention is quantified using the VMR, which is an electromyographic (EMG) recording of the abdominal muscle contraction in response to bladder distention (Ness and Gebhart, 1988, 1991; Ness and Elhefni, 2004). Previous studies have demonstrated a critical role for mGluR5 activation in the right amygdala in the development of somatic pain sensitization (Ji and Neugebauer, 2009; Kolber et al., 2010), but it is not known whether mGlu5 also regulates bladder nociception. To address this question, the selective mGluR1/5 agonist, DHPG, was infused to the right amygdala via an indwelling cannula (Fig. 1B) and the effects of this infusion on the VMR to bladder distention were measured. Bladder distention (20–80 mmHg distention in 10 mmHg steps) reliably produces an evoked VMR that increases with increasing distention pressure (Fig. 1A; C, two-way ANOVA main effect of pressure, F(6,42) = 8.2, p < 0.0001; D, F(6,42) = 26.3, p = 0.0019). When a control vehicle is infused, and the bladder distention protocol is repeated, no changes in the evoked response are observed (Fig. 1D). In contrast, infusion of DHPG (0.1 nmol) significantly increased the evoked responses to bladder distention compared with pretreatment baseline values (Fig. 1C; two-way ANOVA significant main effect of treatment, F(1,42) = 26.3, p < 0.0001; before DHPG vs after DHPG Bonferroni's post hoc test at 50 mmHg, t = 2.9, p < 0.05). Thus, activation of mGluR1/5 in the right CeA results in significant physiological sensitization to bladder distention.
Intra-amygdala mGluR5 antagonism decreases evoked responses to bladder distention
Prior evidence suggested that mGluR5 in the amygdala is involved in the modulation of somatic pain only after injury and that mGluR5 signaling does not affect acute responses to noxious stimulation (Kolber et al., 2010; Li et al., 2011). Whether mGluR5 modulates acute responses to bladder stimulation is not known, however. To address the role of amygdala mGluR5 in the modulation of bladder pain, the mGluR5 antagonist MPEP (or vehicle) was delivered locally to the right amygdala between two rounds of bladder distentions (from 20 to 80 mmHg in 10 mmHg increments), and the effects on the evoked VMR were determined. We found that MPEP infusion to the CeA induced a significant decrease in bladder distention-evoked responses (two-way ANOVA significant main effects of pressure, F(6,35) = 5.9, p = 0.0003, and treatment, F(1,35) = 22.45, p < 0.0001) that was particularly noticeable at the higher noxious pressures (Fig. 2A; before MPEP vs after MPEP Bonferroni's post hoc test at 70 mmHg, t = 4.5, p < 0.001, and at 80 mmHg, t = 6.1, p < 0.001) compared with baseline VMRs. These results were corroborated in a second experiment in which we measured the effect of pretreatment with MPEP on the VMR to a single round of noxious distention of the bladder with no baseline distention. We found that mice that had received infusion of MPEP into the right CeA had significantly decreased VMR to high noxious (80 mmHg) bladder distention when compared with responses in vehicle-infused mice (Fig. 2C; Student's t test, t(11) = 2.3, p = 0.04). These findings provide further support for the notion that mGluR5 activation in the amygdala is required for acute regulation of bladder nociception.
Figure 2.
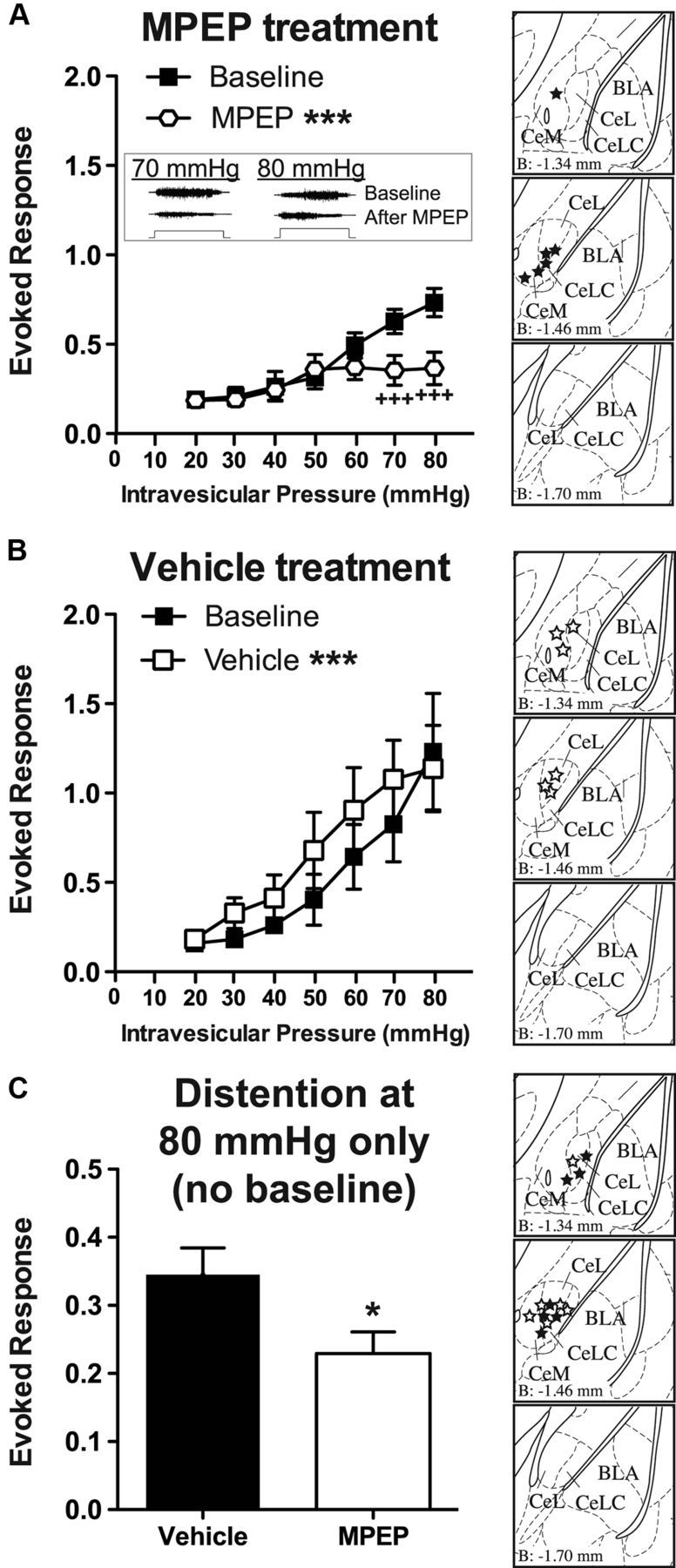
