

Abstract
Melanoma differentiation-associated protein 5 (MDA5) detects viral dsRNA in the cytoplasm. On binding of RNA, MDA5 forms polymers, which trigger assembly of the signaling adaptor mitochondrial antiviral-signaling protein (MAVS) into its active fibril form. The molecular mechanism of MDA5 signaling is not well understood, however. Here we show that MDA5 forms helical filaments on dsRNA and report the 3D structure of the filaments using electron microscopy (EM) and image reconstruction. MDA5 assembles into a polar, single-start helix around the RNA. Fitting of an MDA5 homology model into the structure suggests a key role for the MDA5 C-terminal domain in cooperative filament assembly. Our study supports a signal transduction mechanism in which the helical array of MDA5 within filaments nucleates the assembly of MAVS fibrils. We conclude that MDA5 is a polymerization-dependent signaling platform that uses the amyloid-like self-propagating properties of MAVS to amplify signaling.
Keywords: innate immune receptor, ligand-binding cooperativity, nucleic acid sensor, prion-like switch, DExD/H-box RNA helicase
Innate immune receptors detect broadly conserved microbial structures throughout the cell, inducing a rapid inflammatory response. In the cytoplasm, retinoic acid-inducible gene 1 (RIG-I) recognizes the 5′-triphosphate-capped blunt ends of plus-stranded RNA viruses, whereas melanoma differentiation-associated protein 5 (MDA5) recognizes long cytosolic dsRNA delivered or generated during viral infection (1–3). RIG-I and MDA5 each contain two N-terminal caspase recruitment domains (CARDs), a DExD/H-box helicase (consisting of two RecA-like helicase domains, Hel1 and Hel2, and an insert domain, Hel2i), and a C-terminal domain (CTD). RIG-I adopts a closed inactive conformation in the absence of RNA (4). RNA binding through the helicase domain and CTD (5, 6) releases the CARDs, which then recruit unanchored lysine 63-linked polyubiquitin chains and activate the signaling adaptor mitochondrial antiviral-signaling protein (MAVS, also called interferon-β promoter stimulator 1, or IPS-1) (7). In contrast, MDA5 does not sequester its CARDs (8) and cooperatively assembles into ATP-sensitive filaments on dsRNA (8, 9). Moreover, the MDA5 CTD is required for cooperative filament assembly, but not for RNA binding (8, 10, 11). The MDA5 CARDs have been proposed to nucleate the assembly of MAVS into its active fibril form (8, 12) in a process involving free polyubiquitin chains (13). The prion- or amyloid-like properties of MAVS fibrils amplify signaling, culminating in activation of NF-κB (12).
The recent discovery of MDA5 filament assembly on dsRNA suggests a signaling mechanism in which the regular arrangement of MDA5 CARDs on the outside of the filaments serves as a scaffold for the nucleation of the MAVS fibril assembly. However, a more detailed structural picture of the MDA5-RNA filament is needed to gain a mechanistic understanding at the molecular level of how MDA5 generates an innate immune signal. Here we report the 3D structure of the MDA5-dsRNA filaments detected using EM.
Results and Discussion
MDA5 Forms a Polar Helical Filament with Variable Twist Around dsRNA.
MDA5-RNA filaments have been described previously as rings stacked with a ∼44-Å spacing, but no evidence of a helical arrangement of these rings has been reported (9). We have determined that the apparent lack of helicity stems from the variable twist within the filaments. We used EM to image negatively stained MDA5-dsRNA filaments formed in the presence of either ATP or the slowly hydrolyzable ATP analog ATP-γ-S (Fig. 1 A and B). The average power spectrum from 9,154 overlapping 100-pixel (416 Å) segments of MDA5-dsRNA-ATP-γ-S filaments shows a strong meridional reflection at ∼1/44 Å−1 (Fig. 1D), which arises from the spacing of the rings. A weak layer line is visible at ∼1/55 Å−1. Given the distance of this layer line from the meridian, and considering the diameter of the filaments (∼100 Å), this layer line can arise only from a one-start helix. Thus, these two reflections determine the helical symmetry of these filaments. We generated a reconstruction by the iterative helical real space reconstruction method (14) using all of the segments. We then used this global reconstruction for sorting with different helical rise and twist values.
Fig. 1.

Image reconstruction of MDA5-dsRNA filaments. (A and B) Negatively stained electron micrographs of MDA5 bound to viral genomic dsRNA with ATP (A) and ATP-γ-S (B). (C) ΔCARD-MDA5 with ATP-γ-S. (D) Power spectrum from 9,154 overlapping segments of the ATP-γ-S filaments in B showing a strong meridional layer line (blue arrow) and a weak near-meridional layer line (red arrow). (E) Improved power spectrum after sorting by twist (1,362 segments). (F and G) Reconstructions from this subset (F) and from ΔCARD-MDA5 filaments with ATP-γ-S (G) revealing an asymmetric unit of four domains (labeled 1–4 in F) arranged as a ring around a central RNA cavity (4-σ contour). (H) At the 1-σ contour level, two lobes of density extend from domain 1 in full-length MDA5 (gray) but not in ΔCARD-MDA5 (blue), identifying the approximate position of the CARDs or the CARD–Hel1 linker (black arrows). A difference map shows density at the same two positions (Fig. S2).
