

Abstract
2,4-Oxazole is an important structural motif in various natural products. An efficient modular synthesis of this structure is achieved via a [3+2] annulation between a terminal alkyne and a carboxamide by using a gold-catalyzed oxidation strategy. The postulated reactive intermediate, a terminal α-oxo gold carbene, previously known to be highly electrophilic and hence impropable to be trapped by stoichiometric external nucleophiles, is coerced to react smoothly with a carboxamide en route to the oxazole ring by a P,N- or P,S-bidentate ligand such as Mor-DalPhos; in stark contrast, often used ligands including monodentate phosphines and NHCs are totally ineffective. The role of these bidentate phosphines in this reaction is attributed to the formation of a tricoordinated gold carbene intermediate, which is less electrophilic and hence more chemoselective when reacting with nucleophiles. The success in using bidentate phosphine ligands to temper the reactivities of in-situ generated gold carbenes would likely open many new opportunities to apply the oxidative gold catalysis to the development of novel methods, and the implication of tricoordinated gold intermediates in homogeneous gold catalysis should stimulate further advance in gold catalysis.
2,4-Disubstituted oxazole is a structural motif found in many bioactive natural products including (−)-hennoxazole A1 and phorboxazoles2 (Figure 1). Biosynthetically it is formed via post-translational modifications of serine-containing peptides via sequential cyclization, dehydration and oxidative aromatization. Chemical syntheses3 of this functionality can often be achieved via sequential dehydrative cyclization4 and dehydrogenation5 of substituted N-(2-hydroxyethyl)amides. Though this multi-step linear approach proves to be reliable, the construction of this important motif through a one-step bimolecular annulation6 would offer excellent step economy and desirable synthetic convergence. Several elegant protocols of this nature,7 however, have their own limitations. Herein we disclose a new development featuring gold-catalyzed oxidative [3+2] annulations between readily available terminal alkynes and carboxamides under mild reaction conditions. A key intermediate implicated in the catalytic cycle is a gold(I) complex of tri-coordination, which is rare in gold catalysis.8
Figure 1.

Selected natural products containing 2,4-disubstituted oxazole moieties.
We have recently developed a strategy of gold-catalyzed intermolecular alkyne oxidations (Scheme 1A),6b,9,10 where α-oxo gold carbenes are postulated as key reactive intermediates. A range of versatile synthetic methods have been developed based on this design by us6b,9 and others,11 offering strong support for the intermediacy of these gold carbenes and revealing their potent electrophilicities. While α-oxo metal carbenes/carbenoids can typically be generated via metal-catalyzed dediazotization of α-diazo ketones,12,13 this strategy provides a safe, step-economic and scalable alternative in the case of metal gold by using readily available alkynes as substrates. Unfortunately, all the reactions developed so far have relied on rapid trapping of the gold carbenes via either facile intramolecular processes or by the reaction solvent.6b In the latter case, we developed a rapid synthesis of 2,5-disubstituted oxazoles in a [2+2+1] manner using nitrile as both the solvent and the trapping reagent.6b Otherwise, the reaction tends to be messy, which could be attributed to a lack of chemoselectivity by the α-oxo gold carbene intermediate due to its high electrophilicity. This rationale is further corroborated by the observation that it can abstract chloride from the reaction solvent dichloroethane.9d The highly electrophilic nature of the α-oxo gold carbene moiety, especially for those without additional substitutions at the carbene center, can be understood by invoking the relatively weak back donation by the electronegative gold.14 As a result, it is highly challenging to trapping these highly electrophilic gold carbenes with stoichiometric external nucleophiles.
Scheme 1.
