

Abstract
A negative influence of central chemosensitivity on peripheral chemoreflex response has been demonstrated recently in a decerebrate-vagotomized rat preparation in situ with separate carotid body and brainstem perfusions. Here, we report similar negative influences of hypercapnia on the hypoxic respiratory response in anesthetized, spontaneously breathing rats before and after vagotomy and anesthetized, artificially ventilated rats after vagotomy. Baseline breathing patterns and responsiveness to hypercapnia and hypoxia varied widely between the three respiratory modes. Despite this, the responses in inspiratory amplitude and expiratory duration (and hence respiratory frequency and neural ventilation) to hypoxia varied inversely with the background CO2 level in all three groups. Results demonstrate a hypoadditive hypercapnic-hypoxic interaction in vivo that resembles the hypoadditive central-peripheral chemoreceptor interaction in situ for these respiratory variables in the rat, regardless of differences in vagal feedback, body temperature and ventilation method. These observations stand in contrast to previous reports of hyperadditive peripheral-central chemoreceptor interaction.
Keywords: Peripheral chemoreceptor, Central chemoreceptor, Respiratory control, Hypercapnic-hypoxic interaction, Vagotomy
1. Introduction
Central and peripheral chemoreceptor influences on breathing are traditionally modeled as additive for simplicity although many extant data in human subjects and animal models suggest the possibility of not only additive (Clement et al., 1992, 1995; Heeringa et al., 1979; Mohan and Duffin, 1997; StCroix et al., 1996; Swanson and Bellville, 1974; van Beek et al., 1983) but also hyperadditive (Adams et al., 1978; Cunningham et al., 1986; da Silva et al., 2011; Honda et al., 1981; Loeschcke et al., 1963; Robbins, 1988; Roberts et al., 1995; Tenney and Brooks, 1966; Teppema et al., 2010) or even hypoadditive interaction (Adams and Severns, 1982; Berger et al., 1978; Cragg and Drysdale, 1983; Eldridge et al., 1981; Gesell et al., 1940; Giese et al., 1978; Ou et al., 1976; Smith et al., 1984). A major confound on this issue is that hypercapnia and hypoxia are often used as physiological stimuli to activate the central and peripheral (carotid) chemoreflex loops, respectively, whereas hypercapnia may stimulate both and hence its effect is nonspecific. Another difficulty is that hypercapnic and hypoxic stimuli may interact at multiple sites in the chemoreflex loops and the interaction could vary with stimulus intensities and timing at different sites. For example, hypercapnia and hypoxia are well-known to stimulate carotid chemoreceptors in a multiplicative (i.e., hyperadditive) fashion in adult rats (Kumar, 2009; Pepper et al., 1995; Roy et al., 2000) as in cats (Fitzgerald and Parks, 1971; Lahiri and DeLaney, 1975) but the corresponding central interaction effects are unclear. It has been suggested that hypoxic and hypercapnic stimuli at sufficiently high intensities may cause saturation of neuronal circuits (Eldridge et al., 1981) or exert depressant effects on neuronal activity (Cherniack et al., 1970) especially when presented in combination. Hypoxia may also occasion multiple physiological changes such as decreased whole-body metabolism (Frappell et al., 1992), increased or decreased systemic arterial blood pressure (Kontos et al., 1965; Song and Poon, 2009b; Song et al., 2011) and improved pulmonary ventilation-perfusion matching (Alfaro et al., 2001), all of which may secondarily affect breathing and perturb the interaction of central and peripheral chemoreflexes in a complex manner.
Recently, several authors have tackled this problem by surgically isolating the central from the peripheral chemoreceptor contributions to the interaction. Day and Wilson (2009) using an in situ dual (brainstem and carotid chemoreceptor) perfused preparation in vagotomized-decerebrate rats showed that neural minute ventilation was more responsive to peripheral chemoreflex the lower the brainstem PCO2. This was achieved even when the latter was kept at hypocapnic levels, thus ruling out possible neuronal saturation or depressant effects that might confound the interaction at high CO2 levels. They interpreted their findings as indicative of a hypoadditive (negative) interaction between central and peripheral chemosensitivity in modulating the peripheral chemoreflex. In contrast, Blain et al. (2010) employing a carotid body perfusion preparation in unanesthetized vagal-intact dogs reported that carotid body stimulation augmented central hypercapnic chemoreflex responses in both respiratory frequency and tidal volume, whereas carotid body inhibition (disfacilitation) produced the opposite effects. They concluded that peripheral chemoreceptor stimulation resulted in hyperad-ditive (synergistic) ventilatory responses to central hypercapnia. Both studies were designed to obviate possible confounds in previous studies of hypercapnic and hypoxic chemoreflex inter action, yet divergent outcomes emerged. A possible explanation of this surprising discrepancy is that the interaction of central and peripheral chemoreceptors may be non-commutative: the influence of central chemosensitivity on peripheral chemoreflex (central-peripheral chemoreflex interaction) (Day and Wilson, 2009) may differ from that of peripheral chemosensitivity on central chemoreflex (peripheral-central chemoreflex interaction) (Blain et al., 2010) depending on the sites of interaction. Another possibility is that the hypoadditive interaction that exists in a highly reduced preparation in rats (Day and Wilson, 2009) may morph into a hyperadditive one in the presence of some unknown factors only found in a more intact preparation or in dogs (Blain et al., 2010).
Peculiar to the in situ artificially perfused rat preparation is the relatively low respiratory frequency due to vagotomy and relatively low perfusion temperature (33–34°C). Since the resulting hypoadditive central–peripheral chemoreflex interaction was reportedly confined to the response in respiratory frequency mainly owing to impaired peripheral chemoreflex responsiveness in expiratory duration (TE) at higher brainstem PCO2 levels, it is germane to inquire whether impaired vagal and temperature modulation of TE and respiratory frequency might be the culprit behind the negative interaction. This is of interest since central and peripheral chemosensitivities are dependent on vagal feedback (Kiwull-Schone and Kiwull, 1979; Moreira et al., 2007; Richardson and Widdicombe, 1969; Tonkovic-Capin et al., 2000) and body temperature (Baker et al., 1996; Cummings and Frappell, 2009; Cunningham and O'Riordan, 1957; Kiley et al., 1984; Watanabe et al., 1996). Further, recent studies have shown that the TE responses to acute hypoxia and hypercapnia are specifically modulated by distinct regions of the pneumotaxic center in dorsolateral pons, where central and peripheral chemoreceptor inputs are likely integrated along with vagal inputs (Ezure, 2004; Kubin et al., 2006; Mizusawa et al., 1995; Song and Poon, 2009a,b; Song etal., 2011; St John, 1975). Elucidation of the contribution of TE responsiveness to hypercapnic–hypoxic interaction in vagotomized and vagal–intact animals may shed light on the mechanism of such interaction. However, to our knowledge there has been no study that examines the effects of vagal feedback and body temperature on central and peripheral chemoreflex interaction and the contribution of impaired TE responsiveness to such interaction. Another interesting question is whether such interactions at the chemoreceptor level can be correlated to interactions of hypercapnic and hypoxic chemoreflex responses in animals and humans. This is a pertinent question because the ventilatory responses to hypoxia and hypercapnia are widely studied in animal models and in healthy subjects and patients, in whom artificial brainstem or carotid body perfusion is impracticable.
