

Abstract
Protein translation is essential for all forms of life and is conducted by a macromolecular complex, the ribosome. Evolutionary changes in protein and RNA sequences can affect the three-dimensional organization of structural features in ribosomes in different species. The most dramatic changes occur in animal mitochondria, whose genomes have been significantly reduced and altered. The RNA component of the mitochondrial ribosome (mitoribosome) is reduced in size, with a compensatory increase in protein content. Until recently, it was unclear how these changes affect the three-dimensional structure of the mitoribosome. Here we present a structural model of the large subunit (LSU) of the mammalian mitoribosome developed by combining molecular modeling techniques with cryo-electron microscopic (cryo-EM) studies. The model contains 93% of the mitochondrial rRNA (mito-rRNA) sequence and 16 mitochondrial ribosomal proteins (MRPs) in the large subunit of the mitoribosome. Despite the smaller mitochondrial rRNA, the spatial positions of RNA domains known to be directly involved in protein synthesis are essentially the same as in Bacterial and Archaeal ribosomes. However, the dramatic reduction in rRNA content necessitates evolution of unique structural features to maintain connectivity between RNA domains. The smaller rRNA sequence also limits the likelihood of tRNA binding at E-site of the mitoribosome, and correlates with the reduced size of D- and T-loops in some animal mitochondrial tRNAs, suggesting co-evolution of mitochondrial rRNA and tRNA structures.
Introduction
The mammalian mitochondrial ribosome (mitoribosome) is responsible for synthesis of 13 mitochondrial gene products, which are essential components of the complexes involved in oxidative phosphorylation.1,2 The importance of these genes in generating ATP places a significant burden of accuracy on the mitoribosome. The mitochondrion also plays a crucial role in the initiation of apoptosis3 and mitochondrial defects have been implicated in a wide variety of degenerative diseases, aging, and cancer.4 However, evolutionary pressures to maintain nuclear control of cellular metabolism following endosymbiosis5 may be responsible for the significant reduction in animal mitochondrial ribosomal RNA (mito-rRNA) sequence when compared to Bacteria, Archaea and Eukaryotes. This reduction is compensated by an increase in the size and number of mitochondrial ribosomal proteins (MRPs),6-8 whose genes are under nuclear control.9
Even though mitochondria are believed to have descended from an endosymbiotic eubacterium,10,11 the structural components of their ribosomes are noticeably different. The ratio of protein to rRNA mass in animal mitochondria (2:1) is inverted from the ratio found in Bacterial and Archaeal ribosomes (1:2). A decrease in particle density is therefore observed in sedimentation experiments, yielding a 55S value for the intact bovine mitoribosome compared to 70S in Bacteria and Archaea. The decreased sedimentation coefficient can also be attributed to a more porous structure in mitochondrial ribosomes.12,13
The bovine 55S mitoribosome is comprised of two asymmetric subunits, a small (28S) subunit and large (39S) subunit. The small subunit contains a 12S rRNA (955 nucleotides) with 29 proteins,6,14,15 and the large subunit contains a 16S rRNA (1571 nucleotides) with 48 proteins.6-8 All of the MRPs are encoded by rapidly evolving nuclear genes,16 while the mitochondrial rRNA genes, which are also evolving at a rapid rate,5 are encoded by the mitochondrial genome and are transcribed within the mitochondrion. For comparison, the Bacterial and Archaeal ribosomes are also comprised of two asymmetric subunits, but the small (30S) subunit contains a 16S rRNA (1500 nucleotides on average) with roughly 20 proteins and the large (50S) subunit contains two ribosomal RNA (rRNA) components, 5S rRNA (120 nucleotides on average) and 23S rRNA (3000 nucleotides on average), with more than 30 proteins. tRNAs migrate through three relatively stable binding sites in cytoplasmic ribosomes during translation: the A- (aminoacyl), P- (peptidyl), and E- (exit) sites.
In this article, we focus our attention on the structure of the large subunit of the mammalian mitoribosome. The large subunit (LSU) rRNA is responsible for catalysis of peptide bond formation. Recent studies suggest that the mechanism of peptide bond formation can be attributed to positioning transfer RNA (tRNA) substrates charged with amino acid residues in a specified proximity during the reaction.17,18 The 2′-OH of residue A76 of the P-site tRNA has been proposed as the catalytic component.19 The LSU rRNA domain V, which contains the peptidyl transferase center (PTC), is largely conserved through all organisms.20 Many of the rRNA regions of domain IV that are involved in tRNA and inter-subunit interactions21 are also preserved.20
Furthermore, the ribosomal protein L11-binding domain (L11-BD) within the LSU rRNA domain II and the sarcin-ricin loop within domain VI, which together constitute the GTPase-associated center of the ribosome that interacts with translation cofactors22,23 (EF-Tu, EF-G, RF3, etc.), are also conserved. However, the sequences and structures that connect the central core and these peripheral structural elements are truncated in several mitochondrial LSU rRNAs.24 On the opposing side of the LSU, dynamic motions of the L1 protein and associated rRNA have been proposed to directly affect E-site occupancy on the ribosome.25-27 Together, these functional domains work in concert during prokaryotic translation, and structural studies are beginning to elucidate the mechanisms required for protein synthesis. Even so, it is unclear how the evolving mitoribosome compensates for the large reduction in rRNA sequence while maintaining the precision that protein synthesis demands.
