

Abstract
Gastric H+,K+-ATPase is responsible for gastric acid secretion. ATP-driven H+ uptake into the stomach is efficiently accomplished by the exchange of an equal amount of K+, resulting in a luminal pH close to 1. Because of the limited free energy available for ATP hydrolysis, the stoichiometry of transported cations is thought to vary from 2H+/2K+ to 1H+/1K+ per hydrolysis of one ATP molecule as the luminal pH decreases, although direct evidence for this hypothesis has remained elusive. Here, we show, using the phosphate analog aluminum fluoride (AlF) and a K+ congener (Rb+), the 8-Å resolution structure of H+,K+-ATPase in the transition state of dephosphorylation, (Rb+)E2∼AlF, which is distinct from the preceding Rb+-free E2P state. A strong density located in the transmembrane cation-binding site of (Rb+)E2∼AlF highly likely represents a single bound Rb+ ion, which is clearly different from the Rb+-free E2AlF or K+-bound (K+)E2∼AlF structures. Measurement of radioactive 86Rb+ binding suggests that the binding stoichiometry varies depending on the pH, and approximately half of the amount of Rb+ is bound under acidic crystallization conditions compared with at a neutral pH. These data represent structural and biochemical evidence for the 1H+/1K+/1ATP transport mode of H+,K+-ATPase, which is a prerequisite for generation of the 106-fold proton gradient in terms of thermodynamics. Together with the released E2P-stabilizing interaction between the β subunit’s N terminus and the P domain observed in the (Rb+)E2∼AlF structure, we propose a refined vectorial transport model of H+,K+-ATPase, which must prevail against the highly acidic state of the gastric lumen.
Keywords: electron crystallography, P-type ATPases, membrane proteins, bioenergetics
Like other P-type ATPases (1), the vectorial cation transport of gastric H+,K+-ATPase (2) is accomplished by cyclical conformational changes of the enzyme (abbreviated as E), generally described using an E1/E2 nomenclature based on the Post–Albers scheme for Na+,K+-ATPase (3) (SI Appendix, Fig. S1). In contrast to the closely related Na+,K+-ATPase, which exchanges three Na+ for two K+ ions in an electrogenic transport reaction, gastric H+,K+-ATPase operates electroneutrally, although its transport stoichiometry (2H+/2K+/ATP or 1H+/1K+/ATP) has remained controversial (4, 5). A measurement of the proton transport (5) revealed an H+/ATP ratio of 2 at neutral pH. Accordingly, two K+ ions must be counter transported during a single turnover of the transport cycle, accompanied by the hydrolysis of one ATP molecule (2H+/2K+/ATP). The reported free energy for ATP hydrolysis of the gastric secretory membrane [−13 kcal/mol (4)] provides sufficient energy to achieve a maximum change in pH (ΔpH) of 4.7 units when two H+ ions are transported per hydrolysis of one ATP molecule; thus, the generation of pH 1 (ΔpH > 6 units in the stomach) with a 2H+/2K+/ATP stoichiometry is thermodynamically impossible. Therefore, according to the most widely held hypothesis, the stoichiometry of transported cations per ATP changes from 2 to 1 as the luminal pH decreases (5, 6), or the stoichiometry simply remains 1 independent of pH (4), although direct evidence for this is lacking. To address this issue, we attempted to capture H+,K+-ATPase in a transition state of dephosphorylation with bound counter ion(s) in it, to characterize its structural and functional properties.
Results
Characterization of the Conformational State of H+,K+-ATPase.
We previously characterized the effects of fluorinated phosphate analogs (7–9) on H+,K+-ATPase (10) and found that the addition of Mg2+ to the aluminum fluoride (AlF)-inhibited enzyme induced the dissociation of AlF, resulting in the recovery of its ATPase activity (Fig. 1A). This Mg2+-induced reactivation is, however, strongly suppressed (i.e., ATPase activity remains inhibited) in the presence of K+ (10) or its congener Rb+ (Fig. 1A). Such an allosteric effect of K+ indicates that the AlF-inhibited H+,K+-ATPase can bind K+, and, thus, the enzyme can accumulate in a separate, distinguishable conformational state (Fig. 1A, cartoon; see also SI Appendix, Results and Discussion). The observed stable inhibition allowed us to determine the amount of radioactive 86Rb+ binding to H+,K+-ATPase (Fig. 1B). Our data qualitatively suggest that 86Rb+ binds to the AlF-inhibited H+,K+-ATPase with high affinity (K0.5 = 16 μM), although the determined stoichiometry is only around 0.15 bound Rb+ per ATPase molecule (11), which might be attributable, however, to the exchange of bound 86Rb+ during the washing step of the experiment (see below and SI Appendix, Results and Discussion). The observed high-affinity Rb+ binding indicates that the cation-binding site is in an appropriate conformation for Rb+ (and also for K+) coordination, in contrast to the more than fivefold smaller amount of 86Rb+ binding to the E2P ground-state analog, the beryllium fluoride (BeF)-inhibited enzyme (Fig. 1B, Inset). As shown in Fig. 1A, Mg2+-induced reactivation is almost completely suppressed in the presence of 1 mM Rb+, and, thus, stoichiometric amounts of Rb+ must bind to the enzyme in solution. The substoichiometric amount of detected Rb+ binding is most likely attributable to its dissociation and the following exchange with cold Rb+ during the washing process. The above data, together with other findings (SI Appendix, Results and Discussion and Figs. S2–S5), thus strongly suggest that the combination of AlF and Rb+ induces a conformational state close to (and subsequent in terms of the forward transport cycle) the canonical E2∼P transition state, (Rb+)E2∼AlF.
Fig. 1.

