

Abstract
Hypertrophic cardiomyopathy (HCM) is the most common heritable cardiovascular disease. A recent study showed that male KLF10-encoded TGFβ Inducible Early Gene-1 knock-out mice (TIEG-/-) develop HCM with 13-fold up-regulation of PTTG1-encoded pituitary tumor-transforming gene 1. We hypothesized TIEG1 could be a novel candidate gene in the pathogenesis of genotype negative HCM in humans, possibly through a loss of its repression on PTTG1 expression. A cohort of 923 unrelated patients from two independent HCM centers was analyzed for mutations in TIEG's 4 translated exons using DHPLC and direct DNA-sequencing. Site directed mutagenesis was performed to clone novel variants. The effect of TIEG1 mutations on SMAD7 and PTTG1 promoters was studied using transient transfection and luciferase-assays. Altered expression of PTTG1 in cardiac tissue was studied by immunohistochemistry (IHC) to determine levels of PTTG1 protein in hypertrophic diseases. Six novel TIEG1 missense mutations were discovered in 6 patients (2 males/4 females, mean age at diagnosis 56.2 ± 23 years, MLVWT 20.8 ± 4 mm). Compared to WT TIEG1, 5 TIEG1 mutants significantly increased PTTG1 promoter function similar to TIEG1-/--mice. By IHC, PTTG1-protein expression was significantly increased in multiple models of hypertrophic cardiac disease, including TIEG1-mutation positive HCM compared to normal hearts. This is the first paper to associate mutations in TIEG1 to human disease with the discovery of 6 novel, HCM-associated variants. Functional assays suggest a role for PTTG1 in the pathogenesis of TIEG1-mediated HCM. Up-regulation of PTTG1 seems to be a common pathway in hypertrophic heart disease, including TIEG1-mediated HCM.
Keywords: Hypertrophic cardiomyopathy, genetics, TGFβ, TIEG1
Introduction
In the last two decades, over 24 disease-susceptibility genes have been elucidated for hypertrophic cardiomyopathy (HCM), a disease characterized by unexplained cardiac hypertrophy that affects approximately 1 in 500 individuals (Bos et al., 2009; Maron, 2002). Currently over 80% of reverse-curve HCM and 10% of sigmoidal-HCM is secondary to mutations in genes encoding the myofilaments of the cardiac sarcomere(Binder et al., 2006), making a large portion of sigmoidal HCM genetically elusive. More recently, rare mutations in genes encoding Z-disc proteins and calcium handling proteins have been linked to HCM(Bos et al., 2009), but the search for novel HCM-causing genes continues.
KLF10-encoded TGFβ-inducible early gene-1 (TIEG1) was discovered originally as an early response gene following transforming growth factor-β (TGFβ) treatment of human osteoblast and is now known to be expressed in many tissues, including cardiac myocardium(Subramaniam et al., 1995; Subramaniam et al., 1998). TIEG1 is a member of the Krüppel-like family of transcription factors, which are known to be involved in anti-proliferative and apoptotic inducing functions following TGFβ-induction(Dang et al., 2000; Tachibana et al., 1997). Subsequent studies in engineered TIEG1-knock out (TIEG-/-) mice(Subramaniam et al., 2005) showed that male mice develop HCM with significant, but relatively late-onset cardiac hypertrophy at 16 months of age(Rajamannan et al., 2007). Microscopic examination of TIEG-/--male mice hearts showed characteristic hallmarks of HCM: cardiomyocyte hypertrophy, fibroblast hyperplasia and myocyte disarray(Rajamannan et al., 2007). Furthermore, microarray analysis revealed a significant up-regulation of PTTG1-encoded pituitary tumor transforming gene-1, demonstrating that TIEG1 plays an important role in the repression of proliferative and hypertrophic pathways, possibly through the actions of PTTG1. Based on these findings, we hypothesized that TIEG1 could be a novel HCM-susceptibility gene in humans.
Methods
Study cohort
All studies were performed under protocols approved by each site's Institutional Review Board conform the principles outlined in the Declaration of Helsinki. Our study cohort consisted of 923 unrelated patients with HCM from two large cardiac referral centers – Mayo Clinic (Rochester, MN USA) and the Academic Medical Center (AMC, Amsterdam, The Netherlands) enrolled after signing of informed consent. All patients were genotype negative for mutations in the 9 HCM-associated genes, currently included in commercially available genetic tests (MYBPC3, MYH7, TNNT2, TNNI3, TNNC1, TPM1, MYL2, MYL3, and ACTC). Clinical data were collected on all patients, including pertinent personal and family history (especially with regard to HCM or sudden cardiac death (SCD), and an echocardiogram to determine maximum left ventricular wall thickness (MLVWT) and resting left ventricular outflow tract gradient (LVOT). Clinical diagnosis of HCM was made when subjects had a MLVWT over 13 mm in the absence of hypertrophy inducing conditions such as aortic stenosis or hypertension.
Genetic analysis
DNA of all patients was extracted from peripheral blood lymphocytes (Gentra Inc, Minneapolis, MN). After design of intronic primers, the 4 translated exons of KLF10 (Nm_005646) were amplified by polymerase chain reaction (PCR) in all patient's DNA samples and subsequently analyzed for genetic variations by denaturing high performance liquid chromatography (DHPLC)(WAVE®, Transgenomic, Omaha, NE). Abnormal DHPLC-elution profiles were subjected to direct DNA sequencing (ABI Prism 377, Applied Biosystems, Foster City, CA) to determine the nature of nucleotide substitution. All translated exons were analyzed for 800 Caucasian reference alleles from ostensibly healthy, ethnically-matched controls to distinguish novel HCM-associated mutations from rare or common polymorphisms.
