

Abstract
Our previous target validation studies established that inhibition of methionine aminopeptidases (MtMetAP, type 1a and 1c) from Mycobacterium tuberculosis (Mtb) is an effective approach to suppressing Mtb growth in culture. A novel class of MtMetAP1c inhibitors comprising of N'-hydroxy-N-(4H,5H-naphtho[1,2-d]thiazol-2-yl)methanimidamide (4c) was uncovered through a high-throughput screen (HTS). A systematic structure—activity relationship study (SAR) yielded variants of the hit, 4b, 4h, and 4k, bearing modified A- and B-rings as potent inhibitors of both MtMetAPs. Except methanimidamide 4h that showed a moderate Mtb inhibition, a desirable minimum inhibitory concentration (MIC) was not obtained with the current set of MtMetAP inhibitors. However, the SAR data generated thus far may prove valuable for further tuning of this class of inhibitors as effective anti-tuberculosis agents.
Keywords: Methionine aminopeptidase, Tuberculosis, N’-hydroxy-N-(thiazol-2-yl)methanimidamide
1. Introduction
It is estimated that as much as one-third of the world's population is infected with Mycobacterium tuberculosis (Mtb). Globally mortality due to Mtb infection is second only to HIV/AIDS,1 and an expanding epidemic of multi-drug resistant, extensively drug-resistant, and totally drug-resistant2 tuberculosis (TB) coupled with Mtb (including latent) and HIV co-infection is a serious health threat requiring urgent attention.3 Since the approval of rifampin as a treatment for TB by the US Food and Drug Administration (FDA) in 1971, there has been a long hiatus with no new and significant anti-TB drugs brought to the clinic. Recently there has been an awakening to the urgency of the situation, and now there are several candidate anti-TB drugs in various stages of development.4 Continued emphasis on drug discovery is needed as new drugs with novel modes of action are required to catch up with the rapidly evolving resistant strains of Mtb. To this end we have been actively pursuing inhibitors of Mtb methionine aminopeptidases (MetAPs) as novel targets.
MetAPs are metalloproteases that perform the vital function of removing the initiator methionine from the nascent polypeptides emanating from ribosomes, and hence are expressed and present in every life form. In prokaryotes, and in organelles like mitochondria and plastids the start codon is translated by N-formylmethionine, and a prerequisite step of deformylation by peptide deformylase (PDF) is required.5 The importance of these two enzymes is underscored by the study by Solbiati et el. showing that null mutations in PDF or MetAP gene are lethal to Escherichia coli.6 The genome of the H37Rv strain of Mtb contains two MetAP genes, mapA and mapB encoding for Mtb methionine aminopeptidase 1a (MtMetAP1a) and MtMetAP1c, respectively. We have previously characterized these two isozymes both biochemically and by X-ray crystallography.7 The crystal structure revealed the presence of an SH3 domain at the N-terminal extension (40 amino acids) suggesting a mode of association with the ribosome. MtMetAP1a isozyme is a minimal protein exhibiting optimal enzyme activity at 55 °C whereas MtMetAP1c is fully active at 37 °C. The two isozymes also differ in their specificity towards substrates and metal ion cofactors. Further, Zhang et al.8 determined that the mRNA levels for the two isoforms were vastly different in different growth phases—mapA gene was expressed more in log phase, while mapB transcript levels were higher in stationary phase—suggesting that the two MetAPs are important for Mtb growth in different phases of its unique life cycle. Using a combination of chemical and genetic approaches, we had validated MtMetAPs to be viable targets. MtMetAP1a had emerged as a more sensitive target in the culture setting of Mtb.9 In an effort to identify novel inhibitors of MtMetAPs, we conducted a high-throughput screen (HTS) of a 175,000 compound library (from ASDI, Inc., Newark, DE) against MtMetAP1c. One of the most potent hits against MtMetAP1c was N'-hydroxy-N-(7-methoxy-4H,5H-naphtho[1,2-d]thiazol-2-yl)methanimidamide (4c, see Table 1). This article is an account of our medicinal chemistry effort at optimizing this hit for potent inhibition of both MtMetAPs.
Table 1.
4H,5H-Naphtho[1,2-d]thiazole derivatives—MtMetAP assays
| Entry | Structure | Substituents (when not specified, R# = H) |
IC50 (µM), MtMetAP | |||
|---|---|---|---|---|---|---|
| 1a, Co2+ | 1a, Mn2+ | 1c, Co2+ | 1c, Mn2+ | |||
| 3n | 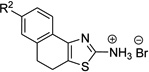 |
– | >50 | |||
| 3o | R2 = OMe | |||||
| 3p | 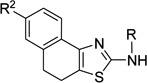 |
R2 = OMe; R = COMe | >50 | |||
| 3q | R = CO(CH2)7Me | |||||
| 3r | R2 = OMe; R = CO(CH2)7Me | |||||
| 3s | R2 = OMe; R = (S)-COCH(OH)Me | |||||
| 3u | R2 = OMe; R = 2-furanyl | |||||
| 3a | 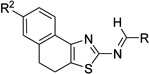 |
R = NMe2 | 28.9 ±5.7 | 27.7 ±13.9 | 36.3 ±9.2 | >50 |
| 3t | R2 = OMe; R = 2-furanyl | >50 | ||||
| 3v | R2 = OMe; R = Ph(2-OH) | |||||
| 4c | 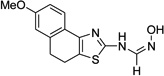 |
R2 = OMe | 5.35 ±1.5 | >50 | 1.33 ±0.24 | >50 |
Note: IC50 values are the average of at least three independent experiments, each consisting of triplicates and ± standard deviation is given.
