

Abstract
Substantial evidence suggests that transient production of reactive oxygen species (ROS) such as hydrogen peroxide (H2O2) is an important signaling event triggered by the activation of various cell surface receptors. Major targets of H2O2 include protein tyrosine phosphatases (PTPs). Oxidation of the active site Cys by H2O2 abrogates PTP catalytic activity, thereby potentially furnishing a mechanism to ensure optimal tyrosine phosphorylation in response to a variety of physiological stimuli. Unfortunately, H2O2 is poorly reactive in chemical terms and the second order rate constants for the H2O2-mediated PTP inactivation are ~10 M−1s−1, which is too slow to be compatible with the transient signaling events occurring at the physiological concentrations of H2O2. We find that hydroxyl radical is produced from H2O2 solutions in the absence of metal chelating agent by the Fenton reaction. We show that hydroxyl radical is capable of inactivating the PTPs and the inactivation is active site directed, through oxidation of the catalytic Cys to sulfenic acid, which can be reduced by low molecular weight thiols. We also show that hydroxyl radical is a kinetically more efficient oxidant than H2O2 for inactivating the PTPs. The second-order rate constants for the hydroxyl radical-mediated PTP inactivation are at least 2–3 orders of magnitude higher than those mediated by H2O2 under the same conditions. Thus, hydroxyl radical generated in vivo may serve as a more physiologically relevant oxidizing agent for PTP inactivation.
Keywords: Protein tyrosine phosphatases, hydrogen peroxide, hydroxyl radical, redox regulation
1. Introduction
Protein tyrosine phosphorylation plays a central role in many cellular processes ranging from growth and metabolism to adhesion and differentiation [1]. Major insights into tyrosine phosphorylation mediated cellular events have been derived from studies of protein tyrosine kinases (PTKs). This is due in part to the fact that many transmembrane receptors for peptide hormones and growth factors possess intrinsic PTK activity. Receptors for cytokines lack intrinsic kinase activity but associate with non-receptor PTKs inside the cell. Consequently it is common to view signaling pathways as cascades of reactions emanating from the PTKs. However, protein tyrosine phosphorylation is a dynamic process controlled by two opposing biochemical reactions catalyzed by PTK and protein tyrosine phosphatases (PTP) [2]. Not surprisingly, disturbance of the normal balance between PTK and PTP activity leads to aberrant tyrosine phosphorylation, which has been linked to a variety of human diseases. Given the reversible nature of protein tyrosine phosphorylation, illumination of the regulatory mechanisms for PTP function is a prerequisite to gaining a complete understanding of the physiological consequences of tyrosine phosphorylation and how such signaling events are abrogated in pathological conditions.
An emerging layer of regulation of the PTP activity is reversible inactivation through stimulus-mediated oxidation. Although initially viewed as toxic byproducts, reactive oxygen species (ROS) such as hydrogen peroxide (H2O2) are increasingly recognized as important regulators of cellular and physiological processes [3,4]. For example, stimulation of cognate cell surface receptors with ligands as diverse as insulin [5], EGF [6], PDGF [7] or TGF-β [8] induces a burst of intracellular production of H2O2 immediately after ligand binding. Importantly, it appears that H2O2 production is required for ligand-mediated PTK activation and tyrosine phosphorylation. A major proposed mechanism by which the ligand-induced H2O2 regulates the tyrosine phosphorylation-dependent signaling is through transient oxidation and inactivation of the PTPs [9–13].
The PTPs constitute a large family of enzymes (>100) that parallel PTKs in their structural diversity and complexity [14]. Unlike protein kinases, where tyrosine specific and serine/threonine specific kinases share sequence identity, the PTPs show no sequence similarity with serine/threonine phosphatases, or the broad specificity phosphatases such as acid or alkaline phosphatases. The hallmark that defines the PTP superfamily is the active site sequence (I/V) HCXAGXGR (S/T), the PTP signature motif. Extensive mechanistic studies have established that members of the PTP family utilize a common mechanism for substrate turnover [15]. A key feature in the PTP catalytic mechanism is utilization of the active site cysteine as the attacking nucleophile to form a thiophosphoryl enzyme intermediate, which is hydrolyzed in the dephosphorylation reaction. Due to the highly positively charged environment in the PTP active site, the sulfhydryl group of this Cys residue has an extremely low pKa (~5) [16,17] in comparison to the typical pKa for a Cys residue in proteins (~8.5). Thus at physiological conditions, the side chain of the PTP active site Cys exists as the thiolate anion, which not only enhances its nucleophilicity but also renders it susceptible to oxidation. Biochemical studies indicate that upon exposure to H2O2, the catalytic Cys is converted into the sulfenic acid form (Cys-SOH), effectively inactivating the PTPs because the oxidized Cys can no longer function as a nucleophile [9,11,18]. This offers a biochemical basis for why ROS generation enhances growth factor-induced tyrosine phosphorylation. The sulfenic acid can be reduced back to the thiolate form through the action of cellular thiols [19], leading to the restoration of PTP activity. Thus, oxidation of the catalytic Cys by ROS is reversible and represents a dynamic mechanism of PTP regulation.
H2O2, being produced in cells downstream of many surface receptors, is thought to be the major ROS for the reversible oxidation of sulfhydryl groups inside the cell. Indeed, the majority of studies examining PTP redox regulation have been performed with H2O2. However, although H2O2 is capable of oxidizing the PTPs, H2O2 is poorly reactive in chemical terms [20]. The rate constants measured for H2O2-mediated PTP inactivation are in the range of 10–20 M−1s−1 [9,21], indicating that the loss of PTP activity will be a very slow process (t1/2 > 10 h) at the physiological concentrations of H2O2 thought to exist during signaling events (0.1–1 μM) [22,23]. Paradoxically, ROS-mediated PTP inactivation during signaling events typically occurs rapidly (2–5 min) which coincides with the transient increase in H2O2 concentration upon growth factor stimulation [5,7,10,13]. The kinetic discrepancy between the observed robust PTP inactivation during cellular signaling events and the apparent sluggish activity of H2O2 toward the PTPs in the test tube may be reconciled if H2O2 undergoes spontaneous or enzymatic conversion to more reactive oxidizing agents that can mediate rapid intracellular PTP inactivation [22–24]. In this study we provide evidence that hydroxyl radical may serve as the more physiologically relevant oxidizing agent for PTP inactivation.