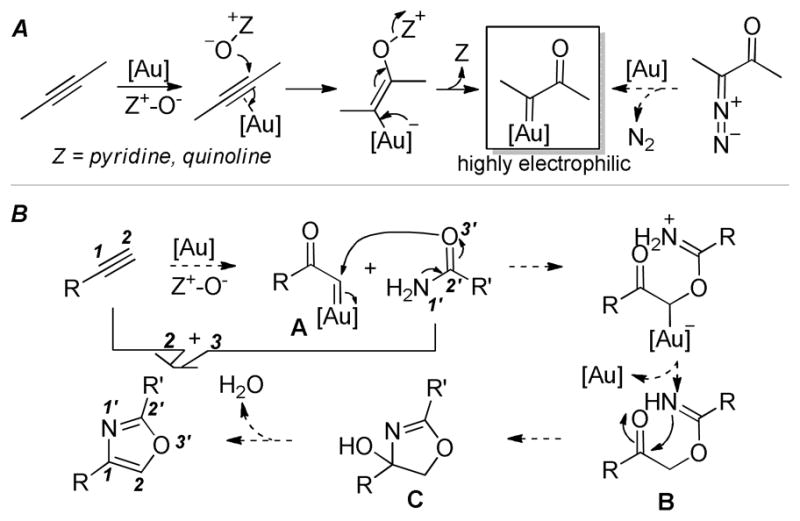
A) alkynes as surrogates of hazardous α-diazo ketones in the generation of α-oxo gold carbene intermediates; B) its application for the synthesis of 2,4-disubstituted oxazoles in a [3+2] annulation
Not deterred, we set out to discover catalysts and conditions that would overcome this daunting challenge. The targeted reaction was a [3+2] annulation between a terminal alkyne and a carboxamides for the formation of 2,4-disubstituted oxazoles. The design is rationalized in Figure 2B: the α-oxo gold carbene intermediate A could be attacked by an amide by its carbonyl oxygen, yielding the imidate B upon protodeauration; cyclization of B would generate the oxazolinol C, the in situ dehydration of which would afford the desired product.
Figure 2.

New efficacious sulfide-functionalized phosphines as ligand for the gold catalysis and the Ortep drawing of of L3AuCl (with 50% ellipsoid probability).
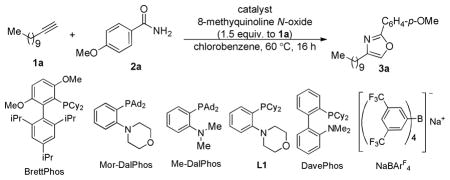
Table 1 shows the reaction discovery and subsequent condition optimization. We chose chlorobenzene as the reaction solvent in order to avoid solvent participation.6b,9d Initially, no desired reaction was detected with various typical gold catalysts of ranging electronic and steric characteristics (entries 1–4). The only exception was BrettPhosAuNTf2,9c,15 but the expected oxazole 3a was formed in a pitiful 4% yield (entry 5). These results highlighted the low selectivities associated with the highly reactive α-oxo gold carbenes. By expanding the types of ligands examined, we fortuitously came upon Mor-DalPhos, a P,N-bidentate ligand developed by Stradiotto.16 With Mor-DalPhosAuNTf2 as the catalyst, the reaction yield, to our amazement, jumped to 58% (entry 6). The effect of the counter anion was examined. While SbF6− (entry 7) and OTf− (entry 8) fared worse, [BArF4]−, a non-coordinating and lipophilic anion developed by Kobayashi,17 improved the reaction yield by a significant 6% (entry 9). The reactive catalyst in this case can be readily generated in situ, driven by the precipitate of insoluble NaCl in PhCl. Other ligands with substituted amino groups were also tested. Me-DalPhos,16 a ligand in the same series as Mor-DalPhos, led to an identical result (entry 10), and L1,16a a ligand different from Mor-DalPhos by having smaller cyclohexyl groups on the phosphorus atom, led to a lower yet acceptable yield (entry 11); however, DavePhos was completely ineffective (entry 12). These data suggested that the position of the amino group is critical for the desired chemistry, the steric congestion is beneficial and the morpholine oxygen is inconsequential. A much higher yield (87%) was achieved when the carboxamide was the limiting reagent and 1.5 equiv of the alkyne was used (entry 13). Other more conventional solvents such as DCE (entry 14) and toluene (entry 15) were less conducive to this reaction presumably due to side reactions involving the solvents. It remains to be pointed out that in order to avoid further oxidation of the gold carbene intermediate by the remaining 8-methylquinoline N-oxide to form α-ketoaldehyde, the oxidant had to be introduced to the reaction flask slowly by a syringe pump so that its concentration remained low during the reaction.
Table 1.