2. Chemistry
Compounds 3n through 3r were from the ASDI compound library and they were verified by ESI-MS analysis to confirm the presence of expected molecular ions. The N'-hydroxy-N-(thiazol-2-yl)methanimidamides were prepared using a three-step protocol starting from ketones 1 (Scheme 1). The ketones unavailable from commercial sources, namely 1f,10 1g,11 1h,12 1j,13 and 1k14 were prepared following literature procedures. Aminothiazoles 2a–2m were prepared using a variation of Hantzsch thiazole synthesis,15 where α-halo ketones generated in situ (from 1a–1m) were reacted with thiourea. Amide 3s was synthesized by coupling a silyl-protected (S)-lactic acid with aminothiazole 2c and by subsequent removal of the silyl protection revealing the free hydroxy group. The Schiff bases 3t and 3v were made from 2c by treating them with furfural and salicylaldehyde, respectively, following a reported procedure.16 The Schiff base 3t was reduced using sodium borohydride to form the alkylated thiazole 3u. The aminothiazoles (2a–2m) were further converted to the intermediates N',N'-dimethylamidines 3a to 3m by refluxing with DMF-dimethylacetal. And finally, the formamidine intermediates (3a to 3m) were transformed into N'-hydroxy-N-(thiazol-2-yl)methanimidamides 4a–4m by treating with hydroxylamine, as per the protocol of Sasaki et al.17 The overall yields of 4a–4m starting from the ketones (1a–1m) ranged from 37–75%.
Scheme 1.

Synthesis of thiazole derivatives. Reagents and Conditions: i) I2/(NH2)2C=S, EtOH, 100 °C, 2–3 h; then free-base with NaHCO3. ii) furfural or salicylaldehyde, anhydrous MgSO4, 1:1 CH2Cl2—MeOH, rt, 6 h; iii) (OMe)2CHNMe2/toluene, reflux, 4–6 h; iv) (S)-(O)-TBDPS-lactic acid, HBTU/i-Pr2NEt, THF, 16 h, then n-Bu4NF/THF; v) 3, NaBH4/MeOH, rt, 16 h; vi) NH2OH·HCl, MeOH, rt, 18 h.
N'-hydroxy-N-(thiazol-2-yl)methanimidamide can exist as two distinct tautomers as shown in Scheme 2. In the case of methanimidamide 4c, we were able to discern by 2D-NOESY experiment that the prevailing tautomer in acetone is 4c-2. This observation is in complete agreement with the literature reports where N'-hydroxy-N-arylmethanimidamides were seen in this tautomeric form in the X-ray structures.18,19
Scheme 2.

Tautomers of N'-hydroxymethanimidamides, exmaple 4c-1 and 4c-2. Evidence form NOESY suggesting a preponderence of 4c-2.
3. Results and discussion
A target-based HTS of a 175,000 compound library from ASDI, Inc., Newark, DE, was performed to identify novel inhibitors of MtMetAP1c using a coupled enzyme assay20 with Met-Pro-pNA as the substrate, Co2+ as the cofactor, and proline aminopeptidase from Bacillus coagulans (BcProAP) as the coupling enzyme.8 The HTS furnished us a hit bearing a novel N'-hydroxy-N-(7-methoxy-4H,5H-naphtho[1,2-d]thiazol-2-yl)methanimidamide structure, 4c with an IC50 of 1.33 µM. We then undertook a systematic structure—activity relationship (SAR) study to fine-tune the structure for inhibition against MtMetAPs. As mentioned previously, each of the isoforms, MtMetAP1a and MtMetAP1c, appears to be important for TB during different growth phases, and hence, we assayed all the compounds against both the isoforms. Also, it is still unclear what the physiologically relevant metal cofactors are for these two isoforms. In the in vitro assay setting, Wang's group demonstrated that highest catalytic activity of MtMetAP1a could be achieved with Co2+ as the cofactor followed by Mg2+ and Mn2+ with nearly two and 3.5 fold less efficiency respectively when compared to Co2+ activation.21 On the other hand, Ye's group reported that the most efficient activators of MtMetAP1a were Ni2+ and Co2+ ions (in that order).22 When it comes to MtMetAP1c, Co2+, Mn2+, Zn2+, and Mg2+ (in that order) were shown to be the effective activators by Wang's group;2 whereas Ye's group established kcat/kM (in M−1,S−1) to be 659, 232, and 107 for Ni2+, Co2+, and Mn2+, respectively.23 Under our assay conditions, Co2+ and Mn2+ (in that order) were the most effective metal ion cofactors for the MtMetAPs and hence we chose to carry out all the in vitro enzyme inhibition assays using these two metal ions. A likelihood of the coupling enzyme (BcProAP) inhibition in the MtMetAP assay was ruled out by performing an additional control experiment.
First we wanted to interrogate the importance of the N'-hydroxymethanimidamide functionality of the hit 4c, and thus we browsed through the ASDI library and tested structurally related analogs. Neither of the simple naphthothiazolyl amine salts 3n and 3o nor their amide derivatives 3p, 3q, and 3r inhibited MtMetAPs (Table 1). At this point we reasoned that the presence of an oxygen atom in α or β position with respect to the nitrogen atom of aminothiazole might be necessary for the inhibitory activity, and we prepared and tested lactamide 3s, 2-furyl derivative 3u, and Schiff bases 3t and 3v. All the analogs listed above failed to inhibit either MtMetAPs. Only methanimidamide 3a showed a weak inhibition of both MtMetAPs. Thus we concluded N'-hydroxy-N-(thiazol-2-yl)methanimidamide to be the optimal pharmacophore and focused our attention on varying substitutions on the A-ring (see 3 in Scheme 1 for ring designation).