A recent cryo-electron microscopic (cryo-EM) study has provided the first detailed structure of the mammalian mitoribosome.13 It has several unique features when compared with structures of cytoplasmic ribosomes from prokaryotic and eukaryotic organisms. Also, the additional protein content does not compensate for the missing RNA sequence, as had previously been proposed.7,16 Instead, many of the additional proteins assume unique positions in the mitoribosome structure, thereby leaving regions of structure vacant where rRNA helices present in bacteria have been deleted in the mitoribosome. Also, in contrast to the characteristics of cytoplasmic ribosomes, no tRNA was found at the putative exit site (E-site) of the 55S mitoribosome, suggesting that the E-site either very weak or non-existent in the mitoribosome, consistent with suggestions available from a comparative analysis of mitochondrial and nuclear-encoded ribosomes.24
In this study, we analyze the structural perturbations in those regions of the mammalian mitoribosome that have significant changes in size and sequence of rRNA and proteins. We utilize a variety of methods, including bioinformatics to study the evolution of RNA and protein sequences, structural biology (cryo-EM), and novel computational tools to unify the results in a detailed three-dimensional model of the mitoribosome. In most cases, the reduction in mito-rRNA sequence does not alter the three-dimensional spatial location of conserved rRNA helical elements. Thus many of the interactions between the mitoribosome and tRNAs and protein cofactors during the translation cycle are preserved. However, some of the reductions in rRNA sequence result in the loss or reorientation of functional domains, indicating structures in the Bacterial and Archaeal ribosomes that are highly variable and may not be essential for translation. We have also modeled the structures and placement for 16 mitochondrial ribosomal proteins (MRP) that have sufficient sequence identity with homologous prokaryotic proteins whose structures have been determined by X-ray crystallography. This work provides a first step towards assigning structure to the protein mass of the mitochondrial ribonucleoprotein structure. Additional structural and biochemical studies are required to predict the placement and conformations of the remaining, unmodeled proteins, especially those that are unique to the mitoribosome.
Results
Homology modeling of mitochondrial rRNA (mito-rRNA)
Based on the assumption that the secondary and tertiary structures for rRNA molecules are mostly conserved for all organisms, we have used comparative sequence analysis to identify sequence conservation and variation and determine similar structural elements that are present in sets of distantly related and closely related rRNA sequences. Previous success in determining the secondary structures for the small and large subunit rRNAs28 lends confidence to the secondary structure model for the mitochondrial LSU rRNA derived from comparative sequence analysis (Fig. 1). Approximately 86% of the Bos taurus mito-rRNA can be assigned to structural features present in the Archaeal Haloarcula marismortui secondary structure, available from the Comparative RNA Website.20 The B. taurus mito-rRNA (Fig. 1a) has many features in common with all nuclear-encoded rRNAs, including the peptidyl transfer center (PTC), the sarcin-ricin loop (SRL), and the L11-binding domain (L11-BD). However, many differences are evident, because the size of the LSU mito-rRNA is reduced by almost half compared to bacteria. Many of the helical structures in domains I and III are lost, as are the A-site finger motif (ASF) in domain II, and the L1- and the 5S rRNA-binding domain (L1-BD and 5S-BD, respectively) in domain V.
Figure 1.

rRNA secondary structure comparison. a. Bos taurus mitochondrial rRNA secondary structure based on comparative sequence analysis. b. Secondary structure of H. marismortui 23S rRNA. Regions that align with the mitochondrial rRNA are highlighted in red, and those that are absent in the mitoribosome are shown in black. Some relevant functional regions are labeled (green), and six domains of the 23S rRNA are identified with roman numerals.
