

Abstract
Reduced representation bisulfite sequencing (RRBS), which couples bisulfite conversion and next generation sequencing, is an innovative method that specifically enriches genomic regions with a high density of potential methylation sites and enables investigation of DNA methylation at single-nucleotide resolution. Recent advances in the Illumina DNA sample preparation protocol and sequencing technology have vastly improved sequencing throughput capacity. Although the new Illumina technology is now widely used, the unique challenges associated with multiplexed RRBS libraries on this platform have not been previously described. We have made modifications to the RRBS library preparation protocol to sequence multiplexed libraries on a single flow cell lane of the Illumina HiSeq 2000. Furthermore, our analysis incorporates a bioinformatics pipeline specifically designed to process bisulfite-converted sequencing reads and evaluate the output and quality of the sequencing data generated from the multiplexed libraries. We obtained an average of 42 million paired-end reads per sample for each flow-cell lane, with a high unique mapping efficiency to the reference human genome. Here we provide a roadmap of modifications, strategies, and trouble shooting approaches we implemented to optimize sequencing of multiplexed libraries on an a RRBS background.
1. Introduction
The emergence of next generation sequencing (NGS) in recent years has revolutionized genetic researchers' ability to interrogate genomes on an unprecedented scale. The sequencing-by-synthesis NGS technologies are evolving rapidly and throughput capacity is increasing exponentially [1, 2]. The Illumina sequencing platform, on which we focus here, involves clonal amplification of adaptor-ligated fragments of genomic DNA that are sequenced using reversible terminator-based chemistry. NGS has a broad scope of applications including de novo genome assembly, transcriptomics, SNP discovery and clinical diagnostics [3–5]. Of particular interest is the potential that NGS has for the field of epigenetics, specifically genome-wide DNA methylation analysis. Catalyzed by DNA methyltransferases (DNMTs), DNA methylation involves the covalent addition of a methyl group (CH3) to the cytosine (C) to make 5-methylcytosine (5mC) [6]. In mammals, this stable modification almost exclusively occurs in the context of CpG dinucleotides [7, 8]. DNA methylation has been associated with gene silencing, tissue differentiation, genomic imprinting, X chromosome inactivation, phenotypic variation, and possibly disease susceptibility [9–13]. Aberrant DNA methylation is implicated in several diseases and has a well-established role in tumorigenesis [14].
Recently, several NGS-based methods have been developed to profile the DNA methylation status of a genome. Among them, reduced representation bisulfite sequencing (RRBS) can be used to generate comprehensive methylomes [15]. Compared to whole genome methylation sequencing, RRBS provides an alternative sequencing technology at a reduced cost [16] and it is used by many groups worldwide [17–22]. By representing a small fraction of the genome (~2.5% of the human genome), it reduces the amount of sequencing required, while enriching for promoter associated CG-rich regions. The method involves bisulfite conversion, which converts unmethylated cytosines to uracil, while leaving methylated cytosines unchanged, thereby generating base-pair resolution DNA methylation profiles.
Alignment of bisulfite-converted sequence reads to a large reference genome brings computational challenges. A thymine (T) in the sequenced read could reflect either an unmethylated C or a T, but not vice versa. Therefore, the asymmetric mapping increases the chances of false-positive matches and the search space for mapping [23]. Nevertheless, several tools and pipelines have been developed that enable molecular biologists to analyze the large volume of methylation data with increasing ease [16, 24, 25].
Recent advances in the Illumina sequencing platform (see Table S1 in Supplementary Material available online at doi: 10.1155/2012/741542) have increased the capacity from 20–30 million reads per lane (Genome Analyzer/GAII) to ~200 million reads per lane (HiSeq 2000). In the case of smaller or reduced representation genomes, such a high number of reads provides ample coverage. Indeed, multiplexed sequencing of these smaller or reduced representation genomes permits considerable cost savings. DNA libraries for multiplexed sequencing are prepared by ligating adaptors containing different 6 bp index sequences to each fragmented DNA sample (Supplementary Figure 1). This allows multiple samples to be combined into a single sequencing reaction and then individually identified and demultiplexed during base-calling analysis.
Meissner's group has previously described the RRBS protocol in several articles [15, 20, 26, 27]. In each of these articles, the protocol was described for the Illumina Genome Analyzer sequencer, where one RRBS library was sequenced per lane. However, due to the increased capacity of the new HiSeq 2000 sequencer (almost 8-fold increase in terms of read numbers), it became inevitable to sequence multiple RRBS libraries in one single flow-cell lane. The library preparation method for multiplexed runs and the sequencing pipeline significantly differ from the described Genome Analyzer workflow. Here we describe the required modifications of the original protocol and the strategies we employed for successful sequencing. Specifically, we demonstrate improved strategies for bisulfite conversion, library purification, and PCR amplification.