Intra-amygdala MPEP is analgesic during bladder distention. A, Infusion of a mGluR5 antagonist, MPEP (0.01 nmol), into the right CeA (n = 6) induced an analgesic response to graded bladder distention in the noxious range compared with baseline (pretreatment) values. The inset shows representative EMG traces to 70 and 80 mmHg distention at baseline and following MPEP treatment (step represents 20 s distention stimulus). Brain atlas insets show cannula targeting with black stars indicative of cannula tip. B, Treatment with intra-amygdala injection of vehicle (aCSF) (n = 6) induced a significant increase in evoked response during bladder distention. However, this change was in the opposite direction of treatment with MPEP and likely represents a small nonspecific effect possibly due to repeated bladder distention. Brain atlas insets show cannula targeting with white stars indicative of cannula tip. C, In a separate cohort, pretreatment with MPEP (n = 7) before a single round of higher noxious pressure bladder distention (i.e., no baseline distention) induced a significant decrease in the evoked response compared with vehicle-treated mice (n = 6). Brain atlas insets show cannula targeting with solid black stars indicative of cannula tip of MPEP-treated mice and white stars indicative of cannula tip of vehicle-treated mice (***p < 0.001, two-way ANOVA main effect of treatment; +++p < 0.001, Bonferroni's post test compared with indicated baseline value; *p < 0.05, Student's t test compared with vehicle-treated group). Error bars indicate SEM.
Surprisingly, when vehicle was infused between two rounds of graded bladder distention, the evoked responses were significantly increased relative to preinfusion baseline measurements (Fig. 2B; two-way ANOVA significant main effects of pressure, F(6,35) = 5.0, p = 0.0009, and treatment, F(1,35) = 15.4, p = 0.0004). These results might suggest that repeated distention to noxious pressures can induce mild sensitization of the bladder. To address this issue, we examined the VMR in control mice that received only a single round (four to five distentions) of 80 mmHg distention (with no baseline UBD). Comparing the VMR after the first distention at 80 mmHg in these mice to the last distention, we found no significant sensitization of the VMR to repeated distentions (first distention, 0.54 ± 0.10 evoked response; last distention, 0.30 ± 0.05 evoked response; paired t test, t(5) = 1.9, p = 0.11). These data suggest that repeated bladder distention does not sensitize the bladder.
Disruption of mGluR5 in the right amygdala reduces responses to bladder distention
The results described above demonstrate that pharmacological activation of mGluR5 in the right CeA enhances the evoked response to bladder distention, whereas inhibition of mGluR5 in the CeA inhibits these responses. These data are consistent with the hypothesis that mGluR5 in the right CeA plays a central role in the endogenous modulation of visceral bladder pain. However, pharmacological manipulations in vivo can always have the potential for off-target effects. To complement our pharmacological studies, we took a genetic approach to test the hypothesis that endogenous mGluR5 activation is required for bladder pain. A conditional knock-out approach was used to specifically disrupt mGluR5 expression in the right CeA using recombinant lentivirus-mediated expression of Cre recombinase (LV-Cre) in floxed mGluR5 mice. Floxed mGluR5 mice injected with LV-Cre (mGluR5CeAKO) exhibited a significant decrease in mGluR5 protein levels when compared with mGluR5 expression in the right amygdala of LV-Cre-injected WT (control) mice, thus confirming deletion of mGluR5 (Fig. 3A; Student's t test, t(9) = 3.8, p = 0.0045). To measure the impact of CeA mGluR5 disruption on bladder pain-related behavior, we compared the responses of mGluR5CeAKO with control mice during graded bladder distention (distention from 20 to 80 mmHg in 10 mmHg steps). We found a significant decrease in the evoked response to bladder distention in the mGluR5CeAKO mice compared with control mice (Fig. 3B; two-way ANOVA significant main effects of pressure, F(6,105) = 7.8, p < 0.0001, and genotype, F(1,105) = 14.4, p = 0.0003). Overall, these data suggest that mGluR5 activity in the CeA modulates ongoing sensitivity to visceral stimulation.
Figure 3.