Optimization of reaction conditions.a
| |||
|---|---|---|---|
| entry | 1a/2a | catalyst | yieldb |
| 1 | 1:1.2 | Ph3PAuNTf2 (5 mol %) | 0c |
| 2 | 1:1.2 | Cy-JohnPhosAuNTf2 (5 mol %) | 0c |
| 3 | 1:1.2 | IPrAuNTf2 (5 mol %) | 0c |
| 4 | 1:1.2 | (4-CF3Ph)3PAuNTf2 (5 mol %) | 0c |
| 5 | 1:1.2 | BrettPhosAuNTf2 (5 mol %) | 4%c |
| 6 | 1:1.2 | Mor-DalPhosAuNTf2 (5 mol %) | 58% |
| 7 | 1:1.2 | Mor-DalPhosAuCl (5 mol %)/AgSbF6 (5 mol %) | 37% |
| 8 | 1:1.2 | Mor-DalPhosAuCl (5 mol %)/AgOTf (5 mol %) | 30% |
| 9 | 1:1.2 | Mor-DalPhosAuCl (5 mol %)/NaBArF4 (10 mol %) | 64% |
| 10 | 1:1.2 | Me-DalPhosAuCl (5 mol %)/NaBArF4 (10 mol %) | 64% |
| 11 | 1:1.2 | L1AuCl (5 mol %)/NaBArF4 (10 mol %) | 52% |
| 12 | 1:1.2 | DavePhosAuCl (5 mol %)/NaBArF 4 (10 mol %) | 0c |
| 13 | 1.5:1 | Mor-DalPhosAuCl (5 mol %)/NaBArF4 (10 mol %) | 87%d |
| 14 | 1.5:1 | Mor-DalPhosAuCl (5 mol %)/NaBArF4 (10 mol %) | 59%e |
| 15 | 1.5:1 | Mor-DalPhosAuCl (5 mol %)/NaBArF4 (10 mol %) | 78%f |
The reaction was run with everything except the oxidant in a vial capped with a septum, and the oxidant was introduced into the reaction mixture slowly using a syringe pump. The initial [1a] = 0.1 M.
Measured by 1H NMR using diethyl phthalate as the internal standard.
<20% of 1-dodecyne left, and the crude 1H NMR mostly messy.
81% Isolated yield.
DCE as solvent.
Toluene as solvent.
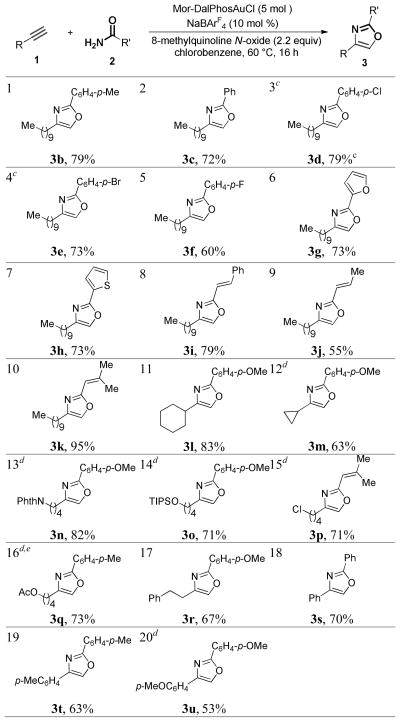
With the optimized reaction conditions in hand, the reaction scope was then examined. First, a range of carboxamides were tested using 1-dodecyne as the alkyne component (entries 1–10). Benzamides with electron-donating (i.e., in the cases of 3a and entry 1) and weakly electron-withdrawing (entries 3–5) para-substituents and without any substitution (entry 2) all underwent the reaction smoothly, affording 2,4-disubstituted oxazoles in accetable to good yields. With furan-2-carboxamide or thiophene-2-carboxamide, the biheteroaryl 3g or 3h were obtained without any event in accidently the same 73% yield (entries 6 and 7). The reaction also worked with α,β-unsaturated carboxamides. While 3j was isolated in only 55% yield by using crotonamide (entry 9), the reaction proceeded efficiently with cinnamamide (entry 8) and especially well with 3,3-dimethylacrylamide (entry 10, 95% yield). The reaction, however, did not work with aliphatic carboxamides,18 and poor yields (<30%) were observed with benzamides with strongly electron-withdrawing para substituents such as acetyl and nitro groups and with ortho substitutions such as Me and F due to their decreased nucleophilicities and/or increased steric hindrance.