Rb+ binding to the H+,K+-ATPase. (A) Suppression effect of Rb+ on Mg2+-induced reactivation. Control (open columns, 1 and 2) or AlF-inhibited (gray columns, 3–6) enzyme preparations were incubated for 5 h in the presence or absence of 10 mM Mg2+ and/or 1 mM Rb+ as indicated in the figure (Reactivation), and the ATPase activities of these samples were determined. The specific ATPase activity of the AlF and Mg2+-free conditioned enzyme was defined as 100%. (Right) Cartoon model of the Mg2+-induced reactivation (green) of the AlF-inhibited enzyme (gray with bound AlF) and its suppression by Rb+ (blue). (B) Rb+-concentration dependence of the amount of 86Rb+ specifically bound to H+,K+-ATPase (closed circles) was determined as the difference between +AlF and –AlF samples (see SI Appendix, Fig. S4A). (Inset) Effect of various phosphate analogs (AlF, aluminum fluoride; BeF, beryllium fluoride; MgF, magnesium fluoride; VO4, orthovanadate) and a potassium-competitive acid suppressant (SCH28080) on the amount of 86Rb+ binding (see SI Appendix, Results and Discussion for details). Data shown are means ± SD of triplicate experiments.
Cryo-EM Structure of H+,K+-ATPase at Eight-Angstrom Resolution.
To study the molecular events induced by counter ion binding to the E2P conformation, the 3D structure of H+,K+-ATPase in the (Rb+)E2∼AlF state was determined at 8-Å resolution by image-based electron crystallography (12) of 2D crystals (Fig. 2A; see also SI Appendix, Results and Discussion, Fig. S6, and Table S1). In combination with the homology model based on the closely related Na+,K+-ATPase structure (13) (PDB ID code 2ZXE), the present electron-microscopic density map (EM map) is able to determine the bound AlF in the P (phosphorylation) domain as a strong density (SI Appendix, Fig. S7), providing an important validation criterion for the reliability of our structure although the resolution is limited to 8 Å. The AlF-bound phosphorylation site at the P domain is covered by the A (actuator) domain, and the N (nucleotide-binding) domain is retracted from the P domain (Movie S1), showing a characteristic E2P-type conformation, like other medium-resolution structures of H+,K+-ATPase (10, 14, 15). Notably, the density responsible for the bound ADP at the N domain as observed previously in the Rb+-free E2AlF structure was absent in the present (Rb+)E2∼AlF structure, which was likely related to subtle changes in the relative orientations of the cytoplasmic domains (SI Appendix, Fig. S7 and Movie S2). Such an allosteric effect of Rb+ also suggests that the present (Rb+)E2∼AlF structure adopts a conformational state distinct from the preceding Rb+-free E2AlF structure (for details, see SI Appendix, Results and Discussion). In contrast to the cytoplasmic domains, the transmembrane (TM) helices appear to be represented by a large continuous density, which might be related to their inherent flexibility and the lack of crystal contacts in this region. As observed in the horizontal sections of the membrane (SI Appendix, Fig. S8 and Movie S3), however, the 11 distinct features at the TM domain allow us to ensure the quality of the fitting of the individual TM helices. The reliability of our fitted model is also confirmed by the sharpened EM map (16), because it restores the high-resolution amplitude (see Methods for details), in which most of the TM helices can be observed as separate cylinders (SI Appendix, Fig. S9).