2. Materials and Methods
2.1. Materials
Catalase (from bovine liver), superoxide dismutase (from E. coli.), glutathione (GSH, 98% purity), and 30% H2O2 solution were from Sigma; 4-chloro-7-nitrobenzo-2-oxa-1,3-diazole (NBD-Cl, 98% purity), 5,5-Dimethyl-1-pyrroline N-oxide (DMPO, 97% purity) were Aldrich products; and DTT was from LabScientific Inc. FeCl2 and MnCl2 were obtained from Sigma-Aldrich.
2.2. Inactivation assay
All PTP inactivation reactions were measured continually by following the change of optical density at 405 nm at 25°C. The reaction was initiated by adding enzyme to the 1 ml reaction system in a cuvette: 50 mM 3,3-dimethylglutarate (pH 6.0), 20 mM p-nitrophenyl phosphate (pNPP), H2O2 of different concentrations, with or without 1 mM EDTA. The final enzyme concentrations were 10 nM for PTP1B, 80 nM for VHR, 32 nM for PTPα, 40 nM for HePTP, and 30 nM for CD45. The observed first-order inactivation rate constant k, was obtained by fitting the data to Abs = (Amp)e−kt + B, where Abs is absorbance at 405 nm, Amp is the change in absorbance, k is the first-order rate constant, t is time, and B is the starting absorption [9]. The second-order rate constant for the inactivation in the presence of EDTA, was obtained by plotting the first-order rate constant versus H2O2 concentration; the second-order rate constant for the inactivation in the absence of EDTA, was derived from fitting the first order rate constant to the Michaelis-Menton formulation.
2.3. NBD-Cl modification of PTP1B
10 μM PTP1B in 50 mM 3,3-dimethylglutarate buffer (pH 6.0) was firstly treated with 1 mM DTT to fully reduce all cysteine residues. The DTT was then removed by Amicon ultra PL-10 (Molecular weight cut-off is 10,000, from Amersham Pharmacia). H2O2 was added to the final concentration of 300 μM for the oxidation of PTP1B in the presence of 1 mM EDTA and 30 μM for the oxidation of PTP1B in the absence of EDTA. After 15 min, 100 units of catalase were added to eliminate the residual H2O2. After 3 min catalase treatment, NBD-Cl was added to a final concentration of 0.6 mM. The modification reactions were then allowed to proceed at room temperature for 30 min. The samples were then spinned with Amicon ultra PL-10 with multiple changes of the 50 mM 3,3-dimethylglutarate buffer (pH 6.0) to remove the residual NBD-Cl. Finally, the sample with a final volume of 0.5 ml was scanned by UV spectrometer [9]
2.4. EPR measurements
The following buffer solutions were freshly made: 50 mM 3,3-dimethylglutarate pH 6.0, without EDTA; 50 mM 3,3-dimethylglutarate pH 6.0, 1 mM EDTA; 20 mM PBS, pH 6.0, without EDTA; 20 mM PBS, pH 6.0, 1 mM EDTA. A stock of 5,5-Dimethyl-1-pyrroline N-oxide (DMPO) was prepared in water (880 mM). DMPO was added to an appropriate buffer with or without 100 μM H2O2 to a final concentration of 88 mM. 50 μl of the incubation mixture was immediately sealed in a capillary tube before the EPR measurement. EPR spectra were obtained with a Bruker 200D X-band spectrometer with ESP 300 upgrade and VT 4111 temperature controller. Instrumental settings were as follows: temperature, 25°C; microwave power, 20 mW; microwave frequency, 9.514 GHz; modulation amplitude, 1.011 G, and receiver gain, 1.60×105.
3. Results and discussion
The term ROS encompasses many species including H2O2, superoxide anion radical, lipid peroxides, nitric oxide and hydroxyl radical. In addition to H2O2, it has been shown that treatment of various PTPs with superoxide radical anion [11], nitric oxide [25], and lipid peroxides [26] all lead to the oxidation of the active site Cys. The majority of studies have analyzed the effects of H2O2 since it is produced upon activation of many cell surface receptors. However, the concept that H2O2 is the most relevant oxidant for the PTPs has been recently challenged based upon kinetic considerations of H2O2 reactivity. Thus, the exact identity of the oxidants which directly mediate PTP oxidation is not known. Although H2O2 is not active, it can be readily converted to much more reactive hydroxyl radical (•OH), either by exposure to UV light or by reduced (lower oxidation states) transition metal ions through the Fenton reaction [20,27]. In the following we provide evidence that hydroxyl radical (•OH) is highly reactive with the PTP and may serve as a physiologically relevant PTP oxidant.
3.1. PTPs can be more rapidly inactivated by H2O2 in the absence of EDTA
The potential for hydroxyl radical as a physiological oxidant for the PTPs has not been considered. This may have stemmed from the fact that in kinetic studies it is a common practice to include EDTA or other chelating agents in the assay buffers in order to protect the active site thiols of the PTPs from reacting with heavy metals as well as metal ion-catalyzed oxidation by molecular O2. We hypothesized that in the absence of EDTA, the rate of H2O2-mediated PTP inactivation may be accelerated due to hydroxyl radical production from H2O2 in the presence of trace amount of free transition metal ions. To test this hypothesis, we initially focused our study on the prototypic member of the PTP family PTP1B [28], which plays important roles in inhibiting insulin and leptin signaling and promoting HER2-mediated breast tumorigenesis [29]. We first determined the effect of H2O2 on the PTP1B-catalyzed hydrolysis of p-nitrophenyl phosphate (pNPP) at 25 °C in 50 mM 3,3-dimethylglutarate pH 6.0 buffer containing 1 mM EDTA. Inactivation of PTP1B was monitored continuously by the formation of p-nitrophenolate absorbance at 405 nm in a UV-visible spectrophotometer. As expected, we found that H2O2 can inactivate PTP1B in a time- and concentration-dependent first order process (Figure 1A). From the linear relationship of the pseudo-first-order rate constant and H2O2 concentration (Figure 1B), the second order rate constant was calculated to be 11.6 ± 0.7 M−1s−1, a value similar to those previously determined for the PTPs including PTP1B [9,21].