Starting with the secondary structure for the large subunit rRNA of the bovine mitoribosome (Fig. 1a), we have used homology modeling29 to generate a structural model for the mito-rRNA of the LSU of the mitoribosome. The X-ray crystallographic structures of large ribosomal subunits from H. marismortui,30 an archaeon, and Deinococcus radiodurans,25 a bacterium, provide homologous rRNA structures to guide the modeling. For our studies, we primarily used the H. marismortui structure (PDB accession code: 1JJ2) as the template for modeling the mitochondrial rRNA sequence. Helices and loop structures that are conserved in mitochondria are modeled based on geometry determined by X-ray crystallography for the archeon (see Methods). Three significant differences were explored: (1) Identical secondary structure elements composed of the same base pairings and unpaired nucleotides are modeled one for one, when the sequences are identical or vary in composition. The nucleotides from H. marismortui are replaced by corresponding nucleotides in the mito-rRNA. These changes do not affect the backbone or sugar orientation of the bases. (2) For canonical base pairs that are replaced by non-canonical (i.e. G•U or G•A) pairs, the latter pair is superposed on the canonical pair. The same method is used for non-canonical pairs that are replaced by canonical pairs. None of these changes in base-pair types severely distorts the helical geometry, and differences in the backbone interactions with neighboring nucleotides are easily resolved with a round of energy minimization. (3) For more dramatic differences between the two secondary structures, including changes in the size of loops and bulges, the mitochondrial sequence is modeled from previously determined X-ray crystallography structures of RNA with similar sequence (available from the RCSB protein databank31).
Structure refinement and validation
The homology model of the mito-rRNA provides a starting structure with helical positions based on similarities to prokaryotic ribosomes as determined by comparative sequence analysis. Information from a cryo-EM study13 provides additional, experimental restraints that can be incorporated using YAMMP,32 our in-house molecular modeling package. The software includes a rigid-body Monte Carlo module with simulated annealing that we use to refine our model. The vector lattice (VLAT) component of YAMMP generates a force field term that defines the electron density from experimental studies as a three-dimensional potential, providing a score for the fit of the model to the density.
Initially, the complete model for the mitochondrial rRNA is treated as a single rigid unit using a reduced representation.33 The starting model contains pseudoatoms representing the phosphate positions of each nucleotide in the RNA homology model (P-atoms). To get an initial placement of the model in the EM density, the rigid unit was subjected to Monte Carlo refinement with simulated annealing (see Methods). A good fit is obtained, suggesting that the structural organization is largely conserved in the mitoribosome, with small changes due to differences in sequence and size of some helices and connecting regions.
The mito-rRNA model was then divided into 52 rigid units, based on conserved helical structures (Fig. 2), and the structure was refined with multiple rigid-body Monte Carlo with simulated annealing. Each rigid unit is topologically connected to neighboring units using one of two bond-types: (1) Strong bonds are used to connect adjacent conserved structures that are separated by distances corresponding to the normal phosphate-phosphate distances between neighboring bases (∼6-7Å). (2) Weak bonds connect nucleotides separated by a gap in the model due to a lack of secondary structure information (grey regions, Fig. 2). The weaker bonds allow greater freedom for the connected structures, while maintaining reasonable connectivity during the Monte Carlo refinement. Non-bond interactions with 7.5Å exclusion diameters were used for every P-atom in the structure to prevent structural overlap.
Figure 2.

Rigid-body refinement of the rRNA model. Fifty-two independent rigid units were defined for refinement of the RNA structure using Monte Carlo with simulated annealing (see methods). Regions are colored to represent the distinct units that were used for the refinement. Unmodeled regions are represented as dashed, grey lines. Helices 13, 50 and 59.1, proposed by comparative sequence analysis (http://rna.icmb.utexas.edu), did not fit the EM density. We propose an alternate structure in the region with helices 50 and 59.1 in domain III (see Figs. 3 and 4).
All-atom models for the 52 units were then superposed on the final reduced representation models fit to the EM density, given that the reduced rRNA units were treated as rigid bodies and maintained their geometry. The all-atom models for each unit were covalently linked, except for the regions where gaps occur (grey dashed lines in Fig. 2), and a round of energy minimization was used to resolve structural discrepancies caused by rearrangements during rigid-body refinement.
Additional and alternate RNA secondary structure predictions
The refined homology model based on the comparative structure model of the rRNA fits the RNA density13 from cryo-EM well. The only helices in the LSU rRNA comparative model that do not fit the EM density are in domains I and III (in Fig. 2, helix 13, the end of helix 50 and helix 59.1 did not find a reasonable fit to the cryo-EM density). We therefore explored the possibility that these two regions of the rRNA have alternative folds using Alifold.34 In contrast to comparative analysis of the rRNAs that identifies base pairs from patterns of variation and covariation in a set of aligned sequences,35 the Alifold34 program combines thermodynamic and comparative analyses to predict RNA secondary structure. A multiple sequence alignment was created for a set of mammalian mitochondrial LSU rRNAs, that includes B. taurus and related organisms (see Methods). The Alifold program identified helices that are thermodynamically stable and present in the set of aligned sequences. This suggests a secondary structure in the region of helices H50 and H59.1 (Fig. 3, green nucleotides) that differs somewhat from that proposed by comparative analysis (Fig. 2). The modeled structure closely matches the EM density (Fig. 4c). This suggests one of two possibilities: (1) that after endosymbiosis the sequence in domain III of the bovine mito-rRNA has evolved to generate a somewhat different three-dimensional structure, or (2) the structure determined by cryo-EM represents a conformational change in this region from the structure predicted by comparative sequence analysis. We cannot rule out either possibility, but our model is based on the structure solved by cryo-EM and the alternative secondary structure (shown in Figure 3). We also examined alternative secondary structures for the unstructured regions of domain I (including helix 13), but we were unable to find secondary structure predictions that could be placed in the EM density with confidence.