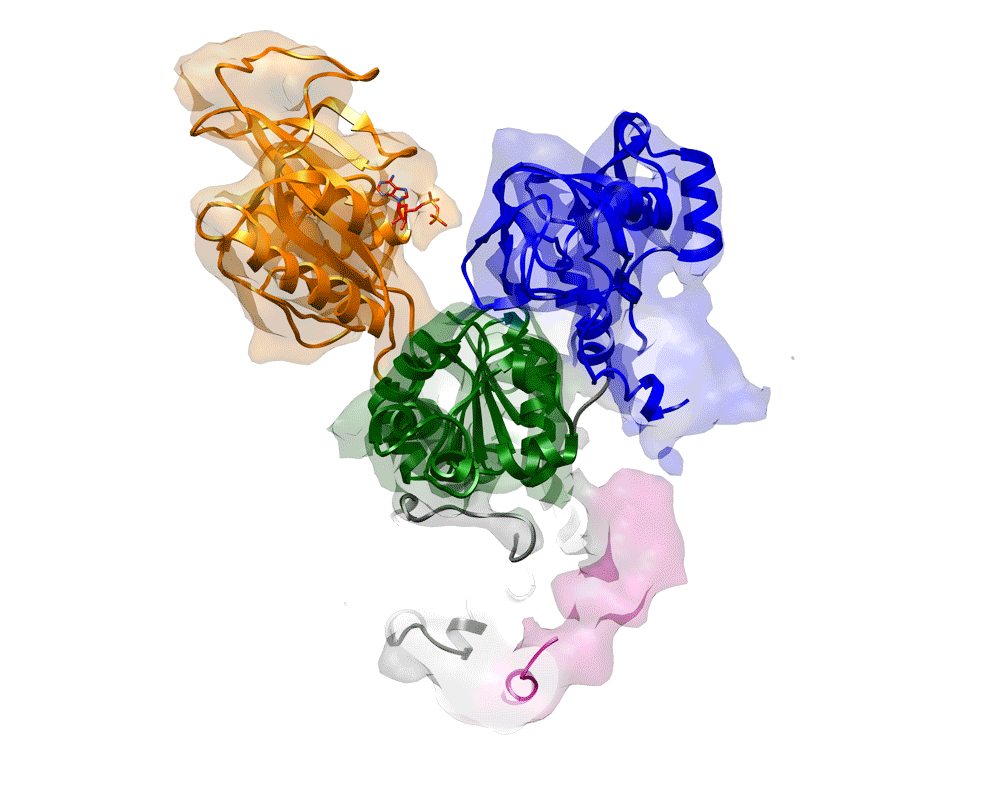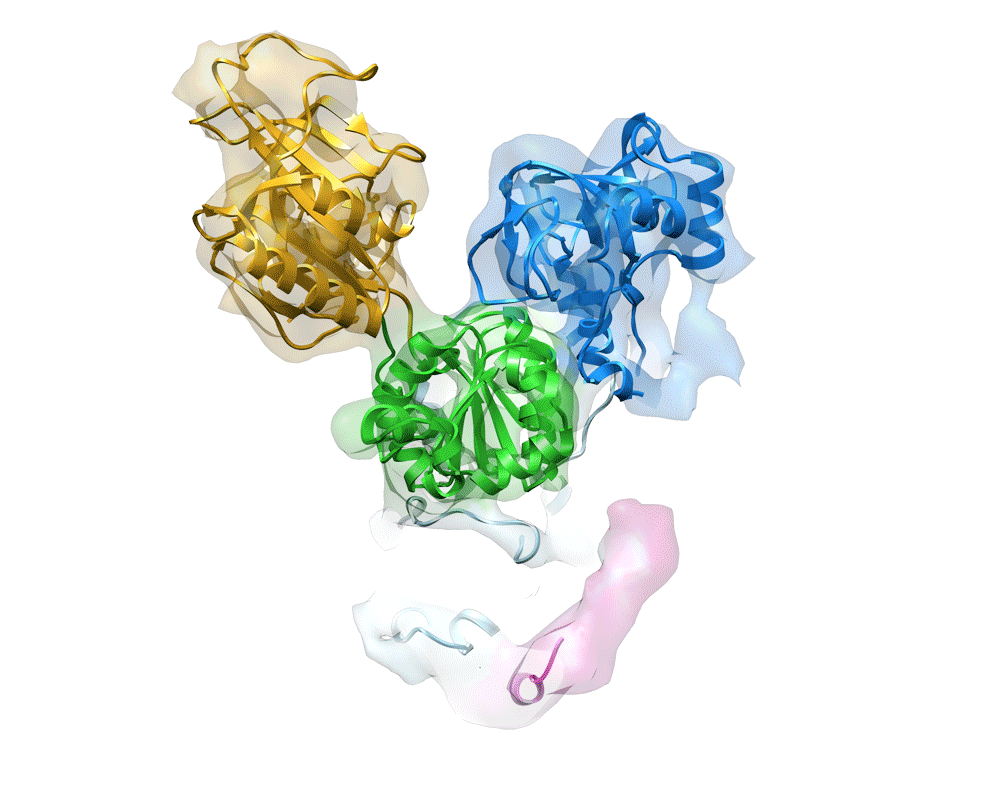
Fig. 2.

Single Rb+ binding to the transmembrane region of H+,K+-ATPase. (A) Molecular surface represented by an EM density map with a 1 σ contour level (light blue) and a superimposed homology model of H+,K+-ATPase (ribbon) in the (Rb+)E2∼AlF conformation. Highly contoured densities (5 σ) are indicated by the red color. Several important structural components, including the βNt and bound AlF and Rb+, are highlighted in the figure. The coloring of the homology model gradually changes from the N (blue) to the C (red) terminus of the α-subunit, and the β-subunit is shown in pink. The wheat-colored box indicates the approximate location of the lipid bilayer. (B and C) Cation-binding site of (Rb+)E2∼AlF from the view point parallel to the membrane normal (cytoplasmic side-up) (B) or from the cytoplasmic side (C). White spheres indicate putative K+-binding sites (site I and II) in our homology model. Green mesh and red surface represent EM densities with 4 or 5 σ contour levels, respectively. (D–F) Cross-sections of the cation-binding sites (the position is indicated as a dotted line in C) of the indicated conformations, viewed as in B. The surface color shows the contour level at the indicated plane, the color of which gradually changes from blue (low) to red (high). Displayed contour levels in each map have been adjusted as described in Methods, for fair comparison. (G) The amount of 86Rb+ binding at different pH levels. The mean value at neutral pH (6.7–7.3) was defined as 100%.
Single Counter Ion Binding.
A remarkable difference is observed at the TM cation-binding site of (Rb+)E2∼AlF compared with the Rb+-free E2AlF structure (Fig. 2 B–F and SI Appendix, Fig. S10). A strong density contoured at 4∼5 σ is observed right between the unwound regions of the TM helices M4 and M6 (Fig. 2 B and C), which notably localized at cation-binding site II rather than at site I of our superimposed homology model based on the Na+,K+-ATPase (K+)2E2MgF state (13). The density is likely to be surrounded by several amino acids involved in cation coordination (SI Appendix, Fig. S11), as determined by mutagenesis (17–22), and most of the homologous residues in Na+,K+-ATPase or SERCA are also involved in cation coordination (21, 23). Among them, E343 is in close proximity to the strong density located at site II in our homology model (SI Appendix, Fig. S11). Because a mutation of E343 in H+,K+-ATPase results in the loss of K+ activation (17, 19), site II is likely to be a primary K+-binding site. Mutation at E327 of Na+,K+-ATPase (24), a residue analogous to E343 in H+,K+-ATPase, resulted in the complete loss of high-affinity K+ occlusion (E327Q) or almost the complete loss of ATPase activity (E327D). These similarities suggest that the two closely related ATPases uses site II as a primary K+-binding site and further support the reliability of the proposed Rb+ binding at site II in our homology model, which is based on the Na+,K+-ATPase structure. The density observed at site I, however, was much weaker than that at site II, suggesting a relatively low occupancy of Rb+ at site I (Fig. 2D).
