

Abstract
Carbonates from approximately 2.3–2.1 billion years ago show markedly positive δ13C values commonly reaching and sometimes exceeding +10‰. Traditional interpretation of these positive δ13C values favors greatly enhanced organic carbon burial on a global scale, although other researchers have invoked widespread methanogenesis within the sediments. To resolve between these competing models and, more generally, among the mechanisms behind Earth’s most dramatic carbon isotope event, we obtained coupled stable isotope data for carbonate carbon and carbonate-associated sulfate (CAS). CAS from the Lomagundi interval shows a narrow range of δ34S values and concentrations much like those of Phanerozoic and modern marine carbonate rocks. The δ34S values are a close match to those of coeval sulfate evaporites and likely reflect seawater composition. These observations are inconsistent with the idea of diagenetic carbonate formation in the methanic zone. Toward the end of the carbon isotope excursion there is an increase in the δ34S values of CAS. We propose that these trends in C and S isotope values track the isotopic evolution of seawater sulfate and reflect an increase in pyrite burial and a crash in the marine sulfate reservoir during ocean deoxygenation in the waning stages of the positive carbon isotope excursion.
Keywords: Lomagundi excursion, Great Oxidation Event, Precambrian
Fluctuations in organic carbon (OC) burial on geological time scales control the redox state of the ocean–atmosphere system and are linked with major geochemical and biological evolutionary events (1, 2). Carbon isotopes in marine carbonate rocks (limestones and dolostones) are generally assumed to track the balance between OC burial and weathering and are therefore the most widely used proxy for carbon cycling through time (3). Isotopic variations recorded in carbonates throughout Earth’s history are typically small (|δ13C| = 5‰) and short-lived [< 20 million years (Myr)], a pattern attributed to the overall stability of the Earth’s biogeochemical carbon cycle (3). However, a significant deviation from this pattern occurred ca. 2.3–2.1 billion years (Ga) ago following the initial rise of atmospheric oxygen. Carbonates of this age have markedly positive δ13C values often reaching +10‰ and peaking above +20‰ (4–8). These markedly positive values—found in over 15 formations worldwide—are traditionally assumed to track the marine dissolved inorganic carbon (DIC) reservoir and to reflect greatly enhanced organic carbon burial (4–8). It has been estimated that 12–22 times the present atmospheric inventory of oxygen was released because of organic carbon burial during this event, commonly referred to as the Lomagundi excursion (LE) (5).
Alternatively, some researchers have attributed the positive δ13C values of Lomagundi-age carbonates to widespread diagenetic carbonate formation—that is, precipitation below the sediment–water interface (e.g., 9, 10). Models suggesting a diagenetic origin for isotopically heavy carbonates often invoke precipitation in the methanic zone of the sediment column to explain the dramatic 13C enrichments. Methanogenesis occurs once energetically more favorable oxidants (e.g., nitrate, Fe and Mn oxides, and sulfate) are exhausted (11). The net result of methanogenesis is a pore-water DIC reservoir with strongly positive isotope values, provided the isotopically depleted methane is not reoxidized back to DIC (12). Although carbonates formed in the methanic zone can have δ13C values larger than +10‰ (13–15), similar to those formed during the LE, carbonates with extreme 13C enrichments are rare in shallow-water settings and are typically found adjacent to carbonates with highly negative carbon isotope values linked to local methane oxidation (e.g., 16).
It is possible that the predominance of positive δ13C values seen in carbonate rocks deposited during the LE reflect an unmatched period of methanogenesis close to the sediment–water interface, and a coupling with Earth’s redox evolution could explain the historical uniqueness of the event. Specifically, the LE might be a product of progressive oxygenation of the oceans in phase with the initial rise of atmospheric oxygen and with a concomitant shift in the primary locus of biological methanogenesis from the water column to the surface sediments where carbonate minerals then precipitated with the unique 13C enrichments (9). The coincidence between the onset of the carbon isotope excursion and the initial rise of atmospheric oxygen could be consistent with such a model (17). In this case, the LE would reflect a transition in the ocean from a predominantly anoxic to an oxic redox state, at which point extensive methanogenesis occurred near the sediment–water interface, rather than in the water column, and drove carbonate precipitation. Such conditions would have been favored if sulfate concentrations in seawater were still very low.
Contrasting models for the LE that pit diagenetic controls against isotopic shifts in the oceanic DIC pool (tied to organic carbon burial) have profoundly different implications for the chemistry of the oceans, biogeochemical cycling of carbon, and our basic understanding of Precambrian redox history. Because carbon and sulfur cycles are intricately linked on both global and local scales (18), sulfur isotope records of seawater and pore-water sulfate can be used to test between these two models. The δ34S values of pore-water sulfate typically increase with depth in sediments due to biological fractionations that favor light sulfur isotopes during bacterial sulfate reduction (BSR). Carbon isotope values of DIC show an initial decrease with depth linked to remineralization of 12C-enriched organic matter with a variety of oxidants (oxygen, Mn4+, Fe3+, nitrate, and sulfate) and then shift to markedly positive values in the methanic zone, where 12C from organic matter is preferentially incorporated into methane (19). Sulfate is structurally substituted into the carbonate lattice during carbonate precipitation (20), and therefore carbonate-associated sulfate (CAS) incorporated during precipitation in the methanic zone should have extremely positive δ34S values. Further, because sulfate must be essentially depleted before the onset of appreciable methanogenesis, CAS concentrations in methanic carbonates should be very low. We would expect subsequent additional diagenetic overprints, such as meteoric recrystallization, to result in lower, rather than higher, CAS concentrations (e.g., 21).