With examples 4a through 4f (see Table 2), we systematically probed the effect of methoxy substitution on the A ring (4b, 4c, and 4d), where compound 4b stood out for its good potency against the two metalloforms of MtMetAPs. Dimethoxy derivative 4e was less potent against MtMetAPs and the methylenedioxy substitution (4f) led to even poorer inhibition. We then altered the B-ring by replacing the benzylic methylene with an oxygen atom (examples 4g, 4h, and 4i). A single methoxy group substitution on the A-ring combined with or without an additional phenyl group substitution on the B-ring (4g or 4i), produced compounds exhibiting moderate inhibition of both the metalloforms of MtMetAP1a, but they did not inhibit MtMetAP1c. Contrary to the case of 4f, methylenedioxy substituted compound 4h turned out to be potent at inhibiting the cobalt-form of MtMetAP1c (730 nM) with good inhibition of MtMetAP1a as well (1.43 µM, Co2+–MtMetAP1a). We then synthesized the B-ring expanded versions 4j and 4k, and gratifyingly, 4k became the most potent MtMetAP inhibitor. Unlike the benzosuberan congener 4j, benzoxepine derivative 4k inhibited both the metalloforms of MtMetAPs while having an IC50 of 1.2 µM and 380 nM for Co2+–MtMetAP1a and Co2+–MtMetAP1c, respectively.
Table 2.
Analogs of N’-Hydroxy-N-(4H,5H-naphtho[1,2-d]thiazol-2-yl)methanimidamide—MtMetAP assays and MICs (Mtb, H37Rv)
| Entry | Structure | Substituents (when not specified, R# = H) |
IC50 (µM), MtMetAP | MIC (µg/mL) |
|||
|---|---|---|---|---|---|---|---|
| 1a, Co2+ | 1a, Mn2+ | 1c, Co2+ | 1c, Mn2+ | ||||
| 4a | 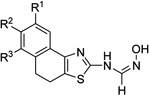 |
– | 11.53 ±3.5 | >50 | 1.05 ±0.39 | >50 | 100 |
| 4b | R1 = OMe | 1.90 ±0.5 | >50 | 0.64 ±0.08 | 2.33 ±0.39 | 200 | |
| 4c | R2 = OMe | 5.35 ±1.5 | >50 | 1.33 ±0.24 | >50 | >200 | |
| 4d | R3 = OMe | 11.26 ±4.1 | 11.67 ±3.2 | 1.89 ±0.3 | 10.30 ±3.9 | >200 | |
| 4e | R1,R2 = OMe | 14.56 ±3.3 | 14.6 ±3.6 | 3.77 ±0.4 | >50 | >200 | |
| 4f | R1–R2 = OCH2O | 15.44 ±4.6 | >20 | >20 | 12.5 ±3.1 | >200 | |
| 4g | 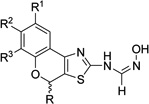 |
R2 = OMe | 4.9 ±1.9 | 4.7 ±1.2 | >50 | >50 | >200 |
| 4h | R1–R2 = OCH2O | 1.43 ±0.38 | >10 | 0.73 ±0.09 | 4.4 ±0.9 | 100 | |
| 4i | R2 = OMe; R = ph | 6.7 ±1.9 | 9.7 ±2.3 | >50 | >50 | >200 | |
| 4j | 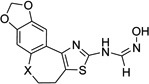 |
X = CH2 | 15.4 ±4.1 | >50 | 1.4 ±0.25 | 14.5 ±3.6 | >200 |
| 4k | X = O | 1.26 ±0.2 | 24.5 ±2.2 | 0.38 ±0.05 | 3.13 ±0.9 | >200 | |
| 4l | 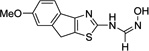 |
– | >50 | >50 | >50 | >50 | 100 |
| 4m | 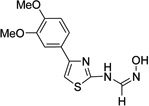 |
– | >20 | >20 | 6.6 ±1.8 | 7.0 ±1.7 | 200 |
| NQa | 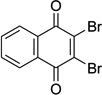 |
– | 1.14 | ND | 0.71 | ND | 25 |
Note: IC50 values are the average of at least three independent experiments, each consisting of triplicates and ± standard deviation is given.
Presented in our earlier work8 and is given here for comparison.
To explore the SAR further about the B-ring, we tested a ring-contracted version, indan derivative 4l, and a ring-excised version 4m. Of the two, 4l was not an inhibitor of MtMetAPs, but 4m did inhibit MtMetAP1c with moderate potency, emphasizing the relevance of B-ring, important perhaps in fixing the orientation of the N'-hydroxymethanimidamide pharmacophore.
Although N'-hydroxymethanimidamide moiety appears to be intuitively acid labile, it has proved to be quite useful in improving the oral bioavailability of methanimidamide containing drug candidates, in particular as a prodrug approach24 (examples: antithrombotic agent sibrafiban18 and a direct thrombin inhibitor called ximelagatran25). Another salient example of an N'-hydroxymethanimidamide is an inhibitor of 20-hydroxyeicosatetraenoic acid, called TS-011 which is being evaluated for the treatment of cerebral ischaemia.26 Considering these successes, the MtMetAP inhibitors we have described here are not likely to defy the druggability criteria.
3.1. Metal complexation studies
We recorded UV-vis spectra of 4g, 4h, and 4k, in the MetAP assay buffer (at 10 µM) with or without 10 µM cobalt or manganese chlorides, which closely emulated the enzyme assay conditions. All three compounds form metal complexes and yet despite such a close structural relationship 4g did not inhibit either metalloforms of MtMetAP1c. It should be noted that salicylaldimine like 3b–u is a well-established metal ion chelator, but it did not inhibit MtMetAPs. Thus, it appears that akin to our earlier experience with the human MetAP inhibitors,27 ability to complex metal ions is not a sufficient or necessary criterion for rationalizing MtMetAP inhibition.