We found that most of the variability was in the twist, making the MDA5-dsRNA filaments similar to polymers such as F-actin that have a regular rise but variable twist (15). After sorting by twist, the power spectrum from a subset of filaments (Fig. 1E) was much improved over the global power spectrum. Differences in the power spectra from three different twist states (74°, 78°, or 84°) explain why the layer lines other than the strong meridional reflection are blurred out in a global average (Movie S1). There was no evidence of discrete twist states, and grouping into three classes was arbitrary. The reconstruction shows that the asymmetric unit is a ring-like set of four domains, designated domains 1–4) (Fig. 1F). Filaments formed with ADP or ATP (Fig. 1A) were similar to filaments formed with ATP-γ-S, although small nucleotide-dependent differences cannot be ruled out (Movie S2). The resolution was judged to be ∼22 Å using the Fourier shell correlation = 0.5 criterion (Fig. S1).
Identification of the MDA5 Hel1 Domain and Its Link to the CARDs in the EM Structure.
To begin assigning the MDA5 domains in the EM structure (Fig. 2A), we prepared filaments on dsRNA with ATP-γ-S using a fragment of MDA5, ΔCARD-MDA5, lacking the two N-terminal CARD domains (Fig. 1C). Although these filaments appeared narrower than those formed by full-length MDA5, the reconstruction was surprisingly similar (Fig. 1G); however, at lower contour levels, the peripheral density in the reconstruction of full-length MDA5 was absent in ΔCARD-MDA5 (Fig. 1H). Our interpretation of this finding is that the two CARD domains are at a larger radius in the filament and are not rigidly oriented with respect to the filament, consistent with solution scattering data for unliganded MDA5 (8). Thus, this peripheral density contributes to the filament width in raw micrographs (Fig. 1 A and B), but is largely lost in the reconstruction by the imposed helical symmetry. This interpretation is confirmed in a statistical difference map between the full-length and ΔCARD reconstructions (Fig. S2). The linkage observed between the peripheral density and domain 1 identifies this domain as Hel1 (Fig. 1H).
Fig. 2.

Atomic model of the MDA5-dsRNA filament with ATP-γ-S present. (A) Domain organization of ΔCARD-MDA5. (B) Atomic homology models fitted into the ΔCARD-MDA5 reconstruction. The C terminus of Hel1 (blue dot) may connect to CTD N termini in the same or adjacent rings (red dots), 20 Å and 38 Å away, respectively. (C and D) Ring contacts are formed by a Hel1–CTD interface (C) and a Hel1–Hel2 interface (with minor Hel1–Hel1 contacts) (D). Note that the C-terminal sequence of the CTD was modeled in an extended conformation (Materials and Methods). (E) The ring-invading Hel1-CTD model results in an intermolecular interface between the Hel2i domain (medium gray) and the CTD (cyan). One ΔCARD-MDA5 molecule is shown as a surface; adjacent molecules are shown as cartoons.
Atomic Homology Model of the MDA5-RNA Filament.
To identify the remaining MDA5 domains in the EM density, we fitted a homology model of ΔCARD-MDA5 based on the structures of MDA5 fragments (8, 10) and of RIG-I bound to A-form RNA and nucleotide (5) into the density. The reconstruction had an enantiomorphic ambiguity, because both the volume shown and its mirror image were equally consistent with the observed projections. The fit was better for one of the two enantiomers (correlation, 0.96 vs. 0.92). In both enantiomers, the Hel1, Hel2, and Hel2i domains and the CTD of MDA5 fit into domains 1–4, respectively. In the best-fitting enantiomer (i.e., one with a right-handed one-start helix connecting adjacent rings), the helix bridging Hel1 and Hel2 was in a density that contacted Hel1 of an adjacent ring (Fig. S3). The right-handed helix of the best-fitting model was confirmed by fast-freeze/deep-etch EM (Fig. S3 C–E) (16). The only major change in relative domain orientations with respect to the RIG-I structure was in the position of the CTD, which obstructed the dsRNA cavity before refinement. Rigid body refinement placed the CTD into domain 4 without significantly altering the positions of the other domains (Fig. S4). The refined model fit well in the EM volume, except for the eight-residue linker between Hel1 and the CTD (Fig. 2B). Assuming an A-form RNA, the 44-Å axial rise per ring was consistent with 14–16 bp per MDA5, and with the 16- to 18-bp footprint of MDA5 determined from RNase protection assays (8). Understanding the details of the interaction with RNA will require higher-resolution studies, however.
Structural Basis of Cooperative Filament Assembly.