Figure 1.
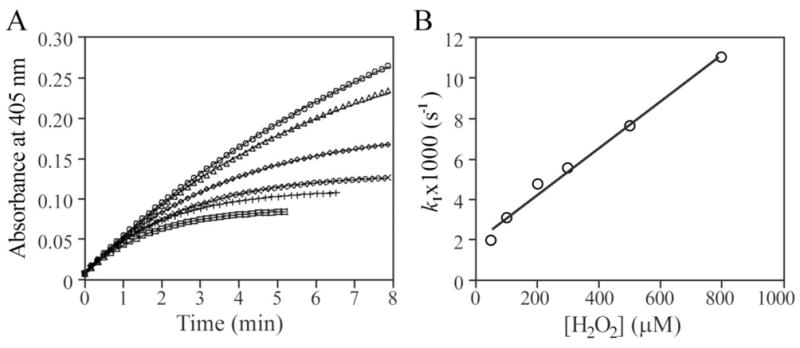
Inactivation kinetics of PTP1B by H2O2 in the presence of EDTA. A. Time and concentration dependence for the H2O2-mediated PTP1B inactivation. PTP1B inactivation was measured continually by following the change of optical density at 405 nm at 25°C. The reaction was initiated by adding PTP1B to the 1 ml reaction system in a cuvette: 50 mM 3,3-dimethylglutarate (pH 6.0), 20 mM pNPP, 1 mM EDTA. The H2O2 concentrations were 50, 100, 300, 500, 800, and 1,000 μM for the time courses from the top to the bottom. B. Concentration dependence of the pseudo-first-order rate constant k1 for H2O2-mediated PTP1B inactivation.
We then studied the effect of H2O2 on PTP1B-catalyzed pNPP hydrolysis in the same buffer without EDTA. Incubation of H2O2 with PTP1B resulted in a time- and concentration-dependent loss of PTP activity at much lower H2O2 concentrations in the absence of EDTA (Figure 2A). This result indicated that, in the absence of EDTA, H2O2 is a significantly more efficient inactivator of PTP1B. Interestingly, unlike the reaction in the presence of EDTA, analysis of the pseudo-first-order rate constant as a function of H2O2 concentration showed that the H2O2-mediated PTP1B inactivation displayed saturation kinetics (Figure 2B), yielding values for the equilibrium binding constant KI and the inactivation rate constant ki of 6.0 ± 0.4 μM and 4.04 ± 0.12 × 10−3 s−1, respectively. Thus the second-order rate constant for the H2O2-mediated PTP1B inactivation in the absence of EDTA is 674 ± 41 M−1s−1, which is nearly sixty fold higher than that in the presence of EDTA.
Figure 2.
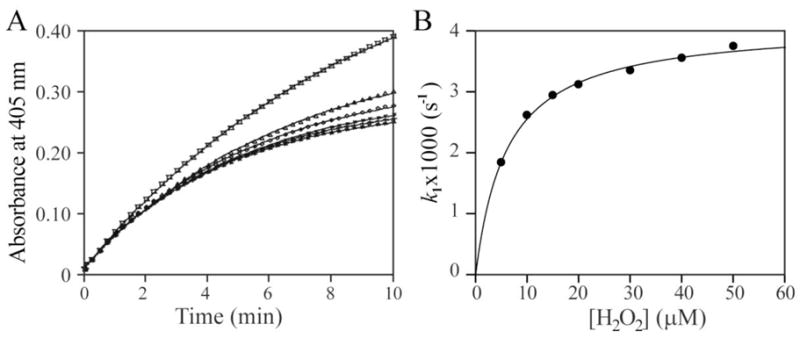
Inactivation kinetics of PTP1B by H2O2 in the absence of EDTA. A. Time and concentration dependence for the H2O2-mediated PTP1B inactivation. PTP1B inactivation was measured continually by following the change of optical density at 405 nm at 25°C. The reaction was initiated by adding PTP1B to the 1 ml reaction system in a cuvette: 50 mM 3,3-dimethylglutarate (pH 6.0), and 20 mM pNPP. The H2O2 concentrations were 5, 10, 20, 30, 40, and 50 μM for the time courses from the top to the bottom. B. Concentration dependence of the pseudo-first-order rate constant k1 for H2O2-mediated PTP1B inactivation.
In order to determine whether the heightened H2O2 reactivity observed in the absence of EDTA was unique to PTP1B, we also examined the ability of H2O2 to inactivate several other PTP family members including the cytosolic HePTP, the receptor-like PTPs, CD45 and PTPα, and the dual specificity phosphatase VHR. Similar to PTP1B, the inactivation of HePTP, CD45, PTPα, and VHR by H2O2 in the presence of EDTA occurred with a second-order rate constant of 2.8 ± 0.3 M−1s−1, 4.7 ± 0.2 M−1s−1, 6.0 ± 0.6 M−1s−1, and 3.1 ± 0.4 M−1s−1, respectively, while in the absence of EDTA, the second order rate constants for H2O2-mediated inactivation of HePTP, CD45, PTPα, and VHR were 973 ± 90 M−1s−1, 1,530 ± 232 M−1s−1, 152 ± 15 M−1s−1, and 212 ± 29 M−1s−1 (Table 1). Taken together, the results show that H2O2 is 2 to 3 orders of magnitude more reactive toward the PTPs in the absence of EDTA.
Table 1.