Further, the previous protocols focused only on library preparation methods, but the downstream data processing and bioinformatics were not described. We show the steps and challenges involved in base calling of multiplexed RRBS libraries due to its unique base composition. We describe a pipeline to evaluate and improve the quality of the data obtained and increase the output from indexed sequencing runs. Additionally, we comment on the different data formats generated by different versions of the sequencing chemistry and the sequence alignments with high mapping efficiency, which demonstrates the effectiveness of the method described. This paper provides a complete workflow starting from library preparation to base calling and to successful mapping of the obtained library.
2. Methods
Previous papers have described in detail the process of generating reduced representation libraries for methylation analysis [15, 26, 27]. Here, we briefly describe the protocol with emphasis on the modifications required to make successful multiplexed libraries for sequencing. Key differences between the previously used paired-end library preparation protocol and the TruSeq (version 2) multiplexing protocol are outlined in Supplementary Table S2.
2.1. Library Preparation
Although the TruSeq protocol recommends 1 μg of input DNA for normal genomic DNA library preparation, we found that for RRBS this amount was not sufficient. For RRBS, 2.5 μg of genomic DNA was digested overnight with MspI (New England Biolabs, Ipswich, MA) using 20 units of enzyme per μg of DNA to ensure complete digestion. Digested DNA was purified on a QIAquick spin column (Qiagen, Hilden, Germany). Then, end repair and addition of 3′A overhangs were performed using the TruSeq DNA kit (Illumina, San Diego, CA). Indexed TruSeq adaptors were ligated according to the manufacturer's protocol and purified with AMPure beads (Agencourt Bioscience, Beverly, MA). For detailed methodology of multiplexed adaptor ligation, see Supplementary Table S3. DNA fragments ranging from 160 to 340 bp (40–220 bp of DNA plus 120 bp of adaptors) were excised from a 3% (w/v) NuSieve GTG agarose gel (Lonza, Basel, Switzerland) and purified using the QIAquick gel extraction protocol.
2.2. Bisulfite Conversion
Previously published methods recommend two rounds of bisulfite conversion with the Qiagen EpiTect kit for complete bisulfite conversion of human RRBS libraries [27]. However, we found that two rounds of conversion and purification resulted in significant loss of the template in the bisulfite-converted library leading to a requirement for high cycle number for library amplification. Also, 5-methylcytosine can undergo deamination, at a slow rate, during prolonged bisulfite treatment [28]. Based on a small number of samples, we achieved more consistent bisulfite conversion of size-selected libraries using the EZ DNA methylation kit (Zymo Research, Irvine, CA). Although the manufacturer's instructions are to incubate DNA samples with bisulfite reagent for 12–16 hours at 50°C, we incubated them for 18–20 hours. This longer incubation period was associated with consistent conversion of human genomic DNA and minimal loss during the process.
2.3. Amplification
The previously described paired-end protocol used the forward and reverse primers P.E. 1.0 and P.E. 2.0 to amplify libraries [27]. The TruSeq protocol has streamlined the library amplification workflow by providing two master-mixed reagents, a PCR master mix and a PCR primer cocktail. However, we found that in contrast to a normal genomic DNA library used as a positive control, this protocol did not amplify bisulfite-converted libraries within 20 or 30 cycles of PCR. We optimized the library amplification step by incorporating the TruSeq primer cocktail into the PCR protocol used by Smith et al. [26]. For amplification of the bisulfite-converted DNA, we used PfuTurbo Cx DNA polymerase (Stratagene, La Jolla, CA). It has been proposed that a high frequency of uracils in bisulfite-converted DNA results in uracil stalling by the DNA polymerase [26]. PfuTurbo efficiently reads through uracils in the template strand, but its enhanced proofreading activity prevents PCR-induced point mutations. The final libraries (1.44 μL of bisulfite-converted DNA per 12 μL PCR reaction) were amplified using 1.45 U PfuTurbo Cx DNA polymerase, 0.3 mM dNTP stock, and 1.44 μL TruSeq PCR primer cocktail (see Supplementary Table S4) with the following thermocycler conditions: 95°C for 2 min, n × (95°C for 30s, 65°C for 30 s, and 72°C for 45 s), 72°C for 7 min. Initially, analytical PCR reactions were performed to determine the optimal number of cycles for amplification of libraries (a higher number of cycles can induce amplification bias). Libraries were amplified with 15–18 cycles of amplification and then visualized on a 4–20% Criterion gradient polyacrylamide TBE gel (BioRad Laboratories, Hercules, CA) stained with SYBR green nucleic acid gel stain (Life Technologies, Grand Island, NY).
2.4. Assessment of of Library Quality
To verify fragment size and quality of the amplified libraries, individual aliquots were run on a 2100 Bioanalyzer (Supplementary Figure S2) using a high-sensitivity DNA chip (Agilent Technologies, Santa Clara, CA). Libraries were quantified using a Qubit 2.0 fluorometer (Life Technologies, Grand Island, NY).
2.5. Sequencing
Libraries were pooled at equimolar concentrations based on Qubit measurements and sequenced on a single flow cell lane of an Illumina HiSeq 2000 sequencer with a paired end, 100 bp run. The initial forward read is termed Read1 and the reverse read is termed Read2. 5% phiX genomic DNA (control) was spiked into the lane. In between these 2 reads, a 6-cycle read is made of the adaptor index sequence to allow for demultiplexing of the pooled samples.
