

Abstract
Traumatic brain injury (TBI) causes substantial morbidity and mortality worldwide. A key component of both mild and severe TBI is diffuse axonal injury. Except in cases of extreme mechanical strain, when axons are torn at the moment of trauma, axonal stretch injury is characterized by early cytoskeletal proteolysis, transport disruption, and secondary axotomy. Calpains, a family of Ca2+-dependent proteases, have been implicated in this pathologic cascade, but direct in vivo evidence is lacking. To test the hypothesis that calpains play a causal role in axonal stretch injury in vivo, we used our rat optic nerve stretch model following adeno-associated viral (AAV) vector-mediated overexpression of the endogenous calpain inhibitor calpastatin in optic nerve axons. AAV vectors were designed for optimal expression of human calpastatin (hCAST) in retinal ganglion cells (RGCs). Calpain inhibition by the expressed protein was then confirmed in primary cortical cultures. Finally, we performed bilateral intravitreal injections of AAV vectors expressing hCAST or the reporter protein ZsGreen 3 weeks prior to unilateral optic nerve stretch. Immediately after stretch injury, Fluoro-Gold was injected into the superior colliculi for assessment of retrograde axonal transport. Rats were euthanized 4 days after stretch injury. Both hCAST and ZsGreen were detected in axons throughout the optic nerve to the chiasm. Calpastatin overexpression partially preserved axonal transport after stretch injury (58.3±15.6% reduction in Fluoro-Gold labeling relative to uninjured contralateral controls in ZsGreen-expressing RGCs, versus 33.8±23.9% in hCAST-expressing RGCs; p=0.038). These results provide direct evidence that axonal calpains play a causal role in transport disruption after in vivo stretch injury.
Key words: axonal injury, axonal transport, head trauma, traumatic brain injury, viral mediated transfection
Introduction
Worldwide, traumatic brain injury (TBI) leading to death or hospitalization is estimated to afflict over 10 million people each year, and it is predicted that TBI will surpass many diseases as the major cause of death and disability by the year 2020.1 Traumatic axonal injury (TAI) is implicated in cognitive dysfunction after mild TBI, as well as coma and mortality after severe TBI.2,3 Blunt brain trauma produces a rapid elongation or deformation of axons that may lead to progressive axolemmal and cytoskeletal damage, culminating in secondary axotomy.4–8 Mechanical deformation of axons results in focal impairment of axonal transport with subsequent accumulation of transported proteins and organelles. Accumulations of amyloid precursor protein (APP), which undergoes fast anterograde transport, are seen as early as 2 h after TBI in humans.9,10 Anterograde transport of intravitreally-injected horseradish peroxidase (HRP), and retrograde transport of Fluoro-Gold™ after its injection into the superior colliculi, are both disrupted after optic nerve stretch, an in vivo model that isolates mechanical strain primarily to axons.8,11,12 Similarly, axonal transport disruption is seen in diffuse brain injury models, such as fluid percussion injury or concussive brain injury.13,14
Post-traumatic calpain activity within axons has been proposed to contribute to cytoskeletal degradation, neurofilament compaction, and transport disruption.15–17 Calpains are a family of non-lysosomal Ca2+-dependent proteases, and are likely involved in signal transduction, gene expression, long-term potentiation, and apoptosis.18 The two ubiquitous calpains, μ- and m-calpain, are heterodimeric peptides, consisting of different catalytic isoforms and a common regulatory subunit. They differ in the Ca2+ concentration required for in vitro activity. The family also consists of calpastatin (CAST), an endogenous inhibitor of both μ- and m-calpain that does not inhibit proteases in any other family.18 Calpain activity measured by immunolabeling for calpain-cleaved α-spectrin (Ab38) is observed in axons in the pyramidal tracts and medial lemnisci 15 min after head acceleration injury in rats.19 Early axonal calpain activity after fluid percussion injury has been reported.20 After optic nerve stretch, calpain-cleaved spectrin is detectable 20–30 min post-injury,8,12 but is typically no longer present by 4 h.8 A delayed secondary phase of calpain-mediated spectrin proteolysis is observed 4 days after injury, which may be related to axonal degeneration.8
Pharmacological inhibition of central nervous system calpain activity remains challenging, and there is presently no pharmacological inhibitor completely specific for calpains.18 Nevertheless, various calpain inhibitors have demonstrated neuroprotection in diffuse TBI models.17 MDL-28170 is perhaps the most well studied calpain inhibitor. Even though it is moderately specific for calpains, it also has significant activity against cathepsin B.21 A single intravenous (IV) bolus (30 mg/kg) of MDL-28170 reduces axonal pathology in diffuse TBI models.16,22,23 In contrast, in the optic nerve stretch model, intensive MDL-28170 therapy does not protect retrograde transport of Fluoro-Gold.12 It is not known whether pharmacological protection seen in the diffuse TBI models occurs at the level of the axons, supra-axonal structures, or both, as neither the injury nor therapy is selective for axons. It is also unclear if peripherally-administered pharmacological inhibitors achieve therapeutic levels in optic nerve axons.
In contrast to pharmacological inhibitors, calpastatin is completely specific for calpains.18 Calpastatin overexpression has been used to protect hippocampal neurons after kainic acid injection,24–26 and transient global ischemia.27 Our strategy was to overexpress calpastatin in retinal ganglion cell (RGC) axons prior to optic nerve stretch. We hypothesized that calpastatin overexpression within axons would protect retrograde transport. Determining the specific role of calpains in isolated axonal injury may have important implications for targeting pathological calpain activity after TBI and spinal cord injury.
Methods
All animal procedures were performed in accordance with National Institutes of Health (NIH) guidelines for the care and use of laboratory animals, and were approved by our institutional animal care and use committee. All appropriate bio-safety procedures were followed in handling the adeno-associated viral (AAV) vectors.
Viral vector generation
Cloning and generating the recombinant AAV vectors (serotype 2) were performed by the Vector Core at the University of Pennsylvania. The human calpastatin (hCAST; clone ID 3878564 from Open Biosystems, Huntsville, AL), or ZsGreen gene (Clontech, Mountain View, CA), was cloned downstream of the CB7 promoter into pENN.AAV.CB7 (an AAV packaging plasmid designed by the University of Pennsylvania Vector Core). CB7 is a chicken β-actin promoter with cytomegalovirus enhancer elements. The viral genomes encoded in both constructs were verified by sequencing analysis prior to vector production. Generation of the AAV vectors has been previously described.28 Genome copy (GC) titers were determined by TaqMan (Life Technologies, Grand Island, NY) analysis by using probes and primers targeting the rabbit β-globin polyA region.
AAV vector testing in primary cortical neuron culture
Cortices from E17-19 Sprague-Dawley rat embryos were trypsinized in Ca2+- and Mg2+-free N-2-hydroxyethylpiperazine-N-2-ethane sulfonic acid (HEPES) buffered saline solution containing 0.027% trypsin for 15 min. The cortices were then washed and placed in media consisting of Dulbecco's modified Eagle's medium (DMEM; BioWhittaker-Lonza, Basel, Switzerland) supplemented with 10% fetal calf serum (Hyclone, Logan, UT). The solution was then repeatedly aspirated using a Pasteur pipette. Dissociated cells were plated at a density of ∼260 cells/mm2 in 24-well plates with glass cover-slips, or ∼620 cells/mm2 in 35-mm dishes. Dishes and cover-slips were coated with poly-L-lysine (Peptides International, Louisville, KY). Cultures were maintained in serum-free Neurobasal medium (Life Technologies) supplemented by B27 (Life Technologies), in a humidified 5% CO2 incubator at 37°C. Mitotic inhibitors and antibiotics were not used.