Kinetic parameters for the H2O2-mediated PTP inactivation in the presence and absence of EDTA
| PTP | With EDTA | Without EDTA | |
|---|---|---|---|
| k2 (M−1s−1) | k2 (M−1s−1) | Ki (μM) | |
| PTP1B | 11.6±0.7 | 674±41 | 6.0±0.4 |
| VHR | 3.1±0.4 | 212±29 | 29.4±4.0 |
| PTPα | 6.0±0.6 | 152±15 | 196±18 |
| HePTP | 2.8±0.3 | 973±90 | 7.5±0.7 |
| CD45 | 4.7±0.2 | 1530±232 | 7.4±1.1 |
3.2. Hydroxyl radical is responsible for the increased reactivity of H2O2 with the PTPs in the absence of EDTA
There should be no free transition metal ions in the presence of the chelator EDTA. Consequently, the observed rates for H2O2-mediated PTP inactivation in the presence of EDTA are intrinsic to H2O2. We speculated that the increased rate of PTP inactivation by H2O2 in the absence of EDTA might be due to the highly reactive hydroxyl radical generated by Fenton chemistry catalyzed by the trace amount of transition metals in the solution. If this were the case, then a further increase in the rate of H2O2-mediated PTP inactivation could be observed upon introduction of free Fe2+ to the reaction in the absence of EDTA. Indeed, as shown in Figure 3, addition of Fe2+ (10 nM) to the reaction further increased the H2O2-mediated PTPB inactivation. Like Fe2+, Mn2+ ion (10 nM) can also increase the H2O2 mediated PTP1B inactivation (data not shown). However, neither Fe2+ (10 nM) nor Mn2+ (10 nM) by itself affected PTP1B activity. Furthermore, PTP1B can be reactivated by DTT, but not EDTA, after it was completely inactivated in the presence of both Fe2+ and H2O2 (Figure 4). The results suggested that the increased inactivation was not caused by metal ion itself, but by metal ion mediated change of oxidation mechanism, likely the production of hydroxyl radicals.
Figure 3.
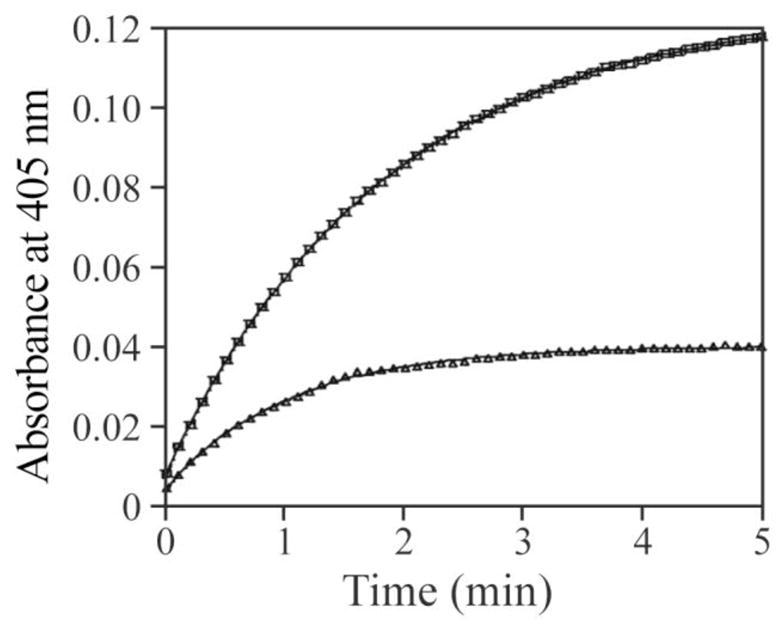
Effect of Fe2+ on H2O2 induced PTP1B inactivation in the absence of EDTA. PTP1B was added to the inactivation system in a 1 ml cuvette which was pre-incubated at 25°C; and the final concentration of PTP1B was 10 nM. The inactivation system was 50 mM 3,3-dimethylglutarate (pH 6.0), 20 mM pNPP, and 100 μM H2O2 without (for the upper curve) or with 10 nM FeCl2 (for the lower curve). The reaction was measured by following the change of the optical density at 405 nm upon the addition of PTP1B.
Figure 4.
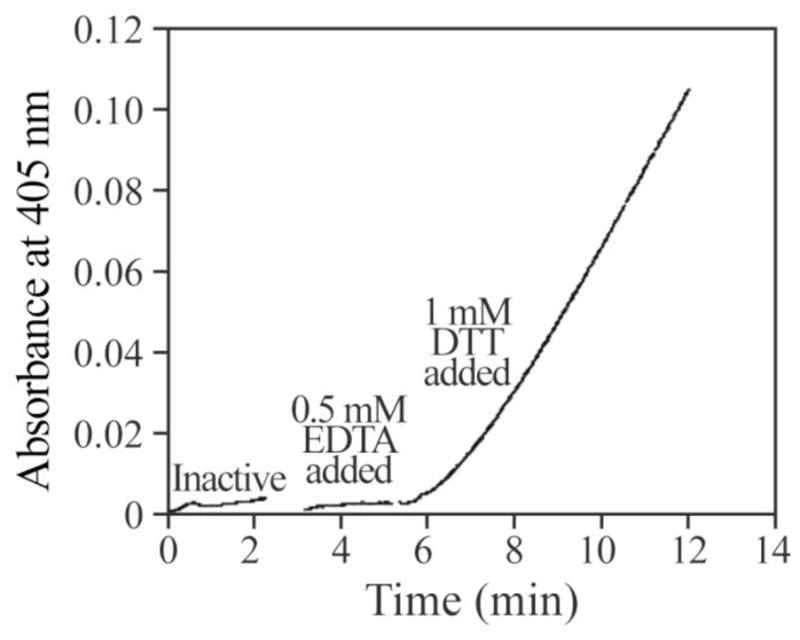
Recovery of PTP1B activity after it was inactivated by H2O2 in the presence of trace amount of Fe2+. PTP1B with final concentration of 2 μM was incubated in 100 μl of 50 mM 3,3-dimethylglutarate buffer (pH 6.0) containing 10 nM FeCl2 50 μM H2O2. When it was completely inactivated, 5 μl the inactivated PTP1B was added to a cuvette containing 1 ml 50 mM 3,3-dimethylglutarate (pH 6.0), 20 mM pNPP and was pre-incubated at 25°C. The final concentration of PTP1B was 10 nM. The change of optical density at 405 nm was measured upon the addition of PTP1B. Three minutes later EDTA was added to the cuvette with final concentration of 0.5 mM. After continually 2 min measurement, DTT was added to 1 mM; and the measurement was allowed to proceed for 5 more minutes.