With respect to the alkyne, both cyclohexylacetylene (entry 11) and cyclopropylacetylene (entry 12) were suitable substrates. In the former case, the oxazole 3l was isolated in a good 85% yield. In contrast, its analog with the anisyl group replaced with a phenyl group was obtained in only 18% yield7h using the Blumlein-Lewy method.19 Linear aliphatic terminal alkynes with remote functional groups such as NPhth (entry 13), TIPS-protected HO group (entry 14), chloro (entry 15) and acetoxy (entry 16) were also allowed. When these groups were placed at propargyl or homopropargyl positions, the reaction became much less efficient, due to their interferences with the gold carbene moiety. Not withstanding, when a weakly nucleophilic phenyl group was present (entry 17), the reaction could still afford an acceptable yield. Phenylacetylene (entry 18) and those containing electron-donating p-substituents (entries 19 and 20) were suitable alkynes as well, and the triaryl products were formed in serviceable yields. On the other hand, p-nitrophenylacetylene led to a mere ~30% yield of the expected oxazole product (R′ = anisyl, data not shown).
To explain why Mor-DalPhos and the related Me-DalPhos were so effective and the role of the neighbouring amino group, we first thought that H-bonding might be at play.16b However, by inspecting the X-ray structure of Mor-DalPhosAuCl16b it is apparent that the nitrogen atom, with its lone electron pair pointing to and thereby shielded by the gold center, is too congested to be a H-bond acceptor (Scheme 2). The ineffectiveness of DavePhos provides some circumvential evidence for this conclusion. In addition, we prepared the related P,S-bidentate ligands, L2 and L3 (see SI for their preparation). To our delight, their corresponding gold complexes promoted the oxazole formation with efficiencies close to that by Mor-DalPhos (Figure 2). The structure of L3AuCl was secured via X-ray diffraction studies and is shown in the Figure as well. As sulfides are not considered as H-bond acceptors, these reaction outcomes effectively ruled out the participation of the ortho-heteroatoms via H-bonding. The dramatic impact by these hetereoatoms (N or S) suggests that they might alternatively be involved in coordination20 to the formally cationic gold center in the postulated metal carbene moiety. As shown in Scheme 2, such a tricoordinated gold center in E/E′ should be less electron-deficient than the dicoordinated one in D, therefore rendering it more capable of back-donating electrons to the carbene center. Consequently, the carbene center would be less cationic due to decreased contribution by the mesomeric cationic form E′. The thus-tempered electrophilicity of the gold carbene is likely the key to achieving its chemoselective trapping by carboxamides for the formation of 2,4-disubstituted oxazoles. The slightly higher yields in the cases of L3 vs. L2 (Figure 2) and Mor-DalPhos vs. L1 are consistent with tricoordinated gold carbene intermediates as the bigger substituents on the heteroatoms of L3 and Mor-DalPhos would better facilitate their formation via steric coercion.9e
Scheme 2.

Rationale for the role of Mor-DalPhos in the gold catalysis
To provide further support for the involvement of the tricoordinated gold carbene E, we resorted to DFT calculations. As shown in Figure 3, the dicoordinated gold carbene D-Me (R in D is Me) was partially optimized by fixing the distance between Au and N at 2.930 A, the same as that found in the X-ray structure of Mor-DalPhosAuCl,16b and the tricoordinated species E-Me (R in E is Me) was fully optimized. The latter was 5.4 kcal/mol more stable than the former, therefore strongly supporting the role of E in the catalysis. Moreover, the summed NBO charge for all the atoms in the indicated box decreases from +0.346 to +0.246 upon nitrogen coordination, which supports our reasoning that the electrophilicity of the carbene center might be attenuated in the process. An additional notable change upon the formatin of E-Me is that the Au-C bond was shortened by 0.034 A, suggesting an increased back donation by the gold center.
Figure 3.