We previously showed that the MDA5 CTD is required for cooperative filament assembly, and proposed that this cooperativity is what gives MDA5 its specificity for long dsRNA ligands (8). Thus, cooperative filament assembly likely originates from intermolecular contacts involving the CTD. There are three main contacts between rings in the structure, between the Hel1 domain and the Hel1 domain, Hel2 domain, and CTD in adjacent rings, respectively (Fig. 2 C and D). Notably, the regions involved in the Hel1–Hel2 and Hel1–CTD intermolecular contacts each contain sequence motifs that are conserved across species in MDA5, but not in RIG-I (Fig. S5), supporting the notion that these regions might have evolved to promote MDA5 filament assembly. However, the lack of connectivity in the EM structure between the Hel1 domain and the CTD within rings, and the presence of density at the Hel1–CTD interface between rings, suggest an alternative mechanism of filament assembly. Instead of occupying an entire ring, each MDA5 molecule may span two rings, with the CTD “invading” an adjacent ring and forming contacts with the Hel2i domain of the adjacent molecule (Fig. 2E). Thus, in this ring-invading model, the Hel2i–CTD interface within each ring of density is an intermolecular interface. In this configuration, the linker between the Hel1 domain and the CTD would lie within the EM density (Fig. 2B, right dashed line). In contrast, the linker would lie outside the density in a noninvading model (Fig. 2B, left dashed line).
In support of the ring-invading model, small-angle X-ray scattering data suggest that Hel2i–CTD contacts are the main intermolecular contacts when two MDA5 molecules bind a 20-bp dsRNA (8), although MDA5 adopts a much more open conformation in this complex than in the filament, presumably owing to the constraints imposed by the short length of the 20-bp ligand, which is shorter than the ∼30 bp of dsRNA contained in two asymmetric units of the filament. The distance between the end of the Hel1 bridging helix and the N terminus of the CTD is 20 Å within rings and 38 Å in the ring-invading model (Fig. 2 B and E). The eight-residue Hel1–CTD linker could bridge a distance of up to 35 Å in a fully extended conformation, consistent with both stacked-ring and ring-invading connectivities. The actual connectivity remains to be confirmed; however, we propose that flexibility of the Hel1–CTD linker, along with the relatively small number of contact between rings, allow for the large variability in twist between rings.
Implications for the MDA5 Signaling Mechanism.
The biological relevance of the MDA5-dsRNA EM structure reported here has experimental support. First, both the Φ6 dsRNA used in the present study and a synthetic Φ6 dsRNA segment activate IFN-β signaling in an MDA5-dependent manner in cell-based luciferase reporter assays (Fig. S6) (17). Second, the MDA5 loop-deletion mutant used in this study (with residues 646–663 deleted) has comparable signaling activity to WT MDA5 (Fig. S6). Third, given that filament formation is inhibited by the ATPase activity of MDA5 (8), and that ATPase-deficient mutants of MDA5 exhibit constitutive signaling in cells (18), filaments—or filament-like structures—may be involved in signaling. Similarly, a natural partial loss-of-function mutant in the MDA5 CTD (I923V) reduces binding cooperativity and filament formation in vitro (9) while reducing signaling in vivo (19, 20), supporting a physiological role for filaments in signaling.
The discovery that MDA5 polymerizes into helical filaments along dsRNA supports a signal transduction mechanism in which the helical array of MDA5 CARDs on the outside of MDA5-RNA filaments recruits MAVS and nucleates its assembly into an active fibril form. The amyloid-like properties of MAVS fibrils then propagate and amplify the signaling cascade (12). Thus, the MDA5 CARDs do not serve not as repressors of the helicase, but rather provide a polymerization-dependent signaling platform. This mechanism follows the ongoing shift in the paradigm of how innate and apoptotic signals are transduced, from prototypical linear signaling cascades to oligomeric platform-based signal amplification (21–23). High-resolution structural studies are needed to understand how MAVS binds the MDA5 CARDs and how MAVS forms fibrils, and how these processes are promoted by unanchored ubiquitin chains.
Materials and Methods
Expression and Purification of MDA5 Constructs.
Full-length MDA5 and ΔCARD-MDA5 were expressed and purified as described previously (8). In brief, amino acid residues 1–1026 and 304–1026 of mouse MDA5 were expressed in Escherichia coli Rosetta(DE3) cells (Novagen). In both constructs, residues 646–663 were deleted. The proteins were purified by nickel-affinity, anion-exchange, cation-exchange, and size-exclusion chromatography (in that order).
IFN Signaling Assay.
HEK293T cells in six-well plates were transfected with 178 ng/mL of firefly luciferase under control of the IFN-β promoter (pIFN-Luc) (24), 30 ng/mL of Renilla luciferase (pRL-TK) (Promega), and 15 ng/mL of empty pCDNA3.1 vector, WT human MDA5 (pEFBos MDA5) (25), or human MDA5-Δ(644-663) [pEFBos MDA5-Δ(644-663) generated from pEFBos MDA5 by site-directed mutagenesis]. All transfections were performed with Lipofectamine 2000 (Invitrogen). After expression for 24 h, cells were transfected with genomic dsRNA from Φ6 (Thermo Scientific) at the indicated concentrations. After 16 h, cell lysates were prepared, and luciferase activity was measured using Promega assay kits in accordance with the manufacturer’s instructions. Firefly luciferase activity was normalized to cotransfected Renilla luciferase under a constitutive promoter.