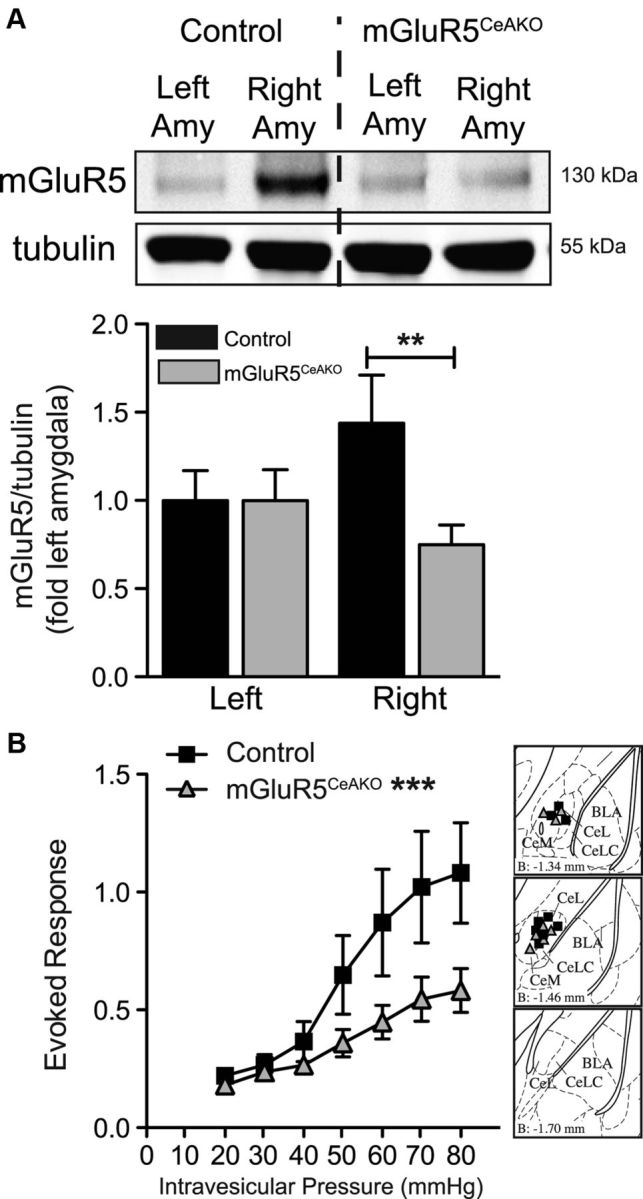
Lentivirus-mediated conditional disruption of mGluR5 in the central amygdala is analgesic. A, Representative Western blot for mGluR5 (top) and β-tubulin (bottom) in mGluR5CeAKO and WT LV-Cre-injected (control) mice. Graph of mGluR5/β-tubulin shows reduced mGluR5 expression in the right amygdala of mGluR5CeAKO mice (n = 8) compared with the right amygdala of control mice (n = 8). B, Disruption of mGluR5 in the right amygdala (n = 8) induced a significant decrease in evoked response during bladder distention compared with control mice (n = 9). Brain atlas insets show cannula targeting with black squares indicative of cannula tips of control mice and gray triangles indicative of cannula tips of mGluR5CeAKO mice (***p < 0.001, two-way ANOVA main effect of genotype; **p < 0.01, Student's t test). Error bars indicate SEM.
Amygdala control of bladder hyperalgesia is associated with ERK activation in the spinal cord
Next, we sought to evaluate the mechanisms by which activation of the CeA might modulate evoked responses to bladder distention. We hypothesized that mGluR5 activation in the CeA could modulate bladder nociception by regulating ERK1/2 activity in the CeA or spinal cord (through modulation of descending pathways). We first tested whether bladder distention in naive mice would induce ERK1/2 phosphorylation. We compared the protein collected from the lumbosacral spinal cord and the CeA in mice distended five times at 80 mmHg with protein from undistended sham mice. We found that bladder distention induced a significant increase in phosphorylated ERK1 and ERK2 in the spinal cord (Fig. 4A,B; Student's t test, pERK1, t(12) = 2.6, p = 0.022; pERK2, t(12) = 3.3, p = 0.0068). In contrast, we found no significant change in activation of ERK1 or ERK2 in the CeA following distention compared with sham mice (Fig. 4C,D).
Figure 4.

Bladder distention induces spinal cord ERK phosphorylation. A, Representative Western blot showing phosphorylated ERK1 and ERK2 bands (top) and total ERK1 and ERK2 (bottom) from spinal cord tissue of undistended control (sham) mice (n = 7) or 80 mmHg bladder distended mice (n = 7). B, Quantification of Western blots showing increased ERK1 and ERK2 phosphorylation in distended mice compared with sham mice. C, Representative Western blot showing phosphorylated ERK1 and ERK2 bands (top) and total ERK1 and ERK2 (bottom) from left and right amygdala of sham mice or 80 mmHg bladder-distended mice. D, Quantification of Western blots showing no significant change ERK1 and ERK2 phosphorylation from right amygdala tissue samples of distended mice compared with sham mice (*p < 0.05, **p < 0.01, unpaired Student's t test compared with sham group). Error bars indicate SEM.
Since bladder distention induced an increase in activated ERK1/2 in the spinal cord and we found a decrease in the evoked response to bladder distention in mGluR5CeAKO mice, we reasoned that mGluR5CeAKO mice might show a decrease in spinal ERK1/2 activation following bladder distention. Indeed, we found that disruption of mGluR5 in the right CeA caused a significant decrease in the phosphorylation of ERK1 and ERK2 in the spinal cord after bladder distention compared with distended control mice (Fig. 5A,B; Student's t test, pERK1, t(14) = 2.3, p = 0.038; pERK2, t(14) = 2.3, p = 0.035). These data suggest the presence of tonic modulation of spinal circuitry driven by mGluR5 activation in the CeA. In contrast, when we compared the phosphorylation of ERK1 and ERK2 by Western analysis in the spinal cord after CeA treatment with MPEP or vehicle followed by a single round of noxious distention, we failed to find any significant differences between groups (data not shown).
Figure 5.

Genetic disruption of mGluR5 in the right amygdala reduces bladder distention-induced ERK phosphorylation. A, Representative Western blot showing phosphorylated ERK1 and ERK2 bands (top) and total ERK1 and ERK2 (bottom) from spinal cord tissue from LV-Cre-injected control mice (n = 9) or mGluR5CeAKO mice (n = 7) following graded bladder distention. B, Quantification of Western blots showing decreased ERK1 and ERK2 phosphorylation in mGluR5CeAKO mice compared with control mice (*p < 0.05, unpaired Student's t test compared with control group). Error bars indicate SEM.