Neuronal cultures 7 days in vitro (DIV) were transduced with 6×109 (24-well plate), or 3.3×1010 genome copies (35-mm dish), of AAV vectors expressing either ZsGreen (AAV.ZsGreen) or hCAST (AAV.hCAST). Fourteen days later, the cultures were fixed with 4% paraformaldehyde, immunolabeled for hCAST (MAB3084, 1:4000; Millipore, Billerica, MA), and counter-stained with 4,6-diamidino-2-phenylindole (DAPI) nuclear label. MAB3084 detects human, but not rat, CAST. Additional cultures for Western blot and calpastatin activity assay were harvested and sonicated in homogenization buffer (25 mM HEPES [pH 7.4], 1 mM EDTA, and 0.1% CHAPS). Protein concentration was quantified using the Bradford assay. Proteins were separated by SDS-PAGE on a 4–20% Tris-glycine gel, transferred to a nitrocellulose membrane, and probed for hCAST (MAB3084; 1:1000), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH; PA1-988, 1:1000; Thermo Scientific, Rockford, IL). The blots were visualized using enhanced chemiluminescence. HRP-conjugated secondary antibodies and enhanced chemiluminescence substrate were purchased from Perkin Elmer (Waltham, MA).
Functional hCAST expression was assessed by measuring calpastatin activity in transduced and nontransduced cell extracts. Cultures (3–5 per condition) were harvested as described above. Protein concentrations of the cell extracts were adjusted to 3 μg/μL. The extracts were heated to 90°C for 5 min, then centrifuged at 16,000g at 4°C for 20 min. Reactions contained 5 μL of supernatant, 10 nM human erythrocyte calpain 1 (EMD, Gibbstown, NJ), 200 μM Suc-Leu-Tyr-MNA (MP Biomedicals, Solon, OH), and 195 μL of the homogenization buffer with 5 mM B-mercaptoethanol. The reaction was started by adding CaCl2 to a final concentration of 5 mM (room temperature). Time-dependent substrate proteolysis, which results in increasing fluorescence, was measured on a microplate reader (Turner Biosystems, Sunnyvale, CA; 365 nm excitation, 410–460 nm emission). All samples were assayed in triplicate. Western blot of each sample was performed to confirm the presence of hCAST in cultures transduced with AAV.hCAST, and its absence in nontransduced cultures and those transduced with AAV.ZsGreen.
Intravitreal injection of AAV vectors
Adult male Long-Evans rats (130–160 g) underwent isoflurane anesthesia, followed by application of 0.5% proparacaine eyedrops. Using an operating microscope (Zeiss Opmi MD, Thornwood, NY), a superotemporal conjunctival incision was made to expose the sclera posterior to the lens. A pilot hole was made with a 30-gauge needle followed by the introduction of the tip of a glass micropipette containing the viral vector. The micropipette was connected to a 5-μL glass syringe by polyethylene tubing filled with mineral oil. Both eyes in the same animal were injected with the same vector (4 μL at a concentration of 1.13×1012 genome copies/mL).
Optic nerve stretch
One investigator performed all surgeries, and was blinded to the viral vector injection that the animals had received. On each operative day, approximately half of the rats were transduced with either AAV.hCAST or AAV.ZsGreen. Three weeks after intravitreal injection, the rats underwent unilateral optic nerve stretch as described previously.12 In brief, the rats were anesthetized with isoflurane and placed in a stereotactic head holder that was positioned on the injury device. Body temperature was maintained at ∼37.5°C and pulse oximetry between 94 and 98%. A flexible thin ring-shaped plastic sling, which had a continuous loop of 4-0 monofilament nylon suture secured to it at four places 90° apart, was placed behind the eye. The monofilament nylon loop was then attached to the optic nerve stretch device. To ensure consistent loading, a 12-gram preload, measured by a force transducer (LFS 270; Cooper Instruments & Systems, Warrenton, VA), was placed on the nerve. The magnitude of the displacement was controlled by a micromanipulator that controls the distance traveled by the solenoid piston (5.5 mm). Immediately after obtaining the preload, the solenoid was triggered to produce a rapid elongation of the nerve. Two animals were excluded as the optic nerves tore, leaving 7 Zsgreen and 8 hCAST rats for analysis. These 15 animals received Fluoro-Gold injections immediately after optic nerve stretch.
After stretch injury, erythromycin ophthalmic ointment was applied to both eyes, and the lids of the injured side were sutured together for 2 days to prevent corneal drying. The contralateral uninjured eye and optic nerve were used as controls.
Assessment of Fluoro-Gold labeling of RGCs
To label RGCs that were capable of retrograde transport, the animals received bilateral injections of Fluoro-Gold (Fluorochrome, Denver, CO) into each superior colliculus as previously described.12 Four days later, the rats were reanesthetized and transcardially perfusion fixed with cold 1× PBS followed by cold 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4). The end point was selected based on previous work.12 Retinas were removed from the eye and post-fixed for 2–3 h in 4% paraformaldehyde (4°C). After washing with 1× PBS, retinal flat mounts were blocked in 10% goat serum, 1% bovine serum albumin (BSA), and 0.2% Triton X-100 in 1× PBS for 1 h at room temperature. The retinas were incubated overnight at 4°C with an antibody directed against neuronal class III β-tubulin (PRB-435P, 1:5000; Covance, Emeryville, CA), which labels RGCs,29 and/or MAB3084 (1:4000). After further washes, the retinas were incubated in Alexa Fluor® secondary antibodies (Life Technologies) for 2 h at room temperature. The retinas were washed and cover-slipped with Fluoromount G (Electron Microscopy Sciences, Hatfield, PA).
The retinas were flat mounted and viewed with a fluorescence microscope (Leica DM4500B; Leica, Buffalo Grove, IL). Camera and microscope settings for Fluoro-Gold were kept constant across all retinas. Images were taken at 400× magnification in three areas—1/6, 3/6, and 5/6 of the retinal radius from the optic disc—in the region with the highest AAV transduction. The “levels” tool in Adobe Photoshop was used in a standardized way to increase hCAST and Fluoro-Gold contrast and brightness. The black and white level sliders were adjusted to outside the image histogram so the histogram was extended to fill the entire tonal range. Scoring was performed by an observer blinded to both injury and treatment using Adobe Photoshop and NIH ImageJ software. Approximately 30–35 transduced RGCs per retinal area were scored for Fluoro-Gold. The percentages determined for the central, middle, and peripheral areas were averaged, and that number is reported here. Preliminary work suggested there would be a lower percentage of hCAST-expressing RGCs than ZsGreen-expressing RGCs that label with Fluoro-Gold (data not shown). To control for this potential baseline difference, we decided a priori to compare the percentage loss of Fluoro-Gold labeling of transduced RGCs compared to its contralateral uninjured side.
Assessment of RGC counts after viral transduction and optic nerve stretch
To assess RGC counts 4 days after optic nerve stretch, immunolabeled retinas were divided into thirds, and images were taken at 200× magnification in three areas—1/6, 3/6, and 5/6 of the retinal radius from the optic disc—in each of the three regions. Two of the three regions were randomly selected, and RGCs (PRB-435P positive cells) were counted by an observer blinded to both injury and treatment group. RGCs obscured by axonal fascicles were not counted. The counts from the six images were summed and reported here. The total area of the retina scored was 1.1 mm2.