2.6. Base Calling
Illumina base calling is usually performed by the real-time analyzer (RTA) running on the sequencer, but in some cases, as described in Section 3, it was necessary to repeat the base-calling step afterwards. The Illumina Off line Basecaller (OLB) v 1.9.4, running on a Redhat Linux server, was used for this. The application applies the same algorithm as the original RTA application, using the compressed image files generated by the machine, but allows a wider range of processing options to be employed [29].
2.7. Generating FASTQ File
FASTQ sequence file generation from the raw base-call data was performed using Illumina's CASAVA v1.8.2 package [30]. CASAVA scripts use the details of each sample, including its lane and index sequence, to identify the contents of each flow-cell lane and to partition multiplexed reads into index-specific FASTQ files. The amplification steps used in flow cell preparation generate a series of clusters of identical sequences at a specific pixel location on the flow-cell surface. Since the position is conserved through the reads (Read1, Read2, and the 6 bp index read), individual reads are identified by the pixel coordinates of the cluster on the flow-cell surface. CASAVA permits various processing options but, for the work described here, the output becomes a directory for each index containing a single gzip-compressed FASTQ file for each of the forward and reverse reads of a paired-end run. The logistics of running a flow cell require that the paired-end chemistry is applied to all samples if any on the cell needs that processing; CASAVA can be directed to ignore the reverse read or the extra data can be disregarded if not required. In the event that CASAVA finds reads for which the index sequence cannot be identified, through sequencing errors, the reads are saved in to a directory of “undetermined indices.”
2.8. Data Processing
Quality evaluation of the sequenced reads, filtering, processing and alignment of each dataset have been performed as previously published [24]. Briefly, the quality of the reads was checked using the FASTQC (version 0.10) program and then adaptor contamination was removed using cleanadaptors [24]. The alignment was performed using Bismark v0.6.4 [25] against the GRCh37.65 build of the human genome. The key aspects of the data processing are described in Section 3.
3. Results and Discussion
We have optimized the RRBS library preparation protocol and typically sequenced five multiplexed libraries on a single flow-cell lane of the Illumina HiSeq 2000. We obtain an average of 42 million paired-end reads per sample, with a high unique mapping efficiency to the reference human genome. Here we discuss the critical aspects of the DNA sample preparation protocol and data processing and comment on relevant troubleshooting approaches on an RRBS background.
The TruSeq protocol recommends the use of AMPure beads for purification of DNA fragments. However, this method is optimized for fragments >100 bp length. Consequently fragments shorter than 100 bp will be lost during the purification process, resulting in a much lower coverage of the genome (see Supplementary Figure S3). For the RRBS protocol, selection of 40–220 bp fragments (preligation) is necessary and therefore, prior to adaptor ligation, we recommend that DNA is purified using columns designed to retain DNA fragments within this size range. After the adaptors are ligated the fragment sizes are modified to 160–340 bp in length.
TruSeq adaptor ligation at a 2.5 : 1 (adaptor: DNA) ratio resulted in a distinct band at 125 bp in all our libraries following PCR amplification (Figure 1), which we had not observed in library preparations using the paired-end sample preparation kit that preceded the TruSeq protocol. Since the band is smaller than the DNA fragments excised from the gel, we concluded this band was a product of PCR amplification. Although we suspected the 125 bp band was due to adaptor-adaptor dimerization, reducing the adaptor, DNA ratio did not eliminate these bands. However, we successfully bypassed this issue by performing a second round of gel size selection following PCR amplification, removing the dimer from the final library.
Figure 1.
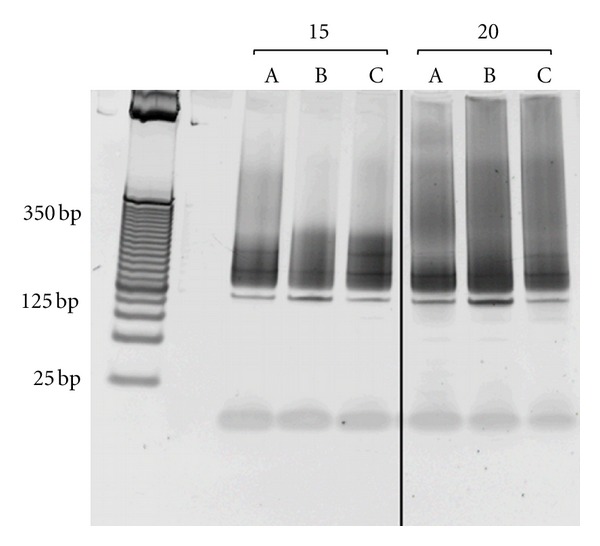
Representative analytical PCR of size-selected RRBS libraries.The 160–340 bp size-selected RRBS libraries (represented by a, b, and c) were amplified with either 15 or 20 cycles of PCR to determine the optimal number of cycles for large-scale amplification. PCR products were visualized on a 4–20% Criterion gradient polyacrylamide TBE gel stained with SYBR green nucleic acid gel stain alongside a 25 bp DNA ladder. For these libraries, 13 (a) and 14 (b and c) cycles were chosen for large-scale amplification. The distinct band at 125 bp in all libraries was possibly due to adaptor-adaptor dimerization.