Figure 3.

Proposed secondary structure for the mammalian mitochondrial LSU rRNA. The structure is presented as modeled with regions predicted by comparative sequence analysis (blue) comprising roughly 86% of the secondary structure. Additional secondary structure was predicted using mfold34 (orange) to extend the structure for regions where comparative analysis does not predict secondary structure. Alifold34 was used to predict an alternate secondary structure in domain III (green) which differs slightly from that predicted by comparative analysis, but more closely matches the cryo-EM density.13
Figure 4.
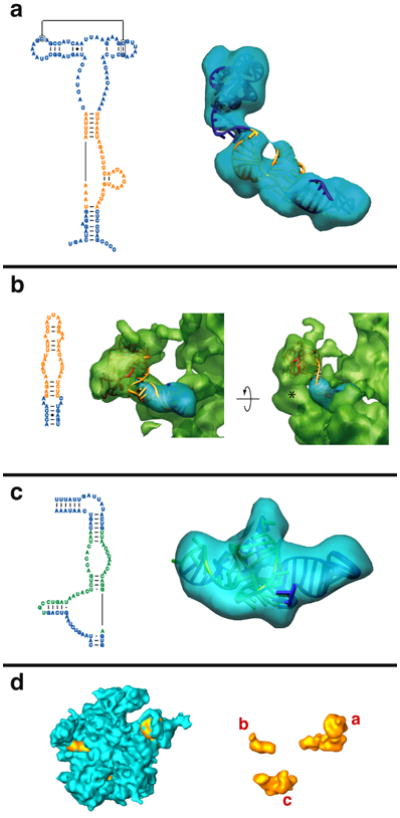
Additional and alternate secondary structures predicted using thermodynamic and comparative methods. The left side of each panel shows the predicted secondary structure, colored as in Fig. 3, and the right side of each panel shows the fit to the corresponding region in the cryo-EM density map.13 a. The L11-BD, where additional structure is predicted for the adjacent sequence (orange) using thermodynamic predictions from mfold.36 b. The L1-BD, where additional pairing is predicted using mfold36 for the rRNA sequence that interacts with MRP-L1 (red). A large globular mass of unidentified protein(s) (marked by * on the semitransparent green density) may restrict the movement of the L1 region in the mitoribosome. c. The domain III region where an alternate secondary was predicted using Alifold.34 This structure differs from that predicted by comparative sequence analysis (Fig. 1a) and fits the corresponding cryo-EM density much better (not shown). d. The cryo-EM density for the large mitoribosome subunit is shown in blue (interface view). The three regions that have been modeled using additional and alternative folding predictions are shown in orange and labeled a, b and c to correspond with the preceding panels.
Part of the region in domain II that connects the L11-BD to the highly conserved core of the LSU (Fig. 1a) has fewer nucleotides in the mammalian mitochondria than the corresponding region in all nuclear-encoded LSU rRNAs. No base pairing and helices that are shared in the mammalian mitochondria were identified with comparative and covariation analysis for this region that is truncated. To examine the possibility of additional secondary structure in this region, we used the mfold36 thermodynamic folding program to predict helices to expand the structural model from comparative analysis. Several thermodynamically stable secondary structure helices were identified, and we tested each of these by examining their fits to the cryo-EM density. From this, we were able to generate a three-dimensional model that matches the cryo-EM density very well in this region (Fig. 4a). This model reveals how, in spite of the drastic reduction in rRNA size, the L11-BD of the mito-rRNA can remain on the periphery of the mitoribosome. A previous modeling study24 proposed a movement of L11-BD closer to the core of the LSU during the course of evolution in the C. elegans mitoribosome, because no previously characterized RNA structure of similar sequence and length was known to span such a large distance. No such movement is necessary in the mammalian mitochondria for two reasons: (1) the sequence is not as reduced in mammalian mitochondria as in C. elegans, and (2) the unique structure found in the mammalian mitoribosome extends from the core of the subunit to the periphery (∼80Å) by restricting base pairing and allowing the RNA strand to stretch without the constraints of helical geometry. In fact, a large portion of the sequence near the L11-BD does not form helical base pairs, because the sequence consists almost entirely of adenine and cytosine bases (one uracil and no guanines, Fig. 3a). Cryo-EM density connecting the L11-BD with the rest of the rRNA is relatively thin, which suggests conformational variability in the region, consistent with non-canonical interactions.