Optogenetic activation of the central amygdala is sufficient to induce pain hypersensitivity with no changes in ERK1/2 phosphorylation
The mechanisms through which mGluR5 in the CeA might modulate responses to bladder distention are unknown. For example, mGluR5 activation in the CeA could suppress a tonically active descending inhibitory pathway to the spinal cord, or conversely could drive increased descending excitatory circuitry. To address the hypothesis that an overall increase in the activity of CeA neurons may be sufficient to induce bladder hyperalgesia, we used an optogenetic approach to drive excitation of CeA neurons and asked whether this increased CeA activity could modulate the VMR elicited by innocuous and noxious bladder distention.
To accomplish this goal, channelrhodopsin-2 or control protein expression was driven in the CeA by targeted injection in the right CeA (Fig. 6A) of HSV vectors expressing YFP only (HSV-YFP control; Fig. 6B) or channelrhodopsin-2 fused to YFP (HSV-ChR2-YFP; Fig. 6C). Four days after viral injection, mice were stimulated with low (30 mmHg) or with higher noxious (60 mmHg) bladder distention at baseline, during, and after laser stimulation (5 ms, 20 Hz pulse with 18 ms spike width). The optical stimulation protocol chosen for these experiments was modeled to mimic the CeA neuronal firing pattern that occurs after injury in rodents (Ji and Neugebauer, 2009). In HSV-YFP control mice, optical stimulation did not alter evoked responses to bladder distention at 30 mmHg (Fig. 6D,F) or 60 mmHg (Fig. 6G,I). In contrast, 30 min of optical stimulation of HSV-ChR2-YFP-treated mice caused a significant increase in bladder distention-evoked responses compared with baseline measurements (Fig. 6E,F; H,I, Bonferroni's post hoc test at 30 mmHg, t = 2.5, p < 0.05, and at 60 mmHg, t = 4.3, p < 0.001) and compared with HSV-YFP control mice (Fig. 6I; Bonferroni's post hoc test at 60 mmHg, t = 3.0, p < 0.05). Interestingly, CeA stimulation induced a lasting hypersensitivity to bladder distention in HSV-ChR2-YFP-treated mice. That is, 15 min after the laser stimulation was terminated, HSV-ChR2-YFP-treated mice still showed increased responses to bladder distention (Fig. 6E,F; Bonferroni's post hoc test at 30 mmHg compared with baseline, t = 2.8, p < 0.05; H,I, at 60 mmHg compared with baseline, t = 4.6, p < 0.001, and at 60 mmHg compared with HSV-YFP control mice, t = 3.1, p < 0.05).
Figure 6.
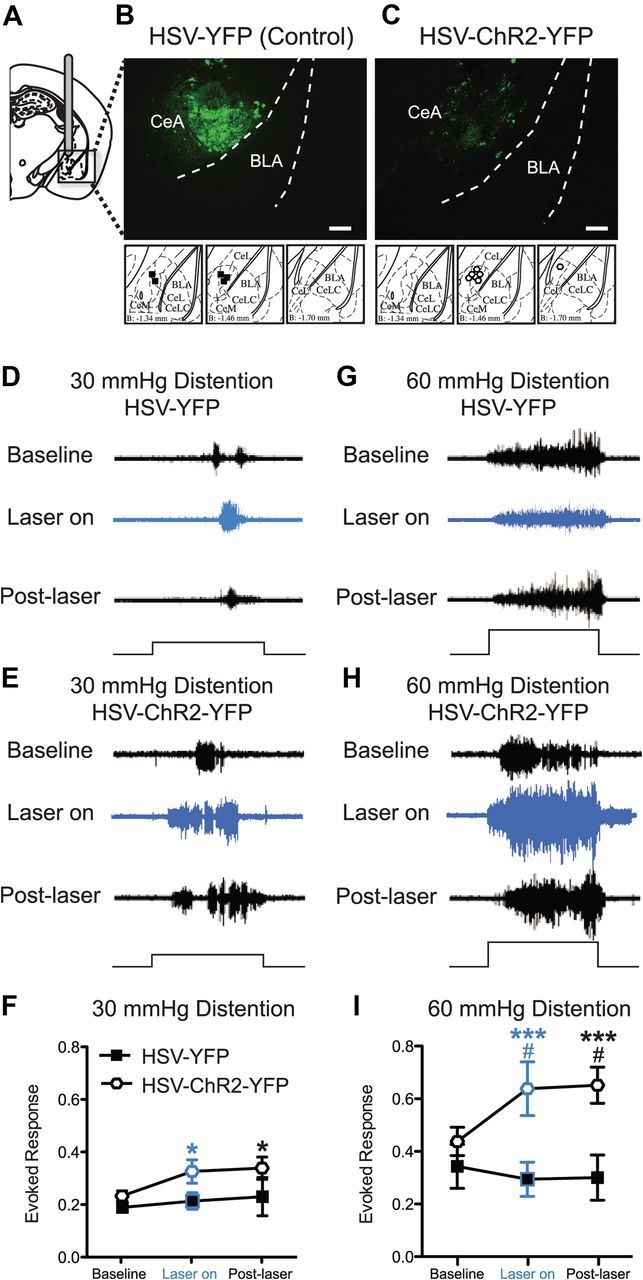
Optogenetic stimulation of right central amygdala induces bladder hyperalgesia. A, Mice are cannulated in the right CeA and HSV vectors are delivered via the cannula. B, C, YFP fluorescence (via immunohistochemistry) is seen in the CeA of both HSV-YFP (control vector) (B) and HSV-ChR2-YFP (optogenetic vector)-injected mice (C). Scale bars, 0.2 mm. Differences in staining pattern for HSV-YFP- versus HSV-ChR2-YFP-treated mice are likely due to the fact that there is probably higher expression of cytoplasmic YFP (i.e., HSV-YFP) compared with the ChR2-YFP fusion protein, which is membrane bound. Brain atlas insets show cannula targeting with black squares indicative of cannula tips of HSV-YFP mice and white hexagons indicative of cannula tips of HSV-ChR2-YFP mice. Representative images of raw EMG traces from HSV-YFP- or HSV-ChR2-YFP-injected mice during 30 mmHg (D, E) and 60 mmHg (G, H) bladder distention. Optic stimulation of HSV-ChR2-YFP-treated mice (n = 7) increases the evoked response to both 30 and 60 mmHg bladder distention compared with baseline (prestimulation) responses (E, H) and increases the evoked response to 60 mmHg bladder distention compared with HSV-YFP-injected mice (n = 5) (I). Laser light alone does not increase the evoked response to bladder distention in HSV-YFP mice (D, F, G, I). The black steps in traces show 20 s bladder distention stimuli (*p < 0.05; ***p < 0.001, Bonferroni's post test compared with baseline for HSV-ChR2-YFP mice; #p < 0.05, Bonferroni's post test compared with HSV-YFP control mice at indicated time points). Error bars indicate SEM.