Immunohistochemistry of optic nerves
After perfusion fixation, the optic nerves were removed from the head and post-fixed for 2–3 h in 4% paraformaldehyde (4°C). The nerves were subsequently cryoprotected in 30% sucrose. The right and left optic nerves of the same animal were frozen in the same OCT block, and were cut longitudinally into 14-μm-thick sections on a cryostat.
After drying, the slides were blocked in 3% goat serum and 0.1% Triton X-100 in 1× PBS (pH 7.2). The tissue sections were incubated overnight at 4°C with MAB3084 (1:1000), sc-20779 (detects both rat and human CAST; 1:250; Santa Cruz Biotechnology, Santa Cruz, CA), and/or SMI-32 (1:1000; Sternberger Monoclonals, Lutherville, MD). SMI-32 reacts with a non-phosphorylated epitope on neurofilament heavy subunit (NFH). Neurofilament accumulation within axons highlights swellings and bulbs.8,30 After further PBS rinses, the slides were incubated with Alexa Fluor secondary antibodies for 1 h at room temperature. The slides were rinsed and cover-slipped with Fluoromount G.
Harvesting of optic nerve homogenates for Western blot
Three weeks after intravitreal injection of the viral vectors, the rats were anesthetized with isoflurane and decapitated. The optic nerves were homogenized in 50 mM Tris, 150 mM NaCl, 1% Triton, and 2 mM EGTA (pH 7.0) with protease inhibitors (Roche, Indianapolis, IN). Western blot for hCAST and GAPDH was performed as described above.
Statistical analysis
One-way analysis of variance (ANOVA) was performed to compare the slopes (fluorescence over time) generated by calpain proteolysis of the fluorogenic substrate. Pairwise comparisons within the interaction space were specified a priori, and were accomplished using the t-test and appropriately pooled variance. There were three pre-planned comparisons: ZsGreen versus non-transduced cultures, hCAST versus ZsGreen cultures, and hCAST versus non-transduced cultures. To adjust for three pairwise comparisons the p value was set at 0.017 for statistical significance. The percentage loss of Fluoro-Gold labeling in ZsGreen- and hCAST-transduced RGCs after optic nerve stretch were compared using Student's t-test. A p value<0.05 was considered statistically significant. Repeated-measures ANOVA was used for all other analyses. A p value<0.05 for main effects and <0.2 for the interaction space were considered statistically significant. For the Fluoro-Gold labeling experiment, there were four pre-planned comparisons: ZsGreen versus hCAST uninjured, ZsGreen injured versus uninjured, hCAST injured versus uninjured, and ZsGreen versus hCAST injured. For four pairwise comparisons a p<0.013 was considered significant. Data are presented as means and standard deviations. All data were analyzed using SAS statistical software (version 9.2; SAS Institute, Cary, NC).
Results
Expression of functional hCAST in primary cortical cultures
AAV vectors were generated to express hCAST or the fluorescent marker protein ZsGreen and were initially characterized in primary cortical neuronal cultures. The cultures were transduced at 7 DIV. Nontransduced cultures served as controls. At 21 DIV, the cultures were analyzed by immunohistochemistry, Western blot, and CAST activity assay. Two weeks after AAV vector transduction, there was robust expression of ZsGreen and hCAST (Fig. 1). To determine if hCAST was functional, we used a fluorogenic substrate microplate assay (Fig. 1B). ZsGreen cultures were not significantly different than nontransduced ones (slopes 17.9±0.7 versus 18.6±0.4, respectively; p=0.15). Lysates from hCAST cultures robustly inhibited exogenous calpain activity compared to ZsGreen and nontransduced cultures (slope for hCAST: 1.0±0.7; p<0.0001). Omitting exogenous calpain, fluorogenic substrate, or CaCl2 resulted in no change in fluorescence (data not shown). These results demonstrated that our AAV vectors effectively transduced primary cortical cultures, resulting in expression of functional hCAST or ZsGreen.
FIG. 1.

Expression of functional hCAST in primary cortical neuronal cultures. (A) AAV vectors expressing ZsGreen or hCAST were used to transduce primary cortical cultures (7 DIV). At 21 DIV, the cultures were immunolabeled for hCAST (MAB3084) and stained for DAPI (scale bar=20 μm). (B) In vitro analysis for calpastatin activity in cell extracts using a fluorogenic substrate assay. Calpain proteolysis of Suc-Leu-Tyr-MNA, which results in increasing fluorescence intensity, was almost completely blocked by hCAST expression. Values are mean±standard deviation for 3–5 cortical neuronal cultures per group (*p<0.0001 versus nontransduced and ZsGreen-expressing cells). Human CAST expression was confirmed in cultures transduced with AAV.hCAST by Western blot (hCAST, human calpastatin; DIV, days in vitro; au, arbitrary units; DAPI, 4,6-diamidino-2-phenylindole; GAPDH, glyceraldehyde 3-phosphate dehydrogenase). Color image is available online at www.liebertonline.com/neu
Expression of hCAST and ZsGreen in RGC somata and axons
After in vivo rat optic nerve stretch, the highest levels of early calpain activity as well as the greatest density of swellings and bulbs occurred near the optic chiasm.12,30 Therefore, we confirmed that expressed hCAST in RGCs undergoes anterograde axonal transport to the prechiasmatic optic nerve.
Three weeks after intravitreal injection of AAV.ZsGreen or AAV.hCAST, rats were sacrificed for immunolabeling of the retina and prechiasmatic optic nerve, as well as immunoblotting of the optic nerve. ZsGreen and hCAST were detected in RGC somata (Fig. 2A), as well as axons near the optic chiasm (Fig. 2B). Western blotting confirmed hCAST in optic nerves of animals that received AAV.hCAST (Fig. 2C).
FIG. 2.