A similar method was used to predict the secondary structure of an rRNA helix in domain V that extends to the L1-binding domain (L1-BD). This region of the LSU rRNA is truncated in the mammalian mitochondria, and no base pairing is predicted at its apex in the mammalian mitochondrial comparative structure model (Fig. 1a). This truncation implies that L1 does not bind to the same RNA site, despite the conservation of L1 in mammalian mitoribosomes.37 Thermodynamic predictions indicate that a single stem-loop structure is feasible (Fig. 3b), and modeling places the RNA in close proximity with the L1 protein (Fig. 4b). For this reason, an RNA:protein interaction is probably maintained, with the large bulge in the hairpin easily fitting the cleft of the L1 protein.38
We attempted to determine energetically stable helices in the remaining regions of the B. Taurus LSU rRNA that did not have helices in the comparative structure model (grey lettering, Fig. 3). Unfortunately, no common structures were found. In fact, no base pairings were predicted by mfold36 for the large loops in domains II and III, because these sequences lack the nucleic acid base diversity required for canonical pairing (no guanines and few uracils). The abundance of adenine and cytosine bases in these unstructured regions suggests that these regions do not contain regular base pairing and helices. The same is true for the single-stranded region in domain VI (Fig. 3, 1 uracil and no guanines). The unmodeled regions in domain I are not completely devoid of guanines, but they are also very G-poor. Neither Alifold nor mfold was able to predict common secondary structures for this sequence. We have therefore elected not to model these regions. Their structures are probably unique to mitoribosomes.
The final model of mito-rRNA
In total, we have modeled 93% of the mito-rRNA sequence in three dimensions (Fig. 5). Much of the 16S mito-rRNA is conserved in domain IV (Fig. 5, green), which lies at the interface between the mitoribosome subunits. Interactions with the penultimate stem of the small ribosomal subunit (SSU helix 44) are maintained near the core of the subunit. The top of helix 44 contains the decoding center, where interactions between the mRNA and the A-site tRNA are examined for fidelity,39 and signals for fidelity may be transmitted through interactions with domain IV of the LSU rRNA during translation. It is not surprising that these interactions are largely conserved, but more peripheral interactions at the subunit interface are replaced by new interactions between MRPs in each subunit.13
Figure 5.
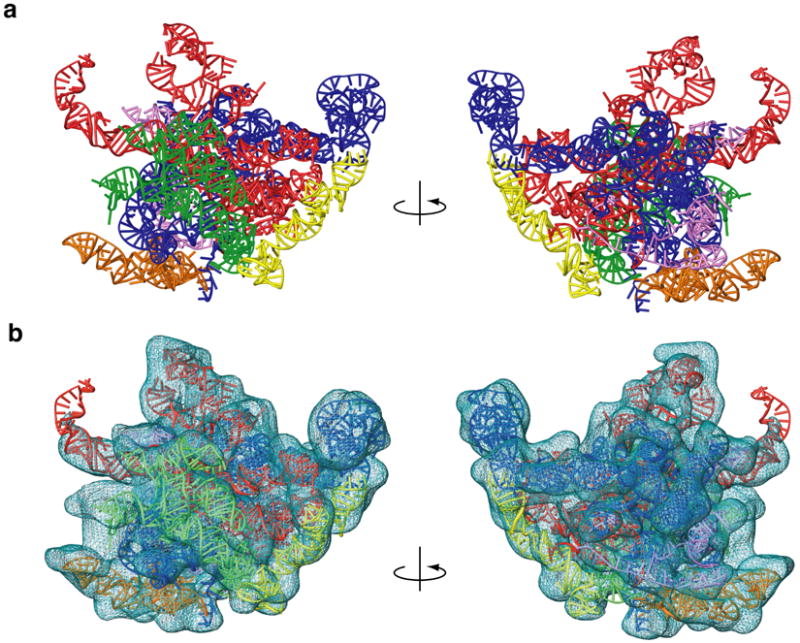
Three-dimensional model for the mitochondrial 16S rRNA. a. The 16S rRNA is represented from the interface and solvent-accessible sides of the structure and colored by domain (I – purple, II – dark blue, III – orange, IV – green, V – red and VI – yellow). b. The model fit to EM density that is attributable to RNA,13 except for the tip of a domain V helix that contacts the L1 protein. However, the model of the extended rRNA segment fits into the complete LSU map (also see Fig. 4b). Coloring is the same as in panel a.