Comparison with the Rb+-free E2AlF or K+-bound (K+)E2∼AlF structures (crystals were grown in the presence of K+ instead of Rb+; SI Appendix, Fig. S12) highlights the different appearance of the EM density with regard to the cation-binding sites (Fig. 2 D–F; see also SI Appendix, Fig. S10). The density distributed around the cation-binding site of the Rb+-free E2AlF structure is weaker than that found in (Rb+)E2∼AlF, and the gravity center of the density (red part in Fig. 2F or SI Appendix, Fig. S10C) was located on the central axis of the M4 helices rather than on the putative location of the cation-binding site. The EM density at site II of the (K+)E2∼AlF structure was also much weaker than that found in the (Rb+)E2∼AlF structure, most likely because of the lower atomic number of K+ compared with Rb+. In contrast to the large impact on the density distribution at site II, site I shows much less variation. To compare the EM densities at the cation-binding sites in each EM map more quantitatively, we conducted a bootstrap resampling analysis with 1,000 replicates (25) (for details, also see Methods), which allowed us to estimate the voxel-wise mean and variance of each EM map (SI Appendix, Fig. S13). In contrast to the almost similar values at cation-binding sites I and II of Rb+-free E2AlF, site II had a significantly higher mean value than site I in (Rb+)E2∼AlF. Similar trends were observed in the (K+)E2∼AlF structure, albeit to a lesser degree. Such differences are not expected if the two cations bind equally well to sites I and II, thus providing further support for the notion that a single Rb+ ion binds at site II.
Another part of the highly contoured density was distributed on the central axes of TM helices M5, M7, and M8, which likely reflects their inherent rigidness and/or stability rather than Rb+ binding (Fig. 2 B–F and SI Appendix, Fig. S10). On the other hand, similar appearances of the density distributions at those TM helices in each EM map (M8 in Fig. 2 D–F and at M5 and M7 in SI Appendix, Fig. S10) also provide a cross-validation for proper comparison of the EM density at the cation-binding sites. In fact, subtraction between EM maps of the Rb+-bound and Rb+-free E2AlF structures in real space accentuates a distinct density at site II without significant densities at M5 and M8 (SI Appendix, Fig. S14).
Because the crystals were grown at the acidic pH of 4.85, such an apparently single Rb+ ion signature at site II prompted us to investigate the pH dependence of the amount of 86Rb+ binding (Fig. 2G). As expected, the amount of Rb+ binding in acidic conditions (pH 4.6 to 5.1) was ∼40% compared with the amount of binding in the neutral pH range. Therefore, we conclude that the observed strong density in the present (Rb+)E2∼AlF structure corresponds to a single bound Rb+ ion at cation-binding site II.
Discussion
As described previously, it has been suggested for gastric H+,K+-ATPase that two H+ and two K+ are transported per hydrolysis of one ATP molecule at neutral pH, whereas for energetic reasons, only one H+ and K+ can be transported under the maximal acidic gradient observed in vivo (5). In conjunction with these energy requirements, one must assume that one cation-binding site has a low pKa for transport at a pH of around 1, whereas a second site has a higher pKa and, thus, retains H+ at a highly acidic external pH to explain the variable transport stoichiometry. Our measurement of 86Rb+ binding at different pH levels clearly revealed a reduced amount of 86Rb+ binding at the acidic pH (Fig. 2G), consistent with the idea of the stoichiometry variation, as well as with the present EM structure, in which one Rb+ is visualized at site II (Fig. 2 B–D). Because the crystals were grown at the acidic pH of 4.85, one Rb+ binding observed in the structure, therefore, represents a situation in which the enzyme functions in an “acidic” mode, probably with one proton occupying the other site at all times. Thus, the regulation of transport stoichiometry by luminal pH, as another unique feature of H+,K+-ATPase, appears reasonable.
In addition to a variation in transport stoichiometry, another key requirement for the generation of a steep proton gradient is that the transport cycle proceeds unidirectionally, avoiding the risk of proton back-flow (26). As described previously based on the Rb+-free E2AlF structure (14), the N-terminal tail of the β subunit (βNt) functions as a “ratchet” that stabilizes the E2P conformation by tethering the P domain in a E2P-specific position (Fig. 3) to prevent a physiologically unfavorable reverse reaction from E2P to E1P (Fig. 4 A and B). In notable contrast to Rb+-free E2AlF, however, the βNt is not in direct contact with the P domain in the present (Rb+)E2∼AlF structure (Fig. 3), indicating that the E2P-stabilizing structural interaction is abolished by Rb+ binding, which drives the transport cycle in the forward direction. If such an E2P-stabilizing effect would persist in the following step of the transport cycle, this would affect the turnover of H+,K+-ATPase. Compared with the wild-type enzyme, however, βNt deletion mutants (in which the E2P-stabilizing effect by βNt is expected to be absent) show apparently no effect on their turnover number in vitro (14) and only a slight effect in vivo (27). These findings suggest that the βNt does not largely interfere with the progress of the transport cycle in the presence of K+. Upon binding of the second transported cation(s) K+ (or Rb+) to the E2P conformation, the βNt is dissociated from the P domain, as substantiated by our present “ratchet released” structure, thus providing a mechanistic rationale for the directional transport achieved by H+,K+-ATPase (Fig. 4 B and C). Because the risk for proton rebinding and subsequent reversal of the transport cycle might be considerably reduced after the cation-binding site is occupied by K+, the finely timed dissociation of this intersubunit interaction is feasible (Fig. 4C). Together with previously reported findings regarding the E2 or E2P preference of H+,K+-ATPase (19–22, 28), the proposed vectorial transport model (Fig. 4) describes how gastric H+,K+-ATPase can generate the highly acidic condition in the gastric lumen.
Fig. 3.

Rb+-induced dissociation of the intersubunit interaction at the N-terminal tail of the β-subunit. EM map of (Rb+)E2∼AlF (color surface) and Rb+-free E2AlF (orange mesh), viewed from the cytoplasmic side. Single arrowhead indicates the intersubunit interaction observed in the E2AlF structure, in contrast to its absence in the (Rb+)E2∼AlF structure. Double arrowhead indicates the EM density responsible for the bound ADP observed in E2AlF, despite its absence in (Rb+)E2∼AlF because of the conformational rearrangement of cytoplasmic domains (SI Appendix, Fig. S7). Color code: A domain, blue; P domain, green; N domain, yellow; TM, light blue; βNt, pink.