Site-directed mutagenesis and luciferase assays
After design of sequence specific primers, identified variants and control variants were created using a Quick Change mutagenesis Kit (Stratagene, CA). The WT TIEG1 cloned into a pCDNA4/TO expression vector was used as a template for the mutagenesis studies. Following the mutagenesis, the plasmid DNA was transformed into XL-10 ultracompetent cells (Quickchange® II, Stratagene, La Jolla, CA). DNA was purified from the bacteria (Miniprep®, Qiagen, Valencia, CA) and all constructs were confirmed by direct DNA sequencing to verify the mutations.
Effects of the various TIEG1 variants on PTTG1-promoter activity (PTTG1-promoter, including the 5′-flanking region (-1,321 to -3) cloned in front of a luciferase reporter, as previously described(Rajamannan et al., 2007)) were studied using luciferase assays. Blinded to the investigator (JMB), the PTTG1-promoter construct (1μg) was transfected into AKR2B mouse embryo fibroblasts along with 1 μg of empty expression vector, wild-type (WT) TIEG1-expression vector or the various mutant TIEG1 expression vectors. Following 24h of transfection, cell lysates were prepared and analyzed for luciferase activity. Luciferase assays were repeated 10 times, normalized to relative expression and expressed as fold-change relative to empty-vector controls.
To assess the effect of variants on the cardiac-expressed SMAD7-promoter, 1 μg of empty expression vector, and either WT-TIEG1 or TIEG1 mutants were transfected into H9c2 rat cardiocytes along with a SMAD7-promoter construct (1 μg). Eight hours after transfection, culture media (DMEM + 10% horse serum (HS)) was replaced with DMEM with 1% HS to induce cardiocyte differentiation. Thirty-six to forty-eight hours after transfection, cells were lysed, and luciferase assays were performed and analyzed as described above.
Immunohistochemistry
To determine the level of PTTG1 expression in diseases associated with cardiac hypertrophy (including TIEG1-mediated disease), we set up an immunohistochemistry analysis comparing paraffin embedded slides of patients with HCM, aortic stenosis, dilated cardiomyopathy (DCM), hypertrophy associated with systemic hypertension as well as healthy controls. Slides were stained with a rabbit monoclonal antibody to PTTG1 (Epitomics, Burlingame, CA). Paraffin embedded cardiac tissue was obtained from both surgical and autopsy cohorts. The surgical cohorts included individuals who underwent subaortic myectomy for either aortic stenosis or obstructive HCM. The autopsy cohorts included basilar septal myocardium from individuals with aortic stenosis, systemic hypertension, DCM and HCM. As retrospective part of this study, autopsy tissue was retrieved under an IRB-approved protocol. Basilar septal myocardium obtained at autopsy from individuals without cardiovascular disease (by clinical and histopathological examination) served as controls. Paraffin-blocks were sectioned at 5 μm for immunohistochemical staining. Deparaffinization with xylene and subsequent rehydration with graded ethanol preceded by heat-induced epitope retrieval with EDTA buffer (pH 8) in a Lab Vision PT Module (Fremont, CA). The staining procedure was carried out by an automated immunohistochemistry-staining machine (DAKO Techmate 500, DAKO, Denmark) using the Envision program. Staining was done for all samples at same time and randomly batched because of number of slides to eliminate bias because of staining. Titration for correct dilution of antibody was performed and after review, a dilution of 1:100 was selected for all assays. Blinded and randomized to the reviewer, cardiac expression of PTTG1 was scored quantitatively for localization, extent of staining (0 – 3) and intensity of staining (0 - 4) by two independent cardiovascular pathologists (JJM and WDE).
Statistical analyses
Statistical analyses were performed using JMP 7.0 statistical software (JMP, Cary, NC) using analysis of variance (ANOVA), Student's t-test for continuous variables and Fisher's exact test for nominal values. A p-value <0.05 was considered statistically significant.
Results
Demographics of the study cohort are summarized in Table 1. Overall, 923 patients with HCM (664 male) were enrolled in this study: 739 from Mayo Clinic (USA) and 184 from Academic Medical Center (AMC, The Netherlands). Patients had an average age at diagnosis of 47.6 ± 18 years and mean MLVWT of 20.0 ± 7 mm. Twenty-six percent of patients reported a family history of HCM and 15% of patients had a family history of SCD. Thirty percent of patients had undergone surgical myectomy and 14% of patients had received an implantable cardioverter defibrillator (ICD). The specific demographics of each institution's cohort (Mayo and AMC) are detailed in Table 1. Overall, patients from the AMC cohort had a lower MLVWT (17.5 ± 5.4 vs. 20.6 ± 8 (p =0.03) but were more likely to have familial HCM (35% vs. 25%, p = 0.005) or SCD (47% vs. 15%, p < 0.001). Patients at Mayo were more likely to have undergone surgical septal myectomy (38% vs, 6%, p<0.001), reflecting the referral bias for Mayo Clinic as a surgical center for treatment of obstructive HCM.