In addition to this residual adaptor contamination, sequencing of size-selected RRBS libraries (160–340 bp) can result in adaptor sequence reads at the 3′ end of the shorter fragments. These adaptor reads will interfere with alignment and possibly contribute to misalignment events and false methylation calls. We recommend removal of potential adaptor contamination by in silico tools (e.g., cleanadaptors) and assessment of the quality of the sequenced reads prior to mapping and further analysis [24].
RRBS libraries are generated after digestion with the methylation insensitive enzyme MspI. Due to the directionality of the Illumina platform and the protocol used, we obtained reads exclusively from MspI digested 5′CGG strands (the recognition motif of MspI is C′CGG). The result of this protocol is that our reads will always start with CGG (when the first C is methylated) or TGG (when the first C is unmethylated and is converted to T after bisulfite conversion and PCR). This nonrandom base composition is a unique property of RRBS libraries, which significantly differs from normal genomic libraries. The Illumina RTA algorithm uses the first 4 sequencing cycles of Read1 to set internal standards for fluorescence throughout the run, a system based on the assumption that a relatively random distribution of bases will be found in at least two of those cycles. We were concerned that MspI fragmentation and the bisulfite chemistry together might confound this scheme as each of our fragment begins with CGG or TGG and therefore cause suboptimal standardization for the run, potentially leading to less accurate base calling.
The base composition features for our libraries are illustrated in Figure 2, which shows the intensities for each base channel over all machine cycles for two lanes: 2A (lane 4) contains regular multiplexed human genomic libraries and 2B (lane 8) contains our multiplexed bisulfite-converted libraries (note that the spikes present at 100 cycles in both lanes correspond to the 6 bp index sequence read). The biased frequencies of C and T in the RRBS samples are evident in Read1, as are the corresponding A and G biases in Read2. This skewed base composition is a specific feature of RRBS libraries as the other non-RRBS multiplexed genomic libraries have a normal distribution of all four bases (Figure 2(a)).
Figure 2.
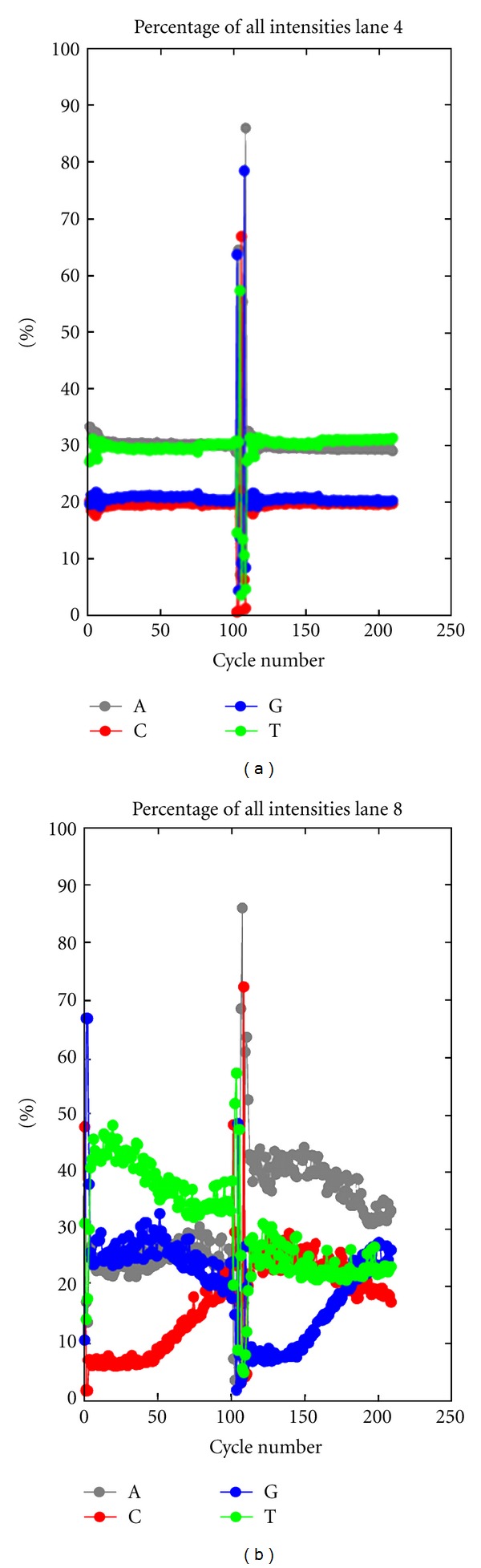
Comparison of base-calling between regular genomic libraries and RRBS libraries. Relative intensities for each base channel are shown across the 200 cycles of a 100 bp paired-end HiSeq 2000 sequencing run. A. Regular multiplexed human genomic libraries, lane 4. B. Multiplexed RRBS libraries, lane 8.