Fig. 4.

Model for the vectorial transport by H+,K+-ATPase. In the H+-occluded E1P-ADP state (A), the P domain is in close proximity to the ADP-bound N domain to form the ADP-aspartylphosphate (Asp–P-ADP) complex. In the proton-transporting step (E1P-ADP to E2P), the P domain is inclined to the βNt, and the Asp-P is covered with the conserved TGES-motif of the A domain to form the E2P state (B). Now, the βNt is tethering the P domain and stabilizes its position (black arrowhead), thus counteracting the reformation of E1P (indicated as a blue dotted arrow in B) and keeping the H+,K+-ATPase resistant to the steep proton gradient, which acts as a strong pressure to drive the transport cycle into the backward direction. Binding of K+ from the luminal side of the membrane, in turn, induces segregation of the βNt-P domain interaction (dotted arrowhead) in the K+-bound E2P transition state (C), releases the P domain, and allows H+,K+-ATPase to proceed with the following transport cycle. Red and blue dotted arrows indicate the forward and reverse reactions of the transport cycle, respectively, and several of important molecular events that accompany the corresponding reaction substeps.
Methods
Preparation of H+,K+-ATPase–Enriched Membrane.
Membrane fractions (G1, G2) containing H+,K+-ATPase were prepared as described previously (29), and further purified (30) with SDS. They were stored at −80 °C in 250 mM sucrose, 0.5 mM EGTA, and 5 mM Hepes (pH 7.0) until use. For 2D crystallization, an SDS-purified G1 fraction was used (31). For 86Rb+-binding and ATPase measurements, both the G1 and SDS-purified G1 fractions were used.
Measurement of the Suppression Effect of Rb+ on Mg2+-Induced Reactivation.
For the AlF inhibition of H+,K+-ATPase, membrane fractions (1 mg/mL) were incubated for 1 h at 37 °C in the presence of 20 mM Hepes/Tris (pH 7.0), 250 mM sucrose in the absence (control) or presence of 0.1 mM AlCl3, 1 mM NaF, and 0.1 mM MgCl2, followed by centrifugation to remove excess inhibitor. The resulting precipitates were suspended in sucrose buffer and used for the following reactivation or 86Rb+-binding experiments.
For the reactivation experiments (Fig. 1A), 1 mg/mL control or AlF-inhibited H+,K+-ATPase preparation was incubated with 20 mM Hepes/Tris (pH 7.0), 250 mM sucrose with or without 10 mM MgCl2, and/or indicated concentrations of RbCl for the indicated time (1–5 h) at 37 °C. ATPase activities [1.5 mM Mg2+-ATP, 10 mM CH3COOK, 250 mM sucrose, 40 mM Hepes/Tris (pH 7.0)] were then measured colorimetrically (32).
Measurement of 86Rb+ Binding.
Membrane preparations of H+,K+-ATPase (2 mg/mL) were incubated with 20 mM Hepes/Tris (pH 7.0), 1 mM MgCl2, and 250 mM sucrose without (control) or with 0.1 mM AlCl3 and 1 mM NaF, for 1 h at 37 °C, and the indicated concentration of 86RbCl (Perkin-Elmer) was added to start the reaction, followed by incubation at 37 °C for 30 min. Aliquots (50 μL) were placed in excess amounts (4 mL) of ice-cold washing buffer [10 mM Hepes/Tris (pH 7.0), 30 mM cold RbCl, 1 mM MgCl2, and 25 mM sucrose] and filtrated under a vacuum using a Millipore filter (HAWP; pore size, 0.45 μm), followed by additional washing with ice-cold washing buffer (33). To measure Rb+ dependence of the amount of 86Rb+ binding (Fig. 1B), samples were incubated for 30 min at 37 °C after the addition of 0.01–2 mM 86RbCl. Even when using an SDS-purified G1 fraction or escin-permeabilized preparation (10) (both preparations are completely leaky, as examined by K+-stimulated ATPase activity), incorporation of 86Rb+ into the samples without AlF treatment increased linearly depending on the concentration of 86RbCl added (SI Appendix, Fig. S4A), which is consistent with the report by Montes et al. (11). Therefore, the amount of 86Rb+ binding was calculated as the difference in the amount of incorporated 86Rb+ between AlF-treated and AlF-free samples, to discriminate specific 86Rb+ binding to the AlF-inhibited H+,K+-ATPase.
To study the effect of inhibitors on 86Rb+ binding (Fig. 1B, Inset), membrane preparations were incubated with 20 mM Hepes/Tris (pH 7.0), 1 mM MgCl2, 250 mM sucrose without (control) or with 0.1 mM AlCl3, and 1 mM NaF (for AlF), or 0.1 mM BeSO4 and 1 mM NaF (for BeF), or 4 mM MgCl2 and 4 mM NaF (for MgF), or 0.2 mM Na3VO4 (for VO4), or 0.1 mM SCH28080 (for SCH), for 1 h at 37 °C. Radioactive 86RbCl with a final concentration of 0.5 mM was added, followed by incubation for 30 min at 37 °C and subsequent separation of the membrane fraction using a Millipore filter. The amount of incorporated 86Rb+ was calculated as the difference between the control (without inhibitor) and each sample with the indicated inhibitor. To study the pH dependence of 86Rb+ binding, a buffer containing propionate (pH 4.6, 5.0), Mes (pH 5.0, 5.5, 5.9), Hepes (pH 6.7, 6.9, 7.1, 7.3), and Tris (pH 7.8, 8.5) was used instead of Hepes/Tris (pH 7.0) buffer.
Two-Dimensional Crystallization and Image Analysis.