The central cavity of the 39S subunit is highly conserved because several essential structures are preserved in domain V (Fig. 5, red), including the peptidyl transferase center (PTC). The 5S-BD is missing, in agreement with the loss of 5S rRNA in the mitochondrial genome. The helices that radiate from the core of the structure to the L1 arm are maintained, but are shorter than in bacterial ribsosomes, making interactions with the L1 protein in the mitoribosome unique. The positions of functional structures near the periphery of the structure (L11-BD and SRL) are conserved, thereby preserving critical interactions with elongation factors (EFs) during the translation cycle.
Protein homology modeling
Of the 48 proteins found in the large subunit of the mitoribosome, 28 are homologous to prokaryotic ribosomal proteins,8 while the remaining 20 are unique to the bovine mitoribosome. All of the bovine MRPs are encoded in the nuclear genome9 and must be translated in the cytoplasm and transported into the mitochondrion.40,41 MRPs are much larger than prokaryotic ribosomal proteins, and they often have insertions at the C-, N-, or both termini. It was originally thought that the increase in protein size compensates for reduction in the mito-rRNA,33 but the cryo-EM structure does not support this theory,13 as only ∼20% of deleted rRNA components are positionally replaced by mitoribosome specific proteins or extensions of homologous proteins.
We compared all 48 MRP sequences6-8 with sequences of ribosomal proteins whose structures have been previously determined by X-ray crystallography. When significant levels of identity and similarity were found, we were able to generate protein homology models based on templates from X-ray crystal structures of ribosomal large subunit complexes.21,25,30,42 We created partial models for 16 proteins (Fig. 6), and a summary of those models is listed in Table 1. (Note that L7 and L12 are identical.)
Figure 6.
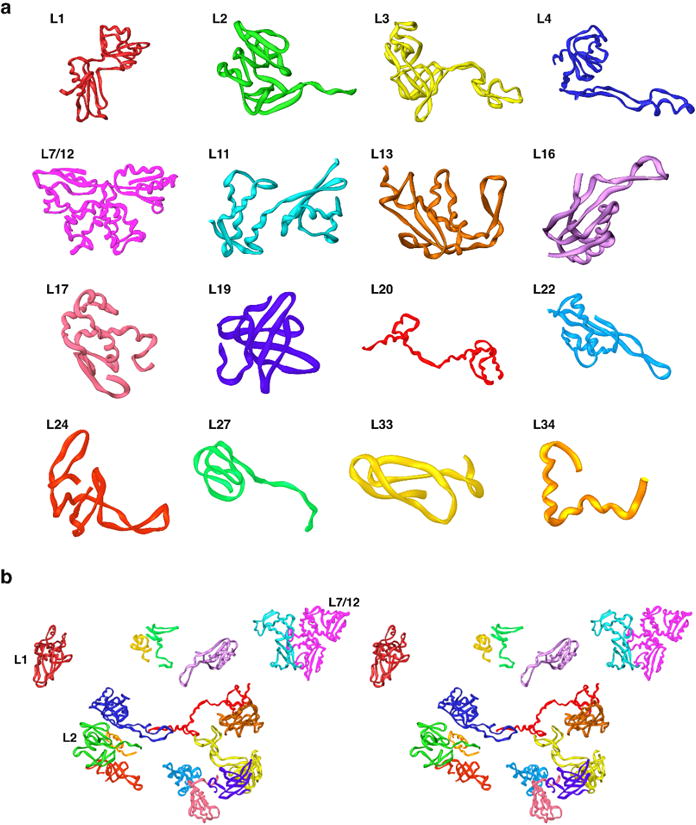
Structural homology models for MRPs. a. Models of the sixteen MRPs that have significant sequence similarity with bacterial ribosomal proteins that have been structurally characterized by X-ray crystallography. b. Structural organization of proteins in the large mitochondrial subunit fit to the cryo-EM density (stereo view).
Table 1. Proteins modeled and their homologs.