3.2. Anti-tuberculosis activity
We determined the minimum inhibitory concentrations (MICs) of methanimidamides 4a–4m in the culture of Mtb (H37Rv strain). Except 4a, 4h, and 4l, which exhibited MICs of a meager 100 µg/mL, the remaining compounds did not affect the growth of Mtb. The positive control on the other hand, 2,3-dibromonaphthoquinone (NQ, IC50s, 1.14 [Co2+–MtMetAP1a], and 0.71 [Co2+–MtMetAP1c) had an MIC of 25 µg/mL matching our previous results.8 On the basis of enzyme inhibition data, we were expecting 4b, 4h, and 4k to manifest superior MICs, but counter to our expectations only 4h had a weak MIC of 100 µg/mL. Although methanimidamide 4l did not display any MtMetAP inhibitory activity, it nevertheless registered an MIC of 100 µg/mL. This result is quite puzzling, because MICs did not correlate with the enzyme inhibition. In the MtMetAP target validation study we conducted previously, NQ was effective in blocking the Mtb growth. A plausible interpretation of this puzzling result of MICs is that the methanimidamides 4a–4m either lack the ability to permeate the bacterial cell or they succumb to an intrinsic efflux mechanism.28,29 A control experiment was conducted to allay suspicion of instability of N’-hydroxymethanimidamide functionality and unsurprisingly compounds 4a–c and 4k were quite stable to the Mtb culturing conditions (see Supplementary data). Curiously, an MtMetAP inhibitor bengamide analog was reported to exhibit a reasonable inhibition of replicating Mtb (IC50s, µM: 6.9 [Mn2+–MtMetAP1a], 0.54 [Mn2+–MtMetAP1c], MIC: 18.5 µg/mL).30
4. Conclusions
Starting from an N’-hydroxy-N-(naphthothiazolyl)methanimidamide 4c as a hit in an HTS, we established N'-hydroxymethanimidamide as the required pharmacophore for the inhibition of the two Mtb MetAP isoforms. Three inhibitors 4b, 4h, and 4k possessing potent activity against both MtMetAP1a and MtMetAP1c were uncovered through an SAR campaign where benzoxepine derivative 4k was identified as the best inhibitor. Unfortunately the MtMetAP inhibitory potency of these methanimidamides did not transpire into good MICs in Mtb culture. Undoubtedly, the N'-hydroxymethanimidamide inhibitors need to be tuned further with more SAR to find analogs that are effective in Mtb culture, and the SAR described herein is an important step towards achieving that goal.
5. Experimental
5.1. Chemistry: general methods
Unless stated otherwise, all non-aqueous reactions were carried out under ambient atmosphere in oven-dried glassware. Indicated reaction temperatures refer to those of the reaction bath, while room temperature (RT) is noted as 25 °C. All the solvents were of reagent grade purchased from Fisher Scientific or VWR and used as received. Commercially available starting materials and reagents were purchased from Acros, Aldrich, or TCI America and were used as received, unless stated otherwise.
Analytical thin layer chromatography (TLC) was performed using Analtech Uniplates (silica gel HLF, W/UV254, 250 µm). Visualization was achieved using 254 nm UV light, or additionally by staining with iodine, or ceric ammonium molybdate stain. The retention factor reported as follows: Rf (eluent system). Crude products were purified by air-flashed column chromatography over silica gel (0.06 – 0.2 mm, 60 Å, from Acros) using indicated eluents. Melting points were recorded on a Mel-Temp II apparatus and are uncorrected. NMR data were collected on a Varian Unity-400 (400 MHz 1H, 100.6 MHz 13C) or Bruker-Spectrospin 400 MHz spectrometers. Chemical shifts are reported in ppm (δ) with the solvent resonance or 0.1% tetramethylsilane contained in the deuterated solvent as the internal reference. Data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, dd = doublet of doublet, m = multiplet, br = broad, app = apparent, exch = exchangeable), coupling constants (J, reported in Hertz, Hz), and number of protons. Low resolution mass spectra were acquired on a Thermo-Finnigan MAT, LCQ Classic ESI-mass spectrometer or Voyager DE-STR, MALDI-TOF (Applied Biosystems) instruments. The MALDI-samples were prepared by mixing the droplets of the sample solutions in chloroform or methanol and 2,5-dihydroxybenzoic acid solution in acetone, where the latter served as the matrix. Data are reported in the form m/z (% abundance, ion).
Samples were analyzed for purity on a JASCO HPLC equipped with a Rainin Microsorb-MV C18 column (5 µm, 4.6 × 100 mm). The mobile phase set at a flow rate of 1 mL/min was programmed for a gradient elution starting from a 75:20:5 to 10:75:15 mixture of H2O—MeCN—MeOH over 10 minutes, and the mixture was held steady at that ratio for six more minutes, and during the next minute it was reverted back to the original ratio of 75:20:5, making each run a total of 17 minutes long. Purity of final compounds was determined to be >95%, using a 10 µL injection (approximately 1 mM in acetonitrile) with quantitation by area under the curve (AUC) at 254, 275, and 296 nm (JASCO Diode Array Detector). The retention time (tR) is reported in minutes with the AUC given in parentheses.
5.1.1. (S)-2-Hydroxy-N-(7-methoxy-4H,5H-naphtho[1,2-d]thiazol-2-yl)propanamide (3s)
7-Methoxy-4H,5H-naphtho[1,2-d]thiazol-2-amine (85 mg, 365 µmol) was coupled with (S)-O-(tert-butyldiphenylsilyl)lactic acid31 using HBTU (152 mg, 1.1 equiv.) and i-Pr2NEt (230 µL, 3.5 equiv.) in THF (10 mL). After stirring the reaction mixture overnight, water (10 mL) was added and the product was extracted into EtOAc (2× 15 mL). The pooled organic phase was evaporated and the product was diluted with THF (10 mL) and treated with n-Bu4NF (1.2 mL, 1 M in THF) at room temperature for 4 h. The reaction mixture was concentrated and the residue was subjected to flash column chromatography over silica gel (eluent, 1:5 EtOAc/hexanes) to yield an off-white solid. Yield: 59 mg, 53% (over two steps); Rf (3:7 EtOAc/hexanes): 0.21 (UV active); tR = 9.080 min. (98%); 1H-NMR (400 MHz, CDCl3): δ, 7.55 (d, J = 9.1, 1H, H-9), 7.37 (br s, 1H, NH), 6.79 (m, 2H, H-6,7), 4.54 (m, 1H, H-1'), 3.83 (s, 3H, OMe), 3.07–2.86 (m, 4H, H-4,5), 1.28 (d, J = 6.7 Hz, 3H, H-3'); MALDI-TOF: m/z, 305 (30%, MH+), 327 (80%, M+Na+).