In the present study, we have therefore re-examined the influences of both hyper- and hypocapnia on the hypoxic respiratory chemoreflex response in both vagotomized and vagal-intact anesthetized but spontaneously breathing rats as well as vagotomized, anesthetized-paralyzed and artificially ventilated rats, all at normal body temperature. To allow direct comparison with Day and Wilson (2009) and to examine (i) how nonspecific central vs. peripheral effects of the hypercapnic stimulus might influence hypercapnic-hypoxic interaction and (ii) whether any saturation/depressant effects at high stimulation intensities might contribute to such interaction, we employed a hypercapnic and hypoxic stimulation and hypocapnic disfacilitation protocol that paralleled the brainstem and carotid chemoreceptor stimulation and disfacilitation sequence in Day and Wilson's study. We found that despite the widely different resting breathing patterns and responsiveness to hypoxia and hypercapnia in these preparations due to differences in vagal feedback and ventilation method, a hypoadditive influence of hypercapnia on hypoxic chemoreflex responses in TE and respiratory frequency (hypercapnic–hypoxic interaction) prevailed in all cases. In addition, we found that such hypoadditive interaction also applied to the inspiratory amplitude (neural tidal volume) component. Our results are in agreement with Day and Wilson (2009) and run counter to Blain et al. (2010).
2. Methods
2.1. Animal preparation
Experiments were performed on 10 adult, male Sprague–Dawley rats (330–380 g, Charles River Laboratories, Wilmington, MA). Experimental protocols were reviewed and approved by the M.I.T. Committee on Animal Care in accordance with published guidelines. Animals were injected with atropine sulfate (0.025 mg, s.c.), then anesthetized with urethane (Sigma, 1.5g/kg, i.p.). Trachea was cannulated. Lactated Ringer's solution was continuously infused (0.05– 0.1 ml/min) through a femoral vein cannula. A femoral artery was cannulated and connected to a blood pressure monitor (BP-100, CWE, Ardmore, PA). Supplemental urethane was given as needed (1/10 original dosage, i.v. or i.p.). Body temperature was maintained at 36.5 ± 0.2°C (TC-831, CWE, Ardmore, PA).
One group of animals (n = 5) breathed O2–enriched (40%) medical air spontaneously. A separate group of animals (n = 5) were paralyzed with pancuronium bromide (Sigma, initial dose 0.5 mg, i.v., supplemented every hour at 0.1 mg, i.v.), vagotomized and arti ficially ventilated with the same gas using a mechanical ventilator (AVS-1, CWE, Ardmore, PA). The O2 and CO2 concentrations of the respired gas were monitored with a Gemini Respiratory Gas Analyzer (CWE, Ardmore, PA). Spontaneously breathing animals were studied before and after bilateral vagotomy. Ventilated animals were studied after bivagotomy only.
2.2. Electrophysiological recording
To monitor respiratory motor output in spontaneously breathing animals, diaphragmatic EMG was recorded by implanting two fine silver wires into the diaphragm. In ventilated animals, the phrenic nerve was isolated and mounted on a bipolar platinum wire electrode (FHC, Bowdoin, ME). The phrenic discharge or diaphragmatic EMG signal was amplified (CyberAmp 380, Axon Instruments, Union City), integrated with an analog Paynter filter (time constant 50 or 100 ms) and sampled (at 10 kHz) into a Dell PC with LabView (National Instruments, Austin, TX).
2.3. Hypoxia tests
Fig. 1 illustrates the experimental protocols for the hypoxia test under varying CO2 backgrounds. In spontaneously breathing animals, the hypoxia was presented under either normocapnic or hypercapnic background. For normocapnic hypoxia, the animal was given 8% O2 (balance N2) for 30 s. For hypercapnic hypoxia, the animal was given 5% CO2 (balance O2) for 4–5 min and then 5% CO2 and 8% O2 (balance N2) for 30 s. After bilateral vagotomy, both hypoxia tests were repeated.
Fig. 1.

Experimental protocol for the hypoxic test under hypocapnic (broken line), normocapnic (solid line) and hypercapnic (dotted line) backgrounds.
In ventilated animals, the hypoxia was given under normocapnic (end-tidal CO2 kept at 5%), hypercapnic (ventilation kept at normocapnic level), or hypocapnic background, and only after bilateral vagotomy. For normocapnic and hypercapnic hypoxia the same protocols were used as described above. For hypocapnic hypoxia, the animal was hyperventilated (on 40% O2) until end-tidal CO2 was lowered to 4% (while rhythmic phrenic discharge remained) for 4–5 min before a gas mixture of 8% O2 (balance N2) was given for 30 s.
In all animals, a recovery period of 15–20 min was allowed after each hypoxia test or vagotomy before another hypoxia test was given.
2.4. Data analyses
Inspiratory duration (TI), TE, respiratory frequency (Freq) and inspiratory amplitude (Amp) were computed for each respiratory cycle from the integrated phrenic or diaphragmatic EMG signal. Inspiratory drive is defined as Amp/TI and neural (min) ventilation as Amp × Freq. For each of these respiratory variables (denoted here as the generic variable Y) the corresponding responses to hypercapnia/hypocapnia and hypoxia were calculated as follows (see Fig. 1 for notations).
YA = averaged value over the last 1 min in hyperoxic normocapnia baseline.
YB = averaged value over the last 1 min of hyperoxic hypercapnia/normocapnia/hypocapnia background.
YC = maximum or minimum value during hypercapnic/normocapnic/hypocapnichypoxia test binned every 5 s.
| (1a) |
| (1b) |
| (2a) |
| (2b) |
For between-group comparisons, the relative hypoxic response was defined as the hypoxic response normalized against the averaged value over all animals in each group under normocapnic background:
| (3) |
where n is the number of animals.
To minimize inter-subject variability, all statistical tests were performed on the normalized data as described above. Within-group effects for breathing pattern responses to hypocapnia/hypercapnia and hypoxia were tested using the two-tailed t-test. Between-group effects were tested with either one-way ANOVA (baseline data) or two-way ANOVA with repeated measures (test data) for dependence on respiratory mode and CO2 background, followed by post hoc analysis using Tukey method for pairwise group comparison.Confidence level of 95% was used throughout.