EM Image Reconstruction.
The MDA5-dsRNA complexes were formed in 25 mM triethanolamine-HCl (Fisher) buffer (pH 7.2) at 37 °C for 10 min, with 2 μM MDA5, Φ6 bacteriophage dsRNA (New England Biolabs) at a ratio of 1:40 (wt/wt) RNA:MDA5, and 1.25 mM ATP-γ-S (Boehringer) or ATP (Sigma-Aldrich). Samples were applied to glow-discharged carbon-coated grids, negatively stained with uranyl acetate [2% (wt/vol)], and imaged with an FEI Tecnai 12 transmission electron microscope at an accelerating voltage of 80 keV. Micrographs were scanned with a Nikon Coolscan 8000 at a raster of 4.16 Å per pixel. Image reconstructions were performed using images from samples containing ATP-γS. The helixboxer routine in EMAN (26) was used to cut filaments from micrographs. The SPIDER software package (27) was used for most of the subsequent processing. For the ΔCARD-MDA5-dsRNA-ATP-γ-S filaments, 8,953 overlapping segments (100 pixels long) were collected, with a shift of 16 pixels between adjacent segments. A global reconstruction was generated, after which segments were sorted by twist and axial rise. This was done by generating three different values for the axial rise and three different values for the twist, and imposing these new helical parameters on the global reconstruction to produce nine different volumes. These nine volumes were used for reference-based sorting via cross-correlations between the actual images and the projections of these volumes. It was found that most of the variability in the images could be accounted for by a variable twist. A subset containing 1,438 segments was used for the reconstruction shown, which had a twist of 83.7° and a rise of 43.5 Å. For the MDA5-dsRNA-ATP-γ-S filaments, the subset used (n = 1,362) had a twist of 82.7° and a rise of 43.6 Å. The EM reconstruction has been deposited in EMDataBank under accession no. EMD-5444.
Homology Modeling and Fitting of the Model into the EM Structure.
A homology model of mouse ΔCARD-MDA5 (304–1026) was generated with MODELER v9.10 (28) using the crystal structures of human MDA5 Hel1 [Protein Data Bank (PDB) ID code 3B6E], mouse MDA5 Hel2i (PDB ID code 3TS9) (8), human MDA5 CTD (PDB ID code 3GA3) (10), and RIG-I bound to ADP-BeF3 and a 14-bp dsRNA (PDB ID code 3TMI) as templates (5). In the MDA5 CTD crystal structure, a motif at the C terminus conserved across species in MDA5 but absent in RIG-I (Fig. S3A) was replaced by a hexahistadine tag and formed an α-helix because of crystal packing. This region was flexible and extended in NMR models of the MDA5 CTD (11), and thus was modeled speculatively as an extended structure here to illustrate its complementary properties to the adjacent surface on Hel1 in the filament (Fig. 2C). The MDA5 homology model was initially fitted to both enantiomers of the reconstructed volume with UCSF Chimera (29). Homology models generated using the other RNA-bound RIG-I structure (PDB ID code 2YKG) (6) resulted in a much poorer fit. Helically symmetric models were then generated and simultaneously fitted to the volume. A filament model with five MDA5 molecules was refined with Situs (30) to iteratively refine domain positions and impose helical symmetry. The energy of the model was then minimized with Chiron (31).
Supplementary Material
Acknowledgments
We thank Robyn Roth (Washington University in St. Louis) for fast-freeze/deep-etch electron microscopy. This work was supported by National Institutes of Health Grants P01 GM022778 (to Y.M.) and R01 GM035269 (to E.H.E.), and by a Burroughs Wellcome Investigator in the Pathogenesis of Infectious Disease Award (to Y.M.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The EM reconstruction has been deposited in EMDataBank, www.emdatabank.org (accession no. EMD-5444).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1212186109/-/DCSupplemental.