The second-order rate constant for superoxide radical anion-mediated PTP1B inactivation is 334 ± 45 M−1s−1 [11], which is comparable to those determined for H2O2 in the absence of EDTA. To determine whether superoxide radical anion was involved in PTP1B inactivation by H2O2 in the absence of EDTA, we included superoxide dismutase in the experiment. Specifically, 5 μl of a PTP1B stock solution was added to a cuvette containing 1 ml 50 mM 3,3-dimethylglutarate (pH 6.0), 20 mM pNPP, 10 μM H2O2, and 60 units of superoxide dismutase to initiate the reaction. The final PTP1B concentration was 10 nM; and the inactivation was measured continually by following the change of optical density change at 405 nm. The first-order rate constant determined in the presence of superoxide dismutase (2.23 ± 0.04 × 10−3s−1) is indistinguishable to that measured in the absence of superoxide dismutase (2.21 ± 0.05 ×10−3s−1). This indicated that there is no superoxide radical anion present in the system.
The hydroxyl radical is highly reactive with a half-life in aqueous solution of less than 1 ns [30]. To provide direct evidence for the existence of hydroxyl radicals in the H2O2 solution without EDTA, electron paramagnetic resonance (EPR) spin trapping experiments with 5,5-dimethyl-1-pyrroline N-oxide (DMPO) were performed. Spin trapping allows the visualization of transient free radical populations by reacting short-lived radicals with a spin trap to produce persistent spin adduct radicals [31]. Figure 5 shows the EPR spectra of 88 mM DMPO in 50 mM 3,3-dimethylglutarate (pH 6.0) buffer with or without 100 μM H2O2 in the presence or absence of EDTA. Formation of hydroxyl radical from H2O2 in the absence of EDTA was supported by the appearance of the typical EPR spectrum of DMPO/•OH adduct [32]. Similar results were obtained in phosphate buffered saline (data not shown). The results clearly demonstrate that hydroxyl radical is generated from H2O2 solution in the absence of EDTA.
Figure 5.
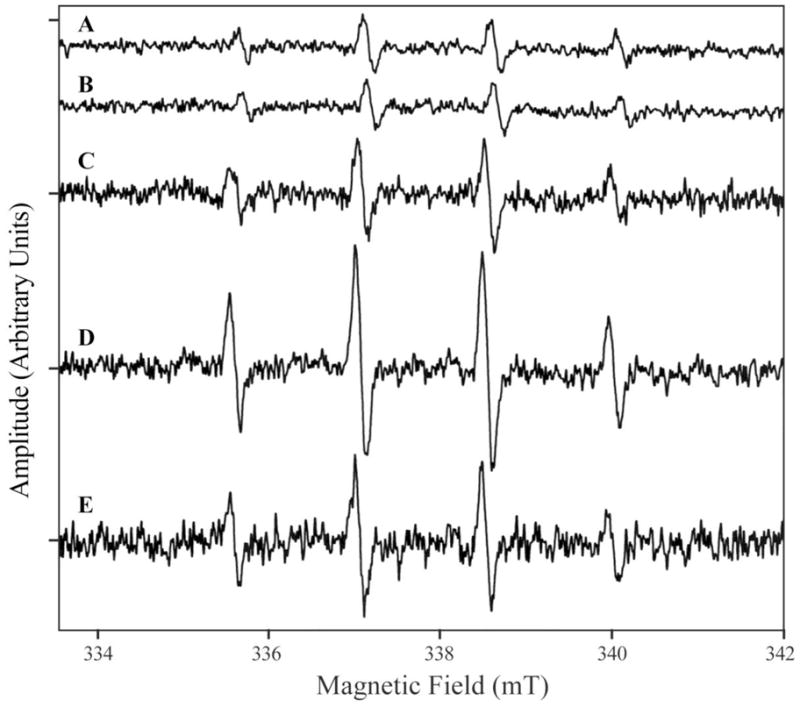
EPR spectra of DMPO/OH formed in the solution of H2O2 and DMPO. All measurements were carried out at 25°C in 50 mM 3,3-dimethylglutarate pH 6.0 and 88 mM DMPO with either 1 mM EDTA and no H2O2 (trace A); no EDTA and no H2O2 (trace B); 1 mM EDTA and 100 μM H2O2 (trace C); or no EDTA and 100 μM H2O2 (trace D). Trace E is the difference spectrum between trace D and C.
3.3. Hydroxyl radical inactivates the PTPs through oxidation of the active site Cys to sulfenic acid
The results presented above suggest that the observed higher reactivity of H2O2 toward the PTPs in the absence of EDTA is likely due to the presence of hydroxyl radical. Given the extremely short half-life of hydroxyl radical and the minute amount of transition metal ions in the solution, we surmise that the amount of hydroxyl radical produced is unlikely stoichiometric to the H2O2 concentration. Consequently the apparent second-order rate constants determined for the H2O2-mediated PTP inactivation in the absence of EDTA probably represent only the low limits for the hydroxyl radical reaction with the PTPs. Nonetheless, we proceeded to determine whether the hydroxyl radical-mediated PTP inactivation is active site directed, reversible, and through the formation of sulfenic acid. The saturation kinetics for PTP inactivation in the absence of EDTA (Figure 2B) suggest that hydroxyl radical is an active site-directed affinity agent whose mode of action likely involves at least two steps: binding to the PTP active site followed by covalent modification of the active site Cys residue. Further evidence in support of the hydroxyl radical-mediated PTP inactivation being directed to the active site included that arsenate, a competitive PTP1B inhibitor [33], was able to protect PTP1B from the H2O2-mediated inactivation in the absence of EDTA (data not shown).