3. Results
Fig. 2 shows the diaphragmatic EMG, airway PCO2 and blood pressure recordings in a spontaneously breathing animal (before and after vagotomy) tested with eucapnic/hypercapnic hypoxia. Fig. 3 shows similar plots for a vagotomized, paralyzed and ventilated animal with phrenic recording and tested with eucapnic, hypercapnic and hypocapnic hypoxia. Baseline breathing pattern for all animals varied widely for the three respiratory modes (i.e., spontaneous breathing vagi-intact, spontaneous breathing bilateral vagotomy, artificial ventilation bilateral vagotomy). Briefly, spontaneous-breathing vagi-intact rats had the fastest respiratory frequency (by ~3×), the shortest TE and TI, and the smallest inspiratory amplitude. After vagotomy, inspiratory amplitude increased and respiratory frequency decreased. Both TE and TI were dramatically increased, with the largest increase in TE in spontaneous-breathing rats and the largest increase in TI in ventilated rats (Table 1).
Fig. 2.

Recordings from a spontaneously breathing rat (1) before and (2) after vagotomy. Data were taken at the end of each experimental period indicated in Fig. 1. Panel A: hyperoxic normocapnia (eucapnia) baseline, hyperoxic normocapnia background, hypoxic normocapnia period. Panel B: hyperoxic normocapnia baseline, hyperoxic hypercapnia background, hypoxic hypercapnia period.
Fig. 3.

Recordings from a vagotomized, paralyzed and artificially ventilated rat. Data were taken at the end of each experimental period indicated in Fig. 1. Panels A and B are as in Fig. 2. Panel C: hyperoxic normocapnia baseline, hyperoxic hypocapnia background, hypoxic hypocapnia period.
Table 1.
Baseline values for respiratory variables in different respiratory modes.
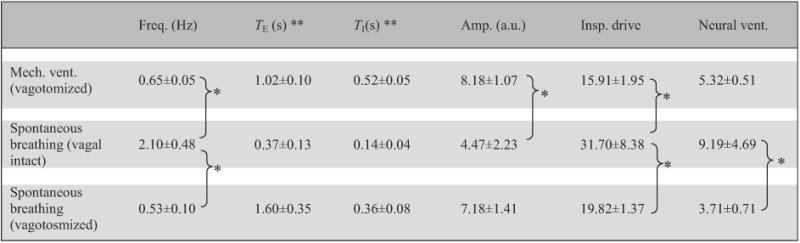
|
Data are means ±SD (n = 5). ** Statistically significant difference between all pairs. * Statistically significant difference between specific pairs only.
As shown in Table 2, the effects of hypocapnia/hypercapnia on breathing pattern in hyperoxia were quite variable within and between the three groups although the response in neural ventilation was not significantly different between groups (p>0.1 one-way ANOVA). Generally, vagal-intact spontaneously breathing animals responded to the hypercapnic stimulus in hyperoxia primarily with an increase in respiratory frequency, whereas vagotomized animals responded primarily with an increase in inspiratory amplitude. Inspiratory drive was significantly increased with hypercapnia in all groups; however, the increase was greatest in the ventilated animals (p<0.05 post hoc) along with a para doxical increase in TE (p<0.05 two-tailed t-test), revealing a latent expiratory-prolonging effect of hypercapnia as recently reported in vagotomized and artificially ventilated rats (Song and Poon, 2009a).
Table 2.
Effects of hypercapnia/hypocapnia in hyperoxia on breathing pattern in different respiratory modes.
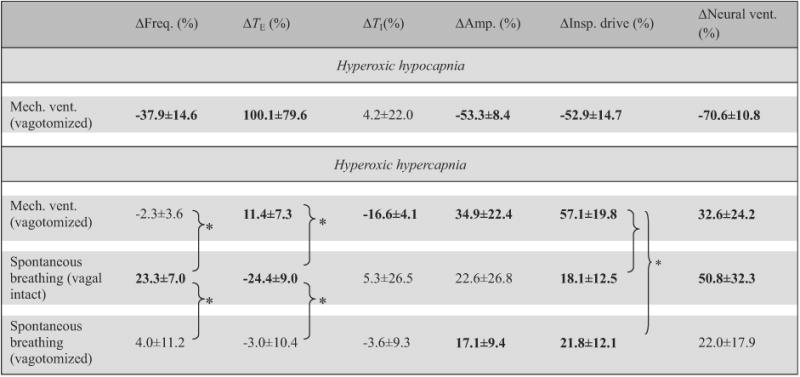
|
Data (means±SD, n = 5) are percentage changes from corresponding hyperoxic–normocapnic baseline values (Eq. (1b)). * Statistically significant difference between specific pairs only. Bolded entries indicate statistically significant differences from zero.
The breathing pattern response to hypoxia under normocapnic background was also distinct for the three respiratory modes (Table 3). Generally, the respiratory frequency and TE responses to hypoxia were stronger in spontaneous-breathing than in ventilated animals, whereas the responses in TI and inspiratory drive showed the opposite trends. Differences in neural ventilation response were not statistically significant between groups (p>0.1, one-way ANOVA).
Table 3.
Breathing pattern response to hypoxia under normocapnic background in different respiratory modes.
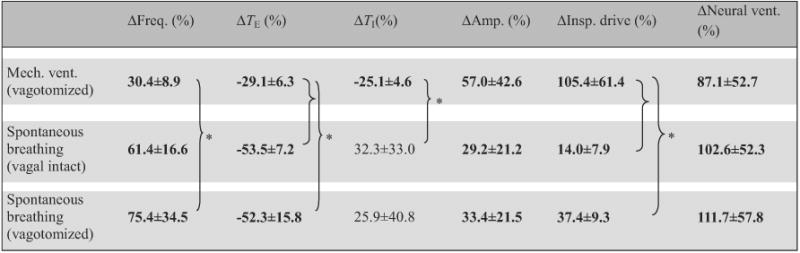
|
Data (means ±SD, n = 5) are percentage changes from corresponding hyperoxic normocapnia background values (Eq. (2b)). * Statistically significant difference between specific pairs only. Bolded entries indicate statistically significant differences from zero.
Despite the widely different baseline breathing patterns and responsiveness to hyperoxic hypercapnia and normocapnic hypoxia, a consistent trend of hypoadditive (negative) hypercapnic-hypoxic interaction is seen for all respiratory variables except TI in all three respiratory modes (Fig. 4). Two-way ANOVA repeated measures analysis of the hypoxic response for all respiratory variables showed that the differences of the trend between respiratory modes were insignificant (p>0.1), whereas there were significant differences between different CO2 back grounds for all respiratory variables (p<0.05) except TI (p>0.1).
Fig. 4.

Averaged hypoxic response forthree different respiratory modes under varying CO2 backgrounds. Data are normalized to normocapnic background (normal CO2, Eq. (3)). Error bars are SE, n = 5 each. Data for each mode are slightly displaced laterally for ease of view. *Significantly different from response under normocapnic background (p<0.05). **Significantly smaller hypoxic TI response under hypercapnic background compared with normocapnic background in artificially ventilated animals (p<0.05).