References
- 1.Kato H, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441(7089):101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- 2.Hornung V, et al. 5′-Triphosphate RNA is the ligand for RIG-I. Science. 2006;314(5801):994–997. doi: 10.1126/science.1132505. [DOI] [PubMed] [Google Scholar]
- 3.Pichlmair A, et al. RIG-I–mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science. 2006;314(5801):997–1001. doi: 10.1126/science.1132998. [DOI] [PubMed] [Google Scholar]
- 4.Kowalinski E, et al. Structural basis for the activation of innate immune pattern-recognition receptor RIG-I by viral RNA. Cell. 2011;147(2):423–435. doi: 10.1016/j.cell.2011.09.039. [DOI] [PubMed] [Google Scholar]
- 5.Jiang F, et al. Structural basis of RNA recognition and activation by innate immune receptor RIG-I. Nature. 2011;479(7373):423–427. doi: 10.1038/nature10537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Luo D, et al. Structural insights into RNA recognition by RIG-I. Cell. 2011;147(2):409–422. doi: 10.1016/j.cell.2011.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zeng W, et al. Reconstitution of the RIG-I pathway reveals a signaling role of unanchored polyubiquitin chains in innate immunity. Cell. 2010;141(2):315–330. doi: 10.1016/j.cell.2010.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berke IC, Modis Y. MDA5 cooperatively forms dimers and ATP-sensitive filaments upon binding double-stranded RNA. EMBO J. 2012;31(7):1714–1726. doi: 10.1038/emboj.2012.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peisley A, et al. Cooperative assembly and dynamic disassembly of MDA5 filaments for viral dsRNA recognition. Proc Natl Acad Sci USA. 2011;108(52):21010–21015. doi: 10.1073/pnas.1113651108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li X, et al. Structural basis of double-stranded RNA recognition by the RIG-I like receptor MDA5. Arch Biochem Biophys. 