5.1.2. N-(Furan-2-ylmethyl)-7-methoxy-4H,5H-naphtho[1,2-d]thiazol-2-amine (3u)
7-Methoxy-4H,5H-naphtho[1,2-d]thiazol-2-amine (325 mg, 1.4 mmol) and freshly distilled furfural (162 µL, 1.7 mmol) were added to a 1:1 CH2Cl2—MeOH mixture containing anhydrous MgSO4 (1 g) and the reaction mixture was stirred under argon for 6 h (the solution turned yellow after 30 min). The mixture was filtered and the filtrate was concentrated. The previously known product (3t) was purified by column chromatography over silica gel (eluent: 1:9 EtOAc/hexanes). Yield: 404 mg, 94%; Rf (3:7 EtOAc/hexanes): 0.75 (UV active); 1H-NMR (400 MHz, acetone-d6): δ, 7.97 (s, 1H, HC=N), 7.79 (d, J = 9.2 Hz, 1H, H-9), 7.35 (d, J = 2.1 Hz,1H, H-5'), 6.85 (m, 3H, H-6,8; H-3'), 6.77 (m, 1H, H-4'), 3.82 (s, 3H, OMe), 2.86–2.82 (m, 4H, H-4,5); MALDI-TOF: m/z, 311 (100%, MH+), 350 (15%, M+K+).
The Schiff base 3t (20 mg, 64.5 µmol) was dissolved in MeOH (8 mL) and NaBH4 (15 mg, 395 µmol) was added and the mixture was stirred overnight. After concentrating, the reaction mixture was purified by flash column chromatography over silica gel (1.5:8.5 EtOAc/hexanes). Yield: 29 mg, 96%; Rf (3:7 EtOAc/hexanes): 0.73 (UV active); tR = 12.400 min. (98%); 1H-NMR (400 MHz, acetone-d6): δ, 7.81 (d, J = 9.2 Hz, 1H, H-9), 7.34 (d, J = 2.1 Hz,1H, H-5'), 6.89 (m, 3H, H-6,8; H-3'), 6.78 (m, 1H, H-4'), 4.66 (br s, 1H, NH), 4.32 (s, 2H, CH2-furyl), 3.85 (s, 3H, OMe), 2.86–2.82 (m, 4H, H-4,5); δ, MALDI-TOF: m/z, 313 (100%, MH+), 335 (20%, M+Na+).
5.1.3. Analogs of N’-hydroxy-N-(4H,5H-naphtho[1,2-d]thiazol-2-yl)methanimidamide
These derivatives were prepared using a three-step protocol starting from an appropriate ketone. As a representative example, synthesis of methanimidamide 4b is described below.
5.1.3.1. Synthesis of 8-methoxy-4H,5H-naphtho[1,2-d]thiazol-2-amine (2b)
Naphthothiazole 2b was prepared using a known procedure.15 Briefly, thiourea (3.88 g, 51 mmol) and iodine (4.75 g, 18.7 mmol) were added to a solution of tetralone 1b (3 g, 17 mmol) in absolute ethanol. The reaction mixture was heated at 100 °C in an open vessel for 3 h, and in the end all the solvent was allowed to evaporate. The residue was washed with ether (3× 15 mL), dissolved in water (50 mL) and heated for 30 min. and cooled. The white solid was filtered, dried, and recrystallized from 9:1 EtOH—H2O and dried under vacuum to afford the hydroidide salt of 2b. Upon a quick free-basing by washing with 5% NaOH and evaporating the dichloromethane layer, the sample was characterized by 1H NMR and MALDI-TOF and it was carried over to the next step.
5.1.3.2. Synthesis of N'-hydroxy-N-(8-methoxy-4H,5H-naphtho[1,2-d]thiazol-2-yl)methanimidamide (4b)
A two step sequence described in the literature17 was applied to making methanimidamide 4b. Aminothiazole 2b (1.4 mmol) from the previous step was heated at 100 °C with N,N-dimethylformamide dimethyl acetal (220 µL, 1.64 mmol) in toluene (15 mL) for 5 h and the solvent was evaporated. The residue was recrystallized from cyclohexane and the resulting solid (4b) was stirred with hydroxylamine hydrochloride (575 mg, 8.3 mmol) in methanol (20 mL) at room temperature for 16 h. The reaction mixture was concentrated and it was neutralized by adding 10% Na2CO3 solution dropwise (pH 9). A brown precipitate was formed and it was filtered and washed with water and recrystallized further from 1,4-dioxane to afford pure methanimidamide 4b.
5.1.3.3. Methanimidamide 4b
Yield: 52% over three steps; Rf (3:7 EtOAc/hexanes): 0.42 (UV active); tR = 8.013 min. (98%); 1H-NMR (400 MHz, acetone-d6): δ, 9.38 (br s, 2H, NH, OH), 9.26 (s, 1H, CH=N), 7.52 (d, J = 8.6 Hz, 1H, H-6), 7.37 (d, J = 1.9 Hz,1H, H-9), 6.85 (dd, J = 8.6, 1.9 Hz, 1H, H-7), 3.75 (s, 3H, OMe), 3.11–2.92 (m, 4H, H-4,5); MALDI-TOF: m/z, 276 (35%, MH+), 298 (20%, M+Na+).