Within this framework, we present a global survey of new, coupled carbonate-carbon and CAS-sulfur isotope data (i.e., from the same sample) from several Lomagundi-age successions. Beyond this general survey, we have focused in greater detail on the Mcheka Formation of the Lomagundi Group, Zimbabwe; the Nash Fork Formation in Wyoming, United States; and the Lower Albanel Formation in Quebec, Canada. Collectively, these formations span the time interval from the peak to the aftermath of the LE and therefore are particularly well-suited to explore the mechanisms responsible for the termination of the event. Importantly, our data are inconsistent with the idea of carbonate precipitation in the methanic zone as the mechanism behind the unparalleled occurrence of long-lived, highly positive δ13C excursion. By default, the onus is put back on organic carbon burial. Accepting that, the LE challenges us to unravel how the marine system sustained unusually high levels of organic carbon burial at the dawn of atmospheric oxygenation.
Materials
We present results from the Lower Albanel, Kona, Lucknow, Silverton, Nash Fork, and Mcheka formations deposited from approximately 2.2 Ga to approximately 2.05 Ga on the Superior, Kaapvaal, Wyoming, and Zimbabwe cratons. The sedimentology, carbon isotope systematics, age constraints, and tectonic setting of the sampled formations have been described previously (4, 22–25). All of the formations were deposited on broad, carbonate platforms and are composed predominantly of fine-grained (micritic) carbonates, but stromatolites and granular units are locally abundant. Similar to other CAS studies (e.g., 26), we have focused on micrite-rich lithologies. Unfortunately, there are no high-resolution age constraints for the units we sampled within a continuous stratigraphic context (Nash Fork, Kona, Mcheka formations), and so it is difficult to estimate their average sedimentation rates with confidence. We therefore estimated average sedimentation rates following the approach of Kah et al. (27). To be conservative in our estimates, we used a very broad range (40–250 m Myr-1). This range is typical of the postcompaction depositional rates over million-year time scales for better constrained Phanerozoic carbonate platform environments (28, 29).
Results
CAS concentrations in carbonates deposited during the LE are higher than those of typical Precambrian carbonates outside the excursion. All our samples from strata deposited during the excursion yield an average CAS concentration of 150 ppm (± 145 ppm, 1 SD). Samples from the Mcheka Formation have an average of 117 ppm (107 ppm, 1 SD), with the uppermost samples typically showing lower concentrations. The Lucknow and Nash Fork formations yield averages of 227 ppm (137 ppm, 1 SD) and 232 ppm (198 ppm, 1 SD), respectively. Carbonates deposited during the final stage of the LE with δ13C ≤ 5‰ in the Mistassini Basin, Quebec, show lower CAS concentrations compared to those deposited during the peak of the excursion (with an average of 46 ppm; ± 14 ppm, 1 SD). Similarly, Lomagundi CAS concentrations are higher than those typical of carbonates (primarily dolomites) deposited during the later Paleoproterozoic (30, 31), Mesoproterozoic (30, 31), and Neoproterozoic (32) at times when the seawater sulfate reservoir was significantly smaller than that of modern and Phanerozoic oceans (32). Lomagundi CAS concentrations are comparable to those of Paleozoic micritic carbonates (SI Text) (33). The inference, then, is that CAS concentrations in Lomagundi-age carbonates fall in the range of typical marine carbonates that precipitated from waters with a significant sulfate concentration.
CAS from each of the studied formations shows a relatively narrow range of typical marine δ34S values (Fig. 1). Combined, CAS from the six formations yields δ34S values ranging from +6 to +29‰ (mean + 14‰, n = 105). These values closely match those from coeval sulfate evaporites (Fig. 1). In the Mcheka Formation, the falling limb of the δ13C excursion is marked by a systematic upsection increase in 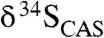 values (Figs. 2 and 3A). There is also an inverse
values (Figs. 2 and 3A). There is also an inverse 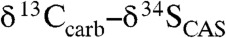 relationship in the Nash Fork Formation, although it is less well-developed.
relationship in the Nash Fork Formation, although it is less well-developed. 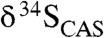 values from carbonates of the Mistassini Basin, deposited during the final stages of the Lomagundi excursion (averaging 29‰, maximum 31‰), and in its aftermath (averaging 33‰, maximum 47‰), are higher than those typical of the LE. These results are consistent with the general trend we see for the event: more positive δ34S values with decreasing δ13C values.
values from carbonates of the Mistassini Basin, deposited during the final stages of the Lomagundi excursion (averaging 29‰, maximum 31‰), and in its aftermath (averaging 33‰, maximum 47‰), are higher than those typical of the LE. These results are consistent with the general trend we see for the event: more positive δ34S values with decreasing δ13C values.
Fig. 1.

A generalized 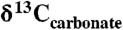 trend and available sulfate δ34S values for the ca. 2.3–2.05-Ga time interval. Light-grey boxes are δ34S values for sulfate evaporite (gypsum and anhydrite), and dark-grey boxes are
trend and available sulfate δ34S values for the ca. 2.3–2.05-Ga time interval. Light-grey boxes are δ34S values for sulfate evaporite (gypsum and anhydrite), and dark-grey boxes are 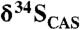 data from this study. Note that
data from this study. Note that 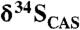 values are essentially the same as those of coeval sulfate evaporites, suggesting that they record the seawater sulfur isotope composition. There is an antithetic relationship between carbonate δ13C and sulfate δ34S values, which is particularly prominent on the falling limb of the Lomagundi carbon isotope excursion (SI Text for additional figure information).
values are essentially the same as those of coeval sulfate evaporites, suggesting that they record the seawater sulfur isotope composition. There is an antithetic relationship between carbonate δ13C and sulfate δ34S values, which is particularly prominent on the falling limb of the Lomagundi carbon isotope excursion (SI Text for additional figure information).