Post hoc analysis showed that hypoxic response was significantly stronger under lower CO2 background, hence a hypoadditive hypercapnic-hypoxic interaction for all three respiratory modes (Fig. 4). Statistical significance was demonstrated for TE, respira tory frequency, inspiratory amplitude, inspiratory drive and neural ventilation (p< 0.05). Response in TI was more variable across animals and hence statistical significance was not evident across all three respiratory modes (p>0.1). However, by considering ventilated animals alone, hypoxic response in TI under hypercapnic background was shown to be significantly smaller (p<0.05, one way ANOVA). For the ventilated animals, hypoadditive interaction is seen not only under high but also low CO2 background (except for TI and inspiratory drive), indicating that the negative interaction was not caused by neuronal saturation or a depressant effect of CO2.
4. Discussion
4.1. Critique of experimental design
Hypercapnia and hypoxia are common abnormalities in disease states such as sleep apnea and respiratory failure; delineating their interaction in the control of breathing is of basic clinical interest. However, relating the interaction of hypercapnic and hypoxic respiratory responses to those of the central and peripheral chemoreflex loops has proved difficult because of the mixed effects of hypercapnia on both central and peripheral chemoreceptors,rendering most previous studies of hypercapnic–hypoxic (influence of hypercapnic stimulation on hypoxic chemoreflex response) or hypoxic-hypercapnic interaction (influence of hypoxic stimulation on hypercapnic chemoreflex response) difficult to interpret. Our experimental design addressed this issue by adopting a hypercapnic and hypoxic stimulation and hypocapnic disfacilitation protocol that paralleled the central–peripheral chemoreceptor stimulation and disfacilitation sequence in Day and Wilson (2009). Rather than relying on elaborate carotid body perfusion to isolate the peripheral chemoreceptor, we used hyperoxia or hypoxia to preferentially suppress or activate peripheral chemosensitivity, respectively, while keeping central chemosensitivity at graded (hypocapnic, normocapnic or hypercapnic) background levels. The inclusion of a hypocapnic background level allowed us to distinguish hypoadditive effects from neuronal saturation or depressant effects. Our keeping of a brief hypoxia duration (30 s) under graded CO2 backgrounds ensured that any resultant decreases in systemic blood pressure (Figs. 2 and 3) and whole-body metabolism during hypoxia should be limited and should equally apply to all CO2 backgrounds, further mitigating these potential confounds compared to previous studies. The use of the same level of hypoxia exposure for all animals and the measurement of the normalized hypoxic response under constant CO2 backgrounds (Eq. (2b)) should help to minimize any potential effects of hypoxia- and hypercapnia-induced changes in ventilation-perfusion distribution on the normalized hypoxic responses. Because CO2–O2 interaction at the level of the carotid chemoreceptors has been shown to be multiplicative in adult rats (Kumar, 2009; Pepper et al., 1995; Roy et al., 2000) as in cats (Fitzgerald and Parks, 1971; Lahiri and DeLaney, 1975), any hypoadditive hypercapnic– hypoxic interaction in our adult rat preparation in vivo would imply a negative central-peripheral chemoreceptor interaction of central origin. These precautions in our experimental design allowed a direct comparison of the present hypercapnic–hypoxic interaction results in vivo with the central–peripheral chemoreceptor interaction results of the Day and Wilson (2009) study in situ.
4.2. Summary of results
With this judicious experimental design, our data showed that the responsiveness in TE (and to a lesser extent TI), respiratory frequency, inspiratory drive and neural ventilation to hypoxia varied inversely with the background CO2 level in all three respiratory modes (spontaneous breathing vagi-intact, spontaneous breathing bilateral vagotomy, artificial ventilation bilateral vagotomy) in the rat in vivo, in agreement with the reported negative interaction of central–peripheral chemoreflex in the rat in situ (Day and Wilson, 2009). In addition, our data showed that part of the negative hypercapnic–hypoxic interaction effect for neural ventilation could also be attributed to the inspiratory amplitude and inspiratory drive. A negative central–peripheral chemoreflex interaction for inspiratory amplitude (neural tidal volume) was also demonstrated in a previous study (Day and Wilson, 2007) although the effect failed to reach statistical significance in (Day and Wilson, 2009). These findings taken together demonstrate a close correlation between the hypoadditive hypercapnic-hypoxic interaction in vivo and hypoadditive central-peripheral chemoreceptor interaction in situ for these respiratory variables in the rat, regardless of differences in vagal feedback, body temperature and ventilation method. This correlation provides a means of studying central–peripheral chemoreflex interaction in terms of hypercapnic–hypoxic interaction, which is more amenable to experimental investigation. The hypoadditive hypercapnic-hypoxic interaction presently demonstrated is most likely of central origin in view of the multiplicative effects of hypercapnia and hypoxia on carotid body chemosensitivity in adult rats (Kumar, 2009; Pepper et al., 1995; Roy et al., 2000) as in cats (Fitzgerald and Parks, 1971; Lahiri and DeLaney, 1975). Such a hypoadditive effect was also manifested under a hypocapnic background; thus it could not be the consequence of neuronal saturation or depressant effects.
4.3. Comparison with previous animal studies
Many previous studies have reported hypoadditive, additive or hyperadditive hypercapnic–hypoxic or hypoxic–hypercapnic interaction for minute ventilation, respiratory frequency and/or tidal volume (or their neural equivalents) in anesthetized or awake animals, subject to the experimental caveats mentioned above (see Section 1 and references cited therein). In particular, Cragg and Drysdale (1983) using various hypercapnic, hypoxic or hypercapnic/hypoxic gas mixtures to stimulate breathing (with 2–10min of exposure from a preceding normoxic and normocapnic back ground) in anesthetized, vagalintact and spontaneous-breathing rats reported similar hypoadditive (occlusive) interaction for minute ventilation and respiratory frequency but additive interaction for tidal volume. However, interpretation of their results is complicated by the mixed effects of hypercapnia on central and peripheral chemosensitivities under a normoxic background, as opposed to a hyperoxic background in our study. Importantly, none of these in vivo animal studies investigated the possible effects of vagal feedback or ventilation method or possible neuronal satu ration/depressant effects on such interaction, and none of them looked into the contributions of respiratory timing components to such interaction as did the present study. These considerations are important because recent evidence indicates that the peripheral chemoreflex response is mediated by discrete central pathways that independently modulate TE, TI and inspiratory amplitude, in a manner that counterpoises vagal modulation of these respiratory variables (Poon, 2004; Song and Poon, 2004; Young et al., 2003). The present demonstration of a hypoadditive hypercapnic–hypoxic interaction for inspiratory amplitude, respiratory frequency, inspiratory drive and TE (and to a lesser extent, TI) responses irrespective of the integrity of vagal feedback or ventilation method and independent of possible multiplicative effects at the carotid chemoreceptors may shed light on the mechanisms and sites of such interactions. Possible sites where central and peripheral chemoreflex pathways are known to converge and hence likely interact include the lateral parabrachial nucleus (LPBN) and Kölliker-Fuse nucleus (KFN) in dorsolateral pons (Mizusawa et al., 1995; Song and Poon, 2009a,b; St John, 1975) and the retro-trapazoid nucleus (RTN) in rostral ventrolateral medulla (Takakura et al., 2006). In particular, the LPBN has been shown to specifically modulate the shortening of TE (but not TI) and increase in respiratory frequency in response to hypoxia or hypercapnia as well as augmentation of inspiratory amplitude and inspiratory drive in response to hypercapnia (Song and Poon, 2009a,b). The selective effects of the LPBN on the responsiveness of these breathing pattern components (except TI) to hypoxic and hypercapnic stimuli are consistent with the hypercapnic–hypoxic interaction components presently demonstrated.