To test one possible downstream effect of optogenetic activation of the amygdala, we evaluated whether optogenetically driven activation of the amygdala (5 ms, 20 Hz pulse with 18 ms spike width) in the absence of bladder distention would induce spinal cord ERK1/2 phosphorylation. Mice received local infusions in the CeA of HSV-ChR2-YFP or HSV-YFP as described above. Four days after viral infusion, mice were anesthetized and stimulated with the laser on for 30 min and were killed after the laser had been off for 15 min. Comparing spinal cord protein samples analyzed by Western or spinal cord sections analyzed for pERK1/2 immunofluorescence, we found that optogenetic stimulation of the CeA failed to induce an increase in ERK1/2 phosphorylation compared with HSV-YFP control mice (Western: Student's t test, pERK1 HSV-YFP, n = 12, 1.0 ± 0.08, vs HSV-ChR2-YFP, n = 10, 1.0 ± 0.09, t = 0.1, p = 0.93; pERK2 HSV-YFP, 1.0 ± 0.07, vs HSV-ChR2-YFP, 1.1 ± 0.07, t = 0.7, p = 0.51) (IHC: Student's t test, deep dorsal commissure, HSV-YFP, n = 3, 40.0 ± 10.6, vs HSV-ChR2-YFP, n = 3, 38.9 ± 5.5, pERK1/2-positive cells, t = 0.10, p = 0.92; spinal parasympathetic nuclei, HSV-YFP, 23.0 ± 8.9, vs HSV-ChR2-YFP, 29.5 ± 2.3, pERK1/2-positive cells, t = 0.82, p = 0.45; lateral dorsal horn, HSV-YFP, 12.6 ± 2.4, vs HSV-ChR2-YFP, 13.2 ± 4.1, pERK1/2-positive cells, t = 0.11, p = 0.91; medial dorsal horn, HSV-YFP, 15.3 ± 6.9, vs HSV-ChR2-YFP, 16.6 ± 3.8, pERK1/2-positive cells, t = 0.18, p = 0.86; total dorsal horn, HSV-YFP, 90.8 ± 25.6, vs HSV-ChR2-YFP, 98.1 ± 12.7, pERK1/2-positive cells, t = 0.28, p = 0.79). Similar to our results with optogenetic activation, treatment of the CeA with DHPG in the absence of bladder distention did not alter ERK1/2 phosphorylation in the spinal cord compared with vehicle-treated mice (data not shown).
Discussion
Our results demonstrate that activation of group I mGluRs in the right amygdala was sufficient to induce an increased pain response to bladder distention. Furthermore, pharmacological inhibition and genetic deletion of mGluR5 in the right amygdala resulted in a reduced response to bladder distention. Optogenetic activation of the right amygdala was sufficient to increase the pain response to bladder distention. Together, these results suggest that mGluR5 in the right CeA modulates bladder pain responses via increased excitation of CeA neurons. Our results are consistent with previous work demonstrating that noxious bladder distention in the absence of injury results in ERK1/2 activation in the lumbosacral spinal cord (Lai et al., 2011). Furthermore, we found that the level of activated ERK1/2 is dependent on the presence of mGluR5 in the right CeA. Our results suggest that distention-evoked ERK1/2 activation in the lumbosacral spinal cord is modulated in part by mGluR5 activity in the right amygdala.
Here, we show for the first time that mGluR5 in the right CeA is necessary for the visceral bladder pain response to noxious stimuli. Our results are consistent with previous work indicating that activation of group I mGluRs in the right amygdala leads to increased somatic pain (Kolber et al., 2010). Specifically, activation of mGluR5 in the right amygdala results in bilateral mechanical hypersensitivity (Kolber et al., 2010). Our results extend this phenomenon to visceral bladder pain and suggest that pharmacological activation of group I mGluRs (mGluR1 and -5) in the right CeA is sufficient to increase the nociceptive responses to bladder distention in naive (noninjured) mice.
Given that previous studies have shown the mGluR1/5 agonist DHPG potentiates the excitatory response of the CeA during visceral colorectal distention (Ji and Neugebauer, 2010), we hypothesized that activation of group I mGluRs in the amygdala could lead to increased excitability as has been reported previously (Li and Neugebauer, 2004), thus resulting in the observed increased response to bladder distention. We demonstrate that optogenetic stimulation of CeA neurons in the right amygdala is sufficient to increase the VMR to urinary bladder distention. Furthermore, this increased VMR is maintained 15 min after the optogenetic activation is turned off. Injury in a colitis model increases the excitability of evoked responses in the CeA (Han and Neugebauer, 2004) and arthritis-induced somatic injury increases the background and evoked responses of CeA neurons (Li and Neugebauer, 2004). The sustained behavioral response we observe after optical excitation suggests that 30 min activation of the CeA may, in effect, model the changes seen after either visceral or somatic injury. Future studies are required to test this hypothesis.