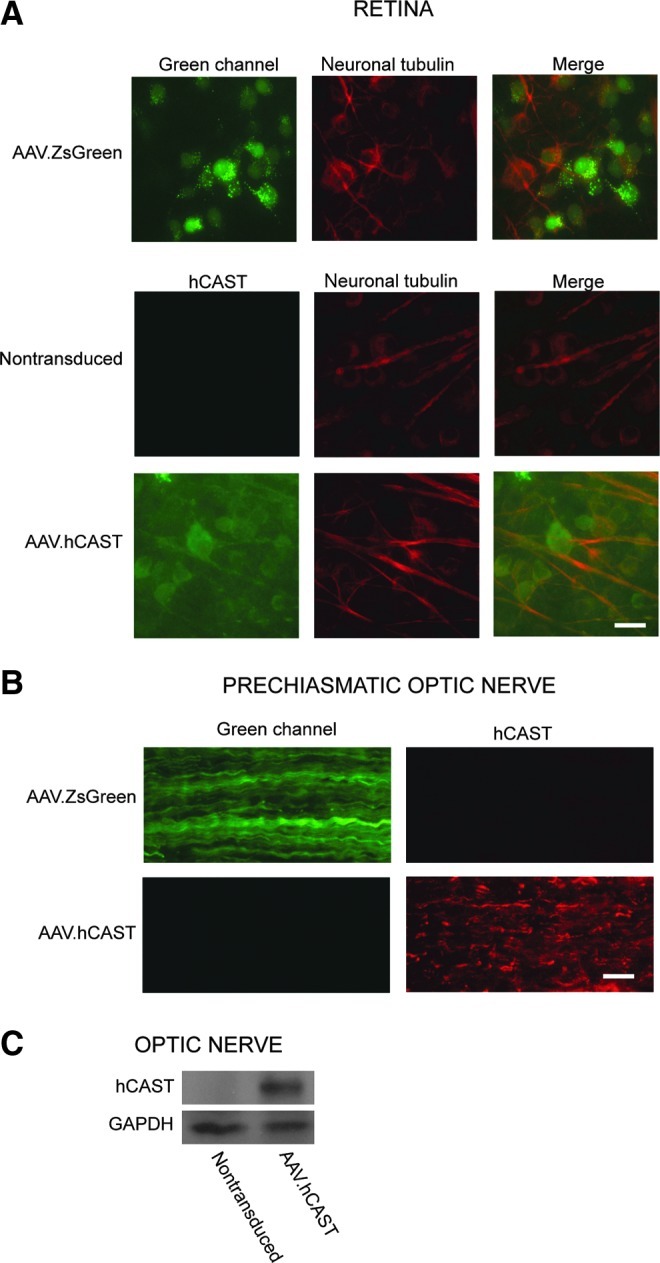
Expression of ZsGreen and hCAST in RGCs and their axons within the optic nerve. Rats were perfusion fixed 3 weeks after intravitreal injection of AAV.ZsGreen or AAV.hCAST. Representative images of retinal flat mounts immunolabeled with MAB3084 (hCAST), and/or an antibody directed against neuron-specific class III β-tubulin that is used to label RGCs (PRB-435P; scale bar=20 μm). (B) Representative images of longitudinal sections of prechiasmatic optic nerves immunolabeled with MAB3084 (scale bar=20 μm). (C) Western blot confirmed hCAST expression in all optic nerves after intravitreal injection of AAV.hCAST (n=10; hCAST, human calpastatin; RGC, retinal ganglion cell; GAPDH, glyceraldehyde 3-phosphate dehydrogenase). Color image is available online at www.liebertonline.com/neu
Rat optic nerve stretch results in axonal swellings and bulbs
Four days after unilateral optic nerve stretch, there were axonal accumulations of ZsGreen and hCAST that morphologically appeared to be swellings and bulbs (Fig. 3). There appeared to be significantly more ZsGreen accumulations than hCAST; however, this difference could not be reliably quantified (see limitations section). Neither type of accumulation was present in the contralateral uninjured optic nerves. We immunolabeled optic nerve sections with SMI-32, a classic marker of swellings and bulbs. A subset of ZsGreen and hCAST accumulations also labeled with SMI-32 (Fig. 3).
FIG. 3.
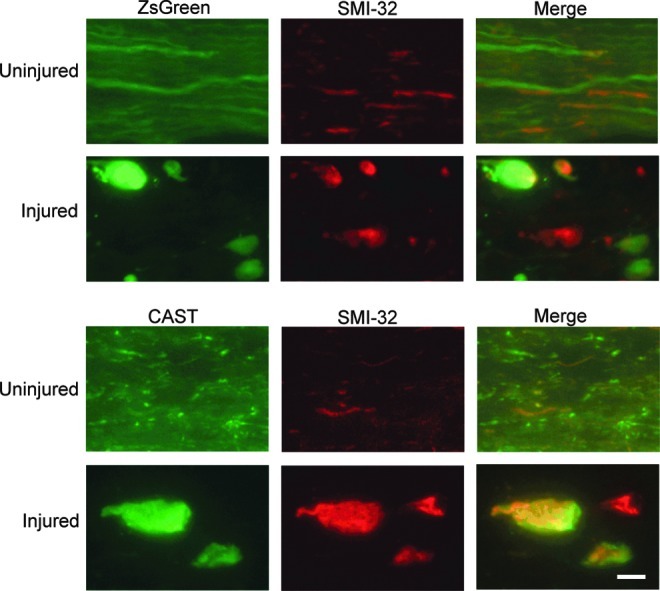
Axonal swellings and bulbs after unilateral optic nerve stretch. Rats received bilateral intravitreal injections of AAV.ZsGreen or AAV.hCAST. After 3 weeks, unilateral optic nerve stretch, immediately followed by Fluoro-Gold injection, was performed. Animals were euthanized by perfusion fixation 4 days later. Longitudinal optic nerve sections were immunolabeled with SMI-32 and/or sc-20779 (CAST). Unilateral optic nerve stretch appeared to disrupt axonal transport, resulting in accumulations of ZsGreen and hCAST. These accumulations frequently label with SMI-32 (scale bar=10 μm; CAST, calpastatin; hCAST, human calpastatin). Color image is available online at www.liebertonline.com/neu
Human calpastatin ameliorates retrograde axonal transport disruption after optic nerve stretch
Axonal stretch injury results in presumably focal disruption of transport at sites of axonal swellings. Therefore our goal was to inhibit local calpain activity within axons by hCAST expression, and examine the effect on axonal transport disruption after stretch injury. Three weeks after intravitreal injection of AAV vectors in both eyes, unilateral optic nerve stretch was performed. This was immediately followed by injection of Fluoro-Gold into the bilateral superior colliculi. Retinal ganglion cells with intact retrograde axonal transport accumulated Fluoro-Gold in their somata. Quantification of RGCs revealed no evidence of RGC loss 4 days after optic nerve stretch, regardless of vector treatment (p=0.55; Fig. 4A and B).
FIG. 4.