Our findings and those of Day and Wilson (2007, 2009) are at variance with Blain et al. (2010), which provides the strongest evidence as yet for a hyperadditive peripheral–central chemoreceptor interaction in awake dogs. Such discrepancy cannot be explained by differences in vagal feedback, body temperature and ventilation mode in light of the present study. Other possible factors such as species and wakefulness differences (decerebrate/anesthetized rats vs. conscious dogs) cannot be ruled out, although presently it is not clear how these factors might influence the interaction of central and peripheral chemoreflexes. There is evidence that hypoxic ventilatory response in rats occurs at a higher arterial PO2 level than in other species such as dogs and humans (Hayashi et al., 1983); hence it may be inferred that the overall response is more likely to saturate even at moderate hypoxic and hypercapnic stimulation intensities. However, this possibility is contradicted by our demonstration that the hypoadditive effect extended to the hypocapnic range (see also Day and Wilson, 2009). Similarly, it has been shown that urethane-anesthetized Sprague–Dawley rats demonstrate EEG profiles that closely resemble periods of wake and slow-wave sleep, with resting ventilation levels and breathing pattern responses to hypoxia and hypercapnia during these periods that mimic those seen in unanesthetized rats cycling between wake and sleep (Boon et al., 2004). Therefore, there is no evidence that respiratory control in urethane-anesthetized rats is different than in unanesthetized rats, sleeping or awake.
On the other hand, it is generally believed that the hypercapnic ventilatory response is higher in wakefulness than in sleep (Phillipson and Bowes, 1987). However, central chemosensitivity to CO2 does not decrease during sleep (Parisi et al., 1992). Indeed, an earlier study using separate brainstem and systemic perfusions in awake goats reported a hypoadditive central-peripheral chemoreceptor interaction (Smith et al., 1984) similar to that shown in (Day and Wilson, 2007, 2009). The divergent results in awake animals when central and peripheral chemoreceptors are independently stimulated (Blain et al., 2010; Smith et al., 1984) raise questions about the suggested role of wakefulness in such interaction. Regarding this discrepancy, in Blain et al. (2010) the authors state that “Hypoadditive effects of an alkaline CSF perfusate (and resultant hypoventilation) on enhancing the transient ventilatory response to systemic cyanide have been reported in the awake goat (Smith et al., 1984); however, these findings were confounded by the potential multiplicative effects of an enhanced background arterial PCO2 on carotid chemoreceptor responsiveness to cyanide”. In awake goats, multiplicative effects of hypercapnic and hypoxic stimuli at the isolated carotid chemoreceptor reportedly resulted in similar hyperadditive interaction for minute ventilation and other breathing pattern variables in the whole animal (Daristotle et al., 1987). In contrast, in (Smith et al., 1984) these authors state that “The combined effects of increased FICO2, with superimposed NaCN on ventilation were studied in three nonperfused goats (Fig. 3). Two-way ANOVA for data of all three goats revealed no significant effect of increased VI on the NaCN dose-response slope despite highly significant effects on VI of CO2 or NaCN alone.” Therefore, the suspected confounding effects as alluded to by Blain et al. (2010) were not confirmed by Smith et al. (1984), in support of the reported hypoadditive central–peripheral chemoreceptor interaction in awake goats. These observations suggest that awake animals may demonstrate both hypoadditive and hyperadditive interactions of central and peripheral chemosensitivities depending on the specific experimental conditions and sites of interaction, independent of any multiplicative effects at the carotid chemoreceptors.
4.4. Comparison with human studies
In human subjects, the interaction of central and peripheral chemoreflexes is reportedly either additive (Clement et al., 1992, 1995; Mohan and Duffin, 1997; StCroix et al., 1996; Swanson and Bellville, 1974) or hyperadditive (Cunningham et al., 1986; Honda et al., 1981; Robbins, 1988; Roberts et al., 1995; Teppema et al., 2010); a hypoadditive effect has not been observed (but see the first method of comparison on p. 565 of Clement et al. (1995)). As mentioned above, interpretation of these results is made difficult by the nonspecific effects of hypercapnia on central and peripheral chemoreflexes. For example, a recent study by Teppema et al. (2010) employed a similar hypercapnic and hypoxic stimulation protocol under hyperoxic background in human subjects and found that the hypoxic sensitivity increased with increasing arterial PCO2 level. However, it is not certain whether such a hyperadditive interaction of hypercapnic and hypoxic stimuli occurred peripherally at the carotid chemoreceptors or centrally, or both. In our experimental design, peripheral multiplicative effects may be ignored in inferring central–peripheral chemoreflex interaction from hypercapnic–hypoxic interaction provided the resultant hypercapnic–hypoxic interaction is hypoadditive, but not when it is hyperadditive. It is possible that the peripheral multiplicative effects are much stronger in humans than in rats, such that any negative interactions centrally are overridden or neutralized peripherally resulting in a net hyperadditive or additive effect. This may explain the lack of hypoadditive hypercapnic-hypoxic inter action in humans. Unfortunately, most previous studies in humans only investigated the interaction of the hypercapnic and hypoxic ventilatory responses and not other breathing pattern responses. Comparison of the corresponding breathing pattern responses in human subjects and in animal models may yield insights into the possible mechanisms and sites of hypercapnic–hypoxic and hypoxic–hypercapnic interactions in humans, in whom isolation of the central and peripheral chemoreceptors is impracticable.
4.5. Multiple mechanisms of hypercapnic–hypoxic/hypoxic–hypercapnic interactions
Based on the present results and the above systematic review of current literature, it appears that multiple hypoadditive and hyperadditive mechanisms of central origins may contribute to the interaction of central and peripheral chemoreflexes. The resultant interaction of hypercapnic and hypoxic stimuli is further compounded by the multiplicative effects at the carotid chemoreceptors peripherally. The relative contributions of the central and peripheral mechanisms to stimulus interaction may differ between species and under different experimental conditions, hence the wide spectrum of observed interactive effects.