| Proteina | Homologsb | Identity | Similarity | Model size | MRP size |
|---|---|---|---|---|---|
| MRP-L1 | 1GIY | 28.6% | 52.4% | 100-289 | 303 aa |
| MRP-L2 | 1NKW | 38.2% | 56.6% | 126-261 | 305aa |
| 1PNU | 36.8% | 54.4% | |||
| 1GIY | 40.4% | 57.4% | |||
| 1JJ2 | 31.6% | 51.5% | |||
| MRP-L3 | 1NKW | 28.4% | 50.7% | 96-306 | 348aa |
| 1PNU | 28.4% | 51.1% | |||
| MRP-L4 | 1PNU | 33.1% | 56.0% | 97-271 | 311aa |
| MRP-L7 | 1GIY | 31.2% | 47.1% | 61-198 | 198aa |
| MRP-L12 | 1GIY | 31.2% | 47.1% | 61-198 | 198aa |
| MRP-L11 | 1GIY | 42.8% | 61.4% | 13-157 | 178aa |
| 1NKW | 35.2% | 57.2% | |||
| 1PNU | 35.2% | 57.2% | |||
| MRP-L13 | 1NKW | 29.1% | 57.4% | 1-148 | 178aa |
| 1PNU | 29.1% | 57.4% | |||
| MRP-L16 | 1NKW | 27.1% | 54.2% | 71-188 | 251aa |
| 1PNU | 27.1% | 54.2% | |||
| MRP-L17 | 1NKW | 33.6% | 59.5% | 11-126 | 175aa |
| 1PNU | 33.6% | 59.5% | |||
| MRP-L18 | 1GIY | 34.9% | 59.3% | 81-170 | 180aa |
| MRP-L19 | 1NKW | 26.5% | 48.0% | 96-193 | 280aa |
| 1PNU | 26.5% | 48.0% | |||
| MRP-L20 | 1NKW | 36.4% | 63.6% | 8-125 | 149aa |
| 1PNU | 36.4% | 63.6% | |||
| MRP-L22 | 1GIY | 29.2% | 47.5% | 69-178 | 206aa |
| 1PNU | 21.7% | 48.3% | |||
| MRP-L24 | 1PNU | 35.4% | 56.3% | 59-154 | 216aa |
| MRP-L27 | 1NKW | 43.5% | 65.2% | 31-99 | 148aa |
| 1PNU | 43.5% | 65.2% | |||
| MRP-L33 | 1NKW | 51.9% | 71.2% | 9-60 | 65aa |
| 1PNU | 51.9% | 71.2% | |||
| MRP-L34 | 1NKW | 44.7% | 65.8% | 53-90 | 92aa |
| 1PNU | 44.7% | 65.8% |
Mitochondrial ribosomal proteins (MRPs) with significant homology to ribosomal proteins that have been previously characterized are listed.
The homologs that were identified are listed by the Protein Data Bank accession code (1GIY is from Thermus thermophilus, 1NKW is from Deinococcus radiodurans, 1PNU is from Escherichia coli, and 1JJ2 is from Haloarcula marismortui).
None of the ribosomal proteins could be modeled completely. Homology between the MRPs and the prokaryotic ribosomal proteins did not extend across the entire length of the protein sequences. Commonly the N-, C-, or both termini were unique. More detailed sequence analysis revealed that these regions are insertions, not mutational differences. This trend has been described previously6-8 and the results from our comparative study confirm those findings.
It had been proposed that these insertions represent the addition of functional domains to compensate for the decreased size of rRNA in the large subunit.6-8 We analyzed these insertions to determine if functional roles could be identified from sequence homology to proteins of known function. Unfortunately, the homology search against the non-redundant (nr) database did not produce any homology matches, suggesting that the sequence insertions in mammalian MRPs are unique.
MRP structure refinement
The homology models for the proteins were fit to the cryo-EM density in much the same way as the RNA helices. During protein fitting, the mito-rRNA model (modeled again with the P-atom reduced representation) was treated as a rigid scaffold that was held fixed. Each protein (modeled with pseudoatoms centered on the Cα coordinates, called C-atoms) was placed manually into the EM density, and rigid-body Monte Carlo with simulated annealing was used to refine protein positions. Refinement included VLAT terms for scoring the fits of the proteins to the density plus a set of restraints based on conserved RNA-protein interactions observed in bacterial crystal structures. These interactions were determined by measuring distances between every nucleotide in the RNA and every amino acid in each protein structure. An interaction threshold of 3Å was used for protein models that had homologous proteins in the H. marismortui30 structure (where all-atom detail is present), whereas a threshold of 6Å was set for protein models with homologous structures available from T. thermophilus21 or D. radiodurans25 (where all-atom detail is not available). If the nucleotide and amino acid involved in the interaction were both conserved in the mitochondrion, harmonic restraints of equivalent length were included between appropriate P- and C-atoms during refinement.
Of the 16 protein models, 13 had conserved interactions with the RNA. The resulting RNA-protein restraints played a critical role in tethering the proteins to their conserved positions in the mitoribosome model during refinement, because much of the protein density remains unfilled, and it would otherwise be difficult to guarantee that the refinement would not move these proteins into inappropriate regions of the density. The positions of the three proteins that did not have conserved interactions were refined, although a lower starting temperature was required during the refinement protocol to prevent large movements. Also, for proteins with long extended loops, minimal rearrangements of some loops were required to prevent steric overlap between the protein and rRNA.