Mild oxidation of Cys by ROS to sulfenic (S-OH) acid is reversible since it can be readily reduced back to the sulfhydryl state by free thiols [19]. In contrast, higher oxidation of the Cys residue to sulfinic (S-O2H) or sulfonic (S-O3H) acid is irreversible. We investigated whether PTP1B inactivation by hydroxyl radical was reversible. PTP1B was first inactivated by H2O2 in the absence of EDTA and the excess of H2O2 was removed by the addition of catalase. Figure 6 shows that both dithiothreitol (DTT) and reduced glutathione (GSH) were capable of reactivating the completely inactivated PTP1B, although reactivation with DTT was significantly faster than with GSH. Similarly, when PTP1B was inactivated by treatment with H2O2 in the presence of EDTA, almost all of the initial activity was recovered by treatment with DTT or GSH. The thiol-reversible nature of the inactivation reaction is consistent with the notion that hydroxyl radical inactivates the PTPs by oxidizing the active site Cys to a sulfenic acid.
Figure 6.
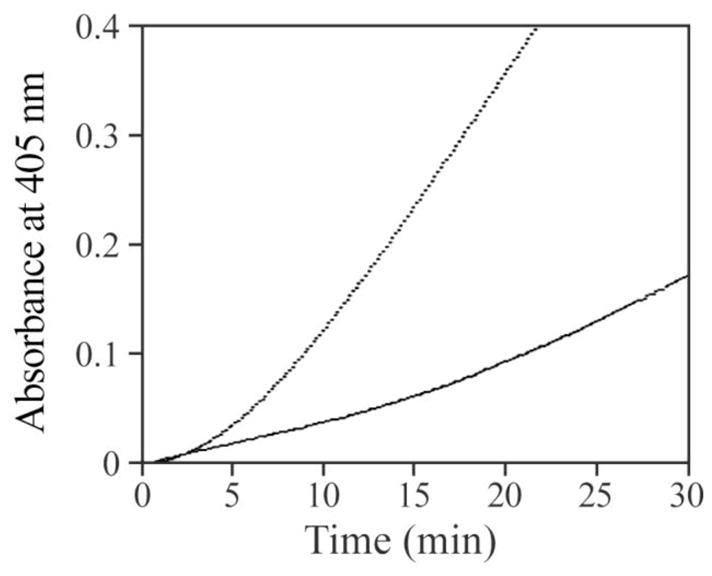
Hydroxyl radical modified PTP1B can be reactivated by thiols. PTP1B with final concentration of 2 μM was incubated in 100 μl 50 mM 3,3-dimethylglutarate buffer (pH 6.0) containing 50 μM H2O2. When it was completely inactivated, 5 μl of the inactivated PTP1B solution was added to the reactivation system in a 1 ml-cuvette which was pre-incubated at 25°C; and the final concentration of PTP1B was 10 nM. The reactivation system was 50 mM 3,3-dimethylglutarate buffer (pH 6.0), 20 mM pNPP, and 10 mM DTT (for the upper curve), or 20 mM GSH (for the lower curve). The reaction was measured by following the change of optical density at 405 nm upon addition of the enzyme at 25°C.
To furnish further evidence that the hydroxyl radical-mediated PTP1B inactivation resulted in the formation of a sulfenic acid, we utilized the sulfenic acid-labeling reagent 7-chloro-2-nitrobenzo-2-oxa-1,3-diazole (NBD-Cl) [9,11,34]. NBD chloride is an electrophilic agent, which reacts with both sulfenic acids and thiols in proteins. The Cys-S-NBD species absorbs maximally at 420 nm whereas the Cys-S(O)-NBD (sulfenic acid) species absorbs maximally at 347 nm, thus enabling clear identification of the two possible adducts. As expected, non-oxidized PTP1B treated with NBD-Cl yielded an λmax at 425 nm (Figure 7). In contrast, incubation of NBD-Cl with H2O2 oxidized PTP1B prepared either in the presence or absence of EDTA gave almost identical spectra with the same λmax at 347 nm (Figure 7). Taken together, our results are consistent with the conclusion that reaction with hydroxyl radical oxidizes the PTP active-site thiolates to sulfenic acid.
Figure 7.
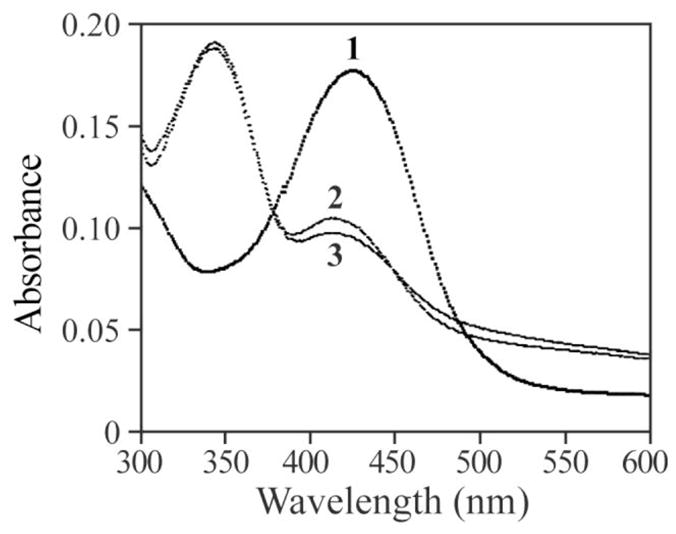
Spectroscopic analysis of NBD-modified PTP1B. 1, spectrum of NBD modified PTP1B (Cys-S-NBD adduct). 2, spectrum of NBD adduct of PTP1B oxidized by H2O2 in the absence of EDTA (Cys-S(O)-NBD). 3, spectrum of PTP1B oxidized by H2O2 in the presence of EDTA (Cys-S(O)-NBD).