The membrane fraction (8 mg/mL protein) was solubilized for 10 min on ice with 6.5–8.5 mg/mL octaethyleneglycol dodecylether (C12E8; Nikko Chemical) in 40 mM Mes, 20 mM Mg(CH3COO)2, 5 mM ATP, 10% (vol/vol) glycerol, and 3 mM DTT, at pH 5.5 adjusted by Tris. The insoluble material was removed by ultracentrifugation, and the supernatant was mixed with dioleoylphosphatidylcholine (Avanti) at a lipid-to-protein ratio (wt/wt) ranging from 1.1 to 1.3. The samples were then placed in 10-μL microdialysis buttons (Hampton Research) using a dialysis membrane with a molecular mass cutoff of 25 kDa (SPECTRA/Pro no. 7; Spectrum Labs) and first dialyzed on ice for 48 h against 300 mL of buffer containing 10 mM Mes, 10% (vol/vol) glycerol, 1 mM ADP, 3 mM DTT, at pH 5.5 adjusted by Tris, with 1 mM MgCl2, 1 mM AlCl3, 4 mM NaF, and 10 mM RbCl. The dialysis buttons were then moved into 5 mL of buffer comprising 20 mM propionate, 1 mM ADP, 3 mM DTT, pH 4.8–4.9 adjusted by Tris, with 1 mM MgCl2, 1 mM AlCl3, 4 mM NaF, and 10 mM RbCl, at 3 °C for 14–18 d. For crystallization of the K+-bound form, 10 mM KCl was used instead of 10 mM RbCl. Samples were negatively stained with 2% (wt/vol) uranyl acetate to screen for crystallization conditions. Specimens for cryo-EM were prepared in a cold room using the carbon sandwich method (34).
Images for the structural analysis were recorded with a JEM-3000SFF electron microscope (JEOL) equipped with a super fluid helium stage (35) on SO-163 film (Carestream). Digitized images were processed with the MRC Image Processing program (36). The crystals were computationally unbent (37), and their initial contrast transfer function parameters were determined for correction (38). The data tilted to 60° were merged using LATLINE (39) at 7-Å resolution, and data to 8-Å resolution were used to calculate a 3D density map to exclude anisotropic structural information at the highest resolution shell of 7 Å (SI Appendix, Fig. S6 and Table S1). EM density maps of (Rb+)E2∼AlF and (K+)E2∼AlF have been deposited in the EMDataBank (http://www.emdatabank.org/; accession code EMD-2219 and EMD-2220), respectively.
Homology Modeling and Structural Analysis.
The homology model (deposited in the PDB under PDB ID code 2YN9) for the H+,K+-ATPase (Rb+)E2∼AlF structure was built with MODELER version 9.7 (40) using the atomic model of the K+-bound E2MgF structure of shark Na+,K+-ATPase (PDB code 2ZXE) as a starting template. The initial manual fitting of the homology model into the density map was achieved using the program O (41). The adjustment for each individual domain and the TM helices with the EM map was performed using SITUS (42). After a positional search, further fine fitting and connecting of the split loop regions were performed manually using O or COOT (43) with regularization refinement. We also generated a sharpened map by applying a B factor of −400 to enhance the weak amplitude at the higher-resolution shell (16). As expected, the resulting sharpened map showed cylindrical densities for TM helices, and most secondary structures found in the cytoplasmic domain were consistent with the fitted homology model (SI Appendix, Fig. S9). The sharpened map was noisier, however, and, thus, the original EM map without amplitude scaling was used for the most of our structural investigations. The real-space subtraction of the EM maps of (Rb+)E2∼AlF and E2AlF (SI Appendix, Fig. S14), and the structure drawings were performed using UCSF Chimera (44).
To compare the density distribution at a high contoured level (4–6 σ) of the TM region of (Rb+)E2∼AlF, (K+)E2∼AlF and Rb+-free E2AlF (Fig. 2 and SI Appendix, Figs. S10, S13, and S14), each density was adjusted to give approximately the same volume at their respective contour levels, according to the relationship between the observed volume and contour level (σ value) in each EM map (SI Appendix, Fig. S15). The adjustment gives ∼0.5% of the volume observed in the 1 σ map, at contour levels of 5, 5.41, and 5.5 σ (Fig. 2 B and C, red surface), and 3.8% for 4, 4.16, and 4.36 σ (Fig. 2 B and C, green mesh) of (Rb+)E2∼AlF, (K+)E2∼AlF and Rb+-free E2AlF structures, respectively.
To estimate the mean and SD of the EM density maps, we performed a bootstrap resampling analysis (24, 45) with 1,000 replicates. Sixty percent of the images were randomly chosen from the original merged dataset, and then a 3D reconstruction for each EM structure was generated during each cycle. The bootstrap-estimated voxel-wise mean and SDs were then calculated. The values plotted in SI Appendix, Fig. S13 were determined at the position of site I, site II, and Oδ2 of D385 (for AlF) in the homology model and scaled according to the trends of the highly contoured density as described above (SI Appendix, Fig. S15).
Supplementary Material
Acknowledgments
We thank Kazumi Kobayashi (JEOL) for technical assistance with the electron microscope, and Kazuya Taniguchi for his scientific advices. This research was supported by Grants-in-Aid for Scientific Research (S), Japan New Energy and Industrial Technology Development Organization (NEDO) (to Y.F.); Grants-in-Aid for Young Scientists (B) (to K.T.); and by the German Research Foundation (Cluster of Excellence “Unifying Concepts in Catalysis”) (T.F.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: EM density maps have been deposited in the EMDataBank, http://www.emdatabank.org/ [accession code EMD-2219 for (Rb+)-E2∼AlF and EMD-2220 for (K+)E2∼AlF]. The homology model of (Rb+)-E2∼AlF have been deposited in the Protein Data Bank, http://www.pdb.org (PDB ID code 2YN9).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1212294109/-/DCSupplemental.