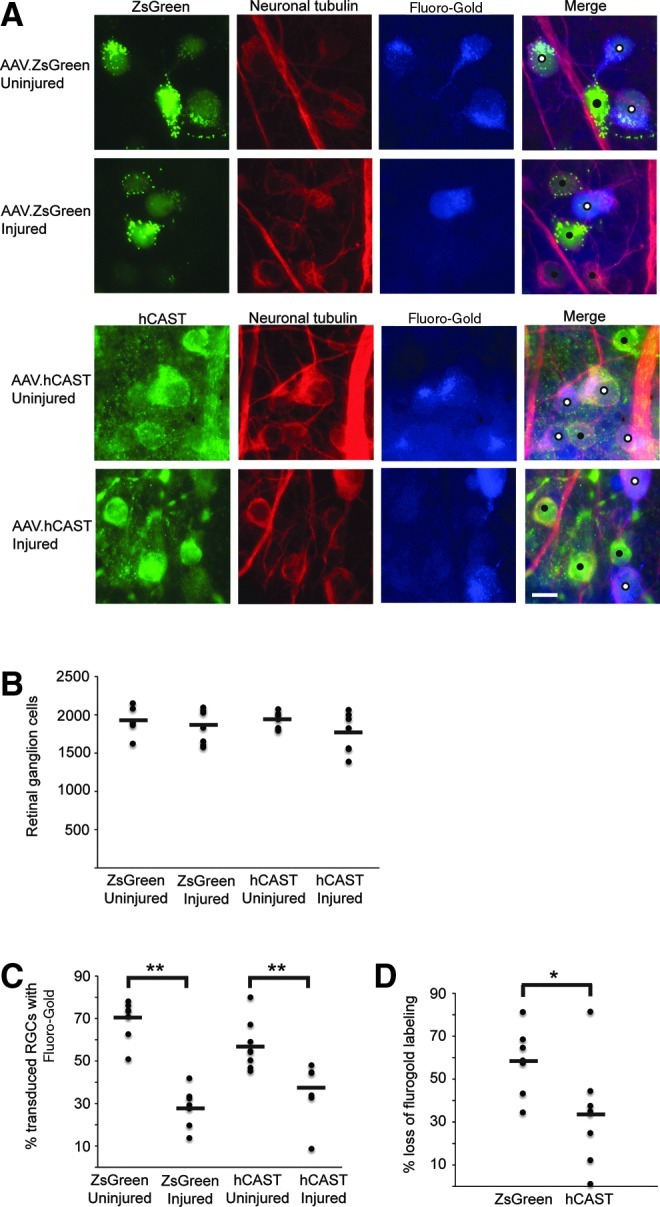
Effect of hCAST on retrograde axonal transport disruption after optic nerve stretch. Rats received bilateral intravitreal injections of AAV.ZsGreen (n=7) or AAV.hCAST (n=8). After 3 weeks, unilateral optic nerve stretch, immediately followed by Fluoro-Gold injection, was performed. The animals were euthanized by perfusion fixation 4 days later. (A) Representative images of retinal flat mounts immunolabeled with an antibody directed against neuron-specific class III β-tubulin (PRB-435P), and/or MAB3084 (hCAST). Transduced RGCs labeled with Fluoro-Gold are marked with white circles, while those without Fluoro-Gold are marked with black circles (scale bar=10 μm). (B) Quantification of RGCs (cells positive for neuron-specific class III β-tubulin) showed no apparent loss 4 days after stretch injury. (C) Percentage of transduced RGCs that labeled with Fluoro-Gold. (D) Percent difference of Fluoro-Gold labeling of transduced RGCs between the injured and contralateral uninjured sides. Human CAST expression ameliorated the disruption of Fluoro-Gold transport after optic nerve stretch. For B, C, and D, each circle represents an individual retina, while horizontal bars represent means (*p<0.05, **p≤0.001; hCAST, human calpastatin; CAST, calpastatin; RGC, retinal ganglion cell). Color image is available online at www.liebertonline.com/neu
To determine the effect of hCAST expression on axonal transport after stretch injury, we quantified the percentage of transduced RGCs (ZsGreen-positive or hCAST-positive) that labeled with Fluoro-Gold. Seven ZsGreen and eight hCAST rats were analyzed. On the uninjured side, 69.4±9.6% and 57.2±11.5% of ZsGreen-positive and hCAST-positive RGCs, respectively, had Fluoro-Gold labeling (p=0.02; Fig. 4C). This result is consistent with our preliminary work that suggested decreased Fluoro-Gold labeling of hCAST-positive RGCs. After stretch injury, 28.3±9.2% of ZsGreen-positive RGCs labeled with Fluoro-Gold (p<0.0001 versus uninjured contralateral retina), while 37.7±12.9% of hCAST-positive RGCs labeled with Fluoro-Gold (p=0.001 versus uninjured contralateral retina). The absolute difference in the percentage of ZsGreen-positive versus hCAST-positive RGCs that label with Fluoro-Gold after optic nerve stretch was not statistically significant (p=0.07). However, when quantified relative to Fluoro-Gold labeling of transduced RGCs in the uninjured contralateral retinas, optic nerve stretch caused a 58.3±15.6% reduction in Fluoro-Gold labeling of ZsGreen-positive RGCs, versus a 33.8±23.9% reduction in hCAST-positive RGCs (p=0.038; Fig. 4D). This statistically significant difference supports our hypothesis that calpains are causally responsible for transport disruption after mechanical stretch of axons.
Discussion
We were able to demonstrate statistically significant protection of retrograde axonal transport after isolated axonal stretch injury with overexpression of the endogenous calpain inhibitor calpastatin. Our current results extend earlier studies that demonstrated axonal protection with pharmacological calpain inhibitors, but were limited by potential off-target effects and the use of TBI models with mixed gray and white matter injury.
Viral vector strategy to study axonal injury mechanisms in vivo
The results of previously published studies designed to elucidate the molecular mechanisms of axonal injury after trauma16,22,23 may be potentially confounded by the off-target effects of pharmacological calpain inhibitors. Particularly when used in vivo, pharmacological inhibitors may have other significant limitations, such as limited blood–brain barrier penetration or drug toxicity. The viral vector approach offers several theoretical advantages. The delivery location, virus type, and promoter, may allow for more selective manipulation of specific neuronal or glial populations. Viral vectors have been previously used to manipulate neurons prior to experimental TBI.31 In our case, because the expressed protein is required to exert local effects in the axonal compartment, we relied on the native axonal transport machinery. For proteins that are normally excluded from axons, selective subcellular targeting of expressed proteins to axons, which has been described by Stowell and Craig,32 may be necessary.
In the brain, it is often difficult to trace back large groups of axons of interest to specific neuronal populations, as large white matter tracts often connect myriad and spatially diverse cell populations, thus limiting experimental manipulation and interpretation. This can be overcome in the optic nerve, which shares similar properties with white matter tracts within the brain, including poor regenerative capacity, myelin sheath structure, and the presence of oligodendrocytes.33,34 The axons are relatively homogenous in size, unidirectional, and have well-defined targets. Manipulating the parent neurons is technically straightforward, and a significant percentage of RGCs can be transduced after a single intravitreal injection.35 In this report, we have demonstrated that delivery of the expressed protein to axons can be readily achieved in the optic nerve. As our injury model preferentially affects axons, this model, in combination with our therapeutic AAV vector strategy, allowed us to explore the mechanisms underlying axonal dysfunction after trauma.
ZsGreen and hCAST accumulates in axonal swellings and bulbs
In our study, we demonstrated that 4 days after optic nerve stretch, there were accumulations of two different exogenous proteins, ZsGreen and hCAST, which occurred at sites where axonal transport is presumably disrupted. Many of these swellings and bulbs labeled with the neurofilament marker SMI-32, which is classically used to detect swellings and bulbs.8,30 Our optic nerve results showing hCAST co-labeling with SMI-32 at axonal swellings are consistent with the incomplete protection seen in our main outcome measure, Fluoro-Gold labeling of RGCs.
Examining optic nerves was useful, as it allowed us to draw several conclusions. First, there was no evidence of injury in unstretched axons containing ZsGreen or hCAST based on the absence of SMI-32 accumulations. Second, all stretched nerves were injured based on the presence of SMI-32-immunoreactive swellings and bulbs. Furthermore, after unilateral stretch, the contralateral uninjured nerves remained free of SMI-32 accumulations.
Pathologic calpain activity contributes to retrograde transport disruption after optic nerve stretch
In the current study, unilateral optic nerve stretch disrupted Fluoro-Gold transport in 58.3±15.6% of ZsGreen-positive RGCs, which is consistent with previous investigations.8,12 After mouse optic nerve stretch, disruption of retrograde axonal transport occurs early and appears to be complete and permanent.8 Stretch injury probably does not simply delay retrograde transport of Fluoro-Gold.
Human CAST expression ameliorated retrograde transport disruption of Fluoro-Gold after stretch injury. The lack of complete protection by hCAST may be due to incomplete calpain inhibition, or the contribution of other injury mechanisms such as the microtubule destabilizing effects of elevated intra-axonal Ca2+ or other proteases.36–39 For example, caspase activity has been detected after experimental TAI.15,40 Although it is also possible that pathological calpain activity affects anterograde transport after stretch injury, the effect of hCAST expression on anterograde transport was not evaluated by the methods used in this study.