Why does the interaction of central and peripheral chemoreceptors appear hypoadditive in some cases and hyperadditive in others? While the mechanisms remain unclear, it should be pointed out that central and peripheral chemoreflexes may not necessarily interact in a reciprocal and mutually exclusive manner; thus hypoadditive and hyperadditive effects could co-exist centrally depending on the sites and modes of interaction. Of note, in (Blain et al., 2010) a hyperadditive peripheral-central chemoreceptor interaction was obtained by abruptly stimulating the central chemoreceptor while holding the carotid chemoreceptor activity constant at varying stimulation or disfacilitation background levels. In contrast, in Smith et al. (1984) as with Day and Wilson (2007, 2009) and the present study, a hypoadditive central–peripheral chemoreceptor interaction was achieved by reversing the order of central and peripheral chemoreceptor stimulation/disfacilitation.This subtle difference in the stimulation sequence could result in very different response dynamics and plasticity in the central circuits that modulate the breathing pattern. For example, acute hypoxia is known to elicit multiple time-dependent responses in breathing pattern during and after the hypoxia period, such as short-term potentiation of inspiratory drive due to corresponding time-dependent augmentation of inspiratory amplitude and shortening of TI, as well as short-term depression of respiratory frequency (post-hypoxic frequency decline) due to corresponding time-dependent prolongation of TE (Day and Wilson, 2005; Dick and Coles, 2000; Poon and Siniaia, 2000; Song and Poon, 2009b). Such peripheral chemoreceptor-induced neuroplasticity may interact with central chemoreceptor input in a complex manner leading to distinct steady-state responses in TI, TE and inspiratory amplitude depending on the timing and magnitude of the central and peripheral chemoreceptor inputs. Further studies are needed in future to elucidate the neural correlates of central-peripheral vis-à-vis peripheral–central chemoreceptor interaction and the potential influence of wakefulness on such interactions in animal models and in human subjects.
Acknowledgments
We thank M. Adams and Y. Ning for technical assistance. C. Tin was supported by an American Heart Association predoctoral fellowship. This work was supported by National Institutes of Health grants HL067966, HL093225 and RR028241.
References
- Adams JM, Attinger FM, Attinger EO. Medullary and carotid chemoreceptor interaction for mild stimuli. Pflug Arch. 1978;374:39–45. doi: 10.1007/BF00585695. [DOI] [PubMed] [Google Scholar]
- Adams JM, Severns ML. Interaction of chemoreceptor effects and its dependence on the intensity of stimuli. J Appl Physiol. 1982;52:602–606. doi: 10.1152/jappl.1982.52.3.602. [DOI] [PubMed] [Google Scholar]
- Alfaro V, Roca-Acin J, Palacios L, Guitart R. Multiple inert gaselimination technique for determining ventilation/perfusion distributions in rat during normoxia, hypoxia and hyperoxia. Clin Exp Pharmacol Physiol. 2001;28:419–424. doi: 10.1046/j.1440-1681.2001.03455.x. [DOI] [PubMed] [Google Scholar]
- Baker JF, Goode RC, Duffin J. The effect of a rise in body temperature on the central-chemoreflex ventilatory response to carbon dioxide. Eur J Appl Physiol Occup Physiol. 1996;72:537–541. doi: 10.1007/BF00242287. [DOI] [PubMed] [Google Scholar]
- Berger W, Berger K, Berndt J, Giese K. Interaction of peripheral and central respiratory drives in cats I. Effects of sodium cyanide as a peripheral chemoreceptor stimulus at different levels of CSF pH. Pflug Arch. 1978;374:205–210. doi: 10.1007/BF00585596. [DOI] [PubMed] [Google Scholar]
- Blain GM, Smith CA, Henderson KS, Dempsey JA. Peripheral chemoreceptors determine the respiratory sensitivity of central chemoreceptors to CO(2) J Physiol. 2010;588:2455–2471. doi: 10.1113/jphysiol.2010.187211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boon JA, Garnett NB, Bentley JM, Milsom WK. Respiratory chemoreflexes and effects of cortical activation state in urethane anesthetized rats. Respir Physiol Neurobiol. 2004;140:243–256. doi: 10.1016/j.resp.2004.03.003. [DOI] [PubMed] [Google Scholar]
- Cherniack NS, Edelman NH, Lahiri S. Hypoxia and hypercapniaas respiratory stimulants and depressants. Respir Physiol. 1970;11:113–126. doi: 10.1016/0034-5687(70)90107-6. [DOI] [PubMed] [Google Scholar]
- Clement ID, Bascom DA, Conway J, Dorrington KL, O'Connor DF, Painter R, Paterson DJ, Robbins PA. An assessment of central–peripheral ventilatory chemoreflex interaction in humans. Respir Physiol. 1992;88:87–100. doi: 10.1016/0034-5687(92)90031-q. [DOI] [PubMed] [Google Scholar]
- Clement ID, Pandit JJ, Bascom DA, Dorrington KL, O'Connor DF, Robbins PA. An assessment of central-peripheral ventilatory chemoreflex interaction using acid and bicarbonate infusions in humans. J Physiol. 1995;485(Pt 2):561–570. doi: 10.1113/jphysiol.1995.sp020752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cragg PA, Drysdale DB. Interaction of hypoxia and hypercapniaon ventilation, tidal volume and respiratory frequency in the anaesthetized rat. J Physiol. 1983;341:477–493. doi: 10.1113/jphysiol.1983.sp014818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings KJ, Frappell PB. Breath-to-breath hypercapnic response in neonatal rats: temperature dependency of the chemoreflexes and potential implications for breathing stability. Am J Physiol Regul Integr Comp Physiol. 2009;297:R124–R134. doi: 10.1152/ajpregu.91011.2008. [DOI] [PubMed] [Google Scholar]
- Cunningham DJ, O'Riordan JL. The effect of a rise in the temperature of the body on the respiratory response to carbon dioxide at rest. Q J Exp Physiol Cogn Med Sci. 1957;42:329–345. doi: 10.1113/expphysiol.1957.sp001278. [DOI] [PubMed] [Google Scholar]