Fig. 2.

Carbonate δ13C and 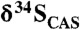 stratigraphic trends in the Mcheka Formation, Zimbabwe. The black circles on the
stratigraphic trends in the Mcheka Formation, Zimbabwe. The black circles on the 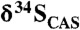 plot are for a duplicate section approximately 200 m along the strike.
plot are for a duplicate section approximately 200 m along the strike. 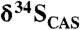 values systematically increase as carbonate δ13C values systematically decrease up-section in the Mcheka Formation.
values systematically increase as carbonate δ13C values systematically decrease up-section in the Mcheka Formation.
Fig. 3.
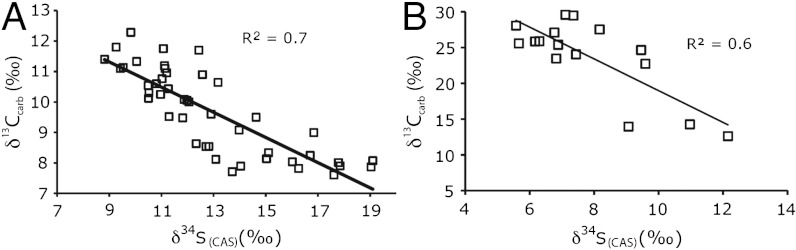
Carbonate δ13C and 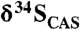 cross-plots for (A) the Mcheka Formation, Zimbabwe, and (B) the Nash Fork Formation, Wyoming, United States.
cross-plots for (A) the Mcheka Formation, Zimbabwe, and (B) the Nash Fork Formation, Wyoming, United States.
Diagenetic Influence on Carbonate Precipitation
A coupled C–S isotope approach can be used to test for a diagenetic influence on carbonate precipitation. Sulfate is structurally substituted into the carbonate lattice during carbonate precipitation, and the concentration of CAS is proportional to ambient sulfate concentrations during carbonate precipitation (20, 34). How CAS concentration specifically scales with seawater sulfate concentration is not well-known and can be influenced by a number of factors (e.g., temperature, mineralogy, and crystal growth rate). Additionally, vital effects can also be important in biogenic phases. However, initial CAS concentrations do, in a more straightforward way, scale with seawater concentrations when abiotic precipitation dominates, as in the case with the samples for this study. Diagenesis can change primary CAS concentrations (21), and the effects of dolomitization on CAS are poorly constrained (35). Nevertheless, although CAS concentrations may shift during diagenesis, they are likely to preserve first-order seawater trends (31). Later diagenetic increases in CAS concentration during recrystallization in the presence of sulfate-rich brines seem unlikely considering that carbonates in our study were collected from different basins, and given the ubiquity of CAS-lean Precambrian calcites and dolomites outside the LE. Therefore, the fact that CAS concentrations in carbonate rocks deposited during the LE fall within the range of typical Phanerozoic marine precipitates (SI Text) is noteworthy. Because sulfate should be essentially depleted before the onset of methanogenesis (11), these values are inconsistent with carbonate formation in the methanic zone.
Given that δ34S values for dissolved sulfate increase with depth in sediments because of fractionations associated with BSR, the very small amounts of CAS, if any, incorporated during precipitation in the methanic zone should have extremely positive δ34S values associated with the high δ13C values. In that light, the observed relatively narrow range of moderate 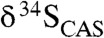 values from our LE carbonates argues against precipitation in the methanic zone. Furthermore,
values from our LE carbonates argues against precipitation in the methanic zone. Furthermore, 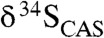 values that closely match those from coeval sulfate evaporites, as we observe, further suggest that our samples record seawater rather than diagenetic C and S isotope signatures (Fig. 3).
values that closely match those from coeval sulfate evaporites, as we observe, further suggest that our samples record seawater rather than diagenetic C and S isotope signatures (Fig. 3).
Changes in Seawater Sulfate Reservoir Size During the Paleoproterozoic
Our results for CAS concentrations point to a large marine sulfate reservoir during the Lomagundi carbon isotope excursion. It has been demonstrated that the extent of temporal isotopic variability in seawater sulfate across Phanerozoic carbon isotope excursions can effectively track the size of the marine sulfate reservoir, and we can employ the same modeling approach here (36). More specifically, sulfate is buffered against isotopic change when the seawater sulfate reservoir is large and sulfate residence time in the ocean is long. The range of isotopic variability in the Mcheka Formation across the wide range of possible sedimentation rates we assumed for its deposition (see above) is less than or equivalent to those observed during Paleozoic positive carbon isotope excursions (36). Similarly, there is extremely muted (< 3‰) 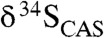 variability in over 300 m of section from the Kona Dolomite. The relatively invariant
variability in over 300 m of section from the Kona Dolomite. The relatively invariant 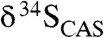 values in LE carbonates and the high CAS concentrations both suggest a large marine sulfate reservoir (Fig. 4). Marine sulfate concentrations were likely < 200 μM in the Archean (37), indicating a orders of magnitude increase during the LE to mM levels—comparable to Phanerozoic levels (Fig. 4). However, significantly higher CAS concentrations in carbonates of the Lomagundi interval compared to those found in Mesoproterozoic and Neoproterozoic carbonates (SI Text) point to a decrease in the size of the marine sulfate reservoir in the aftermath of the excursion. More precisely, low CAS concentrations in carbonates of the Mistassini Basin deposited at the final stages and immediately after the LE suggest that the drawdown of seawater sulfate occurred at the end of or shortly after the excursion. Again, although diagenesis can shift CAS concentrations, we are confident that the first-order trends are preserved.
values in LE carbonates and the high CAS concentrations both suggest a large marine sulfate reservoir (Fig. 4). Marine sulfate concentrations were likely < 200 μM in the Archean (37), indicating a orders of magnitude increase during the LE to mM levels—comparable to Phanerozoic levels (Fig. 4). However, significantly higher CAS concentrations in carbonates of the Lomagundi interval compared to those found in Mesoproterozoic and Neoproterozoic carbonates (SI Text) point to a decrease in the size of the marine sulfate reservoir in the aftermath of the excursion. More precisely, low CAS concentrations in carbonates of the Mistassini Basin deposited at the final stages and immediately after the LE suggest that the drawdown of seawater sulfate occurred at the end of or shortly after the excursion. Again, although diagenesis can shift CAS concentrations, we are confident that the first-order trends are preserved.