In order to counteract the possible bias one could include an internal standard (genomic DNA that can be expected to have random base composition in the first 4 cycles) at a sufficiently high level in the same lane or, as a subsequent operation, to set the standards from a lane containing clusters that are expected to be random. Although the latter operation is an option of the RTA system, we did not perform this step since it is not necessarily obvious in advance which other lanes might contain appropriately random samples or generate good-quality sequence data. For that reason, postrun standardization using a designated standard lane with the Illumina off-line base-calling application (OLB) was performed. OLB applies the same base-calling algorithm as RTA. We used the OLB program to repeat base calling using intensity data derived from genomic DNA (Figure 2(a)) as the standard. Detailed comparisons of base calling performed by RTA and OLB for each sample are illustrated in Table 1. For Read1, OLB showed an increase, ranging from 1.8% to 3.5%, in the proportion of sequence retained after quality trimming to a Phred score of 30 (fastq_quality_trimmer -t 30) compared to RTA. In contrast, Read2 showed more variable results with some libraries giving a decrease in retained bases: the difference ranged from −1.6% to 0.9%. Further, OLB showed an increase in the number of reads returned by CASAVA for both Read1 and Read2 for all RRBS libraries (Table 1). We then checked the quality of the sequencing reads generated by RTA and OLB with FASTQC (version 0.10) and found that the average Phred score value is marginally higher for the reads derived from OLB than RTA (Figure 3).
Table 1.
Comparison between base calling performed by RTA (real-time analyzer) and OLB (off-line basecaller) of multiplexed samples.
| Sample ID | Number of bases after RTA | Number of bases after OLB | Percentage of change (RTA versus OLB) | Number of reads after RTA | Number of reads after OLB | Percentage of change (RTA versus OLB) |
|---|---|---|---|---|---|---|
| Read1 | ||||||
| 1 | 1520286198 | 1548285294 | 1.8 | 16377421 | 16503971 | 0.8 |
| 2 | 2282620124 | 2350814836 | 3.0 | 24702835 | 25201462 | 2.0 |
| 3 | 2753391480 | 2846632920 | 3.4 | 30043201 | 30847092 | 1.7 |
| 4 | 1280388372 | 1325584268 | 3.5 | 13754015 | 14152462 | 2.9 |
| 5 | 1837282806 | 1881566740 | 2.4 | 20849236 | 21178885 | 1.6 |
|
| ||||||
| Read2 | ||||||
| 1 | 1512562937 | 1503732114 | −0.7 | 16377421 | 16503971 | 0.8 |
| 2 | 2214536843 | 2212529249 | −0.1 | 24702835 | 25200712 | 2.0 |
| 3 | 2621636705 | 2631071280 | 0.4 | 30043201 | 30847092 | 2.7 |
| 4 | 1265479416 | 1276840465 | 0.9 | 13754015 | 14152462 | 2.9 |
| 5 | 1516530411 | 1492797800 | −1.6 | 20849236 | 21178885 | 1.6 |
Figure 3.
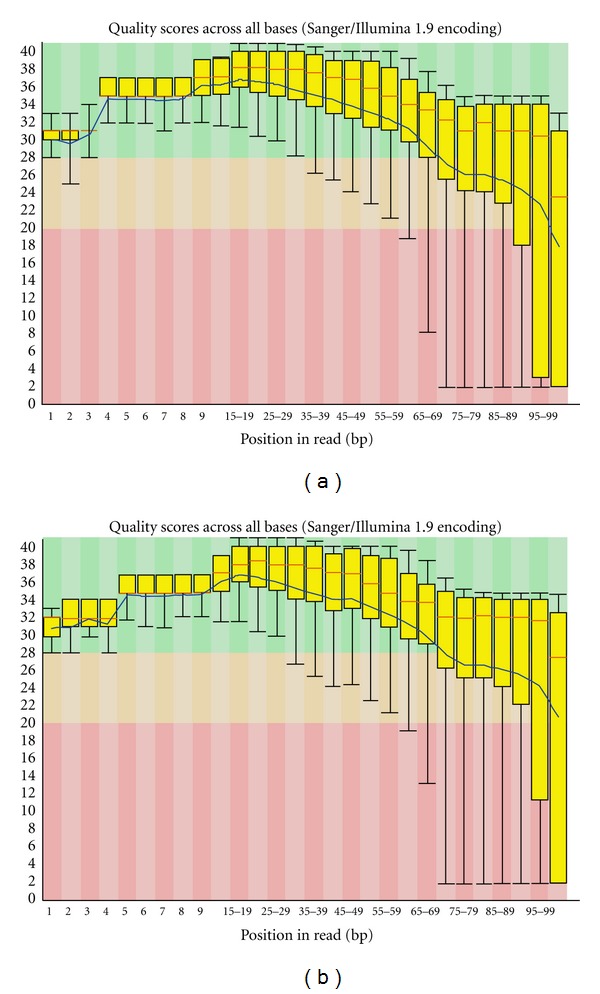
Per base sequence quality of sample 2 as generated by FASTQC for the dataset obtained from RTA base calling (a) and OLB base calling (b). The yellow box plots (red bar: median, box: interquartile ranges 25–75%, and whisker: 10–90% percentile) show the base-calling quality scores across all sequencing reads of sample 2. The blue line indicates the mean quality score. The other samples had similar per base sequence quality.