5.1.3.4. Methanimidamide 4d
Methanimidamide 4d was prepared from the commercially available 5-mehtoxy-1-tetralone. Final compound was purified by flash chromatography over silica gel (eluent: 2% MeOH—DCM). Yield: 61% over 3 steps; Rf (3:7 EtOAc/hexanes): 0.33 (UV active); tR = 9.547 min. (98%); 1H-NMR (400 MHz, acetone-d6): δ, 9.18 (br s, 2H, NH, OH), 9.12 (s, 1H, CH=N), 8.05 (d, J = 8.7 Hz, 1H, H-9), 7.38 (d, J = 8.7 Hz, 1H, H-8), 6.85 (d, J = 8.6 Hz, 1H, H-7), 3.79 (s, 3H, OMe), 3.21–2.97 (m, 4H, H-4,5); MALDI-TOF: m/z, 276 (60%, MH+), 298 (40%, M+Na+).
5.1.3.5. Methanimidamide 4e
This was prepared from the commercially available 6,7-dimehtoxy-1-tetralone (1e). Yield: 48% over 3 steps; Rf (5% MeOH/DCM): 0.28 (UV active); tR = 8.187 min. (96%); 1H-NMR (400 MHz, acetone-d6): δ, 9.61 (br s, 2H, NH, OH), 7.81 (s, 1H, CH=N), 7.39 (s, 1H, H-9), 6.83 (s, 1H, H-6), 3.83 & 3.81 (2× s, 6H, OMe), 3.22–2.85 (m, 4H, H-4,5); MALDI-TOF: m/z, 306 (20%, MH+), 328 (M+Na+), 290 (100%, M+–O).
5.1.3.6. Methanimidamide 4f
This was prepared from 6,7-(methylenedioxy)-1-tetralone (1f).10 Yield: 67% over 3 steps; Rf (3:7 EtOAc/hexanes): 0.39 (UV active); tR = 9.507 min. (97%); 1H-NMR (400 MHz, acetone-d6): δ, 9.55 (br s, 2H, NH, OH), 7.89 (s, 1H, CH=N), 7.25 (s, 1H, H-9), 6.85 (s, 1H, H-6), 6.02 (d, J = 10.3 Hz, 2H, OCH2O), 3.21–2.95 (m, 4H, H-4,5); MALDI-TOF: m/z, 290 (90%, MH+), 312 (30%, M+Na+).
5.1.3.7. Methanimidamide 4g
This was prepared from 7-methoxy-2H,3H,4H-chromen-4-one.11 Yield: 69% over 3 steps; Rf (3:7 EtOAc/hexanes): 0.43 (UV active); tR = 8.943 min. (97%); 1H-NMR (400 MHz, acetone-d6): δ, 9.51 & 9.48 (2× br s, 2H, NH & OH), 9.36 (s, 1H, CH=N), 7.65 (d, J = 9.2 Hz, 1H, H-9), 7.57 (d, J = 9.2, Hz,1H, H-8), 6.70 (d, J = 6 Hz, 1H, H-6), 5.35 (d, J = 9.6 Hz, 2H, H-4); MALDI-TOF: m/z, 278 (76%, MH+), 300 (55%, M+Na+).
5.1.3.8. Methanimidamide 4h
Methanimidamide 4h was prepared from 6,7-dihydro-8H-1,3-dioxolo[4,5-g]benzopyran-8-one.12 Yield: 55% over 3 steps; Rf (3:7 EtOAc/hexanes): 0.36 (UV active); tR = 9.320 min. (96%); 1H-NMR (400 MHz, acetone-d6): δ, 9.64 & 9.55 (2× br s, 2H, NH & OH), 7.79 (s, 1H, CH=N), 7.17 (s, 1H, H-9), 6.51 (s, 1H, H-6), 6.02 (d, J = 10.6 Hz, 2H, OCH2O), 5.33 (d, J = 9.8 Hz, 2H, H-4); MALDI-TOF: m/z, 292 (80%, MH+), 314 (45%, M+Na+).
5.1.3.9. Methanimidamide 4i
This was prepared from the commercially available 7-methoxyflavanone. Yield: 61% over 3 steps; Rf (3:7 EtOAc/hexanes): 0.54 (UV active); tR = 10.403 min. (97%); 1H-NMR (400 MHz, methanol-d4): δ, 8.31 (br s, 2H, NH & OH, partially exchanged), 7.75 (dd, J = 8.8, 1.4 Hz, 1H, H-9), 7.52–7.33 (m, 6H, CH=N, phenyl), 6.55 (dd, J = 8.8 & 2.4 Hz, 1H, H-8), 6.49 (dd, J = 2,4 & 1.4 Hz, 1H, H-6), 5.08 (s, 1H, H-4), 3.78 (s, 3H, OMe); MALDI-TOF: m/z, 354 (65%, MH+), 376 (40%, M+Na+).
5.1.3.10. Methanimidamide 4j
This compound was prepared from 6,7,8,9-tetrahydro-5H-cyclohepta[f]-1,3-benzodioxol-5-one.13 Yield: 37% over 3 steps; Rf (3:7 EtOAc/hexanes): 0.45 (UV active); tR = 9.987 min. (98%); 1H-NMR (400 MHz, acetone-d6): δ, 9.57 (br s, 2H, NH, OH), 7.90 (s, 1H, CH=N), 7.27 (s, 1H, H-10), 6.88 (s, 1H, H-7), 6.05 (d, J = 10.3 Hz, 2H, OCH2O), 3.11–2.85 (m, 4H, H-4,6), 2.28 (m, 2H, H-5); MALDI-TOF: m/z, 304 (70%, MH+), 326 (40%, M+Na+).
5.1.3.11. Methanimidamide 4k
This was prepared from 7H,8H-1,3-dioxolo[4,5-h]benzoxepin-9(6H)-one.14 Yield: 51% over 3 steps; Rf (3:7 EtOAc/hexanes): 0.32 (UV active); tR = 8.640 min. (96%); 1H-NMR (400 MHz, methanol-d4): δ, 7.69 (s, 1H, CH=N), 6.49 (s, 1H, H-10), 5.92 (s, 1H, H-7), 5.91 (d, J = 6 Hz, 2H, OCH2O), 4.28 (t, J = 5.6 Hz, 2H, H-5) 3.18 (t, J = 5.6 Hz, H-4); MALDI-TOF: m/z, 306 (85%, MH+), 328 (50%, M+Na+).