Our previous studies have shown that inhibition of mGluR5 in the right amygdala reduces persistent injury-induced hypersensitivity of the paw, but that inhibition of mGluR5 does not alter acute somatic sensitivity or spontaneous somatic nociceptive responses (Kolber et al., 2010). Here, we find that pharmacological inhibition of mGluR5 following a round of bladder distention is analgesic when compared with the baseline bladder distention. These results could represent either a novel role of mGluR5 in modulating acute visceral sensation or provide additional evidence that mGluR5 modulates sensation after mild injury caused by repeated bladder distentions. We believe that the significant increase in the evoked response in the vehicle-treated mice (Fig. 2B) is likely a statistical aberration as no such effect was seen in a similar experiment (Fig. 1D), and we found that the first VMR is not significantly different from the last VMR during a round of noxious bladder distention with no baseline testing. Nonetheless, to unequivocally determine whether mGluR5 in the CeA modulates baseline (uninjured) responses to bladder distention, we inhibited mGluR5 before noxious stimulation with no baseline distention trials. Providing evidence for the hypothesis that mGluR5 modulates acute bladder nociception, mice pretreated with MPEP have a smaller VMR in response to a single round of 80 mmHg distention when compared with vehicle-treated mice. This hypothesis is further supported by the reduced VMR observed in mice with conditional disruption of mGluR5 in the right amygdala, because these animals receive only a single series of distentions. Interestingly, these data are contrary to previous reports showing that inhibition of mGluR5 in the right amygdala has no effect on vocalizations induced by visceral colorectal distention (Li et al., 2011). One possible explanation for this apparent discrepancy is that there are inherent differences in the responses measured [e.g., vocalizations (Li et al., 2011) vs reflexive abdominal contractions]. Our novel results demonstrate that ongoing mGluR5 activity in the right CeA modulates the visceromotor response to noxious bladder distention in naive (uninjured) mice.
The mechanism of the modulation of distention-evoked bladder pain by CeA-localized mGluR5 seems to be related in part to a modulation of ERK1/2 in the spinal cord. Our previous experiments have demonstrated that ERK1/2 in both the spinal cord and amygdala is involved in the modulation of acute spontaneous somatic pain and behavioral changes after injury (Carrasquillo and Gereau, 2007, 2008; Hu et al., 2007). Furthermore, we know from previous work that ERK1/2 is activated in the dorsal horn neurons of the lumbosacral spinal cord (lamina I, II, V, X) following bladder distention in mice with and without bladder inflammation (Lai et al., 2011). Our results suggest that noxious bladder distention in lightly anesthetized mice immediately activates ERK1/2 in the lumbosacral spinal cord, but not in the amygdala. Furthermore, genetic deletion of mGluR5 in the right CeA reduced activation of ERK1/2 in the lumbosacral spinal cord following bladder distention. In contrast, pharmacologic inhibition of mGluR5 with MPEP did not change the activation of ERK1/2 following noxious distention. This difference may be related to the long-term disruption that exists in the knock-out mice compared with the acute inhibition in the MPEP experiment, or a difference in the extent of mGluR5 inhibition achieved by the two approaches. Nonetheless, these data suggest the presence of a functional connection between the amygdala and the spinal cord nociceptive neurons.
To further test this connection, we activated the amygdala (via optogenetic or pharmacologic techniques) in the absence of bladder distention and measured ERK1/2 phosphorylation in the spinal cord. Finding no effect of amygdala activation on pERK1/2 in the spinal cord, these results suggest either that ERK1/2 modulation occurs only in the context of nociceptive input (e.g., bladder distention) and/or that there are other changes that occur in the spinal cord during amygdala activation. Nociceptive neurons in the CeA project to the PAG and then to the rostral ventromedial medulla (RVM) (Neugebauer et al., 2004). Descending inputs from the RVM exert facilitatory and inhibitory influence on nociceptive transmission in the spinal cord dorsal horn (Millan, 2002; Porreca et al., 2002; Ren and Dubner, 2002). Modulation of visceral nociception between the amygdala and the spinal cord could occur at the sensory fiber/spinal cord synapse (in the spinal cord dorsal horn), on modulatory interneurons, or potentially on motor neurons that mediate abdominal contractions during bladder distention. Future studies will be necessary to determine the precise location and mechanism of this interaction.
Overall, our results suggest that increased CeA output, driven by mGluR5 activation, in combination with bladder distention leads to modulation of ERK activation in the spinal dorsal horn. These results highlight the important role that limbic brain structures play in the regulation of peripheral responses to noxious stimuli. Furthermore, our results showing that mGluR5 inhibition modulates acute visceral nociception provide evidence for a novel role of the amygdala in the modulation of ongoing nociception in the absence of injury or external stress.
Footnotes
This work was supported by the Hope Center for Neurological Disorders and National Institutes of Health Grants F30 DK089969-02 (L.W.C.), F32 NS067761 (B.J.K.), R00DA025182 (M.R.B.), as well as R01 NS48602, and National Institute of Diabetes and Digestive and Kidney Diseases Multidisciplinary Approach to the Study of Chronic Pelvic Pain Network Grant DK82315 (R.W.G.).
The authors declare no competing financial interests.