Table 1. Demographics of study cohort.
| Total | Mayo Clinic | AMC | |
|---|---|---|---|
| N | 923 | 739 | 184 |
| Sex (male/female) | 664/359 | 441/298 | 123/61 |
| Age at diagnosis, years | 47.6 ± 18 | 47.4 ± 18 | 48.3 ± 19 |
| Septal thickness, mm | 20.0 ± 7 | 20.6 ± 8 | 17.5 ± 5.4* |
| LVOT gradient, mm Hg | 45.1 ± 40 | 45.2 ± 44 | 44.7 ± 29 |
| Family history of HCM, n (%) | 249 (26) | 184 (25) | 65 (35)* |
| Family history of SCD, n (%) | 199 (22) | 113 (15) | 86 (47)* |
| Myectomy, n (%) | 289 (31) | 281 (38) | 8 (6)* |
| ICD, n (%) | 126 (14) | 103 (14) | 23 (14) |
HCM, hypertrophic cardiomyopathy; ICD, implantable cardioverter defibrillator; LVOT, left ventricular outflow tract; SCD, sudden cardiac death.
p<0.05
Genetic results
Genetic analysis of TIEG1's open reading frame revealed 6 novel missense variants (A12T, M27T, E137K, A204T, T216A and S225N) in 6 patients with HCM (Figure 1A). Each mutation was absent in 800 reference alleles. One novel variant (Q10H) was discovered in HCM patients as well as healthy controls at similar frequencies (allelic frequency 0.4%). One previously reported rare polymorphism (S249F, rs4734653) was seen in 3 HCM-patients, but not in our cohort of 400 ethnically matched controls. This variant, however, was previously described in 0.8% of healthy Europeans. All variants and surrounding amino acids found in patients with HCM (Figure 1B) were variably conserved across species. Mutated amino acids were not seen in other species, except for the A12T-variant, where the threonine (T) is also seen in the cow.
Figure 1. Topology of TIEG1-protein and sequence conservation.
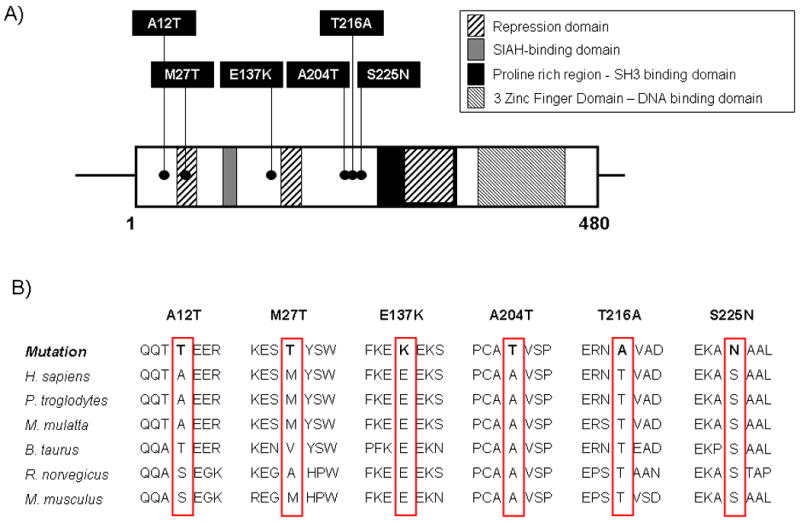
(A) Schematic representation of the 480-amino acid containing TIEG1 protein indicating the identified missense variants implicated with HCM
(B) Conservation across species of novel, HCM-associated TIEG1-variants.
Patient characteristics
Patient characteristics for the 6 TIEG1-positive patients are summarized in Table 2. Each mutation was found once in 2 male and 4 female patients. All patients were of Caucasian ethnicity. Overall, there did not seem to be a specific phenotype associated with TIEG1-mediated HCM, although in most cases – except for case 2 - HCM was late onset (mean age at diagnosis 56.2 ± 23 years). The mean MLVWT of TIEG1-positive patients was 20.8 ± 4 mm. Three patients had undergone surgical septal myectomy for relief of symptoms (cases 2, 3, 5), a number relatively high compared to the annual average rate of myectomy in HCM (∼5-10%). One patient (Cases 3) had a family history of HCM as her mother died of complications of the disease at age 84. The most severely affected patient (case 2) was a man with M27T-TIEG1. He was diagnosed at 15 years of age, with extreme hypertrophy (MLVWT, 26mm) and obstruction (117 mmHg gradient) for which a surgical myectomy was performed. Careful examinations of family histories revealed relative mild to no expression of HCM thereby limiting the ability for co-segregation analyses. Patients with a reported family history of HCM have been contacted, but have thusfar declined participation.
Table 2. Patient characteristics.
| Case | Cohort | Nucleotide change | Mutation | Sex | Age at Dx (yrs) |
MLVWT (mm) |
LVOT gradient (mm Hg) |
FH HCM | FH SCD | Treatment | Other |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | AMC | c.GCG>ACG | p.A12T | M | 65 | 17 | 13 | No | No | - | - |
| 2 | Mayo | c.ATG>ACG | p.M27T | M | 15 | 26 | 117 | No | No | Myectomy | Parents + most siblings echo negative |
| 3 | Mayo | c.GAA>AAA | p.E137K | F | 48 | 20 | 55 | Yes | No | Myectomy, ICD | Two sons echo neg |
| 4 | Mayo | c.GCT>ACT | p.A204T | F | 80 | 20 | 49 | No | No | - | |
| 5 | AMC | c.ACA>GCA | p.T216A | F | 58 | 16 | 20 | No | No | Myectomy | Father died suddenly at age 63 unknown cause |
| 6 | Mayo | c.AGT>AAT | p.S225N | F | 71 | 26 | 16 | No | No | - | Patient deceased at age 76 of HF and COPD |
COPD, chronic obstructive pulmonary disease; Dx, diagnosis; HCM, hypertrophic cardiomyopathy; HF, heart failure; ICD, implantable cardioverter defibrillator; LVOT, left ventricular outflow tract; SCD, sudden cardiac death.