- Cunningham DJC, Robbins PA, Wolff CB. Integration of respiratory response to changes in alveolar partial pressures in CO2 and O2 and in arterial pH. In: Cherniack NS, Widdicombe JC, editors. Handbook of Physiology Section 3: The Respiratory System. American Physiological Society; Bethesda, MD: 1986. pp. 475–528. [Google Scholar]
- da Silva GS, Giusti H, Benedetti M, Dias MB, Gargaglioni LH, Branco LG, Glass ML. Serotonergic neurons in the nucleus raphe obscurus contribute to interaction between central and peripheral ventilatory responses to hypercapnia. Pflug Arch. 2011;462:407–418. doi: 10.1007/s00424-011-0990-x. [DOI] [PubMed] [Google Scholar]
- Daristotle L, Berssenbrugge AD, Bisgard GE. Hypoxic-hypercapnic ventilatory interaction at the carotid body of awake goats. Respir Physiol. 1987;70:63–72. doi: 10.1016/s0034-5687(87)80032-4. [DOI] [PubMed] [Google Scholar]
- Day TA, Wilson RJ. Specific carotid body chemostimulation is sufficient to elicit phrenic poststimulus frequency decline in a novel in situ dual-perfused rat preparation. Am J Physiol Regul Integr Comp Physiol. 2005;289:R532–R544. doi: 10.1152/ajpregu.00812.2004. [DOI] [PubMed] [Google Scholar]
- Day TA, Wilson RJ. Brainstem PCO2 modulates phrenic responses to specific carotid body hypoxia in an in situ dual perfused rat preparation. J Physiol. 2007;578:843–857. doi: 10.1113/jphysiol.2006.119594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day TA, Wilson RJ. A negative interaction between brainstem and peripheral respiratory chemoreceptors modulates peripheral chemoreflex magnitude. J Physiol. 2009;587:883–896. doi: 10.1113/jphysiol.2008.160689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick TE, Coles SK. Ventrolateral pons mediates short-term depression of respiratory frequency after brief hypoxia. Respir Physiol. 2000;121:87–100. doi: 10.1016/s0034-5687(00)00121-3. [DOI] [PubMed] [Google Scholar]
- Eldridge FL, Gill-Kumar P, Millhorn DE. Input–output relationships of central neural circuits involved in respiration in cats. J Physiol. 1981;311:81–95. doi: 10.1113/jphysiol.1981.sp013574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezure K. Respiration-related afferents to parabrachial pontine regions. Respir Physiol Neurobiol. 2004;143:167–175. doi: 10.1016/j.resp.2004.03.017. [DOI] [PubMed] [Google Scholar]
- Fitzgerald RS, Parks DC. Effect of hypoxia on carotid chemoreceptor response to carbon dioxide in cats. Respir Physiol. 1971;12:218–229. doi: 10.1016/0034-5687(71)90054-5. [DOI] [PubMed] [Google Scholar]
- Frappell P, Lanthier C, Baudinette RV, Mortola JP. Metabolism and ventilation in acute hypoxia: a comparative analysis in small mammalian species. Am J Physiol. 1992;262:R1040–R1046. doi: 10.1152/ajpregu.1992.262.6.R1040. [DOI] [PubMed] [Google Scholar]
- Gesell R, Lapides J, Levin M. The interaction of central and peripheral chem ical control of breathing. Am J Physiol —Legacy Content. 1940;130:155–170. [Google Scholar]
- Giese K, Berndt J, Berger W. Interaction of central and peripheral respiratory drives in cats II. Peripheral and central interaction of hypoxia and hypercapnia. Pflug Arch. 1978;374:211–217. doi: 10.1007/BF00585597. [DOI] [PubMed] [Google Scholar]
- Hayashi F, Yoshida A, Fukuda Y, Honda Y. The ventilatory response to hypoxia in the anesthetized rat. Pflug Arch. 1983;396:121–127. doi: 10.1007/BF00615516. [DOI] [PubMed] [Google Scholar]
- Heeringa J, Berkenbosch A, de Goede J, Olievier CN. Relative contribution of central and peripheral chemoreceptors to the ventilatory response to CO2 during hyperoxia. Respir Physiol. 1979;37:365–379. doi: 10.1016/0034-5687(79)90082-3. [DOI] [PubMed] [Google Scholar]
- Honda Y, Hata N, Sakakibara Y, Nishino T, Satomura Y. Central hypoxic-hypercapnic interaction in mild hypoxia in man. Pflug Arch. 1981;391:289–295. doi: 10.1007/BF00581509. [DOI] [PubMed] [Google Scholar]
- Kiley JP, Eldridge FL, Millhorn DE. The effect of hypothermia on central neural control of respiration. Respir Physiol. 1984;58:295–312. doi: 10.1016/0034-5687(84)90006-9. [DOI] [PubMed] [Google Scholar]
- Kiwull-Schone H, Kiwull P. The role of the vagus nerves in the ventilatory response to lowered PaO2 with intact and eliminated carotid chemoreflexes. Pflug Arch. 1979;381:1–9. doi: 10.1007/BF00582324. [DOI] [PubMed] [Google Scholar]
- Kontos HA, Mauck HP, Jr, Richardson DW, Patterson JL., Jr Mechanism of circulatory responses to systemic hypoxia in the anesthetized dog. Am J Physiol. 1965;209:397–403. doi: 10.1152/ajplegacy.1965.209.2.397. [DOI] [PubMed] [Google Scholar]
- Kubin L, Alheid GF, Zuperku EJ, McCrimmon DR. Central pathways of pulmonary and lower airway vagal afferents. J Appl Physiol. 2006;101:618–627. doi: 10.1152/japplphysiol.00252.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P. Peripheral chemoreceptors in mammals: structure, function and transduction. In: Glass M, Wood S, editors. Cardio-RespiratoryControl inVerte brates. Springer-Verlag; Berlin: 2009. pp. 451–473. [Google Scholar]
- Lahiri S, DeLaney RG. Stimulus interaction in the responses of carotid body chemoreceptor single afferent fibers. Respir Physiol. 1975;24:249–266. doi: 10.1016/0034-5687(75)90017-1. [DOI] [PubMed] [Google Scholar]
- Loeschcke HH, Mitchell RA, Katsaros B, Perkins JF, Konig A. Interaction of intracranial chemosensitivity with peripheral afferents to the respiratory centers. Ann N Y Acad Sci. 1963;109:651–660. doi: 10.1111/j.1749-6632.1963.tb13495.x. [DOI] [PubMed] [Google Scholar]
- Mizusawa A, Ogawa H, Kikuchi Y, Hida W, Shirato K. Role of the parabrachial nucleus in ventilatory responses of awake rats. J Physiol. 1995;489(Pt 3):877–884. doi: 10.1113/jphysiol.1995.sp021100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohan R, Duffin J. The effect of hypoxia on the ventilatory response to carbon dioxide in man. Respir Physiol. 1997;108:101–115. doi: 10.1016/s0034-5687(97)00024-8. [DOI] [PubMed] [Google Scholar]