A number of other studies have suggested that calpains play a role in TAI. Rats given an IV bolus of the calpain inhibitor MDL-28170 (30 mg/kg) 30 min prior to fluid percussion or impact head acceleration injury demonstrate reduced axonal swellings and neurofilament compaction in brainstem fiber tracts,16 improved axolemmal integrity in the corticospinal tract,23 and structural and electrophysiological protection of the corpus callosum.22 Interpretation of these results requires caution, as MDL-28170 also inhibits proteases other than calpains.21 As injury to supra-axonal structures and physiological derangements after TBI may secondarily cause axonal dysfunction and degeneration, it is not clear if the protective effect of the calpain inhibitor occurs directly within axons. We attempted to address this question in an earlier investigation by using a model that primarily isolates mechanical strain to axons.12 Rats received a 30-mg/kg IV bolus of MDL-28170 30 min prior to unilateral optic nerve stretch. After the bolus, the animals were infused the drug at 15 mg/kg/h over the following 2.5 h. Despite the early intensive therapy, which was designed to match the time course of early calpain activity,8 we were unable to protect retrograde axonal transport measured 4 days after optic nerve stretch. It was not established if the duration of treatment or the intra-axonal concentration of MDL-28170 was sufficient to block pathological calpain activity.
The mechanism by which calpains disrupt axonal transport has not been clearly established. The ultrastructural pathology seen after TAI is microtubule loss,5 and neurofilament misalignment and compaction.41,42 Microtubule loss has been hypothesized to impair axonal transport.8,43,44 Maxwell and Graham5 demonstrated that 15 min after guinea pig optic nerve stretch, there was a dramatic loss of microtubules throughout axons. By 2 h, internodal axonal swellings containing significantly reduced numbers of microtubules had developed. Calpains may directly proteolyze tubulin and tau,45–47 which is important for microtubule stability.48 It is not known if calpains contribute to microtubule loss in vivo, as elevated Ca2+ has been shown to depolymerize microtubules directly or via activation of calmodulin.36–38,49 Axonal stretch injury results in an acute rise in intra-axonal Ca2+.12,50,51
Neurofilament misalignment and compaction have been suggested to impair axonal transport.41,42 Disruption of the ordered arrangement of neurofilaments occurs in human diffuse axonal injury.41 RMO14 is a commonly used marker of early axonal injury, presumably due to calpain proteolysis of sidearms on the neurofilament medium subunits.16 RMO14 immunoreactivity is prominent in the corticospinal tract and medial longitudinal fasciculus in rodent head impact acceleration injury.16,52 In the corticospinal tract, RMO14-immunoreactive axons do not consistently stain with APP, which is a marker of swellings and bulbs, nor do they have evidence of organelle pooling, suggesting that neurofilament sidearm modifications may not directly impair axonal transport.52 The functional consequence of neurofilament sidearm modifications remains unclear.
Limitations
There was a trend toward differential Fluoro-Gold labeling of uninjured ZsGreen and hCAST RGCs (69.4±9.6% versus 57.2±11.5%; p=0.02), but the difference did not meet our strict criteria of statistical significance (p<0.013 was considered significant due to multiple comparisons). As this discrepancy may have obscured a treatment effect if absolute numbers were compared, we decided a priori to use the contralateral uninjured side in each animal as an internal control for baseline Fluoro-Gold labeling of transduced RGCs. The potential baseline difference in Fluoro-Gold labeling of ZsGreen-positive and hCAST-positive RGCs may be due to an intrinsic property of hCAST. Calpains may regulate key aspects of retrograde axonal transport. E-64 and leupeptin, which inhibit various proteases including calpains, prevent the anterograde-retrograde conversion of axonally-transported vesicles.53 Differential Fluoro-Gold labeling may also result from a more generalizable consequence of protein overexpression. For example, overexpressed proteins may require different molecular machinery for their own axonal transport, thereby “crowding out” other proteins that rely on the same transport mechanisms.
Quantification of ZsGreen and hCAST swellings and bulbs was not performed, as it was not feasible to reliably quantify the differences between groups. The transduction efficiency of RGCs after intravitreal injection of AAV vectors is variable, and it would be difficult to control for this variability in the optic nerve. Individual transduced axons could not be reliably counted in longitudinal optic nerve sections. Contributing factors include the small size of the axons, whose diameters are approximately 1 μm, and the wavy course of axons in the longitudinal axis. Moreover, poor antibody penetration into myelinated tissue could cause an underestimation of hCAST-containing axons relative to ZsGreen-containing axons. Even if we could reliably control for transduction efficiency, comparing the numbers of ZsGreen and hCAST accumulations in optic nerves may be of questionable usefulness, as their turnover rates within axons and anterograde transport speeds may differentially affect their accumulation after stretch injury.
The variable and low transduction efficiency of the AAV vectors was a significant limitation when developing strategies to quantify the direct effects of the two expressed proteins on in vivo calpain activity after optic nerve stretch. Western blot for calpain-cleaved α-spectrin (Ab38) of optic nerve homogenates would not have been sensitive enough given the low transduction efficiency. In a previous study, we used Ab38 immunohistochemistry to demonstrate increased intra-axonal calpain activity 30 min after optic nerve stretch.12 However, it is not likely to be feasible to quantify the Ab38 signal in individual transduced axons after optic nerve stretch. Given these limitations, we did not address whether hCAST at the concentration present within axons could inhibit calpains in vivo using a more direct readout of calpain activity.
Conclusions
In summary, the present findings support our hypothesis that strain-induced calpain activity within axons directly impairs retrograde transport. By using viral vector-mediated transduction of RGCs and the native anterograde transport machinery, we were able to deliver the specific endogenous calpain inhibitor calpastatin to the site of greatest injury (prechiasmatic optic nerve axons). With further optimization, this approach could become a powerful tool to explore the molecular mechanisms of TAI.
Acknowledgments
We thank Xinran Wang for helpful technical assistance, Margaret Maronski for preparing cultured cortical neurons, and David Meaney for the use of his optic nerve stretch device. We would like to acknowledge the University of Pennsylvania Preclinical Vector Core facility for providing vector production services, and partial funding through the Penn Vector Core NIDDK P30 Molecular Therapy Center Grant. Additional support was provided by the National Institutes of Health grant NS055880 (M.M.).