The partially optimized structure D with fixed Au-N distance of 2.930 A and the fully optimized structure E. The selected bond lengths are in angstroms, bond angles are in degrees. The relative energies ΔE are in kcal/mol. Calculated at PBE1PBE/6-311+G** level
Tricoordinated gold complexes, such as (Ph3P)2AuCl21 and XantPhosAuCl,22 though amply documented in literature, have surprisingly had little relevance in homogeneous gold(I) catalysis. The only exception we are aware of is a dehydrogenative silylation reaction between alcohols and R3SiH involving XantPhosAuCl.22–23 Considering the vast majority of gold catalysis involving π systems, this revelation that tricoordinated gold(I) species can attenuate the electrophilicity of carbene centers and hence enable new reactions will likely spur further development in homogeneous gold(I) catalysis via the use of designed effacacious bidentate ligands.
In summary, we have developed a gold(I)-catalzyed modular synthesis of 2,4-disubstituted oxazoles via [3+2] annulations between readily available terminal alkynes and aromatic/alkenic carboxamides under mild reaction conditions. The key reaction intermediate is an α-oxo gold carbene, generated via gold-promoted oxidation of a terminal alkyne. Contrary to our previous observations that this type of intermediates with various monodentate phosphines or NHC as the ligand to the metal center is highly electrophilic and challenging to be efficiently trapped by stoichiometric external nucleophiles, we discovered for the first time that the use of a P,N- or P,S-bidentate ligand especially Mor-DalPhos significantly tempered its reactivities, thereby permitting efficient attack by a carboxamide en route to the formation of the oxazole ring. The nature of this reactivity modulation is attributed to the formation of a tricoordinated gold carbene spieces by engaging the non-phosphorus heteroatom, which is strongly supported by DFT calculations. The rare involvment of tricoordinated gold complexes in homogeneous gold catalysis and their role in modulating reactivities of cationic gold intermediates makes this discovery important and should stimulate new advance in this intensely researched field.
Supplementary Material
Table 2.
|
1/2 = 1.5/1; initially [2] = 0.1 M; the oxidant was introduced to the reaction vial by a syringe pump.
Isolated yield.
2 equiv. of the alkyne and 3 equiv. of the oxides used.
The alkyne was added along with the oxide by a syringe pump.
Reaction temperature: 100 °C.
Acknowledgments
We are grateful for the generous financial support by NIH (R01 GM084254) and NSF (CAREER CHE-0969157). LZ dedicates this work to Professor Masato Koreeda on the occasion of his 70th birthday.
Footnotes
Supporting Information Available: Experimental procedures, compound characterization data and computational details are available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Ichiba T, Yoshida WY, Scheuer PJ, Higa T, Gravalos DG. J Am Chem Soc. 1991;113:3173–3174. [Google Scholar]
- 2.Searle PA, Molinski TF. J Am Chem Soc. 1995;117:8126–8131. [Google Scholar]
- 3.For selected reviews, see: Boyd GV. Sci Synth. 2001;11:383–480.Yeh VSC. Tetrahedron. 2004;60:11995–12042.
- 4.(a) Wipf P, Miller CP. Tetrahedron Lett. 1992;33:907–910. [Google Scholar]; (b) Phillips AJ, Uto Y, Wipf P, Reno MJ, Williams DR. Org Lett. 2000;2:1165–1168. doi: 10.1021/ol005777b. [DOI] [PubMed] [Google Scholar]; (c) Crosignani S, Young AC, Linclau B. Tetrahedron Lett. 2004;45:9611–9615. [Google Scholar]
- 5.(a) Williams DR, Lowder PD, Gu YG, Brooks DA. Tetrahedron Lett. 1997;38:331–334. [Google Scholar]; (b) Barrish JC, Singh J, Spergel SH, Han WC, Kissick TP, Kronenthal DR, Mueller RH. J Org Chem. 1993;58:4494–4496. [Google Scholar]; (c) Meyers AI, Tavares F. Tetrahedron Lett. 1994;35:2481–2484. [Google Scholar]
- 6.For one-step modular synthesis of 2,5-disubstituted oxazoles, see: Jiang H-F, Huang H-W, Cao H, Qi C-R. Org Lett. 2010;12:5561–5563. doi: 10.1021/ol1023085.He W, Li C, Zhang L. J Am Chem Soc. 2011:8482–8485. doi: 10.1021/ja2029188.