References
- Baldoni F, Ercolani M, Baldaro B, Trombini G. Stressful events and psychological symptoms in patients with functional urinary disorders. Percept Mot Skills. 1995;80:605–606. doi: 10.2466/pms.1995.80.2.605. [DOI] [PubMed] [Google Scholar]
- Bernard JF, Besson JM. The spino(trigemino)pontoamygdaloid pathway: electrophysiological evidence for an involvement in pain processes. J Neurophysiol. 1990;63:473–490. doi: 10.1152/jn.1990.63.3.473. [DOI] [PubMed] [Google Scholar]
- Bernard JF, Bester H, Besson JM. Involvement of the spino-parabrachio-amygdaloid and -hypothalamic pathways in the autonomic and affective emotional aspects of pain. Prog Brain Res. 1996;107:243–255. doi: 10.1016/s0079-6123(08)61868-3. [DOI] [PubMed] [Google Scholar]
- Berry SH, Elliott MN, Suttorp M, Bogart LM, Stoto MA, Eggers P, Nyberg L, Clemens JQ. Prevalence of symptoms of bladder pain syndrome/interstitial cystitis among adult females in the United States. J Urol. 2011;186:540–544. doi: 10.1016/j.juro.2011.03.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgeais L, Gauriau C, Bernard JF. Projections from the nociceptive area of the central nucleus of the amygdala to the forebrain: a PHA-L study in the rat. Eur J Neurosci. 2001;14:229–255. doi: 10.1046/j.0953-816x.2001.01640.x. [DOI] [PubMed] [Google Scholar]
- Burstein R, Potrebic S. Retrograde labeling of neurons in the spinal cord that project directly to the amygdala or the orbital cortex in the rat. J Comp Neurol. 1993;335:469–485. doi: 10.1002/cne.903350402. [DOI] [PubMed] [Google Scholar]
- Carrasquillo Y, Gereau RW., 4th Activation of the extracellular signal-regulated kinase in the amygdala modulates pain perception. J Neurosci. 2007;27:1543–1551. doi: 10.1523/JNEUROSCI.3536-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrasquillo Y, Gereau RW., 4th Hemispheric lateralization of a molecular signal for pain modulation in the amygdala. Mol Pain. 2008;4:24. doi: 10.1186/1744-8069-4-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castroman P, Ness TJ. Vigor of visceromotor responses to urinary bladder distension in rats increases with repeated trials and stimulus intensity. Neurosci Lett. 2001;306:97–100. doi: 10.1016/s0304-3940(01)01886-9. [DOI] [PubMed] [Google Scholar]
- Cervero F, Laird JM. Visceral pain. Lancet. 1999;353:2145–2148. doi: 10.1016/S0140-6736(99)01306-9. [DOI] [PubMed] [Google Scholar]
- Covington HE, 3rd, Lobo MK, Maze I, Vialou V, Hyman JM, Zaman S, LaPlant Q, Mouzon E, Ghose S, Tamminga CA, Neve RL, Deisseroth K, Nestler EJ. Antidepressant effect of optogenetic stimulation of the medial prefrontal cortex. J Neurosci. 2010;30:16082–16090. doi: 10.1523/JNEUROSCI.1731-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrakov J, Kroenke K, Steers WD, Berde C, Zurakowski D, Freeman MR, Jackson JL. Pharmacologic management of painful bladder syndrome/interstitial cystitis: a systematic review. Arch Intern Med. 2007;167:1922–1929. doi: 10.1001/archinte.167.18.1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwood-Van Meerveld B, Gibson M, Gunter W, Shepard J, Foreman R, Myers D. Stereotaxic delivery of corticosterone to the amygdala modulates colonic sensitivity in rats. Brain Res. 2001;893:135–142. doi: 10.1016/s0006-8993(00)03305-9. [DOI] [PubMed] [Google Scholar]
- Han JS, Neugebauer V. Synaptic plasticity in the amygdala in a visceral pain model in rats. Neurosci Lett. 2004;361:254–257. doi: 10.1016/j.neulet.2003.12.027. [DOI] [PubMed] [Google Scholar]
- Hu HJ, Alter BJ, Carrasquillo Y, Qiu CS, Gereau RW., 4th Metabotropic glutamate receptor 5 modulates nociceptive plasticity via extracellular signal-regulated kinase-Kv4.2 signaling in spinal cord dorsal horn neurons. J Neurosci. 2007;27:13181–13191. doi: 10.1523/JNEUROSCI.0269-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda R, Takahashi Y, Inoue K, Kato F. NMDA receptor-independent synaptic plasticity in the central amygdala in the rat model of neuropathic pain. Pain. 2007;127:161–172. doi: 10.1016/j.pain.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Ji G, Neugebauer V. Hemispheric lateralization of pain processing by amygdala neurons. J Neurophysiol. 2009;102:2253–2264. doi: 10.1152/jn.00166.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji G, Neugebauer V. Reactive oxygen species are involved in group I mGluR-mediated facilitation of nociceptive processing in amygdala neurons. J Neurophysiol. 2010;104:218–229. doi: 10.1152/jn.00223.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji G, Fu Y, Ruppert KA, Neugebauer V. Pain-related anxiety-like behavior requires CRF1 receptors in the amygdala. Mol Pain. 2007;3:13. doi: 10.1186/1744-8069-3-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR. Peripheral and central mechanisms of inflammatory pain, with emphasis on MAP kinases. Curr Drug Targets Inflamm Allergy. 2004;3:299–303. doi: 10.2174/1568010043343804. [DOI] [PubMed] [Google Scholar]