After base calling we obtained, for example, a total 55.3 Gb of sequence from one lane and 32.8, 49.4, 60.0, 27.5, and 41.7 million paired-end sequence reads for each of the five libraries, which ensures high coverage of CpG sites in the reduced representation genome (Table 2). The variation in read numbers led us to investigate the number of undetermined indices during base calling. We found that a total of 13.2 million reads had undetermined indices (for both Read1 and Read2), being 10.9% of the total reads obtained. The genomic DNA control lane used by OLB had a total of 5.5% undetermined reads, which could suggest that the RRBS library contributes to more misreads during the index sequence read cycles, perhaps through more mispriming. The extent to which index sequence misreading occurs with different libraries may vary and may contribute to variable numbers of reads generated for each. Moreover, accurate quantification of the libraries and pooling the different libraries in equimolar ratio could play an important role in multiplexed runs to achieve similar sequencing yields for each indexed sample.
Table 2.
Details of data generated for multiplexed RRBS libraries.
| Sample ID | Adaptor index ID |
Raw data including cif files (Gb)1 | After RTA base calling (Gb)1 | Sequence volume (Gb)2 | Uncompressed, 2 reads (Gb) | Paired-end reads (106) |
|---|---|---|---|---|---|---|
| 1 | 1 | 3.30 | 8.6 | 32.8 | ||
| 2 | 3 | 4.99 | 12.9 | 49.4 | ||
| 3 | 8 | 320 | 55.3 | 6.06 | 15.7 | 60.0 |
| 4 | 9 | 2.78 | 7.2 | 27.5 | ||
| 5 | 10 | 4.21 | 10.9 | 41.7 |
1RTA uses the cif files to perform the base calling and produce. bcl files; the samples are not demultiplexed at this stage.
2CASAVA performs the demultiplexing and uses the. bcl files to generate FASTQ files for each of the samples.
Previously, we described a pipeline for the efficient processing of RRBS data for methylation analysis [24]. Following a similar strategy, we preprocessed the data and aligned the reads against the complete human reference genome (NCBI GRCh37 build) using the bisulfite aligner Bismark [25] and contrasted the mapping performance of both RTA and OLB derived data (Table 3). We did not observe major differences in mapping efficiencies between the datasets, except decreased mapping for sample 3 on the OLB data. This analysis shows that it is worthwhile assessing the benefits of running OLB for each sequencing run for libraries that have significantly nonrandom base composition (e.g., RRBS) to ensure the quality of the data obtained. However, the choice of which dataset will be processed for further analysis will be based on the extent of improvement in the number of high-quality reads retained and the mapping efficiency. Our results suggest that comparing the performance of each sample (on OLB and RTA datasets) and then choosing the samples with higher quality and better mapping in combination from both datasets will maximize the data returned from an individual library after demultiplexing.
Table 3.
Comparison of mapping performance between RTA and OLB datasets.
| Sample ID | RTA | OLB | ||
|---|---|---|---|---|
| Unique mapping (%) | Uniquely aligned reads | Unique mapping (%) | Uniquely aligned reads | |
| 1 | 71.2 | 11513092 | 71.9 | 11681235 |
| 2 | 59.4 | 13998639 | 58.3 | 13945432 |
| 3 | 66.5 | 18785673 | 55.3 | 15940503 |
| 4 | 71.4 | 9603345 | 71.9 | 9907812 |
| 5 | 63.4 | 11296038 | 63.8 | 11508956 |
1The mapping runs were performed on a Mac Pro with 64 bit duo quad core Intel Xeon processors and with 22 Gb RAM running Mac OS 10.6. The samples were mapped using Bismark v0.6.4 against the GRCh37.65 build of the human genome.
Progressive refinements in the instruments and sequencing chemistry have extended the lengths of reads and these changes have been accompanied by updates to the analytical software. Consequently changes have been made in the header lines of the FASTQ output files: the tile numbering has been changed and the quality codes have switched from an Illumina-defined scheme to that of Sanger [31]. Examples of FASTQ files from two different generations of machines, chemistry, and software are shown in Supplementary Figure S4. While these changes may seem minor, they can pose issues with specific tools for further analysis of the data, for example, graphical quality checks according to tiles (SolexaQA [32], or the methylation analysis of CpG sites. Differences in header lines between different files can confound programs used in further data processing. Hence, it becomes necessary that appropriate changes are made in scripts that interface with programs or that adequate flexibility is written into programs or scripts in order that they work with the data generated from the different versions of the chemistry and software.