SMAD7 promoter activity in H9c2-cardiocytes
Previously, we have demonstrated that TIEG potentiates TGF-B/Smad signaling by repressing the inhibitory SMAD7 gene in fibroblasts (14). Since Smad7 is one of the targets for TIEG, we wanted to determine the effect of TIEG mutants on SMAD7 promoter expression. As expected, WT TIEG1 repressed SMAD7-promoter activity by ∼70%. Four of the 6 HCM-associated variants as well as the two control variants exhibited normal TIEG1-like function relative to SMAD7 (Figure 2). In contrast, two putative TIEG1-HCM variants – A12T and S225N – altered SMAD7-promoter expression with S225N-TIEG1 showing significantly increased activation of SMAD7 promoter activity as compared to wild-type (Figure 3), whereas the controls variants did not differ from WT levels.
Figure 2. SMAD7-promoter activity in H9c2-cardiocytes.
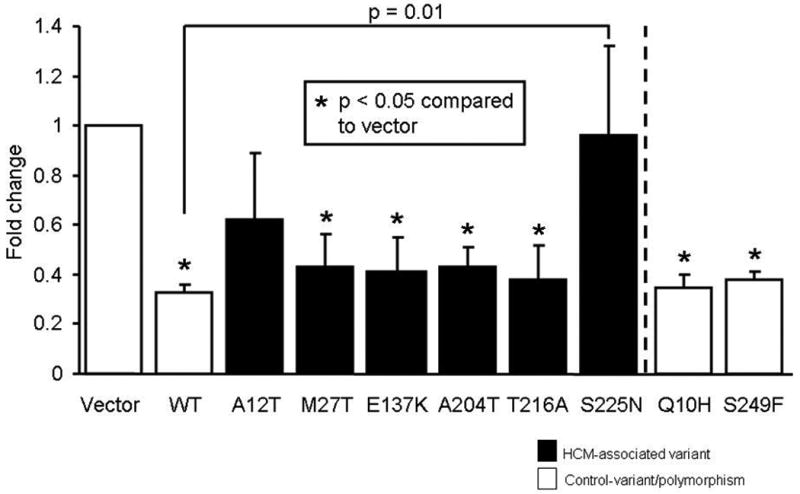
Bar diagram showing SMAD7-promoter activity in H9c2-cardiocytes. Wild-type TIEG1 (WT) repressed SMAD7-expression. Four of the six variants as well as the control polymorphisms behave like WT with respect to TIEG1's effect on SMAD7 promoter activity, whereas the S225N-TIEG1 eliminates TIEG1's ability to repress SMAD7 promoter activity. Fold-change is the relative expression of the luciferase compared to empty vector control.
Figure 3. PTTG1-promoter activity in AKR2B-fibroblasts.
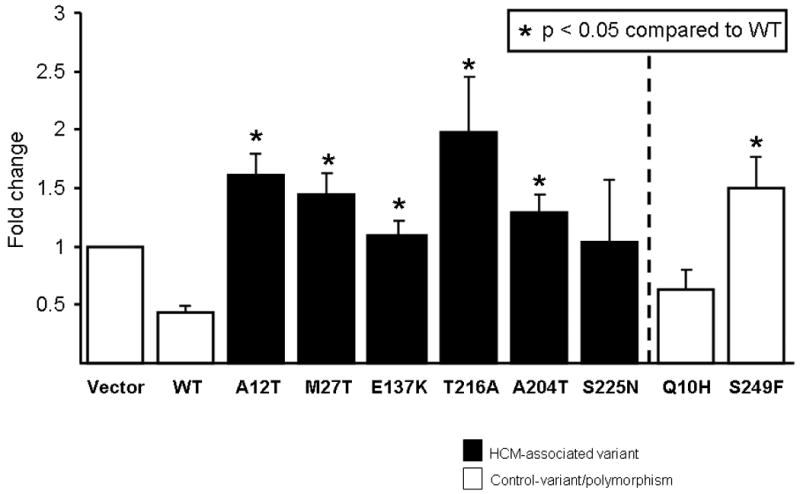
Bar diagram showing PTTG1-promoter activity in AKR2B-fibroblasts. Wild-type TIEG1 (WT) represses PTTG1-promoter, whereas 5 of 6 TIEG1-variants exhibited altered PTTG1 function significantly with regard to its promoter regulation. Fold-change is the relative expression of the luciferase compared to empty vector control.