- Kolber BJ, Roberts MS, Howell MP, Wozniak DF, Sands MS, Muglia LJ. Central amygdala glucocorticoid receptor action promotes fear-associated CRH activation and conditioning. Proc Natl Acad Sci U S A. 2008;105:12004–12009. doi: 10.1073/pnas.0803216105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolber BJ, Montana MC, Carrasquillo Y, Xu J, Heinemann SF, Muglia LJ, Gereau RW., 4th Activation of metabotropic glutamate receptor 5 in the amygdala modulates pain-like behavior. J Neurosci. 2010;30:8203–8213. doi: 10.1523/JNEUROSCI.1216-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai HH, Qiu CS, Crock LW, Morales ME, Ness TJ, Gereau RW., 4th Activation of spinal extracellular signal-regulated kinases (ERK) 1/2 is associated with the development of visceral hyperalgesia of the bladder. Pain. 2011;152:2117–2124. doi: 10.1016/j.pain.2011.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Neugebauer V. Differential roles of mGluR1 and mGluR5 in brief and prolonged nociceptive processing in central amygdala neurons. J Neurophysiol. 2004;91:13–24. doi: 10.1152/jn.00485.2003. [DOI] [PubMed] [Google Scholar]
- Li Z, Ji G, Neugebauer V. Mitochondrial reactive oxygen species are activated by mGluR5 through IP3 and activate ERK and PKA to increase excitability of amygdala neurons and pain behavior. J Neurosci. 2011;31:1114–1127. doi: 10.1523/JNEUROSCI.5387-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macaulay AJ, Stern RS, Holmes DM, Stanton SL. Micturition and the mind: psychological factors in the aetiology and treatment of urinary symptoms in women. Br Med J (Clin Res Ed) 1987;294:540–543. doi: 10.1136/bmj.294.6571.540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millan MJ. Descending control of pain. Prog Neurobiol. 2002;66:355–474. doi: 10.1016/s0301-0082(02)00009-6. [DOI] [PubMed] [Google Scholar]
- Myers B, Greenwood-Van Meerveld B. Elevated corticosterone in the amygdala leads to persistent increases in anxiety-like behavior and pain sensitivity. Behav Brain Res. 2010;214:465–469. doi: 10.1016/j.bbr.2010.05.049. [DOI] [PubMed] [Google Scholar]
- Ness TJ, Elhefni H. Reliable visceromotor responses are evoked by noxious bladder distention in mice. J Urol. 2004;171:1704–1708. doi: 10.1097/01.ju.0000116430.67100.8f. [DOI] [PubMed] [Google Scholar]
- Ness TJ, Gebhart GF. Colorectal distension as a noxious visceral stimulus: physiologic and pharmacologic characterization of pseudaffective reflexes in the rat. Brain Res. 1988;450:153–169. doi: 10.1016/0006-8993(88)91555-7. [DOI] [PubMed] [Google Scholar]
- Ness TJ, Gebhart GF. Interactions between visceral and cutaneous nociception in the rat. I. Noxious cutaneous stimuli inhibit visceral nociceptive neurons and reflexes. J Neurophysiol. 1991;66:20–28. doi: 10.1152/jn.1991.66.1.20. [DOI] [PubMed] [Google Scholar]
- Ness TJ, Richter HE, Varner RE, Fillingim RB. A psychophysical study of discomfort produced by repeated filling of the urinary bladder. Pain. 1998;76:61–69. doi: 10.1016/s0304-3959(98)00023-2. [DOI] [PubMed] [Google Scholar]
- Ness TJ, Lewis-Sides A, Castroman P. Characterization of pressor and visceromotor reflex responses to bladder distention in rats: sources of variability and effect of analgesics. J Urol. 2001;165:968–974. [PubMed] [Google Scholar]
- Neugebauer V. The amygdala: different pains, different mechanisms. Pain. 2007;127:1–2. doi: 10.1016/j.pain.2006.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neugebauer V, Li W, Bird GC, Han JS. The amygdala and persistent pain. Neuroscientist. 2004;10:221–234. doi: 10.1177/1073858403261077. [DOI] [PubMed] [Google Scholar]
- Neve RL, Neve KA, Nestler EJ, Carlezon WA., Jr Use of herpes virus amplicon vectors to study brain disorders. BioTechniques. 2005;39:381–391. doi: 10.2144/05393PS01. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. Ed 2. San Diego: Academic; 2001. [Google Scholar]
- Porreca F, Ossipov MH, Gebhart GF. Chronic pain and medullary descending facilitation. Trends Neurosci. 2002;25:319–325. doi: 10.1016/s0166-2236(02)02157-4. [DOI] [PubMed] [Google Scholar]
- Qin C, Greenwood-Van Meerveld B, Foreman RD. Spinal neuronal responses to urinary bladder stimulation in rats with corticosterone or aldosterone onto the amygdala. J Neurophysiol. 2003;90:2180–2189. doi: 10.1152/jn.00298.2003. [DOI] [PubMed] [Google Scholar]
- Ren K, Dubner R. Descending modulation in persistent pain: an update. Pain. 2002;100:1–6. doi: 10.1016/s0304-3959(02)00368-8. [DOI] [PubMed] [Google Scholar]
- Traub RJ, Silva E, Gebhart GF, Solodkin A. Noxious colorectal distention induced-c-Fos protein in limbic brain structures in the rat. Neurosci Lett. 1996;215:165–168. doi: 10.1016/0304-3940(96)12978-5. [DOI] [PubMed] [Google Scholar]
- Warren JW, Howard FM, Cross RK, Good JL, Weissman MM, Wesselmann U, Langenberg P, Greenberg P, Clauw DJ. Antecedent nonbladder syndromes in case-control study of interstitial cystitis/painful bladder syndrome. Urology. 2009;73:52–57. doi: 10.1016/j.urology.2008.06.031. [DOI] [PubMed] [Google Scholar]
- Xu J, Zhu Y, Contractor A, Heinemann SF. mGluR5 has a critical role in inhibitory learning. J Neurosci. 2009;29:3676–3684. doi: 10.1523/JNEUROSCI.5716-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Gradinaru V, Adamantidis AR, Durand R, Airan RD, de Lecea L, Deisseroth K. Optogenetic interrogation of neural circuits: technology for probing mammalian brain structures. Nat Protoc. 2010;5:439–456. doi: 10.1038/nprot.2009.226. [DOI] [PMC free article] [PubMed] [Google Scholar]