5.1.3.12. Methanimidamide 4l
This prepared from the commercially available 5-mehtoxy-1-indanone. Yield: 71% over 3 steps; Rf (3:7 EtOAc/hexanes): 0.34 (UV active); tR = 8.040 min. (97%); 1H-NMR (400 MHz, DMSO-d6): δ, 10.51 (br s, 1H, NH or OH), 10.33 (s, 1H, CH=N), 7.63 (s, 1H, NH or OH), 7.40 (d, J = 8.8 Hz, 1H, H-8), 7.18 (d, J = 1.4 Hz, 1H, H-5), 6.88 (d, J = 8.8 Hz, 1H, H-6), 3.79 (s, 3H, OMe), 3.76 (d, J = 11.2 Hz, 2H, H-4); MALDI-TOF: m/z, 262 (66%, MH+), 284 (20%, M+Na+).
5.1.3.13. Methanimidamide 4m
This was prepared from the commercially available 3,4-methylenedioxyacetophenone. Yield: 75% over 3 steps; Rf (5% MeOH/DCM): 0.42 (UV active); tR = 7.187 min. (97%); 1H-NMR (400 MHz, acetone-d6): δ, 8.67 (br s, 2H, NH & OH), 7.18 (s, 1H, CH=N), 6.91 (d, J = 8.8 Hz, 1H, H-6), 6.62 (d, J = 2.4 Hz,1H, H-10), 6.41 (s, 1H, H-5), 6.03 (d, J = 8.8 Hz, 1H, H-7), 2.94 & 2.91 (2× s, 6H, OMe); MALDI-TOF: m/z, 280 (70%, MH+), 263 (MH+–OH).
5.2. Biological Assays
5.2.1. MetAP Enzyme Assays
The protein expression and purification of C-terminal 6×His-tagged MtMetAP1a and N-terminal 6×His-tagged MtMetAP1c were carried out as described before. HTS was also conducted as was described in our earlier work.9 However the MtMetAP assays in the present study were performed with slight modification as explained underneath.
Eleven different concentrations of naphthothiazole derivatives (50, 25, 12.5, 6.25, 3.125, 1.563, 0.781, 0.391, 0.195, 0.098, 0.049 µM) were pipetted into a 96-well plate and they were incubated for 20 min at room temperature with an assay buffer (40 mM HEPES [pH = 7.5], 100 mM NaCl and 10 µM CoCl2 or MnCl2). The assay buffer also contained either 200 nM MtMetAP1a or 100 nM MtMetAP1c, and 1.5 U/mL proline aminopeptidase (from Bacillus coagulans, BcProAP) as the coupling enzyme.32 After 20 min preincubation with the compounds, the enzyme reaction was initiated by adding methionylprolyl-p-nitroanilide as the substrate (20 µL, 3 mM; such that a final concentration of 600 µM was attained in a 100 µL of total reaction volume in each well) and the change in absorbance at 405 nm was measured using a microplate reader (BMG Labtech, Offenburg, Germany) for a period of 20 min. Reaction progress was computed against the data from the wells containing DMSO alone taken for uninhibited reactions. In the control experiments to rule out the possibility of the inhibition of BcProAP itself, the assay protocol was modified where prolyl-p-nitroanilide was used as the substrate (obviously MtMetAP was eliminated). EC50 values were determined by plotting the data using nonlinear regression analysis in the GraphPad Prism 4.0 (La Jolla, CA) and the data from at least three independent experiments (each consisting of a triplicate) and IC50s reported are the average while the error indicated is the standard deviation.
5.2.2. Determination of MICs
Compound concentrations prepared directly in the medium were 0, 0.8, 1.6, 3.2, 6.3, 12.5, 25, 50, 100, 200 and 400 µg/mL. A 100 µL volume of Middlebrook 7H9 broth (Difco, USA) with compound in different concentrations was dispensed into each well of a 96-well cell culture plate (BD). A standard log-phase growth bacterial suspension (Mycobacterium tuberculosis, H37Rv strain) was prepared and diluted to ≈106 CFU/mL in 7H9 broth and a 100 µL inoculum was used to inoculate each well of the plate. A sterile control without inoculum was also included. Plates were sealed and incubated at 37 °C for two weeks. The MIC was read as the minimum compound concentration at which the well was clear indicating that no bacterial growth had occurred.
Supplementary Material
Acknowledgments
This work was supported in part by the Department of Pharmacology, Johns Hopkins School of Medicine and the Keck Foundation. We are grateful to ASDI, Inc. for the provision of the compound library. We thank Mr. Steve Fernandes for technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary data
Supplementary data associated with this article can be found, in the online version at, doi:. These data include representative MtMetAP inhibition curves, representative UV-vis spectra recorded as part of the metal complexation studies, and HPLC chromatograms attesting to the stability of N’-hydroxymethanimidamides (4a, 4b, 4c, and 4k) under Mtb culturing conditions.