PTTG1 promoter activity in AKR2B-fibroblasts
Previously, we have demonstrated that TIEG1-/- murine heart tissues expressed an increased PTTG1 mRNA and protein levels, thereby implicating PTTG1 as the target gene for the development of cardiac hypertrophy[8]. Similarly to previous studies [8], WT TIEG1 repressed PTTG1-promoter activity by ∼55% (Figure 3). Akin to data from TIEG1-/- -mice, 5 of the 6 putative TIEG1-HCM variants resulted in luciferase activity that was significantly higher than that of WT (p < 0.05 compared to WT), and in the case of T216A-TIEG1, up to 2-fold higher than vector control (Figure 3). In contrast, the PTTG1-promoter activity, in the setting of Q10H-TIEG1 control variant, was identical to WT TIEG1 effect, while on the other hand, S249F-TIEG1 showed increased regulation of the PTTG1 promoter.
PTTG1 protein expression in cardiac tissue
In order to determine the expression of PTTG1 protein in HCM patients, we performed immunohistochemical analysis on paraffin embedded tissue sections derived from patients with HCM (n =15), including two patients with TIEG1-HCM (cases 2 and 3), as well as tissue from patients with other diseases that exhibit cardiac hypertrophy including aortic stenosis (n = 21), DCM (n = 13), and systemic hypertension (n = 18, Figure 4). Overall, normal heart tissue showed either no or mild PTTG1 expression (Figure 4A). In contrast, PTTG1 protein levels were increased dramatically in tissues of patients with aortic stenosis, DCM and systemic hypertension (Figure 4B, C, and D respectively). A similar increase was observed in both autopsy (Figure 4E) and surgical (Figure 4F) HCM specimens. PTTG1 protein was localized primarily within the sarcoplasm of myocytes. Scored and quantified in a blinded manner by cardiovascular pathologists (JJM and WDE), the extent of staining was not significantly different between all groups (data not shown). However, as shown in Figure 4G, the intensity of staining was significantly higher in patients with aortic stenosis, DCM, and HCM compared to controls. Patients with systemic hypertension exhibited increased PTTG1 staining intensity as well, but this did not reach statistical significance. There was no difference in staining between the tissues from autopsy HCM compared to surgical HCM tissue. Although a statistical comparison was not possible because of the low number, the two patients with TIEG1-mediated HCM showed the highest intensity score for PTTG1 staining.
Figure 4. Immunohistochemical staining of PTTG1 in cardiac tissue.
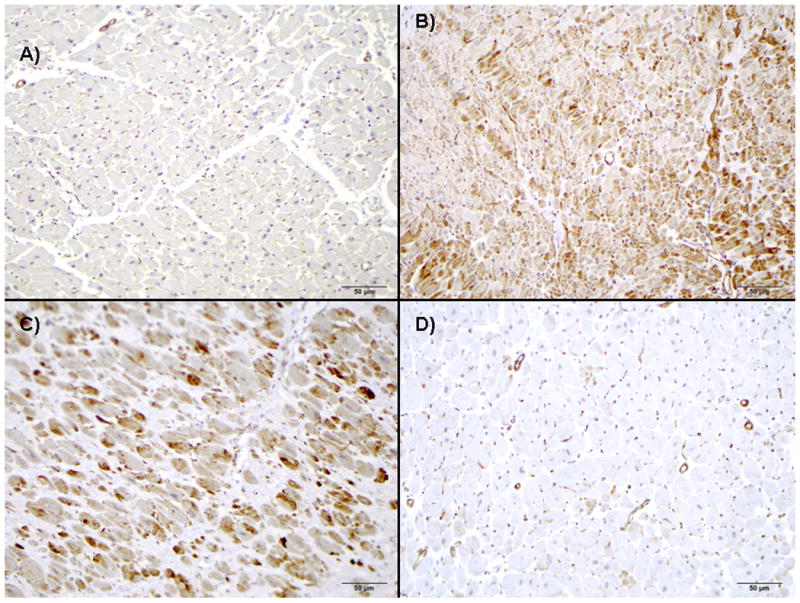
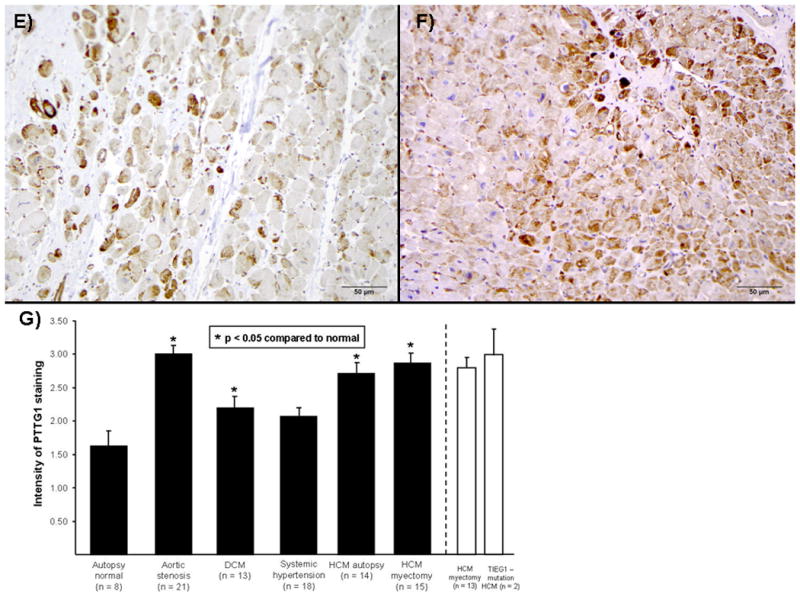
Immunohistochemical staining for PTTG1 protein levels demonstrates little PTTG1-protein expression in autopsy normal hearts (A) but marked increases of PTTG1 expression in tissue of patients with either aortic stenosis (B), dilated cardiomyopathy (C), systemic hypertension (D), autopsy-derived HCM (E), or myectomy-derived HCM (F). (G) This observation is quantitatively shown in bar diagram with significantly higher expression of PTTG1 detected in all hypertrophic models, except hypertrophy in the setting of systemic hypertension. Bars in white demonstrate intensity of two TIEG1-myectomy patients vs. the remaining myectomy samples.