Fig. 4.

Estimates of seawater sulfate concentrations through time. Data are from this study and literature sources (SI Text). Estimates for Paleoproterozoic sulfate concentrations are based on 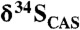 variability for data from the Mcheka Formation (using the approach in ref. 36) and the Kona Dolomite (using the approach in ref. 27).
variability for data from the Mcheka Formation (using the approach in ref. 36) and the Kona Dolomite (using the approach in ref. 27).
Our conclusions about the evolution of the marine sulfate reservoir—that is, an increase and then decrease across the LE—are consistent with the available evaporite record. Extensive sulfate deposition during the Lomagundi excursion preceded halite precipitation in the evaporite mineral sequence, as occurs in the Phanerozoic, suggesting that marine sulfate concentrations were higher than approximately 2.5 mM (38). Although 2.5 mM is much less than modern levels (28 mM), this level is within the lowest range of estimates for sulfate concentrations for some intervals of the Phanerozoic (39), and this is a minimum estimate for the LE. Interestingly, this evaporite mineral sequence is not observed in 1.95-Ga rocks (40), where halite precipitated without gypsum, suggesting a return to lower sulfate concentration following the Lomagundi excursion, consistent with low values inferred for the mid-Proterozoic (41). Therefore, the evaporite record also suggests that there was a rise and then fall in the size of the seawater sulfate reservoir in the Paleoproterozoic (Fig. 4).
Sulfur Cycle During the Lomagundi Carbon Isotope Excursion
The observed carbon and sulfur isotope trends indicate a dramatic change in the sulfur cycle during the falling limb of the LE. The inverse stratigraphic relationship for 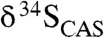 and
and 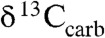 values in the Mcheka Formation (Figs. 2 and 3), which we assume to be tracking seawater sulfate and DIC, may at first blush seem contradictory because the burial rates of reduced carbon and sulfur are positively coupled in modern marine environments through BSR, which is fueled by organic matter and associated pyrite formation (18). Our understanding of their biogeochemical cycles, however, also allows for the possibility of an inverse isotopic relationship in the sulfate and DIC reservoirs (42–44). Indeed, there is a well-documented antithetic relationship between sulfate δ34S and carbonate δ13C values during some long-lived Phanerozoic carbon isotope excursions [e.g., during the Carboniferous (42, 45) and the Phanerozoic, in general, on very long time scales (107- to 108-year time scales)]. This relationship has been linked to the mass balance between oxidized and reduced C and S compounds (18, 43, 46–48) through the equation:
values in the Mcheka Formation (Figs. 2 and 3), which we assume to be tracking seawater sulfate and DIC, may at first blush seem contradictory because the burial rates of reduced carbon and sulfur are positively coupled in modern marine environments through BSR, which is fueled by organic matter and associated pyrite formation (18). Our understanding of their biogeochemical cycles, however, also allows for the possibility of an inverse isotopic relationship in the sulfate and DIC reservoirs (42–44). Indeed, there is a well-documented antithetic relationship between sulfate δ34S and carbonate δ13C values during some long-lived Phanerozoic carbon isotope excursions [e.g., during the Carboniferous (42, 45) and the Phanerozoic, in general, on very long time scales (107- to 108-year time scales)]. This relationship has been linked to the mass balance between oxidized and reduced C and S compounds (18, 43, 46–48) through the equation:
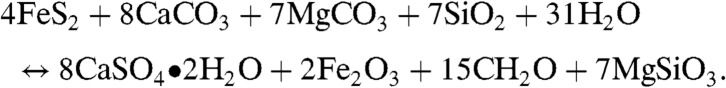 |
This framework has been used to model oxygen levels in the ocean–atmosphere system over Phanerozoic time, and in the simplest sense suggests that the inverse relationship of sulfate δ34S and carbonate δ13C is driven by a coupling between the burial of reduced carbon and oxidized sulfur on a global scale. It is, however, difficult to use the same theoretical framework to understand the observed inverse δ34S–δ13C trends in the Paleoproterozoic. Isotope mass balance arguments (49) suggest that pyrite dominated the sedimentary sulfur burial flux until the Phanerozoic, when the development of bioturbation significantly changed the global S cycle, and burial of sulfate as gypsum and anhydrite became quantitatively important (49). Therefore, long-term marine sulfate δ34S isotopic shifts in the Phanerozoic—but not in the Paleoproterozoic—were strongly influenced by changes in the ratio of reduced to oxidized sulfur buried in marine systems.