4. Conclusions
Although the original RRBS methodology is well described, multiplexing such libraries is relatively new and the unique challenges associated with it have not been previously described. It will become increasingly desirable to do multiplexed (indexed) runs in the future for reduced representation and smaller genomes as the coverage and output from the sequencing run is increasing rapidly. However, methylation sequencing is more complex compared to other methods, so modification of the standard protocol is necessary. We have illustrated a successful strategy to generate high-quality methylation data from multiplexed runs. The challenge will however remain for molecular biologists to analyze and interpret the data as the volume of the data will also increase exponentially and computational tools will need to be updated in parallel with the advances in sequencing platforms and chemistry.
Supplementary Material
Supplementary material contains- schematic of barcoded adaptor ligation to MspI mitigated fragments (Figure S1), Bioanalyzer quality trace of the RRBS library (Figure S2), Purification gel of smaller fragments (Figure S3), Example of different data format generated by various sequencer versions (Figure S4). Feature comparison of Illumina GAII/Genome Analyser and HiSeq2000 sequencers (Table S1), Comparison of overall library preparation workflow between Illumina Paired-End and TruSeq protocols (Table S2), Step-wise recipe of multiplexed adaptor ligation of RRBS libraries (Table S3) and Recipe of PCR amplification step of the indexed RRBS libraries (Table S4).
Authors' Contributions
A. Chatterjee and E. J. Rodger contributed equally to this work.
Conflict of Interests
The authors declare no competing interests.
Acknowledgments
The authors acknowledge the funding and support provided by the National Research Centre for Growth and Development (NRCGD) and the Health Research Council (HRC), New Zealand. A. Chatterjee is supported by a postgraduate scholarship from NRCGD. The authors are also grateful for the support from NZGL (New Zealand Genomics Limited) and Dr. Robert Day, Dr. Rebecca Laurie, and Les McNoe at the Department of Biochemistry, University of Otago.
Abbreviations
- RRBS:
Reduced representation bisulfite sequencing
- NGS:
Next generation sequencing
- RTA:
Real time analyzer
- OLB:
Off-line base-calling application.
References
- 1.Werner T. Next generation sequencing in functional genomics. Briefings in Bioinformatics. 2010;11(5):499–511. doi: 10.1093/bib/bbq018. [DOI] [PubMed] [Google Scholar]
- 2.Metzker ML. Sequencing technologies the next generation. Nature Reviews Genetics. 2010;11(1):31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- 3.Voelkerding KV, Dames SA, Durtschi JD. Next-generation sequencing: from basic research to diagnostics. Clinical Chemistry. 2009;55(4):641–658. doi: 10.1373/clinchem.2008.112789. [DOI] [PubMed] [Google Scholar]
- 4.Meaburn E, Schulz R. Next generation sequencing in epigenetics: insights and challenges. Seminars in Cell & Developmental Biology. 2011;23(2):192–199. doi: 10.1016/j.semcdb.2011.10.010. [DOI] [PubMed] [Google Scholar]
- 5.Ku CS, Naidoo N, Wu M, Soong R. Studying the epigenome using next generation sequencing. Journal of Medical Genetics. 2011;48:721–730. doi: 10.1136/jmedgenet-2011-100242. [DOI] [PubMed] [Google Scholar]
- 6.Bird AP. CpG-rich islands and the function of DNA methylation. Nature. 1986;321(6067):209–213. doi: 10.1038/321209a0. [DOI] [PubMed] [Google Scholar]
- 7.Bestor T, Laudano A, Mattaliano R, Ingram V. Cloning and sequencing of a cDNA encoding DNA methyltransferase of mouse cells. The carboxyl-terminal domain of the mammalian enzymes is related to bacterial restriction methyltransferases. Journal of Molecular Biology. 1988;203(4):971–983. doi: 10.1016/0022-2836(88)90122-2. [DOI] [PubMed] [Google Scholar]
- 8.Bestor TH. Activation of mammalian DNA methyltransferase by cleavage of a Zn binding regulatory domain. The EMBO Journal. 1992;11(7):2611–2617. doi: 10.1002/j.1460-2075.1992.tb05326.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carrel L, Willard HF. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature. 2005;434(7031):400–404. doi: 10.1038/nature03479. [DOI] [PubMed] [Google Scholar]
- 10.Rollins RA, Haghighi F, Edwards JR, et al. Large-scale structure of genomic methylation patterns. Genome Research. 2006;16(2):157–163. doi: 10.1101/gr.4362006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Suzuki MM, Bird A. DNA methylation landscapes: provocative insights from epigenomics. Nature Reviews Genetics. 2008;9(6):465–476. doi: 10.1038/nrg2341. [DOI] [PubMed] [Google Scholar]