Discussion
Over the past two decades, multiple genes encoding proteins involved in various processes in the cell have been implicated in the pathogenesis of HCM. Since the discovery of the first gene, MYH7-encoded β-myosin heavy chain for HCM in 1989, over 24 HCM-susceptibility genes have been reported and commercial genetic testing is now available for a large subset of these genes(Bos et al., 2009; Geisterfer-Lowrance et al., 1990). However, for many patients, the underlying genetic causes remain elusive and research continues to discover novel HCM-associated genes.
Recently, Rajamannan et al. reported that male TIEG1-/- mice exhibited features of late-onset HCM, including asymmetric cardiac hypertrophy, increased ventricular size at 16 months of age, increased heart weight to body weight ratio, increased fibrosis and increased wall thickness compared to WT mice(Rajamannan et al., 2007). Furthermore, Masson's Trichrome-staining demonstrated evidence of myocyte disarray and fibrosis which led us to hypothesize that TIEG1 could be a candidate gene in the pathogenesis of HCM. In this study, 923 unrelated patients with HCM from the USA and the Netherlands were analyzed. After comprehensive analysis of all translated regions of TIEG1, we discovered 6 novel, HCM-associated missense variants in patients which were absent in 400 ethnically-matched Caucasian healthy control subjects. Furthermore, we discovered a novel control variant. The Q10H-variant was seen in our patients as well as in our 800 reference alleles (allelic frequency 0.4%). Notably, S249F-TIEG1 (rs4734653) seen in our patients (allelic frequency 0.5%), was absent in our reference alleles; but was present in 0.8% of European controls.
Overall, no TIEG1-specific phenotype seemed to be associated with human TIEG1-HCM, although similar to the TIEG -/- mice, the patient's cardiac hypertrophy seemed to be of late-onset and obstructive in most cases. While HCM-phenotype was seen exclusively in male-TIEG -/- mice, no gender predilection could be demonstrated among the small cohort of patients with putative TIEG1-HCM. Female TIEG1-/- mice are known to have skeletal defects which have been characterized as osteopenia(Hawse et al., 2008). Here, careful examination of the TIEG1-mutation positive patients' written and electronic medical records revealed no specific skeletal problems in our patients. Unfortunately relatively low penetrance of HCM in families as well as lack of participation has permitted us from performing co-segregation studies to prove causality. We therefore moved forward with functional assays to determine the effect of novel, human TIEG1-variants in vitro.
TGFβ is a key mediator of cardiac adaptations to hemodynamic overload and plays a critical role in induction of cardiac hypertrophy, heart failure and fibrosis(Brand and Schneider, 1995). This is caused by TGFβ-induced expression of collagen mRNA and subsequent collagen deposition in fibroblasts, induced expression of factors of the fetal gene program (MYH7 and ACTC) in cardiomyocytes, and through activation of the MAPK signaling pathway and p38-induced transcription factors(Heineke and Molkentin, 2006; Parker et al., 1990). Recently, TGFβ-signaling provided a pivotal mechanism in non-myocyte driven fibrosis in mouse models of myofilament/sarcomeric-HCM(Teekakirikul et al.). TIEG1 mediates TGFβ signaling in multiple cell types. First, expression of TIEG1 is increased within 30 minutes following TGFβ treatment in osteoblast cells(Subramaniam et al., 1995). In addition, TIEG1 binds to and thereby represses the inhibitory Smad7 promoter which in turn potentiates the TGF-β/Smad signaling in cells(Johnsen et al., 2002a; Johnsen et al., 2002b; Johnsen et al., 2002c).
SMAD7 is a member of the SMAD-family of proteins involved in TGFβ-signaling, and with SMAD6, comprises the subgroup of inhibitory SMADS that antagonize TGFβ-family members (Heldin et al., 1997; Shi and Massague, 2003). While most mice devoid of the indispensible MH2-domain of SMAD7 die in utero, surviving mice have impaired cardiac functions (such as ejection – and shortening fraction) and cardiac arrhythmias(Chen et al., 2009). Because of its known role in TIEG1-TGFβ-signaling, as well as the mutant mouse phenotype, we sought to examine the effects of our novel HCM-associated TIEG1-variants on the activity of the SMAD7-promoter. We found that only one of the variants (S225N-TIEG1) significantly altered TIEG1's regulation of the SMAD7 promoter compared to WT suggesting a minor role for the TIEG1-SMAD7 inhibition in this disease.