The observed rise in sulfate δ34S values and the inverse C–S isotope relationship during the falling limb of the LE can be linked to increased pyrite burial during a global deoxygenation event. Increased pyrite burial, without a corresponding decrease in gypsum burial, at the end of the LE would have triggered a dramatic drop in the size of the marine sulfate reservoir and an increase in the marine sulfate δ34S value. If the markedly positive carbonate δ13C values that characterize the LE are linked to enhanced organic carbon burial (cf. 5), it would follow that there would be a drop in the oxygen levels of the ocean–atmosphere system (caused by lower organic carbon burial and a smaller O2 flux) at the end of the LE. The switch to a more reducing ocean–atmosphere system may also be the consequence of oxidation of uplifted organic carbon-rich rocks buried during the early stages of the LE (50). A global increase in pyrite burial should accompany the switch to a more reducing ocean—because sulfide reoxidation will decrease as marine anoxia becomes more expansive (e.g., 51). Given this framework, the rise in sulfate δ34S values during the end of the LE reflects a drop in oceanic oxygen levels that promoted enhanced pyrite burial (and thus more burial of isotopically light sulfur). At the core of this model is the requirement that large swaths of the ocean were ventilated during the LE and became less oxygenated in its wake.
The observed inverse C–S relationship between sulfate δ34S and carbonate δ13C values in the LE may also be tied to decreases in the extent of continental pyrite oxidation and concomitantly in the magnitude of the continental sulfate flux (52). Limited terrestrial sulfide oxidation in the Archean would have led to higher-than-modern levels of reduced sulfur in the upper crust available for terrestrial weathering (52, 53). With an anoxic atmosphere, the majority of sulfides during weathering would be transported from continental settings to shallow marine environments without being oxidized—leading to burial of detrital pyrites. It has been proposed that first exposure of upper continental crust developed under essentially anoxic conditions to an oxygenated world would fuel intense pyrite oxidation, enhanced sulfate delivery to the oceans, and growth of the marine sulfate reservoir (52, 53). A larger marine sulfate reservoir and a fully oxidative continental sulfur cycle is likely to yield a decrease in the ratio of sulfur buried on the continental margin relative to deep-sea environments. This shift is significant because sediments on the margin have a much higher potential than deep-sea environments to escape subduction and instead be exhumed and subjected to continental weathering. On geologic time scales (tectonic-recycling time scales), increasing deep-sea sulfur burial may result in a decrease in the average amount of sulfide present in exhumed marine sedimentary rocks. The termination of the LE is about one to two sedimentary rock half-lives (somewhere between 100 and 300 Myr—e.g., ref. 54) after the rise of atmospheric oxygen, which is the time required for tectonic processes to replace Archean pyrite-rich sedimentary rocks with the less pyrite-rich sedimentary rocks formed in the wake of the Great Oxidation Event (53). Therefore, it is possible that the sulfur isotope behavior observed at the end of the LE corresponded with a decrease in the continental sulfate flux, which (along with increased pyrite burial) could also drive a shift to more positive sulfate δ34S values.
This decrease in the amount of sulfide present in the upper crust may also be linked to the anomalous carbonate δ13C values that characterize the LE (53). Increased pyrite oxidation in continental weathering environments would have increased acid generation, which could have fueled increased apatite dissolution and amplified the continental phosphorus flux to the oceans. A stronger continental phosphate flux would have enhanced organic carbon burial on a global scale, ultimately leading to the shift to positive carbonate δ13C values (53). High rates of sulfide oxidation would have also increased the flux of some bioessential metals (e.g., molybdenum), which may have been biolimiting up to that point (e.g., 55).
A simple, global S cycle box modeling approach (e.g., 33, 56) can be used to evaluate the relative roles that increased pyrite burial and a decreased continental sulfate flux played in controlling sulfate δ34S trends during the tail end of the LE (SI Text for model description). A strong increase in pyrite burial (approximately two- to threefold increase from the global modern pyrite burial flux) is needed to reproduce the observed shift in δ34S values at the end of the LE (SI Text). This shift in burial is equivalent to estimates for increased burial during Phanerozoic ocean anoxic events (OAEs) (e.g., 33), suggesting relatively dramatic shifts in sulfur burial and the global redox landscape during the end of the LE. Given a reasonable set of model parameters (SI Text), it is not possible to drive the rise in sulfate δ34S values observed at the tail end of the LE exclusively through decreases in the continental sulfate flux. However, it is possible to reproduce the observed trend via a decrease from 115% to 100% of the modern continental sulfate input, as long as there is also a correspondingly strong (three- to fourfold) increase in the amount of marine pyrite burial. Use of much higher continental sulfate inputs (e.g., 130% of modern) results in unrealistically high sulfate concentrations or requires unreasonable pyrite burial fluxes and fractionation factors (SI Text). Implicit in this modeling approach is that pyrite dominated the marine S burial flux in the Paleoproterozoic (cf. 49). A key question is if an approximately 10% to 20% increase in the amount of weathering-mediated pyrite oxidation during the peak of the LE would be able to alter significantly the continental phosphorus flux and drive enhanced organic carbon burial (53).
Conclusions
Approximately 2.3- to 2.1-Ga carbonate rocks have markedly positive δ13C values, commonly reaching beyond +10‰. Because C and S isotopes of dissolved inorganic carbon and sulfate follow predictable trends in diagenetic settings, we can use these isotopic systems to identify or rule out diagenetic carbonate precipitation. Specifically, sulfate δ34S values increase with burial depth because of biological fractionations associated with BSR, whereas DIC isotope values show an initial decrease followed by a shift to markedly positive values in the methanic zone (16). In contrast to the trend predicted for early diagenesis, we found a narrow range of moderate S isotope values in each of the studied carbonate units deposited between approximately 2.3 and 2.1 Ga that is essentially the same as that of the coeval sulfate evaporites, which is inconsistent with carbonate formation in the methanic zone. Lomagundi-excursion carbonates appear to record seawater C and S isotope signatures, and can thus contribute to our understanding of Paleoproterozoic biogeochemical cycles.