2009;488(1):23–33. doi: 10.1016/j.abb.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 11.Takahasi K, et al. Solution structures of cytosolic RNA sensor MDA5 and LGP2 C-terminal domains: Identification of the RNA recognition loop in RIG-I-like receptors. J Biol Chem. 2009;284(26):17465–17474. doi: 10.1074/jbc.M109.007179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hou F, et al. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell. 2011;146(3):448–461. doi: 10.1016/j.cell.2011.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jiang X, et al. Ubiquitin-induced oligomerization of the RNA sensors RIG-I and MDA5 activates antiviral innate immune response. Immunity. 2012;36(6):959–973. doi: 10.1016/j.immuni.2012.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Egelman EH. A robust algorithm for the reconstruction of helical filaments using single-particle methods. Ultramicroscopy. 2000;85(4):225–234. doi: 10.1016/s0304-3991(00)00062-0. [DOI] [PubMed] [Google Scholar]
- 15.Egelman EH, Francis N, DeRosier DJ. F-actin is a helix with a random variable twist. Nature. 1982;298(5870):131–135. doi: 10.1038/298131a0. [DOI] [PubMed] [Google Scholar]
- 16.Heuser J. Preparing biological samples for stereomicroscopy by the quick-freeze, deep-etch, rotary-replication technique. Methods Cell Biol. 1981;22:97–122. doi: 10.1016/s0091-679x(08)61872-5. [DOI] [PubMed] [Google Scholar]