- 7.For examples of one-step bimolecular annulations of oxazoles, see: Theilig G. Chem Ber. 1953;86:96–109.Huisgen R, Blaschke H. Tetrahedron Lett. 1964;5:1409–1413.Hammar WJ, Rustad MA. J Heterocycl Chem. 1981;18:885–8.Schuh K, Glorius F. Synthesis. 2007;2007:2297–2306.Shi B, Blake AJ, Lewis W, Campbell IB, Judkins BD, Moody CJ. J Org Chem. 2009;75:152–161. doi: 10.1021/jo902256r.Zhang X, Teo WT, Chan PWH. J Organomet Chem. 2011;696:331–337.Ritson DJ, Spiteri C, Moses JE. J Org Chem. 2011;76:3519–3522. doi: 10.1021/jo1025332.Kuwano R, Kameyama N, Ikeda R. J Am Chem Soc. 2011;133:7312–7315. doi: 10.1021/ja201543h.
- 8.For selected reviews, see: Furstner A, Davies PW. Angew Chem, Int Ed. 2007;46:3410–3449. doi: 10.1002/anie.200604335.Hashmi ASK. Chem Rev. 2007;107:3180–3211. doi: 10.1021/cr000436x.Arcadi A. Chem Rev. 2008;108:3266–3325. doi: 10.1021/cr068435d.Jimenez-Nunez Es, Echavarren AM. Chem Rev. 2008;108:3326–3350. doi: 10.1021/cr0684319.Li Z, Brouwer C, He C. Chem Rev. 2008;108:3239–3265. doi: 10.1021/cr068434l.Abu Sohel SM, Liu R-S. Chem Soc Rev. 2009;38:2269–2281. doi: 10.1039/b807499m.Wang S, Zhang G, Zhang L. Synlett. 2010:692–706.Xiao J, Li X. Angew Chem, Int Ed. 2011;50:7226–7236. doi: 10.1002/anie.201100148.
- 9.(a) Ye L, Cui L, Zhang G, Zhang L. J Am Chem Soc. 2010;132:3258–3259. doi: 10.1021/ja100041e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ye L, He W, Zhang L. J Am Chem Soc. 2010;132:8550–8551. doi: 10.1021/ja1033952. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ye L, He W, Zhang L. Angew Chem, Int Ed. 2011;50:3236–3239. doi: 10.1002/anie.201007624. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) He W, Xie L, Xu Y, Xiang J, Zhang L. Org Biomol Chem. 2012;10:3168–3171. doi: 10.1039/c2ob25235j. [DOI] [PubMed] [Google Scholar]; (e) Wang Y, Ji K, Lan S, Zhang L. Angew Chem, Int Ed. 2012;51:1915–1918. doi: 10.1002/anie.201107561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.For selected exampels of gold-catalyzed intramolecular alkyne oxidation, see: Shapiro ND, Toste FD. J Am Chem Soc. 2007;129:4160–4161. doi: 10.1021/ja070789e.Li G, Zhang L. Angew Chem, Int Ed. 2007;46:5156–5159. doi: 10.1002/anie.200701449.Yeom HS, Lee JE, Shin S. Angew Chem, Int Ed. 2008;47:7040–7043. doi: 10.1002/anie.200802802.Hashmi AS, Buhrle M, Salathe R, Bats J. Adv Synth Catal. 2008;350:2059–2064.Davies PW, Albrecht SJC. Angew Chem, Int Ed. 2009;48:8372–8375. doi: 10.1002/anie.200904309.Li CW, Lin GY, Liu RS. Chem Eur J. 2010;16:5803–5811. doi: 10.1002/chem.201000009.