Further studies of TIEG1-/- male mice demonstrated that these animals develop characteristic features of HCM during aging with a marked upregulation of PTTG1(Rajamannan et al., 2007). PTTG1 is typically overexpressed in a variety of endocrine-related tumors, especially pituitary, thyroid, breast, ovarian, and uterine tumors as well as non-endocrine tumors. PTTG1 functions in cell replication, proliferation, DNA damage/repair, organ development, and metabolism (reviewed in (Vlotides et al., 2007)). Luciferase assays by Rajamannan et al. (Rajamannan et al., 2007) demonstrated that TIEG1 acts directly on the promoter of PTTG1 causing a 60-70% drop in PTTG1's promoter activity, suggesting that the observed myocyte hypertrophy and fibrosis in male TIEG-/- mice may be mediated by removal of TIEG1's normal inhibition of PTTG1 and consequential accentuation in PTTG1 gene expression (Rajamannan et al., 2007).
Concordant with these observations from mice, our current study showed that 5 of our 6 TIEG1 variants resulted in a significant increase in PTTG1 promoter activity relative to WT TIEG1. Interestingly, protein levels of PTTG1 were increased markedly not only in surgically resected, hypertrophic myocardium of patients with TIEG1-HCM, but in various models of hypertrophic heart disease, such as aortic stenosis and even dilated cardiomyopathy, and less markedly hypertrophy in the setting of systemic hypertension. These data suggest that these putative HCM-associated TIEG1 variants result in dysregulation of TIEG1's normal repressive control over either SMAD7 and/or PTTG1 and that PTTG1 protein expression might be a pathway biomarker in maladaptive cardiac remodeling, including HCM (Bowles et al., 2000; Olivotto et al., 2009). Further studies are therefore needed to dissect the novel pathways elucidated by variations in TIEG1 as well as the biological role of PTTG1. Conceivably, gain-of-function PTTG1 mutations in humans or transgenic overexpression of PTTG1 in mice could precipitate cardiac hypertrophy, or more specifically, HCM.
Conclusions
This is the first report to associate variants in the TIEG1 gene with human disease. We have identified 6 novel, HCM associated TIEG1 missense variants and have demonstrated that a number of these variants have abnormal function with regard to mutant TIEG1's ability to regulate either the PTTG1 or SMAD7 promoters, two genes known to be associated with hypertrophic pathways. Furthermore, tissue expression of PTTG1 was accentuated in not only TIEG1-mediated HCM but also in all models of cardiac hypertrophy suggesting a large role for PTTG1 in its development. These studies have implicated TIEG1 in human HCM, and more over additional in vitro and in vivo functional studies have elucidated novel TGFβ-associated pathways in the pathogenesis of HCM and cardiac hypertrophy in general. Further studies are needed to examine the potential role of PTTG1 as a biomarker in the pathogenesis of HCM.
Acknowledgments
The authors would like to thank Elizabeth S. Bruinsma for her help with cloning the TIEG1 mutants.
Funding Sources: This work was supported by grants from the National Institutes of Health (HL94291 (MJA) and DE14036 (TCS)) and the Mayo Clinic Windland Smith Rice Sudden Cardiac Death Program.
Abbreviations
- HCM
Hypertrophic cardiomyopathy
- IHC
Immunohistochemistry
- KLF10
Krüppel-like factor 10
- MLVWT
Maximum left ventricular wall thickness
- PTTG1
Pituitary tumor-transforming gene-1
- TGFβ
Transforming growth factor-β
- TIEG1
TGFβ-inducible early gene -1
- SCD
Sudden cardiac death
Footnotes
Disclosures: None.
References
- Binder J, Ommen SR, Gersh BJ, Van Driest SL, Tajik AJ, Nishimura RA, Ackerman MJ. Echocardiography-guided genetic testing in hypertrophic cardiomyopathy: septal morphological features predict the presence of myofilament mutations. Mayo Clin Proc. 2006;81:459–467. doi: 10.4065/81.4.459. [DOI] [PubMed] [Google Scholar]
- Bos JM, Towbin JA, Ackerman MJ. Diagnostic, prognostic, and therapeutic implications of genetic testing for hypertrophic cardiomyopathy. J Am Coll Cardiol. 2009;54:201–211. doi: 10.1016/j.jacc.2009.02.075. [DOI] [PubMed] [Google Scholar]
- Bowles NE, Bowles KR, Towbin JA. The “final common pathway” hypothesis and inherited cardiovascular disease. The role of cytoskeletal proteins in dilated cardiomyopathy. Herz. 2000;25:168–175. doi: 10.1007/s000590050003. [DOI] [PubMed] [Google Scholar]
- Brand T, Schneider MD. The TGF beta superfamily in myocardium: ligands, receptors, transduction, and function. J Mol Cell Cardiol. 1995;27:5–18. doi: 10.1016/s0022-2828(08)80003-x. [DOI] [PubMed] [Google Scholar]