- 12.Igarashi J, Muroi S, Kawashima H, et al. Quantitative analysis of human tissue-specific differences in methylation. Biochemical and Biophysical Research Communications. 2008;376(4):658–664. doi: 10.1016/j.bbrc.2008.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chatterjee A, Morison IM. Monozygotic twins: genes are not the destiny? Bioinformation. 2011;7(7):369–370. doi: 10.6026/97320630007369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baylin S, Bestor TH. Altered methylation patterns in cancer cell genomes: cause or consequence? Cancer Cell. 2002;1(4):299–305. doi: 10.1016/s1535-6108(02)00061-2. [DOI] [PubMed] [Google Scholar]
- 15.Meissner A, Mikkelsen TS, Gu H, et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 2008;454(7205):766–770. doi: 10.1038/nature07107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xi Y, Bock C, Muller F, et al. RRBSMAP: a fast, accurate and user-friendly alignment tool for reduced representation bisulfite sequencing. Bioinformatics. 2012;28(3):430–432. doi: 10.1093/bioinformatics/btr668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baranzini SE, Mudge J, Van Velkinburgh JC, et al. Genome, epigenome and RNA sequences of monozygotic twins discordant for multiple sclerosis. Nature. 2010;464(7293):1351–1356. doi: 10.1038/nature08990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bock C, Kiskinis E, Verstappen G, et al. Reference maps of human es and ips cell variation enable high-throughput characterization of pluripotent cell lines. Cell. 2011;144(3):439–452. doi: 10.1016/j.cell.2010.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gertz J, Varley KE, Reddy TE, et al. Analysis of DNA methylation in a three-generation family reveals widespread genetic influence on epigenetic regulation. PLoS Genetics. 2011;7(8) doi: 10.1371/journal.pgen.1002228.e1002228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gu H, Bock C, Mikkelsen TS, et al. Genome-scale DNA methylation mapping of clinical samples at single-nucleotide resolution. Nature Methods. 2010;7(2):133–136. doi: 10.1038/nmeth.1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smallwood SA, Tomizawa SI, Krueger F, et al. Dynamic CpG island methylation landscape in oocytes and preimplantation embryos. Nature Genetics. 2011;43(8):811–814. doi: 10.1038/ng.864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Steine EJ, Ehrich M, Bell GW, et al. Genes methylated by DNA methyltransferase 3b are similar in mouse intestine and human colon cancer. The Journal of Clinical Investigation. 2011;121(5):1748–1752. doi: 10.1172/JCI43169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xi Y, Li W. BSMAP: whole genome bisulfite sequence MAPping program. BMC Bioinformatics. 2009;10, article 232 doi: 10.1186/1471-2105-10-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chatterjee A, Stockwell PA, Rodger EJ, et al. Comparison of alignment software for genome-wide bisulphite sequence data. Nucleic Acids Research. 2012;40(10, article e79) doi: 10.1093/nar/gks150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 2011;27(11):1571–1572. doi: 10.1093/bioinformatics/btr167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smith ZD, Gu H, Bock C, Gnirke A, Meissner A. High-throughput bisulfite sequencing in mammalian genomes. Methods. 2009;48(3):226–232. doi: 10.1016/j.ymeth.2009.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gu H, Smith ZD, Bock C, Boyle P, Gnirke A, Meissner A. Preparation of reduced representation bisulfite sequencing libraries for genome-scale DNA methylation profiling. Nature Protocols. 2011;6(4):468–481. doi: 10.1038/nprot.2010.190. [DOI] [PubMed] [Google Scholar]
- 28.Warnecke PM, Stirzaker C, Song J, Grunau C, Melki JR, Clark SJ. Identification and resolution of artifacts in bisulfite sequencing. Methods. 2002;27(2):101–107. doi: 10.1016/s1046-2023(02)00060-9. [DOI] [PubMed] [Google Scholar]
- 29.Off-Line Basecaller V1. 9 User Guide. Illumina; 2010. [Google Scholar]
- 30.Wright CS, Alden RA, Kraut J. Structure of subtilisin BPN′ at 2.5 Å resolution. Nature. 1969;221(5177):235–242. doi: 10.1038/221235a0. [DOI] [PubMed] [Google Scholar]
- 31.Cock PJA, Fields CJ, Goto N, Heuer ML, Rice PM. The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants. Nucleic Acids Research. 2009;38(6):1767–1771. doi: 10.1093/nar/gkp1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cox MP, Peterson DA, Biggs PJ. SolexaQA: at-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinformatics. 2010;11, article 485 doi: 10.1186/1471-2105-11-485. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material contains- schematic of barcoded adaptor ligation to MspI mitigated fragments (Figure S1), Bioanalyzer quality trace of the RRBS library (Figure S2), Purification gel of smaller fragments (Figure S3), Example of different data format generated by various sequencer versions (Figure S4). Feature comparison of Illumina GAII/Genome Analyser and HiSeq2000 sequencers (Table S1), Comparison of overall library preparation workflow between Illumina Paired-End and TruSeq protocols (Table S2), Step-wise recipe of multiplexed adaptor ligation of RRBS libraries (Table S3) and Recipe of PCR amplification step of the indexed RRBS libraries (Table S4).