High [CAS] and relatively low variability of CAS δ34S values in LE carbonates relative to younger Proterozoic equivalents point to a large coeval marine sulfate reservoir followed by a decrease in seawater concentrations. The falling limb of the Lomagundi excursion is marked by an increase in 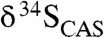 and a strong decrease in [CAS]. We propose that these changes reflect an increase in pyrite burial and a crash in the marine sulfate reservoir during ocean deoxygenation in the waning stages of the positive carbon isotope excursion. A simple global mass balance model for sulfur suggests that there was at least a two- to threefold increase in global pyrite burial at the end of the LE—equivalent to the magnitude of increases associated with the Phanerozoic OAEs. This large shift in pyrite burial suggests the end of the LE was marked by a significant redox shift and points toward a dramatic rise and fall in oxygen levels in the ocean atmosphere system during the Paleoproterozoic. It seems likely that deoxygenation of the ocean–atmosphere system at the end of the LE was an inevitable consequence of decreased organic carbon burial and parallel O2 loss via oxidation of exhumed organic carbon-rich sediments buried during the peak of the LE. There is mounting evidence that, in contrast to the traditional view of a unidirectional oxygen rise, there was significant variability in the redox state of Earth’s surface during the Precambrian (50, 53).
and a strong decrease in [CAS]. We propose that these changes reflect an increase in pyrite burial and a crash in the marine sulfate reservoir during ocean deoxygenation in the waning stages of the positive carbon isotope excursion. A simple global mass balance model for sulfur suggests that there was at least a two- to threefold increase in global pyrite burial at the end of the LE—equivalent to the magnitude of increases associated with the Phanerozoic OAEs. This large shift in pyrite burial suggests the end of the LE was marked by a significant redox shift and points toward a dramatic rise and fall in oxygen levels in the ocean atmosphere system during the Paleoproterozoic. It seems likely that deoxygenation of the ocean–atmosphere system at the end of the LE was an inevitable consequence of decreased organic carbon burial and parallel O2 loss via oxidation of exhumed organic carbon-rich sediments buried during the peak of the LE. There is mounting evidence that, in contrast to the traditional view of a unidirectional oxygen rise, there was significant variability in the redox state of Earth’s surface during the Precambrian (50, 53).
Methods
We used a CAS extraction method modified from traditional approaches to ensure that sulfide oxidation during extraction is negligible. In summary, 20 to 100 g of powder was exposed to successive NaCl and H2O2 treatments with two distilled-water rinses following each step. The intent is to remove sulfates and sulfides (foremost HCl-soluble iron monosulfides) that might skew the primary CAS record. The powders were then dissolved through slow addition of a 5% SnCl2 4N HCl solution. Addition of SnCl2, a reductant, prevented sulfide oxidation by reaction with any Fe(III) liberated during the HCl addition, thus minimizing contamination to the CAS signal. Liberated primary CAS was precipitated through addition of BaCl to induce barite precipitation. Sulfur isotope measurements were made after on-line barite combustion using a ThermoFinnigan DeltaV Plus continuous-flow stable isotope ratio mass spectrometer at University of California, Riverside. Reproducibility was better than 0.2‰ based on single-run and long-term standard monitoring. CAS concentrations were measured using an Agilent inductively coupled plasmonic MS in the Xe collision-cell mode to reduce oxide interferences, and reproducibility was better than 88%. We estimated Paleoproterozoic sulfate concentrations based on maximum 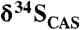 variability following the approach in ref. 36 for the Mcheka Formation, and using the approach in ref. 27 for the Kona Dolomite. Error bars reflect the range of estimates for the duration the examined sequences. Global sulfur cycle box modeling was done following the approach in ref. 32. Model parameters and sensitivity tests are presented in refs. 32 and 53 and SI Text.
variability following the approach in ref. 36 for the Mcheka Formation, and using the approach in ref. 27 for the Kona Dolomite. Error bars reflect the range of estimates for the duration the examined sequences. Global sulfur cycle box modeling was done following the approach in ref. 32. Model parameters and sensitivity tests are presented in refs. 32 and 53 and SI Text.
Supplementary Material
ACKNOWLEDGMENTS.
We are grateful to Ben Gill, Bill Gilhooly, Chris Reinhard, and Don Canfield for formative discussions; Galen Halverson and anonymous reviewers for constructive comments; S. Bates for assistance with the analyses; and the Geology Department of the University of Zimbabwe for logistical support. This work was supported by National Science Foundation Graduate Research Fellowship program and American Philosophical Society (N.J.P.); National Science Foundation Division of Earth Sciences and National Aeronautics and Space Administration Exobiology Program (T.W.L.); Natural Sciences and Engineering Research Council of Canada (A.B.); and the National Research Foundation of South Africa (A.H.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1120387109/-/DCSupplemental.