References
- 1.Fauci AS. J. Infect. Dis. 2008;197:1493. doi: 10.1086/587904. [DOI] [PubMed] [Google Scholar]
- 2.Rowland K. Natur. 2012 Jan 13; (News section). [Google Scholar]
- 3.Lawn SD. Medicne. 2009;37:654. [Google Scholar]
- 4.Check E. Nature Med. 2007;13:266. doi: 10.1038/nm0307-266. [DOI] [PubMed] [Google Scholar]
- 5.Kramer G, Boehringer D, Ban N, Bakau B. Nature Struct. Biol. 2009;16:589. doi: 10.1038/nsmb.1614. [DOI] [PubMed] [Google Scholar]
- 6.Solbiati J, Chapman-Smith A, Miller JL, Miller CG, Cronan CG., Jr J. Mol. Biol. 1999;290:607. doi: 10.1006/jmbi.1999.2913. [DOI] [PubMed] [Google Scholar]
- 7.Addlagatta A, Quillin ML, Omotoso O, Liu JO, Matthews BW. Biochemisty. 2005;44:7166. doi: 10.1021/bi0501176. [DOI] [PubMed] [Google Scholar]
- 8.Zhang X, Chen S, Hu Z, Zhang L, Wang H. Curr. Microbiol. 2009;59:520. doi: 10.1007/s00284-009-9470-3. [DOI] [PubMed] [Google Scholar]
- 9.Olaleye O, Raghunand TR, Bhat S, He J, Tyagi S, Lamichhane G, Gu P, Zhou J, Zhang Y, Grosset J, Bishai WR, Liu JO. Chem. Biol. 2010;17:86. doi: 10.1016/j.chembiol.2009.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beugelmans R, Chastanet J, Ginsburg H, Quintero-Cortes L, Roussi G. J. Org. Chem. 1985;50:4933. [Google Scholar]
- 11.Vu AT, Campbell Alison N, Harris HA, Unwalla RJ, Manas ES, Mewshaw RE. Bioorg. Med. Chem. Lett. 2007;17:4053. doi: 10.1016/j.bmcl.2007.04.068. [DOI] [PubMed] [Google Scholar]
- 12.Cueva JP, Giorgioni G, Grubbs RA, Chemel BR, Watts VJ, Nichols DE. J. Med. Chem. 2006;49:6848. doi: 10.1021/jm0604979. [DOI] [PubMed] [Google Scholar]
- 13.Dallacker F, Tumbrink L. Z. Naturforsch. [B] 1984;39B(7):925. [Google Scholar]
- 14.Freedman J, Stewart KT. J. Heterocycl. Chem. 1989;26:1547. [Google Scholar]
- 15.Chordia MD, Murphree LJ, Macdonald TL, Linden J, Olsson RA. Bioorg. Med. Chem. Lett. 2002;12:1563. doi: 10.1016/s0960-894x(02)00236-6. [DOI] [PubMed] [Google Scholar]
- 16.Popova LA, Yurashevich NY, Knizhnikov VA. Russ. J. Org. Chem. 2004;40:311. [Google Scholar]
- 17.Sasaki K, Zhang Y-X, Okuda K, Hirota T. J. Heterocycl. Chem. 2001;38:425. [Google Scholar]
- 18.Wu SY, McNae I, Kontopidis G, McClue SJ, McInnes C, Stewart KJ, Wang S, Zheleva DI, Marriage H, Lane DP, Taylor P, Fischer PM, Walkinshaw MD. Structure. 2003;11:399. doi: 10.1016/s0969-2126(03)00060-1. [DOI] [PubMed] [Google Scholar]
- 19.Weller T, Alig L, Beresini M, Blackburn B, Bunting S, Hadvary P, Muller MH, Knopp D, Levet-Trafit B, Lipari MT, Modi NB, Muller M, Refino CJ, Schmitt M, Schonholzer P, Weiss S, Steiner B. J. Med. Chem. 1996;39:3139. doi: 10.1021/jm9509298. [DOI] [PubMed] [Google Scholar]
- 20.Zhou Y, Guo XC, Yi T, Yoshimoto T, Pei D. Anal. Biochem. 2000;280:159. doi: 10.1006/abio.2000.4513. [DOI] [PubMed] [Google Scholar]
- 21.Zhang X, Chen S, Hu Z, Zhang L, Wang H. Curr. Microbiol. 2009;59:520. doi: 10.1007/s00284-009-9470-3. [DOI] [PubMed] [Google Scholar]
- 22.Lu J-P, Ye Q-Z. Bioorg. Med. Chem. Lett. 2010;20:2776. doi: 10.1016/j.bmcl.2010.03.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lu J-P, Chai SC, Ye Q-Z. J. Med. Chem. 2010;53:1329. doi: 10.1021/jm901624n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Clement B, Mau S, Deters S, Havemeyer A. Drug. Metab. Dispos. 2005;33:1740. doi: 10.1124/dmd.105.005249. [DOI] [PubMed] [Google Scholar]
- 25.Hirsch J, O'Donnell M, Eikelboom JW. Circulation. 2007;116:552. doi: 10.1161/CIRCULATIONAHA.106.685974. [DOI] [PubMed] [Google Scholar]
- 26.Marumo T, Eto K, Wake H, Omura T, Nabekura J. Brit. J. Pharmacol. 2010;161:1391. doi: 10.1111/j.1476-5381.2010.00973.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bhat S, Shim JS, Zhang F, Chong CR, Liu JO. Org. Biomol. Chem. 2012 [Google Scholar]
- 28.Nguyen L, Pieters J. Ann. Rev. Pharmacol. Toxicol. 2009;49:427. doi: 10.1146/annurev-pharmtox-061008-103123. [DOI] [PubMed] [Google Scholar]
- 29.De Rossi E, Ainsa JA, Riccardi G. FEMS Microbiol. Rev. 2006;30:36. doi: 10.1111/j.1574-6976.2005.00002.x. [DOI] [PubMed] [Google Scholar]
- 30.Lu J-P, Yuan X-H, Yuan H, Wang W-L, Wan B, Franzblau SG, Ye Q-Z. ChemMedChem. 2011;6:1041. doi: 10.1002/cmdc.201100003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Faure S, Piva-Le-Blanc S, Bertrand C, Pete J-P, Faure R, Piva O. J. Org. Chem. 2002;67:1061. doi: 10.1021/jo001631e. [DOI] [PubMed] [Google Scholar]
- 32.Kitazono A, Kitano A, Tsuru D, Yoshimoto T. J. Biochem. 1994;116:818. doi: 10.1093/oxfordjournals.jbchem.a124601. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.