- 17.Jiang M, et al. Innate immune responses in human monocyte-derived dendritic cells are highly dependent on the size and the 5′ phosphorylation of RNA molecules. J Immunol. 2011;187(4):1713–1721. doi: 10.4049/jimmunol.1100361. [DOI] [PubMed] [Google Scholar]
- 18.Bamming D, Horvath CM. Regulation of signal transduction by enzymatically inactive antiviral RNA helicase proteins MDA5, RIG-I, and LGP2. J Biol Chem. 2009;284(15):9700–9712. doi: 10.1074/jbc.M807365200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chistiakov DA, Voronova NV, Savost’Anov KV, Turakulov RI. Loss-of-function mutations E6 27X and I923V of IFIH1 are associated with lower poly(I:C)-induced interferon-β production in peripheral blood mononuclear cells of type 1 diabetes patients. Hum Immunol. 2010;71(11):1128–1134. doi: 10.1016/j.humimm.2010.08.005. [DOI] [PubMed] [Google Scholar]
- 20.Shigemoto T, et al. Identification of loss of function mutations in human genes encoding RIG-I and MDA5: Implications for resistance to type I diabetes. J Biol Chem. 2009;284(20):13348–13354. doi: 10.1074/jbc.M809449200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Park HH, et al. Death domain assembly mechanism revealed by crystal structure of the oligomeric PIDDosome core complex. Cell. 2007;128(3):533–546. doi: 10.1016/j.cell.2007.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin SC, Lo YC, Wu H. Helical assembly in the MyD88-IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature. 2010;465(7300):885–890. doi: 10.1038/nature09121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mace PD, Riedl SJ. Molecular cell death platforms and assemblies. Curr Opin Cell Biol. 2010;22(6):828–836. doi: 10.1016/j.ceb.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fredericksen B, et al. Activation of the interferon-beta promoter during hepatitis C virus RNA replication. Viral Immunol. 2002;15(1):29–40. doi: 10.1089/088282402317340215. [DOI] [PubMed] [Google Scholar]