References
- 1.Holland HD. The Chemical Evolution of the Atmosphere and Oceans. Princeton: Princeton Univ Press; 1984. p. 598. [Google Scholar]
- 2.Schidlowski M. Application of stable carbon isotopes to early biochemical evolution on Earth. Annu Rev Earth Planet Sci. 1987;15:47–72. [Google Scholar]
- 3.Schidlowski M. Carbon isotopes as biogeochemical recorders of life over 3.8 Ga of Earth history: Evolution of a concept. Precambrian Res. 2001;106:117–134. [Google Scholar]
- 4.Bekker A, Karhu JA, Kaufman AJ. Carbon isotope record for the onset of the Lomagundi carbon isotope excursion in the Great Lakes area, North America. Precambrian Res. 2006;148:145–180. [Google Scholar]
- 5.Karhu JA, Holland HD. Carbon isotopes and the rise of atmospheric oxygen. Geology. 1996;24:867–870. [Google Scholar]
- 6.Maheshwari A, et al. Global nature of the Paleoproterozoic Lomagundi carbon isotope excursion: A review of occurrences in Brazil, India, and Uruguay. Precambrian Res. 2010;182:274–299. [Google Scholar]
- 7.Melezhik VA, Fallick AE. A widespread positive delta C-13(carb) anomaly at around 2.33–2.06 Ga on the Fennoscandian Shield: A paradox? Terra Nova. 1996;8:141–157. [Google Scholar]
- 8.Schidlowski M, Eichmann R, Junge CE. Carbon isotope geochemistry of Precambrian Lomagundi Carbonate Province, Rhodesia. Geochim Cosmochim Acta. 1976;40:449–455. [Google Scholar]
- 9.Hayes JM, Waldbauer JR. The carbon cycle and associated redox processes through time. Philos Trans R Soc B. 2006;361:931–950. doi: 10.1098/rstb.2006.1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yudovich YE, Makarikhin VV, Medvedev PV, Sukhanov NV. Carbon isotope anomalies in carbonates of the Karelian Complex. Geochem Int. 1991;28:56–62. [Google Scholar]
- 11.Canfield DE, Kristensen E, Thamdrup B. Aquatic Geomicrobiology. Amsterdam: Elsevier; 2005. [DOI] [PubMed] [Google Scholar]
- 12.Claypool GE, Kaplan IR. The origin and distribution of methane in marine sediments. In: Kaplan IR, editor. Natural Gases in Marine Sediments. New York: Plenum; 1974. pp. 99–139. [Google Scholar]
- 13.Hennessy J, Knauth LP. Isotopic variations in dolomite concretions from the Monterey Formation, California. J Sediment Petrol. 1985;55:120–130. [Google Scholar]
- 14.Irwin H, Curtis C, Coleman M. Isotopic evidence for source of diagenetic carbonates formed during burial of organic-rich sediments. Nature. 1977;269:209–213. [Google Scholar]
- 15.Talbot MR, Kelts K. Primary and diagenetic carbonates in the anoxic sediments of Lake Bosumtwi, Ghana. Geology. 1986;14:912–916. [Google Scholar]
- 16.Maynard JB. Extension of Berner’s new geochemical classification of sedimentary environments to ancient sediments. J Sediment Petrol. 1982;52:1325–1331. [Google Scholar]
- 17.Guo QJ, et al. Reconstructing Earth’s surface oxidation across the Archean-Proterozoic transition. Geology. 2009;37:399–402. [Google Scholar]
- 18.Berner RA. Biogeochemical cycles of carbon and sulfur and their effect on atmospheric oxygen over Phanerozoic time. Global Planet Change. 1989;75:97–122. [Google Scholar]
- 19.Böttcher ME. Carbon, sulfur and oxygen isotope geochemistry in interstitial waters from the western Mediterranean. Proc Ocean Drill Program. 1999;161:413–422. [Google Scholar]
- 20.Pingitore NE, Meitzner G, Love KM. Identification of sulfate in natural carbonates by X-ray absorption spectroscopy. Geochim Cosmochim Acta. 1995;59:2477–2483. [Google Scholar]
- 21.Gill BC, Lyons TW, Frank TD. Behavior of carbonate-associated sulfate during meteoric diagenesis and implications for the sulfur isotope paleoproxy. Geochim Cosmochim Acta. 2008;72:4699–4711. [Google Scholar]
- 22.Bekker A, et al. Fractionation between inorganic and organic carbon during the Lomagundi (2.22–2.1 Ga) carbon isotope excursion. Earth Planet Sci Lett. 2008;271:278–291. [Google Scholar]
- 23.Bekker A, Karhu JA, Eriksson KA, Kaufman AJ. Chemostratigraphy of Paleoproterozoic carbonate successions of the Wyoming Craton: Tectonic forcing of biogeochemical change? Precambrian Res. 2003;120:279–325. [Google Scholar]
- 24.Bekker A, Kaufman AJ, Karhu JA, Eriksson KA. Evidence for Paleoproterozoic cap carbonates in North America. Precambrian Res. 2005;137:167–206. [Google Scholar]
- 25.Master S, Bekker A, Hofmann A. A review of the stratigraphy and geological setting of the Palaeoproterozoic Magondi Supergroup, Zimbabwe—Type locality for the Lomagundi carbon isotope excursion. Precambrian Res. 2010;182:254–273. [Google Scholar]
- 26.Kampschulte A, Strauss H. The sulfur isotopic evolution of Phanerozoic seawater based on the analysis of structurally substituted sulfate in carbonates. Chem Geol. 2004;204:255–286. [Google Scholar]
- 27.Kah LC, Lyons TW, Frank T. Low marine sulphate and protracted oxygenation of the Proterozoic biosphere. Nature. 2004;431:834–837. doi: 10.1038/nature02974. [DOI] [PubMed] [Google Scholar]
- 28.Bosscher H, Schlager W. Accumulation rates of carbonate platforms. J Geol. 1993;101:345–355. [Google Scholar]
- 29.McNeill DF. Accumulation rates from well-dated late Neogene carbonate platforms and margins. Sediment Geol. 2005;175:73–87. [Google Scholar]