- 11.(a) Davies PW, Cremonesi A, Martin N. Chem Commun. 2011;47:379–381. doi: 10.1039/c0cc02736g. [DOI] [PubMed] [Google Scholar]; (b) Qian D, Zhang J. Chem Commun. 2011;47:11152–11154. doi: 10.1039/c1cc14788a. [DOI] [PubMed] [Google Scholar]; (c) Vasu D, Hung HH, Bhunia S, Gawade SA, Das A, Liu RS. Angew Chem, Int Ed. 2011;50:6911–6914. doi: 10.1002/anie.201102581. [DOI] [PubMed] [Google Scholar]; (d) Liu RS, Dateer RB, Pati KK. Chem Commun. 2012;48:7200–7202. doi: 10.1039/c2cc33030j. [DOI] [PubMed] [Google Scholar]; (e) Bhunia S, Ghorpade S, Huple DB, Liu RS. Angew Chem, Int Ed. 2012;51:2939–2942. doi: 10.1002/anie.201108027. [DOI] [PubMed] [Google Scholar]; (f) Qian D, Zhang J. Chem Commun. 2012;48:7082–7084. doi: 10.1039/c2cc31972a. [DOI] [PubMed] [Google Scholar]
- 12.(a) Doyle MP, McKervey MA, Ye T. Modern Catalytic Methods for Organic Synthesis with Diazo Compounds: From Cyclopropanes to Ylides. Wiley; New York: 1998. [Google Scholar]; (b) Taber DF. In: In Carbon-Carbon Σ-Bond Formation. Pattenden G, editor. Vol. 3. Pergamon Press; Oxford, England; New York: 1991. pp. 1045–1062. [Google Scholar]; (c) Davies HML, Beckwith REJ. Chem Rev. 2003;103:2861–2903. doi: 10.1021/cr0200217. [DOI] [PubMed] [Google Scholar]
- 13.For earlier reportes with α-diazoester substrates, see: Fructos MR, Belderrain TR, de Fremont P, Scott NM, Nolan SP, Diaz-Requejo MM, Perez PJ. Angew Chem Int Ed. 2005;44:5284–5288. doi: 10.1002/anie.200501056.Fructos MR, de Fremont P, Nolan SP, Diaz-Requejo MM, Perez PJ. Organometallics. 2006;25:2237–2241.Prieto A, Fructos MR, Mar Diaz-Requejo M, Perez PJ, Perez-Galan P, Delpont N, Echavarren AM. Tetrahedron. 2009;65:1790–1793.Noey EL, Luo Y, Zhang L, Houk KN. J Am Chem Soc. 2012;134:1078–1084. doi: 10.1021/ja208860x.Pagar VV, Jadhav AM, Liu RS. J Am Chem Soc. 2011;133:20728–20731. doi: 10.1021/ja209980d.
- 14.Seidel G, Mynott R, Furstner A. Angew Chem, Int Ed. 2009;48:2510–2513. doi: 10.1002/anie.200806059. [DOI] [PubMed] [Google Scholar]
- 15.Fors BP, Watson DA, Biscoe MR, Buchwald SL. J Am Chem Soc. 2008;130:13552–13554. doi: 10.1021/ja8055358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.(a) Lundgren RJ, Peters BD, Alsabeh PG, Stradiotto M. Angew Chem, Int Ed. 2010;49:4071–4074. doi: 10.1002/anie.201000526. [DOI] [PubMed] [Google Scholar]; (b) Hesp KD, Stradiotto M. J Am Chem Soc. 2010;132:18026–18029. doi: 10.1021/ja109192w. [DOI] [PubMed] [Google Scholar]
- 17.Nishida H, Takada N, Yoshimura M, Sonoda T, Kobayashi H. Bull Chem Soc Jpn. 1984;57:2600–2604. [Google Scholar]
- 18.Instead observed were the related carboxymethyl ketones, formed due to hydrolysis of the imidate intermediates of type B (R = alkyl).
- 19.(a) Blumlein FO. Ber. 1884;17:2578–2581. [Google Scholar]; (b) Lewy M. Ber. 1887;20:2576–2580. [Google Scholar]
- 20.For a case where a pendant aryl group can interact with cationic gold center, see: Perez-Galan P, Delpont N, Herrero-Gomez E, Maseras F, Echavarren AM. Chem Eur J. 2010;16:5324–5332. doi: 10.1002/chem.200903507.
- 21.Bowmaker GA, Dyason JC, Healy PC, Engelhardt LM, Pakawatchai C, White AH. J Chem Soc, Dalton Trans. 1987:1089–1097. [Google Scholar]
- 22.Ito H, Takagi K, Miyahara T, Sawamura M. Org Lett. 2005;7:3001–3004. doi: 10.1021/ol050979z. [DOI] [PubMed] [Google Scholar]
- 23.Ito H, Saito T, Miyahara T, Zhong C, Sawamura M. organometallics. 2009;28:4829–4840. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.