References
- 1.Palmgren MG, Axelsen KB. Evolution of P-type ATPases. Biochim Biophys Acta. 1998;1365(1-2):37–45. doi: 10.1016/s0005-2728(98)00041-3. [DOI] [PubMed] [Google Scholar]
- 2.Ganser AL, Forte JG. K+ -stimulated ATPase in purified microsomes of bullfrog oxyntic cells. Biochim Biophys Acta. 1973;307(1):169–180. doi: 10.1016/0005-2736(73)90035-7. [DOI] [PubMed] [Google Scholar]
- 3.Post RL, Kume S, Tobin T, Orcutt B, Sen AK. Flexibility of an active center in sodium-plus-potassium adenosine triphosphatase. J Gen Physiol. 1969;54(1):306–326. doi: 10.1085/jgp.54.1.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reenstra WW, Forte JG. H+/ATP stoichiometry for the gastric (K+ + H+)-ATPase. J Membr Biol. 1981;61(1):55–60. doi: 10.1007/BF01870752. [DOI] [PubMed] [Google Scholar]
- 5.Rabon EC, McFall TL, Sachs G. The gastric [H,K]ATPase:H+/ATP stoichiometry. J Biol Chem. 1982;257(11):6296–6299. [PubMed] [Google Scholar]
- 6.Shin JM, Munson K, Vagin O, Sachs G. The gastric HK-ATPase: Structure, function, and inhibition. Pflugers Arch. 2009;457(3):609–622. doi: 10.1007/s00424-008-0495-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Danko S, Yamasaki K, Daiho T, Suzuki H. Distinct natures of beryllium fluoride-bound, aluminum fluoride-bound, and magnesium fluoride-bound stable analogues of an ADP-insensitive phosphoenzyme intermediate of sarcoplasmic reticulum Ca2+-ATPase: Changes in catalytic and transport sites during phosphoenzyme hydrolysis. J Biol Chem. 2004;279(15):14991–14998. doi: 10.1074/jbc.M313363200. [DOI] [PubMed] [Google Scholar]
- 8.Toyoshima C, Nomura H, Tsuda T. Lumenal gating mechanism revealed in calcium pump crystal structures with phosphate analogues. Nature. 2004;432(7015):361–368. doi: 10.1038/nature02981. [DOI] [PubMed] [Google Scholar]
- 9.Morth JP, et al. Crystal structure of the sodium-potassium pump. Nature. 2007;450(7172):1043–1049. doi: 10.1038/nature06419. [DOI] [PubMed] [Google Scholar]
- 10.Abe K, Tani K, Fujiyoshi Y. Structural and functional characterization of H+, K+-ATPase with bound fluorinated phosphate analogs. J Struct Biol. 2010;170(1):60–68. doi: 10.1016/j.jsb.2009.12.008. [DOI] [PubMed] [Google Scholar]
- 11.Montes MR, et al. Rb+ occlusion stabilized by vanadate in gastric H+,K+-ATPase at 25°C. Biochim Biophys Acta. 2011;1808(1):316–322. doi: 10.1016/j.bbamem.2010.08.022. [DOI] [PubMed] [Google Scholar]
- 12.Fujiyoshi Y. The structural study of membrane proteins by electron crystallography. Adv Biophys. 1998;35:25–80. doi: 10.1016/s0065-227x(98)90004-1. [DOI] [PubMed] [Google Scholar]
- 13.Shinoda T, Ogawa H, Cornelius F, Toyoshima C. Crystal structure of the sodium-potassium pump at 2.4 A resolution. Nature. 2009;459(7245):446–450. doi: 10.1038/nature07939. [DOI] [PubMed] [Google Scholar]
- 14.Abe K, Tani K, Nishizawa T, Fujiyoshi Y. Inter-subunit interaction of gastric H+,K+-ATPase prevents reverse reaction of the transport cycle. EMBO J. 2009;28(11):1637–1643. doi: 10.1038/emboj.2009.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abe K, Tani K, Fujiyoshi Y. Conformational rearrangement of gastric H+,K+-ATPase induced by an acid suppressant. Nat Commun. 2011;2:155. doi: 10.1038/ncomms1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Havelka WA, Henderson R, Oesterhelt D. Three-dimensional structure of halorhodopsin at 7 A resolution. J Mol Biol. 1995;247(4):726–738. doi: 10.1006/jmbi.1995.0176. [DOI] [PubMed] [Google Scholar]
- 17.Asano S, Furumoto R, Tega Y, Matsuda S, Takeguchi N. Mutational analysis of the putative K(+)-binding site on the fourth transmembrane segment of the gastric H+,K+-ATPase. J Biochem. 2000;127(6):993–1000. doi: 10.1093/oxfordjournals.jbchem.a022716. [DOI] [PubMed] [Google Scholar]
- 18.Swarts HGP, Hermsen HPH, Koenderink JB, Schuurmans Stekhoven FMAH, De Pont JJHHM. Constitutive activation of gastric H+,K+-ATPase by a single mutation. EMBO J. 1998;17(11):3029–3035. doi: 10.1093/emboj/17.11.3029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Swarts HGP, Koenderink JB, Hermsen HPH, Willems PHGM, De Pont JJHHM. K+-independent gastric H+,K+-ATPase activity. Dissociation of K+-independent dephosphorylation and preference for the E1 conformation by combined mutagenesis of transmembrane glutamate residues. J Biol Chem. 2001;276(40):36909–36916. doi: 10.1074/jbc.M103945200. [DOI] [PubMed] [Google Scholar]
- 20.Swarts HGP, Koenderink JB, Willems PHGM, Krieger E, De Pont JJHHM. Asn792 participates in the hydrogen bond network around the K+-binding pocket of Gastric H,K-ATPase. J Biol Chem. 2005;280(12):11448–11494. doi: 10.1074/jbc.M412321200. [DOI] [PubMed] [Google Scholar]
- 21.Koenderink JB, Swarts HG, Willems PH, Krieger E, De Pont JJHHM. A conformation-specific interhelical salt bridge in the K+ binding site of gastric H,K-ATPase. J Biol Chem. 2004;279(16):16417–16424. doi: 10.1074/jbc.M400020200. [DOI] [PubMed] [Google Scholar]