- 30.Chu XL, Zhang TG, Zhang QR, Lyons TW. Sulfur and carbon isotope records from 1700 to 800 Ma carbonates of the Jixian section, northern China: Implications for secular isotope variations in Proterozoic seawater and relationships to global supercontinental events. Geochim Cosmochim Acta. 2007;71:4668–4692. [Google Scholar]
- 31.Gellatly AM, Lyons TW. Trace sulfate in mid-Proterozoic carbonates and the sulfur isotope record of biospheric evolution. Geochim Cosmochim Acta. 2005;69:3813–3829. [Google Scholar]
- 32.Hurtgen MT, Arthur MA, Halverson GP. Neoproterozoic sulfur isotopes, the evolution of microbial sulfur species, and the burial efficiency of sulfide as sedimentary pyrite. Geology. 2005;33:41–44. [Google Scholar]
- 33.Gill BC, et al. Geochemical evidence for widespread euxinia in the Later Cambrian ocean. Nature. 2011;469:80–83. doi: 10.1038/nature09700. [DOI] [PubMed] [Google Scholar]
- 34.Pavlov AA, Hurtgen MT, Kasting JF, Arthur MA. Methane-rich Proterozoic atmosphere? Geology. 2003;31:87–90. [Google Scholar]
- 35.Staudt WJ, Schoonen MAA. Sulfate incorporation into sedimentary carbonates. In: Vairavamurthy MA, Schoonen MAA, editors. Geochemical Transformations of Sedimentary Sulfur. Washington, DC: American Chemical Society; 1995. pp. 332–345. [Google Scholar]
- 36.Gill BC, Lyons TW, Saltzman MR. Parallel, high-resolution carbon and sulfur isotope records of the evolving Paleozoic marine sulfur reservoir. Palaeogeogr Palaeoclimatol Palaeoecol. 2007;256:156–173. [Google Scholar]
- 37.Habicht KS, Gade M, Thamdrup B, Berg P, Canfield DE. Calibration of sulfate levels in the Archean Ocean. Science. 2002;298:2372–2374. doi: 10.1126/science.1078265. [DOI] [PubMed] [Google Scholar]
- 38.Schröeder S, Bekker A, Beukes NJ, Strauss H, van Niekerk HS. Rise in seawater sulphate concentration associated with the Paleoproterozoic positive carbon isotope excursion: Evidence from sulphate evaporites in the similar to 2.2–2.1 Gyr shallow-marine Lucknow Formation, South Africa. Terra Nova. 2008;2:108–117. [Google Scholar]
- 39.Lowenstein TK, Timofeeff MN, Brennan ST, Hardie LA, Demicco RV. Oscillations in Phanerozoic seawater chemistry: Evidence from fluid inclusions. Science. 2001;294:1086–1088. doi: 10.1126/science.1064280. [DOI] [PubMed] [Google Scholar]
- 40.Pope MC, Grotzinger JP. Paleoproterozoic Stark Formation, Athapuscow Basin, Northwest Canada: Record of cratonic-scale salinity crisis. J Sediment Res. 2003;73:280–295. [Google Scholar]
- 41.Canfield DE, Farquhar J, Zerkle AL. High isotope fractionations during sulfate reduction in a low-sulfate euxinic ocean analog. Geology. 2010;38:415–418. [Google Scholar]
- 42.Carpenter SJ, Lohmann KC. Carbon isotope ratios of Phanerozoic marine cements: Re-evaluating the global carbon and sulfur systems. Geochim Cosmochim Acta. 1997;61:4831–4846. [Google Scholar]
- 43.Garrels RM, Lerman A. Coupling of the sedimentary sulfur and carbon cycles: An improved model. Am J Sci. 1984;284:989–1007. [Google Scholar]
- 44.Walker JCG. Global geochemical cycles of carbon, sulfur and oxygen. Mar Geol. 1986;70:159–174. doi: 10.1016/0025-3227(86)90093-9. [DOI] [PubMed] [Google Scholar]
- 45.Veizer J, Holser WT, Wilgus CK. Correlation of C-13-C-12 and S-34-S-32 secular variations. Geochim Cosmochim Acta. 1980;44:579–587. [Google Scholar]
- 46.Berner RA, Raiswell R. Burial of organic carbon and pyrite sulfur in sediments over Phanerozoic time: A new theory. Geochim Cosmochim Acta. 1983;47:855–862. [Google Scholar]
- 47.Carpenter SJ, Lohmann KC. Reply to the comment by S. T. Petsch on carbon isotope ratios of Phanerozoic marine cements: Re-evaluating global carbon and sulfur systems. Geochim Cosmochim Acta. 1999;63:761–766. [Google Scholar]
- 48.Kump LR, Garrels RM. Modeling atmospheric O2 in the global sedimentary redox cycle. Am J Sci. 1986;286:337–360. [Google Scholar]
- 49.Canfield DE, Farquhar J. Animal evolution, bioturbation, and the sulfate concentration of the oceans. Proc Natl Acad Sci USA. 2009;106:8123–8127. doi: 10.1073/pnas.0902037106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kump LR, et al. Isotopic evidence for massive oxidation of organic matter following the Great Oxidation Event. Science. 2011;334:1694–1696. doi: 10.1126/science.1213999. [DOI] [PubMed] [Google Scholar]
- 51.Canfield DE. A new model for Proterozoic ocean chemistry. Nature. 1998;396:450–453. [Google Scholar]
- 52.Holland HD. Volcanic gases, black smokers, and the Great Oxidation Event. Geochim Cosmochim Acta. 2002;66:3811–3826. [Google Scholar]
- 53.Bekker A, Holland HD. Oxygen overshoot and recovery during the early Paleoproterozoic. Earth Planet Sci Lett. 2012;317:295–304. [Google Scholar]
- 54.Garrels RM, McKenzie F. Evolution of Sedimentary Rocks. New York: Norton; 1971. p. 397. [Google Scholar]
- 55.Planavsky NJ, et al. The evolution of the marine phosphate reservoir. Nature. 2010;467:1088–1090. doi: 10.1038/nature09485. [DOI] [PubMed] [Google Scholar]
- 56.Gill BC, Lyons TW, Jenkyns HC. A global perturbation to the sulfur cycle during the Toarcian oceanic anoxic event. Earth Planet Sci Lett. 2011;312:484–496. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.