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Hyder A.A. Wunderlich C.A. Puvanachandra P. Gururaj G. Kobusingye O.C. The impact of traumatic brain injuries: a global perspective. NeuroRehabilitation. 2007;22:341–353. [PubMed] [Google Scholar]
- 2.Gennarelli T.A. Thibault L.E. Adams J.H. Graham D.I. Thompson C.J. Marcincin R.P. Diffuse axonal injury and traumatic coma in the primate. Ann. Neurol. 1982;12:564–574. doi: 10.1002/ana.410120611. [DOI] [PubMed] [Google Scholar]
- 3.Medana I.M. Esiri M.M. Axonal damage: a key predictor of outcome in human CNS diseases. Brain. 2003;126:515–530. doi: 10.1093/brain/awg061. [DOI] [PubMed] [Google Scholar]
- 4.Jafari S.S. Maxwell W.L. Neilson M. Graham D.I. Axonal cytoskeletal changes after non-disruptive axonal injury. J. Neurocytol. 1997;26:207–221. doi: 10.1023/a:1018588114648. [DOI] [PubMed] [Google Scholar]
- 5.Maxwell W.L. Graham D.I. Loss of axonal microtubules and neurofilaments after stretch-injury to guinea pig optic nerve fibers. J. Neurotrauma. 1997;14:603–614. doi: 10.1089/neu.1997.14.603. [DOI] [PubMed] [Google Scholar]
- 6.Povlishock J.T. Katz D.I. Update of neuropathology and neurological recovery after traumatic brain injury. J. Head Trauma Rehabil. 2005;20:76–94. doi: 10.1097/00001199-200501000-00008. [DOI] [PubMed] [Google Scholar]
- 7.Povlishock J.T. Marmarou A. McIntosh T. Trojanowski J.Q. Moroi J. Impact acceleration injury in the rat: evidence for focal axolemmal change and related neurofilament sidearm alteration. J. Neuropathol. Exp. Neurol. 1997;56:347–359. [PubMed] [Google Scholar]
- 8.Saatman K.E. Abai B. Grosvenor A. Vorwerk C.K. Smith D.H. Meaney D.F. Traumatic axonal injury results in biphasic calpain activation and retrograde transport impairment in mice. J. Cereb. Blood Flow Metab. 2003;23:34–42. doi: 10.1097/01.WCB.0000035040.10031.B0. [DOI] [PubMed] [Google Scholar]
- 9.Blumbergs P.C. Scott G. Manavis J. Wainwright H. Simpson D.A. McLean A.J. Topography of axonal injury as defined by amyloid precursor protein and the sector scoring method in mild and severe closed head injury. J. Neurotrauma. 1995;12:565–572. doi: 10.1089/neu.1995.12.565. [DOI] [PubMed] [Google Scholar]
- 10.McKenzie K.J. McLellan D.R. Gentleman S.M. Maxwell W.L. Gennarelli T.A. Graham D.I. Is β-APP a marker of axonal damage in short-surviving head injury? Acta Neuropathol. 1996;92:608–613. doi: 10.1007/s004010050568. [DOI] [PubMed] [Google Scholar]
- 11.Gennarelli T.A. Thibault L.E. Tipperman R. Tomei G. Sergot R. Brown M. Maxwell W.L. Graham D.I. Adams J.H. Irvine A. Gennarelli L.M. Duhaime A.C. Boock R. Greenberg J. Axonal injury in the optic nerve: a model simulating diffuse axonal injury in the brain. J. Neurosurg. 1989;71:244–253. doi: 10.3171/jns.1989.71.2.0244. [DOI] [PubMed] [Google Scholar]
- 12.Ma M. Li L. Wang X. Bull D.L. Shofer F.S. Meaney D.F. Neumar R.W. Short-duration treatment with the calpain inhibitor MDL-28170 does not protect axonal transport in an in vivo model of traumatic axonal injury. J. Neurotrauma. 2012;29:445–451. doi: 10.1089/neu.2011.2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Creed J.A. DiLeonardi A.M. Fox D.P. Tessler A.R. Raghupathi R. Concussive brain trauma in the mouse results in acute cognitive deficits and sustained impairment of axonal function. J. Neurotrauma. 2011;28:547–563. doi: 10.1089/neu.2010.1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Povlishock J.T. Becker D.P. Cheng C.L.Y. Vaughan G.W. Axonal change in minor head injury. J. Neuropathol. Exp. Neurol. 1983;42:225–242. doi: 10.1097/00005072-198305000-00002. [DOI] [PubMed] [Google Scholar]
- 15.Büki A. Povlishock J.T. All roads lead to disconnection?—Traumatic axonal injury revisited. Acta Neurochir. (Wien) 2006;148:181–194. doi: 10.1007/s00701-005-0674-4. [DOI] [PubMed] [Google Scholar]
- 16.Büki A. Farkas O. Doczi T. Povlishock J.T. Preinjury administration of the calpain inhibitor MDL-28170 attenuates traumatically induced axonal injury. J. Neurotrauma. 2003;20:261–268. doi: 10.1089/089771503321532842. [DOI] [PubMed] [Google Scholar]
- 17.Saatman K.E. Creed J. Raghupathi R. Calpain as a therapeutic target in traumatic brain injury. Neurotherapeutics. 2010;7:31–42. doi: 10.1016/j.nurt.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goll D.E. Thompson V.F. Li H. Wei W. Cong J. The calpain system. Physiol. Rev. 2003;83:731–801. doi: 10.1152/physrev.00029.2002. [DOI] [PubMed] [Google Scholar]
- 19.Büki A. Siman R. Trojanowski J.Q. Povlishock J.T. The role of calpain-mediated spectrin proteolysis in traumatically induced axonal injury. J. Neuropathol. Exp. Neurol. 1999;58:365–375. doi: 10.1097/00005072-199904000-00007. [DOI] [PubMed] [Google Scholar]
- 20.Saatman K.E. Bozyczko-Coyne D. Marcy V. Siman R. McIntosh T.K. Prolonged calpain-mediated spectrin breakdown occurs regionally following experimental brain injury in the rat. J. Neuropathol. Exp. Neurol. 1996;55:850–860. doi: 10.1097/00005072-199607000-00010. [DOI] [PubMed] [Google Scholar]
- 21.Markgraf C.G. Velayo N.L. Johnson M.P. McCarty D.R. Medhi S. Koehl J.R. Chmielewski P.A. Linnik M.D. Six-hour window of opportunity for calpain inhibition in focal cerebral ischemia in rats. Stroke. 1998;29:152–158. doi: 10.1161/01.str.29.1.152. [DOI] [PubMed] [Google Scholar]
- 22.Ai J. Liu E. Wang J. Chen Y. Yu J. Baker A.J. Calpain inhibitor MDL-28170 reduces the functional and structural deterioration of corpus callosum following fluid percussion injury. J. Neurotrauma. 2007;24:960–978. doi: 10.1089/neu.2006.0224. [DOI] [PubMed] [Google Scholar]
- 23.Czeiter E. Büki A. Bukovics P. Farkas O. Pál J. Kövesdi E. Dóczi T. Sándor J. Calpain inhibition reduces axolemmal leakage in traumatic axonal injury. Molecules. 2009;14:5115–5123. doi: 10.3390/molecules14125115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Higuchi M. Tomioka M. Takano J. Shirotani K. Iwata N. Masumoto H. Maki M. Itohara S. Saido T.C. Distinct mechanistic roles of calpain and caspase activation in neurodegeneration as revealed in mice overexpressing their specific inhibitors. J. Biol. Chem. 2005;280:15229–15237. doi: 10.1074/jbc.M500939200. [DOI] [PubMed] [Google Scholar]
- 25.Rao M.V. Mohan P.S. Peterhoff C.M. Yang D.-S. Schmidt S.D. Stavrides P.H. Campbell J. Chen Y. Jiang Y. Paskevich P.A. Cataldo A.M. Haroutunian V. Nixon R.A. Marked calpastatin (CAST) depletion in Alzheimer's disease accelerates cytoskeleton disruption and neurodegeneration: neuroprotection by CAST overexpression. J. Neurosci. 2008;28:12241–12254. doi: 10.1523/JNEUROSCI.4119-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Takano J. Tomioka M. Tsubuki S. Higuchi M. Iwata N. Itohara S. Maki M. Saido T.C. Calpain mediates excitotoxic DNA fragmentation via mitochondrial pathways in adult brains: evidence from calpastatin mutant mice. J. Biol. Chem. 2005;280:16175–16184. doi: 10.1074/jbc.M414552200. [DOI] [PubMed] [Google Scholar]