In summary, we discovered that the H2O2-mediated PTP inactivation proceeded more rapidly when EDTA was removed from the reaction. We provided kinetic and biophysical evidence that hydroxyl radical is produced in H2O2 solutions in the absence of metal chelating agent EDTA by the Fenton reaction. We also showed that hydroxyl radical is capable of inactivating the PTPs and the inactivation is active site directed, through oxidation of the catalytic Cys to sulfenic acid, which can be reduced by low molecular weight thiols. We found that hydroxyl radical is a kinetically more efficient oxidant than H2O2 for oxidative inhibition of the PTPs. The differences in the observed kinetic parameters (k2 and KI in Table 1) for the hydroxyl radical-mediated inactivation of PTPs may reflect the intrinsic properties of the different phosphatase active sites. In the case of the receptor-like PTPs, the D2 domain may also influence hydroxyl radical accessibility to the D1 domain active site thus causing differences in reactivity and affinity as observed for CD45 and PTPα. The second-order rate constants for the hydroxyl radical-mediated PTP inactivation are at least 2–3 orders of magnitude higher than those mediated by H2O2 under the same conditions. Thus the half-life for the hydroxyl radical-mediated PTP inactivation at the physiological concentration of 1 μM H2O2 will be closer in time scale typical for signaling events, which occur rapidly (2–5 min) upon growth factor stimulation. Taken together, the results suggest that hydroxyl radical generated in vivo may serve as a more physiologically relevant oxidizing agent for PTP inactivation.
Highlights.
ROS are implicated as mediators of cell-signaling responses by targeting the PTPs
The exact identity of the oxidants that inactivate the PTPs is unknown
We show that hydroxyl radical is capable of inactivating the PTPs
We also show that hydroxyl radical is a kinetically more efficient oxidant than H2O2
Hydroxyl radical is a more physiologically relevant oxidant for PTP inactivation.
Acknowledgments
This work was supported by National Institutes of Health Grant CA69202. We wish to thank Dr. Philip Aisen at the Albert Einstein College of Medicine for assistance in collecting the EPR spectra.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hunter T. Tyrosine phosphorylation: thirty years and counting. Curr Opin Cell Biol. 2009;21:140–146. doi: 10.1016/j.ceb.2009.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tonks NK. Protein tyrosine phosphatases: from genes, to function, to disease. Nat Rev Mol Cell Biol. 2006;7:833–846. doi: 10.1038/nrm2039. [DOI] [PubMed] [Google Scholar]
- 3.Finkel T. Signal transduction by reactive oxygen species. J Cell Biol. 2011;194:7–15. doi: 10.1083/jcb.201102095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rhee SG. Cell signaling. H2O2, a necessary evil for cell signaling. Science. 2006;312:1882–1883. doi: 10.1126/science.1130481. [DOI] [PubMed] [Google Scholar]
- 5.Mahadev K, Zilbering A, Zhu L, Goldstein BJ. Insulin-stimulated hydrogen peroxide reversibly inhibits protein-tyrosine phosphatase 1b in vivo and enhances the early insulin action cascade. J Biol Chem. 2001;276:21938–21942. doi: 10.1074/jbc.C100109200. [DOI] [PubMed] [Google Scholar]
- 6.Bae YS, Kang SW, Seo MS, Baines IC, Tekle E, Chock PB, Rhee SG. Epidermal growth factor (EGF)-induced generation of hydrogen peroxide. Role in EGF receptor-mediated tyrosine phosphorylation. J Biol Chem. 1997;272:217–221. [PubMed] [Google Scholar]
- 7.Sundaresan M, Yu ZX, Ferrans VJ, Irani K, Finkel T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science. 1995;270:296–299. doi: 10.1126/science.270.5234.296. [DOI] [PubMed] [Google Scholar]
- 8.Thannickal VJ, Day RM, Klinz SG, Bastien MC, Larios JM, Fanburg BL. Ras-dependent and -independent regulation of reactive oxygen species by mitogenic growth factors and TGF-beta1. FASEB J. 2000;14:1741–1748. doi: 10.1096/fj.99-0878com. [DOI] [PubMed] [Google Scholar]
- 9.Denu JM, Tanner KG. Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide, evidence for a sulfenic acid intermediate and implications for redox regulation. Biochemistry. 1998;37:5633–5642. doi: 10.1021/bi973035t. [DOI] [PubMed] [Google Scholar]
- 10.Lee SR, Kwon KS, Kim SR, Rhee SG. Reversible inactivation of protein-tyrosine phosphatase 1B in A431 cells stimulated with epidermal growth factor. J Biol Chem. 1998;273:15366–15372. doi: 10.1074/jbc.273.25.15366. [DOI] [PubMed] [Google Scholar]
- 11.Barrett WC, DeGnore JP, Keng YF, Zhang ZY, Yim MB, Chock PB. Roles of superoxide radical anion in signal transduction mediated by reversible regulation of protein-tyrosine phosphatase 1B. J Biol Chem. 1999;274:34543–34546. doi: 10.1074/jbc.274.49.34543. [DOI] [PubMed] [Google Scholar]
- 12.Xu D, Rovira II, Finkel T. Oxidants painting the cysteine chapel: redox regulation of PTPs. Dev Cell. 2002;2:251–252. doi: 10.1016/s1534-5807(02)00132-6. [DOI] [PubMed] [Google Scholar]
- 13.Meng TC, Fukada T, Tonks NK. Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol Cell. 2002;9:387–399. doi: 10.1016/s1097-2765(02)00445-8. [DOI] [PubMed] [Google Scholar]