- Moreira TS, Takakura AC, Colombari E, West GH, Guyenet PG. Inhibitory input from slowly adapting lung stretch receptors to retrotrapezoid nucleus chemoreceptors. J Physiol. 2007;580:285–300. doi: 10.1113/jphysiol.2006.125336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou LC, Miller MJ, Tenney SM. Hypoxia and carbon dioxide as separate and interactive depressants of ventilation. Respir Physiol. 1976;28:347–358. doi: 10.1016/0034-5687(76)90029-3. [DOI] [PubMed] [Google Scholar]
- Parisi RA, Edelman NH, Santiago TV. Central respiratory carbon diox ide chemosensitivity does not decrease during sleep. Am Rev Respir Dis. 1992;145:832–836. doi: 10.1164/ajrccm/145.4_Pt_1.832. [DOI] [PubMed] [Google Scholar]
- Pepper DR, Landauer RC, Kumar P. Postnatal development of CO2–O2 interaction in the rat carotid body in vitro. J Physiol. 1995;485(Pt 2):531–541. doi: 10.1113/jphysiol.1995.sp020749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillipson EA, Bowes G. Control of breathing during sleep. In: Cherniack NS, Widdicombe JG, editors. Handbook of Physiology Section 3: The Respiratory System Am Physiol Soc. Bethesda, MD: 1987. pp. 649–689. [Google Scholar]
- Poon CS. Organization of central pathways mediating the Hering-Breuer reflex and carotid chemoreflex. Adv Exp Med Biol. 2004;551:95–100. doi: 10.1007/0-387-27023-x_15. [DOI] [PubMed] [Google Scholar]
- Poon CS, Siniaia MS. Plasticity of cardiorespiratory neural processing: classification and computational functions. Respir Physiol. 2000;122:83–109. doi: 10.1016/s0034-5687(00)00152-3. [DOI] [PubMed] [Google Scholar]
- Richardson PS, Widdicombe JG. The role of the vagus nerves in the ventilatory responses to hypercapnia and hypoxia in anaesthetized and unanaesthetized rabbits. Respir Physiol. 1969;7:122–135. doi: 10.1016/0034-5687(69)90073-5. [DOI] [PubMed] [Google Scholar]
- Robbins PA. Evidence for interaction between the contributions to ventilation from the central and peripheral chemoreceptors in man. J Physiol. 1988;401:503–518. doi: 10.1113/jphysiol.1988.sp017175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts CA, Corfield DR, Murphy K, Calder NA, Hanson MA, Adams L, Guz A. Modulation by “central” PCO2 of the response to carotid body stimulation in man. Respir Physiol. 1995;102:149–161. doi: 10.1016/0034-5687(95)00067-4. [DOI] [PubMed] [Google Scholar]
- Roy A, Rozanov C, Mokashi A, Lahiri S. PO2-PCO2 stimulus interaction in [ca2+]i and CSN activity in the adult rat carotid body. Respir Physiol. 2000;122:15–26. doi: 10.1016/s0034-5687(00)00116-x. [DOI] [PubMed] [Google Scholar]
- Smith CA, Jameson LC, Mitchell GS, Musch TI, Dempsey JA. Central-peripheral chemoreceptor interaction in awake cerebrospinal fluid-perfused goats. J Appl Physiol. 1984;56:1541–1549. doi: 10.1152/jappl.1984.56.6.1541. [DOI] [PubMed] [Google Scholar]
- Song G, Poon CS. Functional and structural models of pontine modulation of mechanoreceptor and chemoreceptor reflexes. Respir Physiol Neurobiol. 2004;143:281–292. doi: 10.1016/j.resp.2004.05.009. [DOI] [PubMed] [Google Scholar]
- Song G, Poon CS. Lateral parabrachial nucleus mediates shortening of expiration and increase of inspiratory drive during hypercapnia. Respir Physiol Neurobiol. 2009a;165:9–12. doi: 10.1016/j.resp.2008.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song G, Poon CS. Lateral parabrachial nucleus mediates shortening of expiration during hypoxia. Respir Physiol Neurobiol. 2009b;165:1–8. doi: 10.1016/j.resp.2008.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song G, Xu H, Wang H, Macdonald SM, Poon CS. Hypoxia-excited neurons in NTS send axonal projections to Kolliker–Fuse/parabrachial complex in dorsolateral pons. Neuroscience. 2011;175:145–153. doi: 10.1016/j.neuroscience.2010.11.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St John W. Differing responses to hypercapnia and hypoxia following pneumotaxic centerablation. Respir Physiol. 1975;23:1–9. doi: 10.1016/0034-5687(75)90066-3. [DOI] [PubMed] [Google Scholar]
- StCroix CM, Cunningham DA, Paterson DH. Nature of the interaction between central and peripheral chemoreceptor drives in human subjects. Can J Physiol Pharm. 1996;74:640–646. doi: 10.1139/cjpp-74-6-640. [DOI] [PubMed] [Google Scholar]
- Swanson GD, Bellville JW. Hypoxic–hypercapnic interaction in human respiratory control. J Appl Physiol. 1974;36:480–487. doi: 10.1152/jappl.1974.36.4.480. [DOI] [PubMed] [Google Scholar]
- Takakura AC, Moreira TS, Colombari E, West GH, Stornetta RL, Guyenet PG. Peripheral chemoreceptor inputs to retrotrapezoid nucleus (RTN) CO2-sensitive neurons in rats. J Physiol. 2006;572:503–523. doi: 10.1113/jphysiol.2005.103788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenney SM, Brooks JG., 3rd Carotid bodies, stimulus interaction, and ventilatory control in unanesthetized goats. Respir Physiol. 1966;1:211–224. doi: 10.1016/0034-5687(66)90018-1. [DOI] [PubMed] [Google Scholar]
- Teppema LJ, van Dorp EL, Dahan A. Arterial [H+] and the ventilatory response to hypoxia in humans: influence of acetazolamide-induced metabolic acidosis. Am J Physiol Lung Cell Mol Physiol. 2010;298:L89–L95. doi: 10.1152/ajplung.00255.2009. [DOI] [PubMed] [Google Scholar]
- Tonkovic-Capin M, Zuperku EJ, Stuth EA, Bajic J, Dogas Z, Hopp FA. Effect of central CO2 drive on lung inflation responses of expiratory bulbospinal neurons in dogs. Am J Physiol Regul Integr Comp Physiol. 2000;279:R1606–R1618. doi: 10.1152/ajpregu.2000.279.5.R1606. [DOI] [PubMed] [Google Scholar]
- van Beek JH, Berkenbosch A, de Goede J, Olievier CN. Influence of peripheral O2 tension on the ventilatory response to CO2 in cats. Respir Physiol. 1983;51:379–390. doi: 10.1016/0034-5687(83)90030-0. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Kumar P, Hanson MA. Effect of ambient temperature on respiratory chemoreflex in unanaesthetized kittens. Respir Physiol. 1996;106:239–246. doi: 10.1016/s0034-5687(96)00076-x. [DOI] [PubMed] [Google Scholar]
- Young DL, Eldridge FL, Poon CS. Integration-differentiation and gating of carotid afferent traffic that shapes the respiratory pattern. J Appl Physiol. 2003;94:1213–1229. doi: 10.1152/japplphysiol.00639.2002. [DOI] [PubMed] [Google Scholar]