- 27.Cao G. Xing J. Xiao X. Liou A.K.F. Gao Y. Yin X.-M. Clark R.S.B. Graham S.H. Chen J. Critical role of calpain I in mitochondrial release of apoptosis-inducing factor in ischemic neuronal injury. J. Neurosci. 2007;27:9278–9293. doi: 10.1523/JNEUROSCI.2826-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gao G.-P. Alvira M.R. Wang L. Calcedo R. Johnston J. Wilson J.M. Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc. Natl. Acad. Sci. USA. 2002;99:11854–11859. doi: 10.1073/pnas.182412299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cui Q. Yip H.K. Zhao R.C.H. So K.-F. Harvey A.R. Intraocular elevation of cyclic AMP potentiates ciliary neurotrophic factor-induced regeneration of adult rat retinal ganglion cell axons. Mol. Cell. Neurosci. 2003;22:49–61. doi: 10.1016/s1044-7431(02)00037-4. [DOI] [PubMed] [Google Scholar]
- 30.Ma M. Matthews B.T. Lampe J.W. Meaney D.F. Shofer F.S. Neumar R.W. Immediate short-duration hypothermia provides long-term protection in an in vivo model of traumatic axonal injury. Exp. Neurol. 2009;215:119–127. doi: 10.1016/j.expneurol.2008.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Conte V. Raghupathi R. Watson D.J. Fujimoto S. Royo N.C. Marklund N. Stocchetti N. McIntosh T.K. TrkB gene transfer does not alter hippocampal neuronal loss and cognitive deficits following traumatic brain injury in mice. Restor. Neurol. Neurosci. 2008;26:45–56. [PMC free article] [PubMed] [Google Scholar]
- 32.Stowell J.N. Craig A.M. Axon/dendrite targeting of metabotropic glutamate receptors by their cytoplasmic carboxy-terminal domains. Neuron. 1999;22:525–536. doi: 10.1016/s0896-6273(00)80707-2. [DOI] [PubMed] [Google Scholar]
- 33.Cho K.-S. Yang L. Lu B. Ma H.F. Huang X. Pekny M. Chen D.F. Re-establishing the regenerative potential of central nervous system axons in postnatal mice. J. Cell Sci. 2005;118:863–872. doi: 10.1242/jcs.01658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hirano A. Lena J.F. Morphology of central nervous system axons, in: The Axon: Structure, Function, and Pathophysiology. In: Waxman S.G., editor; Kocsis J.D., editor; Stys P.K., editor. Oxford University Press; New York: 1995. pp. 49–67. [Google Scholar]
- 35.Martin K.R.G. Quigley H.A. Gene therapy for optic nerve disease. Eye (Lond.) 2004;18:1049–1055. doi: 10.1038/sj.eye.6701579. [DOI] [PubMed] [Google Scholar]
- 36.Gaskin F. Cantor C.R. Shelanski M.L. Biochemical studies on the in vitro assembly and disassembly of microtubules. Ann. NY Acad. Sci. 1975;253:133–146. doi: 10.1111/j.1749-6632.1975.tb19197.x. [DOI] [PubMed] [Google Scholar]
- 37.Job D. Fischer E.H. Margolis R.L. Rapid disassembly of cold-stable microtubules by calmodulin. Proc. Natl. Acad. Sci. USA. 1981;78:4679–4682. doi: 10.1073/pnas.78.8.4679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Keith C. DiPaola M. Maxfield F.R. Shelanski M.L. Microinjection of Ca++-calmodulin causes a localized depolymerization of microtubules. J. Cell Biol. 1983;97:1918–1924. doi: 10.1083/jcb.97.6.1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maxwell W.L. Povlishock J.T. Graham D.L. A mechanistic analysis of nondisruptive axonal injury: a review. J. Neurotrauma. 1997;14:419–440. doi: 10.1089/neu.1997.14.419. [DOI] [PubMed] [Google Scholar]
- 40.Büki A. Okonkwo D.O. Wang K.K.W. Povlishock J.T. Cytochrome c release and caspase activation in traumatic axonal injury. J. Neurosci. 2000;20:2825–2834. doi: 10.1523/JNEUROSCI.20-08-02825.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Christman C.W. Grady M.S. Walker S.A. Holloway K.L. Povlishock J.T. Ultrastructural studies of diffuse axonal injury in humans. J. Neurotrauma. 1994;11:173–186. doi: 10.1089/neu.1994.11.173. [DOI] [PubMed] [Google Scholar]
- 42.Smith D.H. Meaney D.F. Axonal damage in traumatic brain injury. Neuroscientist. 2000;6:483–495. [Google Scholar]
- 43.Maxwell W.L. Histopathological changes at central nodes of Ranvier after stretch-injury. Microsc. Res. Tech. 1996;34:522–535. doi: 10.1002/(SICI)1097-0029(19960815)34:6<522::AID-JEMT4>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 44.McCracken E. Hunter A.J. Patel S. Graham D.I. Dewar D. Calpain activation and cytoskeletal protein breakdown in the corpus callosum of head-injured patients. J. Neurotrauma. 1999;16:749–761. doi: 10.1089/neu.1999.16.749. [DOI] [PubMed] [Google Scholar]
- 45.Billger M. Wallin M. Karlsson J.-O. Proteolysis of tubulin and microtubule-associated proteins 1 and 2 by calpain I and II. Difference in sensitivity of assembled and disassembled microtubules. Cell Calcium. 1988;9:33–44. doi: 10.1016/0143-4160(88)90036-x. [DOI] [PubMed] [Google Scholar]
- 46.Johnson G.V.W. Jope R.S. Binder L.I. Proteolysis of tau by calpain. Biochem. Biophys. Res. Commun. 1989;163:1505–1511. doi: 10.1016/0006-291x(89)91150-9. [DOI] [PubMed] [Google Scholar]
- 47.Siman R. Noszek J.C. Excitatory amino acids activate calpain I and induce structural protein breakdown in vivo. Neuron. 1988;1:279–287. doi: 10.1016/0896-6273(88)90076-1. [DOI] [PubMed] [Google Scholar]
- 48.Amos L.A. Schlieper D. Microtubules and MAPs. Adv. Protein Chem. 2005;71:257–298. doi: 10.1016/S0065-3233(04)71007-4. [DOI] [PubMed] [Google Scholar]
- 49.O'Brien E.T. Salmon E.D. Erickson H.P. How calcium causes microtubule depolymerization. Cell Motil. Cytoskeleton. 1997;36:125–135. doi: 10.1002/(SICI)1097-0169(1997)36:2<125::AID-CM3>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 50.Iwata A. Stys P.K. Wolf J.A. Chen X.-H. Taylor A.G. Meaney D.F. Smith D.H. Traumatic axonal injury induces proteolytic cleavage of the voltage-gated sodium channels modulated by tetrodotoxin and protease inhibitors. J. Neurosci. 2004;24:4605–4613. doi: 10.1523/JNEUROSCI.0515-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Staal J.A. Dickson T.C. Gasperini R. Liu Y. Foa L. Vickers J.C. Initial calcium release from intracellular stores followed by calcium dysregulation is linked to secondary axotomy following transient axonal stretch injury. J. Neurochem. 2010;112:1147–1155. doi: 10.1111/j.1471-4159.2009.06531.x. [DOI] [PubMed] [Google Scholar]
- 52.Stone J.R. Singleton R.H. Povlishock J.T. Intra-axonal neurofilament compaction does not evoke local axonal swelling in all traumatically injured axons. Exp. Neurol. 2001;172:320–331. doi: 10.1006/exnr.2001.7818. [DOI] [PubMed] [Google Scholar]
- 53.Sahenk Z. Lasek R.J. Inhibition of proteolysis blocks anterograde-retrograde conversion of axonally transported vesicles. Brain Res. 1988;460:199–203. doi: 10.1016/0006-8993(88)91224-3. [DOI] [PubMed] [Google Scholar]