- Chen Q, Chen H, Zheng D, Kuang C, Fang H, Zou B, Zhu W, Bu G, Jin T, Wang Z, Zhang X, Chen J, Field LJ, Rubart M, Shou W, Chen Y. Smad7 is required for the development and function of the heart. J Biol Chem. 2009;284:292–300. doi: 10.1074/jbc.M807233200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang DT, Pevsner J, Yang VW. The biology of the mammalian Kruppel-like family of transcription factors. Int J Biochem Cell Biol. 2000;32:1103–1121. doi: 10.1016/s1357-2725(00)00059-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisterfer-Lowrance AA, Kass S, Tanigawa G, Vosberg H, McKenna W, Seidman CE, Seidman JG. A molecular basis for familial hypertrophic cardiomyopathy: A beta cardiac myosin heavy chain gene missense mutation. Cell. 1990;62:999–1006. doi: 10.1016/0092-8674(90)90274-i. [DOI] [PubMed] [Google Scholar]
- Hawse JR, Iwaniec UT, Bensamoun SF, Monroe DG, Peters KD, Ilharreborde B, Rajamannan NM, Oursler MJ, Turner RT, Spelsberg TC, Subramaniam M. TIEG-null mice display an osteopenic gender-specific phenotype. Bone. 2008;42:1025–1031. doi: 10.1016/j.bone.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol. 2006;7:589–600. doi: 10.1038/nrm1983. [DOI] [PubMed] [Google Scholar]
- Heldin CH, Miyazono K, ten Dijke P. TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature. 1997;390:465–471. doi: 10.1038/37284. [DOI] [PubMed] [Google Scholar]
- Johnsen SA, Subramaniam M, Janknecht R, Spelsberg TC. TGFbeta inducible early gene enhances TGFbeta/Smad-dependent transcriptional responses. Oncogene. 2002a;21:5783–5790. doi: 10.1038/sj.onc.1205681. [DOI] [PubMed] [Google Scholar]
- Johnsen SA, Subramaniam M, Katagiri T, Janknecht R, Spelsberg TC. Transcriptional regulation of Smad2 is required for enhancement of TGFbeta/Smad signaling by TGFbeta inducible early gene. J Cell Biochem. 2002b;87:233–241. doi: 10.1002/jcb.10299. [DOI] [PubMed] [Google Scholar]
- Johnsen SA, Subramaniam M, Monroe DG, Janknecht R, Spelsberg TC. Modulation of transforming growth factor beta (TGFbeta)/Smad transcriptional responses through targeted degradation of TGFbeta-inducible early gene-1 by human seven in absentia homologue. J Biol Chem. 2002c;277:30754–30759. doi: 10.1074/jbc.M204812200. [DOI] [PubMed] [Google Scholar]
- Maron BJ. Hypertrophic cardiomyopathy: A systematic review. JAMA. 2002;287:1308–1320. doi: 10.1001/jama.287.10.1308. [DOI] [PubMed] [Google Scholar]
- Olivotto I, Cecchi F, Poggesi C, Yacoub MH. Developmental origins of hypertrophic cardiomyopathy phenotypes: a unifying hypothesis. Nat Rev Cardiol. 2009;6:317–321. doi: 10.1038/nrcardio.2009.9. [DOI] [PubMed] [Google Scholar]
- Parker TG, Packer SE, Schneider MD. Peptide growth factors can provoke “fetal” contractile protein gene expression in rat cardiac myocytes. J Clin Invest. 1990;85:507–514. doi: 10.1172/JCI114466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajamannan NM, Subramaniam M, Abraham TP, Vasile VC, Ackerman MJ, Monroe DG, Chew TL, Spelsberg TC. TGFbeta inducible early gene-1 (TIEG1) and cardiac hypertrophy: Discovery and characterization of a novel signaling pathway. J Cell Biochem. 2007;100:315–325. doi: 10.1002/jcb.21049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- Subramaniam M, Gorny G, Johnsen SA, Monroe DG, Evans GL, Fraser DG, Rickard DJ, Rasmussen K, van Deursen JM, Turner RT, Oursler MJ, Spelsberg TC. TIEG1 null mouse-derived osteoblasts are defective in mineralization and in support of osteoclast differentiation in vitro. Mol Cell Biol. 2005;25:1191–1199. doi: 10.1128/MCB.25.3.1191-1199.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramaniam M, Harris SA, Oursler MJ, Rasmussen K, Riggs BL, Spelsberg TC. Identification of a novel TGF-beta-regulated gene encoding a putative zinc finger protein in human osteoblasts. Nucleic Acids Res. 1995;23:4907–4912. doi: 10.1093/nar/23.23.4907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramaniam M, Hefferan TE, Tau K, Peus D, Pittelkow M, Jalal S, Riggs BL, Roche P, Spelsberg TC. Tissue, cell type, and breast cancer stage-specific expression of a TGF-beta inducible early transcription factor gene. J Cell Biochem. 1998;68:226–236. [PubMed] [Google Scholar]
- Tachibana I, Imoto M, Adjei PN, Gores GJ, Subramaniam M, Spelsberg TC, Urrutia R. Overexpression of the TGFbeta-regulated zinc finger encoding gene, TIEG, induces apoptosis in pancreatic epithelial cells. J Clin Invest. 1997;99:2365–2374. doi: 10.1172/JCI119418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teekakirikul P, Eminaga S, Toka O, Alcalai R, Wang L, Wakimoto H, Nayor M, Konno T, Gorham JM, Wolf CM, Kim JB, Schmitt JP, Molkentin JD, Norris RA, Tager AM, Hoffman SR, Markwald RR, Seidman CE, Seidman JG. Cardiac fibrosis in mice with hypertrophic cardiomyopathy is mediated by non-myocyte proliferation and requires Tgf-beta. J Clin Invest. 120:3520–3529. doi: 10.1172/JCI42028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlotides G, Eigler T, Melmed S. Pituitary tumor-transforming gene: physiology and implications for tumorigenesis. Endocr Rev. 2007;28:165–186. doi: 10.1210/er.2006-0042. [DOI] [PubMed] [Google Scholar]