- 14.Alonso A, Sasin J, Bottini N, Friedberg I, Friedberg I, Osterman A, Godzik A, Hunter T, Dixon JE, Mustelin T. Protein tyrosine phosphatases in the human genome. Cell. 2004;117:699–711. doi: 10.1016/j.cell.2004.05.018. [DOI] [PubMed] [Google Scholar]
- 15.Zhang ZY. Mechanistic studies on protein tyrosine phosphatases. Prog Nucleic Acid Res & Mol Biol. 2003;73:171–220. doi: 10.1016/s0079-6603(03)01006-7. [DOI] [PubMed] [Google Scholar]
- 16.Zhang ZY, Dixon JE. Active site labeling of the Yersinia protein tyrosine phosphatase: The determination of the pKa of the active site cysteine and the function of the conserved histidine 402. Biochemistry. 1993;32:9340–9345. doi: 10.1021/bi00087a012. [DOI] [PubMed] [Google Scholar]
- 17.Lohse DL, Denu JM, Santoro N, Dixon JE. Roles of aspartic acid-181 and serine-222 in intermediate formation and hydrolysis of the mammalian protein-tyrosine-phosphatase PTP1. Biochemistry. 1997;36:4568–4575. doi: 10.1021/bi963094r. [DOI] [PubMed] [Google Scholar]
- 18.Salmeen A, Andersen JN, Myers MP, Meng TC, Hinks JA, Tonks NK, Barford D. Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate. Nature. 2003;423:769–773. doi: 10.1038/nature01680. [DOI] [PubMed] [Google Scholar]
- 19.Claiborne A, Yeh JI, Mallett TC, Luba J, Crane EJ, Charrier V, Parsonage D. Protein-sulfenic acids: diverse roles for an unlikely player in enzyme catalysis and redox regulation. Biochemistry. 1999;38:15407–15416. doi: 10.1021/bi992025k. [DOI] [PubMed] [Google Scholar]
- 20.Halliwell B, Clement MV, Long LH. Hydrogen peroxide in the human body. FEBS Lett. 2000;486:10–13. doi: 10.1016/s0014-5793(00)02197-9. [DOI] [PubMed] [Google Scholar]
- 21.Sun JP, Wang WQ, Yang H, Liu S, Liang F, Fedorov AA, Almo SC, Zhang ZY. Structure and Biochemical Properties of PRL-1, a Phosphatase Implicated in Cell Growth, Differentiation, and Tumor Invasion. Biochemistry. 2005;44:12009–12021. doi: 10.1021/bi0509191. [DOI] [PubMed] [Google Scholar]
- 22.Stone JR, Yang S. Hydrogen peroxide: a signaling messenger. Antioxid Redox Signal. 2006;8:243–270. doi: 10.1089/ars.2006.8.243. [DOI] [PubMed] [Google Scholar]
- 23.Winterbourn CC. Reconciling the chemistry and biology of reactive oxygen species. Nat Chem Biol. 2008;4:278–286. doi: 10.1038/nchembio.85. [DOI] [PubMed] [Google Scholar]
- 24.Zhou H, Singh H, Parsons ZD, Lewis SM, Bhattacharya S, Seiner DR, LaButti JN, Reilly TJ, Tanner JJ, Gates KS. The biological buffer bicarbonate/CO2 potentiates H2O2-mediated inactivation of protein tyrosine phosphatases. J Am Chem Soc. 2011;133:15803–15805. doi: 10.1021/ja2077137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Caselli A, Chiarugi P, Camici G, Manao G, Ramponi G. In vivo inactivation of phosphotyrosine protein phosphatases by nitric oxide. FEBS Lett. 1995;374:249–252. doi: 10.1016/0014-5793(95)01120-4. [DOI] [PubMed] [Google Scholar]
- 26.Conrad M, Sandin A, Forster H, Seiler A, Frijhoff J, Dagnell M, Bornkamm GW, Radmark O, Hooft van Huijsduijnen R, Aspenstrom P, Bohmer F, Ostman A. 12/15-lipoxygenase derived lipid peroxides control receptor tyrosine kinase signaling through oxidation of protein tyrosine phosphatases. Proc Natl Acad Sci USA. 2010;107:15774–15779. doi: 10.1073/pnas.1007909107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wardman P, Candeias LP. Fenton chemistry: an introduction. Radiat Res. 1996;145:523–531. [PubMed] [Google Scholar]
- 28.Tonks NK. PTP1B: From the sidelines to the front lines! FEBS Lett. 2003;546:140–148. doi: 10.1016/s0014-5793(03)00603-3. [DOI] [PubMed] [Google Scholar]
- 29.Lessard L, Stuible M, Tremblay ML. The two faces of PTP1B in cancer. Biochim Biophys Acta. 2010;1804:613–619. doi: 10.1016/j.bbapap.2009.09.018. [DOI] [PubMed] [Google Scholar]
- 30.Pastor N, Weinstein H, Jamison E, Brenowitz M. A detailed interpretation of OH radical footprints in a TBP DNA complex reveals the role of dynamics in the mechanism of sequence specific binding. J Mol Biol. 2000;304:55–68. doi: 10.1006/jmbi.2000.4173. [DOI] [PubMed] [Google Scholar]
- 31.Taniguchi H, Madden KP. An in Situ Radiolysis Time-Resolved ESR Study of the Kinetics of Spin Trapping by 5,5-Dimethyl-1-pyrroline-N-oxide. J Am Chem Soc. 1999;121:11875–11879. [Google Scholar]
- 32.Ivanova J, Salama G, Clancy RM, Schor NF, Nylander KD, Stoyanovsky DA. Formation of nitroxyl and hydroxyl radical in solutions of sodium trioxodinitrate: effects of pH and cytotoxicity. J Biol Chem. 2003;278:42761–42768. doi: 10.1074/jbc.M305544200. [DOI] [PubMed] [Google Scholar]
- 33.Sarmiento M, Zhao Y, Gordon SJ, Zhang ZY. Molecular basis for substrate specificity of protein-tyrosine phosphatase 1B. J Biol Chem. 1998;273:26368–26374. doi: 10.1074/jbc.273.41.26368. [DOI] [PubMed] [Google Scholar]
- 34.Ellis HR, Poole LB. Novel application of 7-chloro-nitrobenzo-2-oxa-1,3-diazole to identify cysteine sulfenic acid in the AhpC component of alkyl hydroperoxide reductase. Biochemistry. 1997;36:15013–15018. doi: 10.1021/bi972191x. [DOI] [PubMed] [Google Scholar]