- 22.Dürr KL, Seuffert I, Friedrich T. Deceleration of the E1P-E2P transition and ion transport by mutation of potentially salt bridge-forming residues Lys-791 and Glu-820 in gastric H+/K+-ATPase. J Biol Chem. 2010;285(50):39366–39379. doi: 10.1074/jbc.M110.133470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Munson K, Garcia R, Sachs G. Inhibitor and ion binding sites on the gastric H,K-ATPase. Biochemistry. 2005;44(14):5267–5284. doi: 10.1021/bi047761p. [DOI] [PubMed] [Google Scholar]
- 24.Nielsen JM, Pedersen PA, Karlish SJD, Jorgensen PL. Importance of intramembrane carboxylic acids for occlusion of K+ ions at equilibrium in renal Na,K-ATPase. Biochemistry. 1998;37(7):1961–1968. doi: 10.1021/bi972524q. [DOI] [PubMed] [Google Scholar]
- 25.Cheng A, Yeager M. Bootstrap resampling for voxel-wise variance analysis of three-dimensional density maps derived by image analysis of two-dimensional crystals. J Struct Biol. 2007;158(1):19–32. doi: 10.1016/j.jsb.2006.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nissen P. One way for the gastric proton pump. EMBO J. 2009;28(11):1535–1536. doi: 10.1038/emboj.2009.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dürr KL, Abe K, Tavraz NN, Friedrich T. E2P state stabilization by the N-terminal tail of the H,K-ATPase β-subunit is critical for efficient proton pumping under in vivo conditions. J Biol Chem. 2009;284(30):20147–20154. doi: 10.1074/jbc.M109.005769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dürr KL, Tavraz NN, Dempski RE, Bamberg E, Friedrich T. Functional significance of E2 state stabilization by specific α/β-subunit interactions of Na,K- and H,K-ATPase. J Biol Chem. 2009;284(6):3842–3854. doi: 10.1074/jbc.M808101200. [DOI] [PubMed] [Google Scholar]
- 29.Sachs G, et al. A nonelectrogenic H+ pump in plasma membranes of hog stomach. J Biol Chem. 1976;251(23):7690–7698. [PubMed] [Google Scholar]
- 30.Yeh LA, Cosgrove P, Holt WF. SDS purification of porcine H,K-ATPase from gastric mucosa. Membr Biochem. 1990;9(2):129–140. doi: 10.3109/09687689009025835. [DOI] [PubMed] [Google Scholar]
- 31.Nishizawa T, Abe K, Tani K, Fujiyoshi Y. Structural analysis of 2D crystals of gastric H+,K+-ATPase in different states of the transport cycle. J Struct Biol. 2008;162(2):219–228. doi: 10.1016/j.jsb.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 32.Chifflet S, Torriglia A, Chiesa R, Tolosa SA. A method for the determination of inorganic phosphate in the presence of labile organic phosphate and high concentrations of protein: Application to lens ATPases. Anal Biochem. 1988;168(1):1–4. doi: 10.1016/0003-2697(88)90002-4. [DOI] [PubMed] [Google Scholar]
- 33.Yokoyama T, et al. Acid-labile ATP and/or ADP/P(i) binding to the tetraprotomeric form of Na/K-ATPase accompanying catalytic phosphorylation-dephosphorylation cycle. J Biol Chem. 1999;274(45):31792–31796. doi: 10.1074/jbc.274.45.31792. [DOI] [PubMed] [Google Scholar]
- 34.Gyobu N, et al. Improved specimen preparation for cryo-electron microscopy using a symmetric carbon sandwich technique. J Struct Biol. 2004;146(3):325–333. doi: 10.1016/j.jsb.2004.01.012. [DOI] [PubMed] [Google Scholar]
- 35.Fujiyoshi Y, et al. Development of a superfluid helium stage for high-resolution electron microscopy. Ultramicroscopy. 1991;38(3–4):241–251. [Google Scholar]
- 36.Crowther RA, Henderson R, Smith JM. MRC image processing programs. J Struct Biol. 1996;116(1):9–16. doi: 10.1006/jsbi.1996.0003. [DOI] [PubMed] [Google Scholar]
- 37.Henderson R, Baldwin JM, Downing KH, Lepault J, Zemlin F. Structure of purple membrane from Halobacterium halobium: Recording, measurement and evaluation of electron micrographs at 3.5 Å resolution. Ultramicroscopy. 1986;19(2):147–178. [Google Scholar]
- 38.Tani K, Sasabe H, Toyoshima C. A set of computer programs for determining defocus and astigmatism in electron images. Ultramicroscopy. 1996;65(1–2):31–44. [Google Scholar]
- 39.Agard DAA. A least-squares method for determining structure factors in three-dimensional tilted-view reconstructions. J Mol Biol. 1983;167(4):849–852. doi: 10.1016/s0022-2836(83)80114-4. [DOI] [PubMed] [Google Scholar]
- 40.Šali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234(3):779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 41.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A. 1991;47(Pt 2):110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 42.Wriggers W, Milligan RA, McCammon JA. Situs: A package for docking crystal structures into low-resolution maps from electron microscopy. J Struct Biol. 1999;125(2-3):185–195. doi: 10.1006/jsbi.1998.4080. [DOI] [PubMed] [Google Scholar]
- 43.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 12 Pt 1):2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 44.Pettersen EF, et al. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 45.Efron B, Tibshirani RJ. 1993. An Introduction to the Bootstrap, Monographs on Statistics & Applied Probability (Chapman & Hall/CRC, New York), Vol 57.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}