- 25.Rothenfusser S, et al. The RNA helicase Lgp2 inhibits TLR-independent sensing of viral replication by retinoic acid-inducible gene-I. J Immunol. 2005;175(8):5260–5268. doi: 10.4049/jimmunol.175.8.5260. [DOI] [PubMed] [Google Scholar]
- 26.Ludtke SJ, Baldwin PR, Chiu W. EMAN: Semiautomated software for high-resolution single-particle reconstructions. J Struct Biol. 1999;128(1):82–97. doi: 10.1006/jsbi.1999.4174. [DOI] [PubMed] [Google Scholar]
- 27.Frank J, et al. SPIDER and WEB: Processing and visualization of images in 3D electron microscopy and related fields. J Struct Biol. 1996;116(1):190–199. doi: 10.1006/jsbi.1996.0030. [DOI] [PubMed] [Google Scholar]
- 28.Eswar N, et al. Comparative protein structure modeling using Modeller. Curr Protoc Bioinformatics. 2006;15:5.6.1–5.6.30. doi: 10.1002/0471250953.bi0506s15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pettersen EF, et al. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 30.Wriggers W. Using Situs for the integration of multi-resolution structures. Biophys Rev. 2010;2(1):21–27. doi: 10.1007/s12551-009-0026-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ramachandran S, Kota P, Ding F, Dokholyan NV. Automated minimization of steric clashes in protein structures. Proteins. 2011;79(1):261–270. doi: 10.1002/prot.22879. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
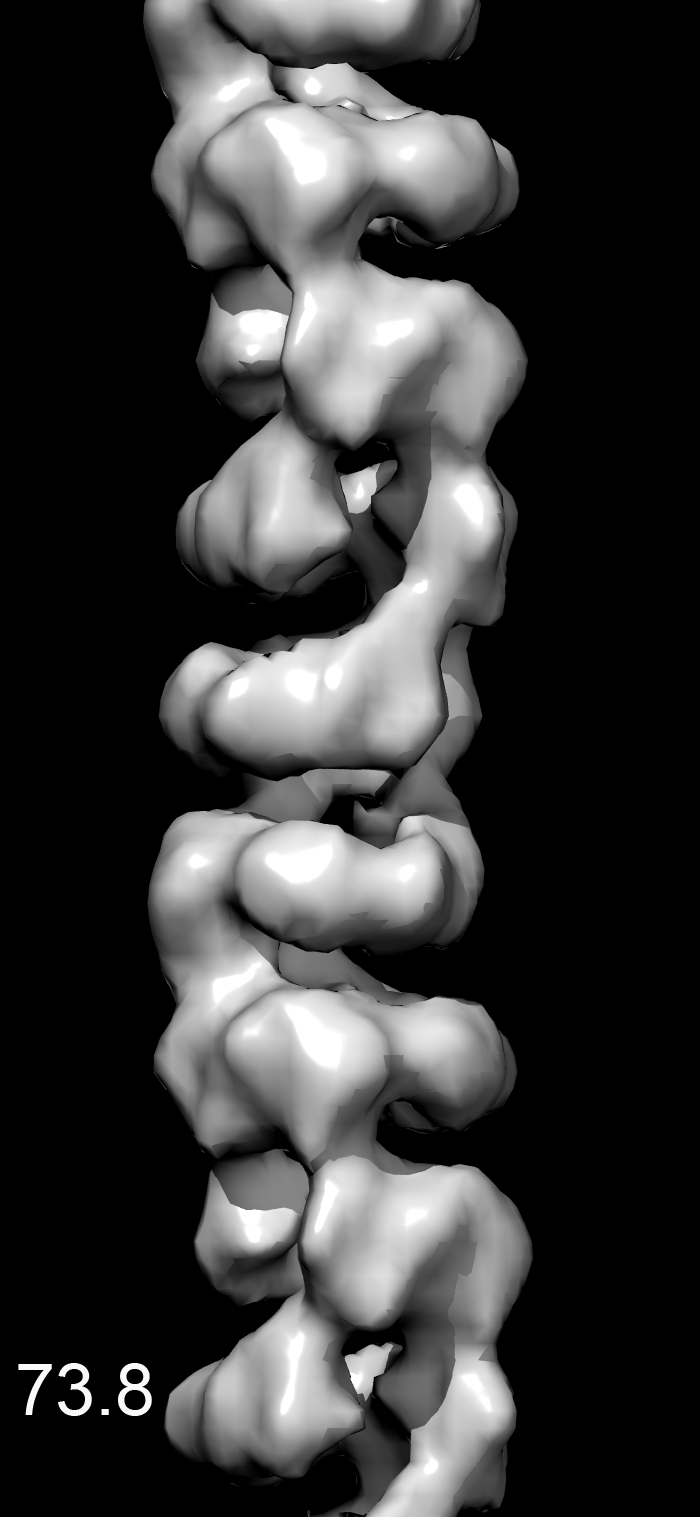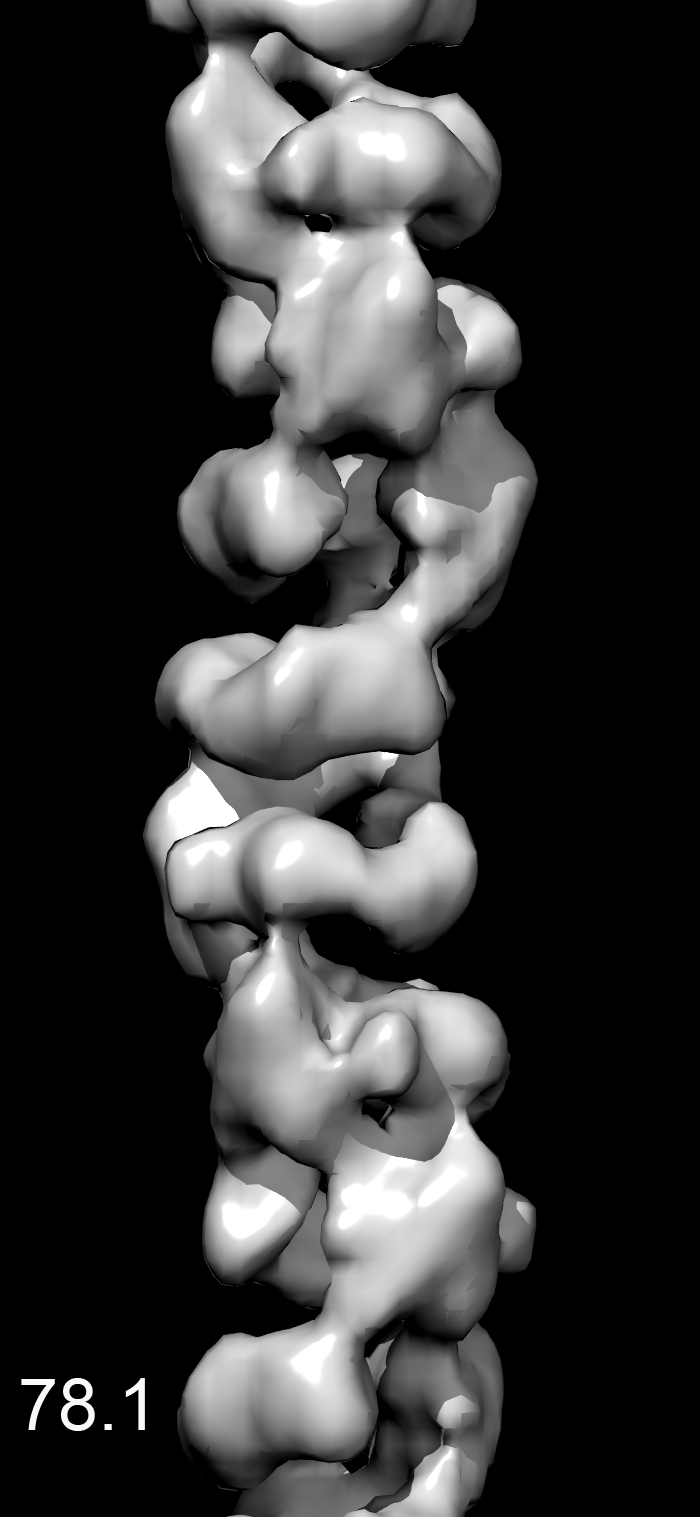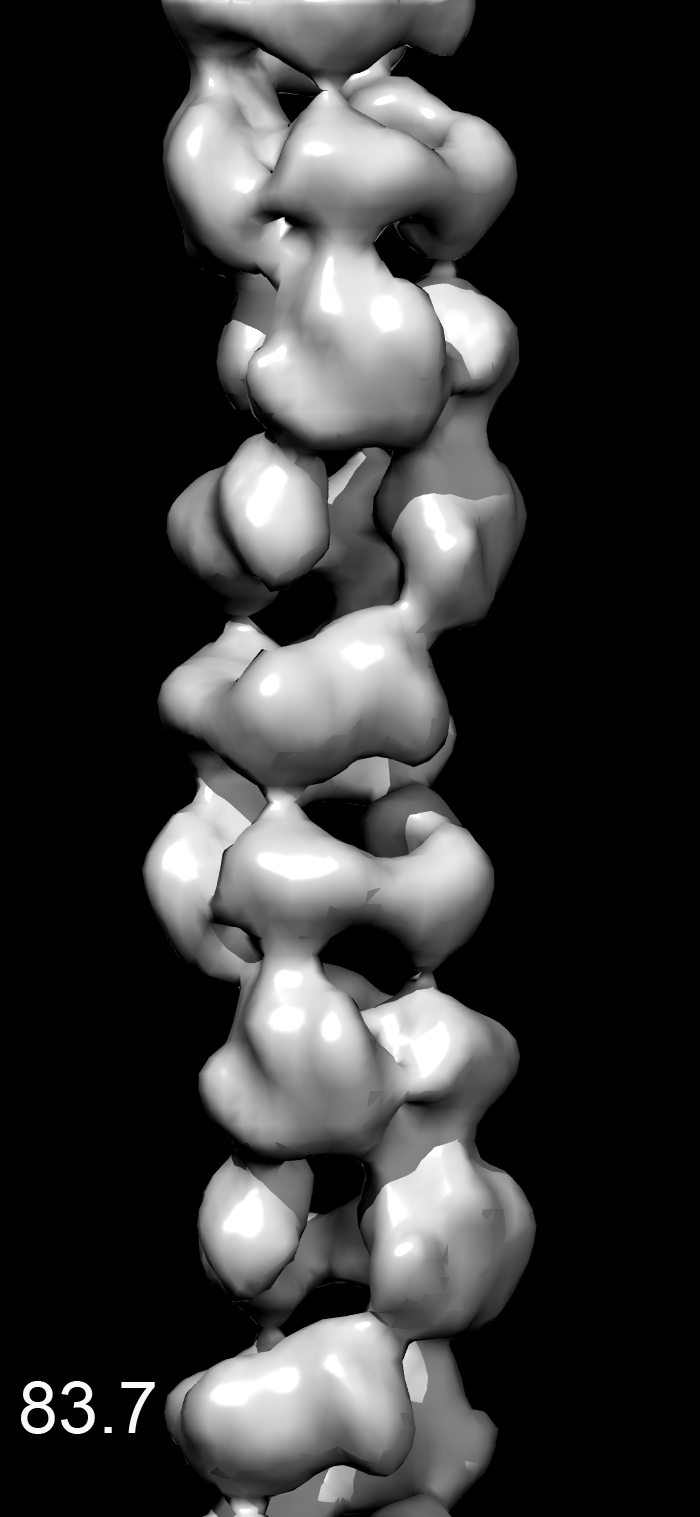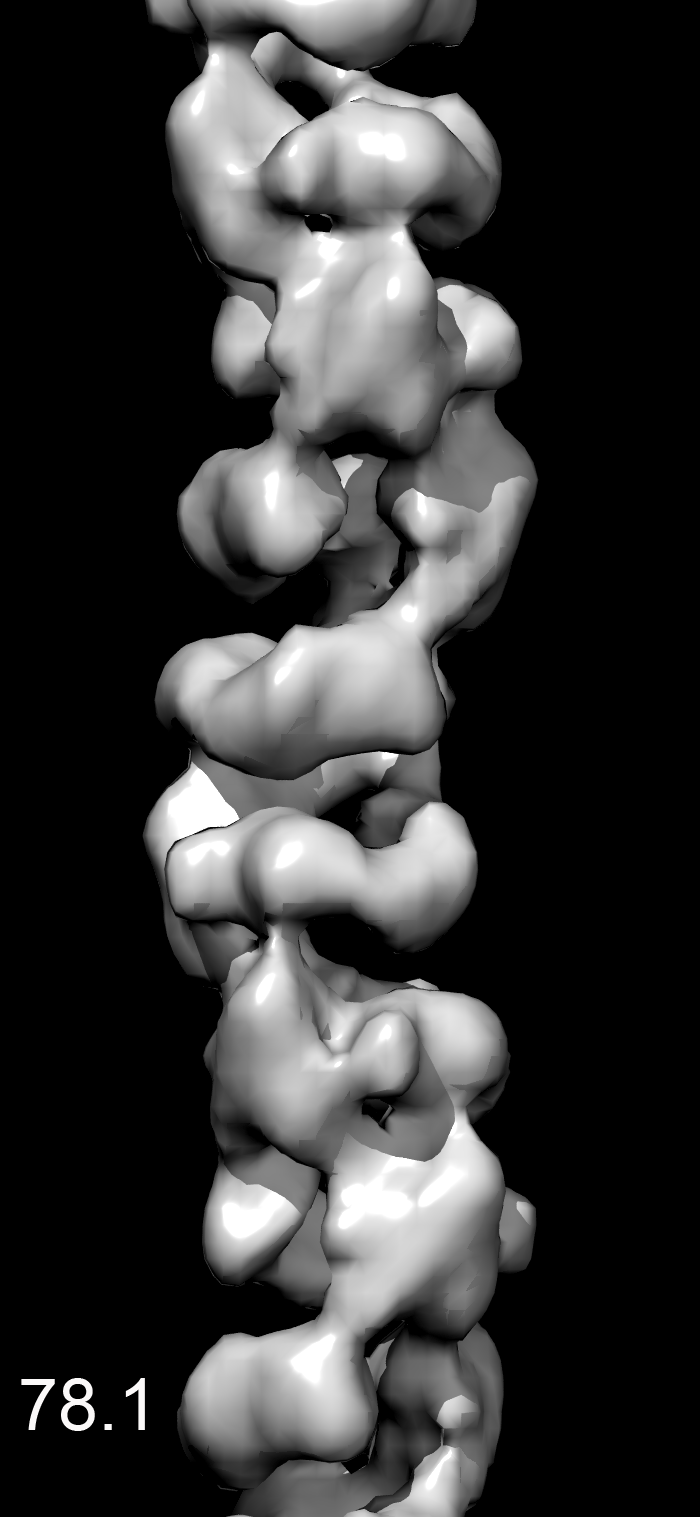{kind=link}