All 16 proteins are in positions very similar to those found in the prokaryotic structures (Fig. 6b). In most cases where restraints are available, the nucleotide binding sites for the modeled proteins are conserved. For example, the binding site for protein L2 is conserved in domain IV, which contains helices responsible for positioning the protein in the same relative orientation as in the bacterial ribosome. L11 and L7/12 are also in positions very similar to those in bacteria, so the geometries of their interactions with protein cofactors and the incoming A-site tRNA in mitochondria should be similar to those in bacteria. However, the rRNA binding site for L1 (L1-BD) is very different in the mito-rRNA than in bacterial rRNA. L1 is repositioned further from the putative E-site, which may be absent in the mitoribosome.13,24 Some of this difference is presumably due to the drastic truncation of the L1-BD in mito-rRNA, but it may also reflect the larger flexibility of the L1 arm of the large subunit25-27 when compared with nuclear-encoded ribosomes (see Discussion).
The final model of the LSU mitosibosome
The final model for the LSU of the mammalian mitoribosome fits the cryo-EM density nicely (Fig. 7). We have placed 93% of the mito-rRNA sequence as well as 16 MRP models with significant sequence similarity to prokaryotic ribosomal proteins. The L1-BD of the RNA that extends out of the density attributed to RNA does fit the density of the complete subunit, and the placement of the L1 protein suggests that an interaction between the RNA and protein is possible. Our placement of the L7/12 dimer is based on an earlier X-ray crystallographic study,21 and the protein does slightly protrude from the EM density. Since this region is known to be flexible in bacterial ribosomal particles,43,44 similar flexibility in mitochondria could explain the weaker density for the protein in this region. Moreover, in a recent study45 it has been suggested that the traditionally assigned stalk of the LSU actually represents the protein L10 and multiple copies of L7/12. If this scenario is also true in the mitoribosome, our placement of L7/12 would need further refinement.
Figure 7.
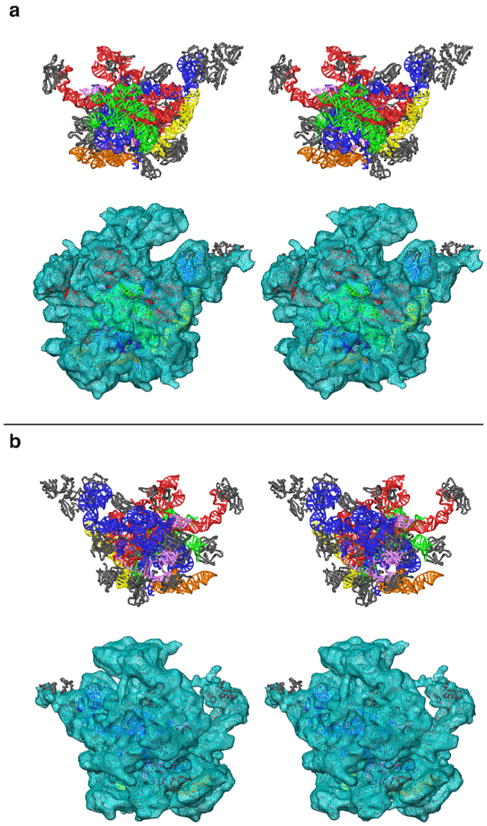
Stereo-view representation of the three-dimensional model of the 39S subunit of the mitochondrial ribosome. a. The interface view of the subunit shows that the conserved interface of the mitochondrial ribosome is still dominated by rRNA structure (colored as in Fig. 5). b. The homologous MRPs (grey) are predominantly located towards the solvent-accessible side of the particle. Upper and lower panels in both sections show the modeled structure (rRNA and proteins), and its fitting into the cryo-EM map,13 respectively.
Discussion
Our model of the large subunit of the mammalian mitoribosome suggests that the reduction in rRNA size compared to bacteria does little to alter the spatial organization of structural and functional domains common to the LSU of mammalian mitoribosomes and prokaryotic ribosomes. However, some conserved interactions, including parts of the tRNA binding sites, are absent in the mitoribosome. The interactions between the large subunit and tRNAs at the A-, P- and E-sites of the bacterial ribosome have been characterized by X-ray crystallography studies.21 Figure 8 compares the interactions predicted by our model with those observed in bacterial ribosomes.
Figure 8.

Interactions with the tRNA binding sites of the mitoribosome. The A-, P- and E-site tRNAs are represented with the structure from yeast tRNA-Phe.66 Interactions with rRNA sequence that were found in the X-ray crystal structure of the T. thermophilus 70S particle21 are represented as either being conserved (red) or absent (yellow) in the mitochondrial ribosome.
rRNA segments interacting with the P-site are highly conserved between mitochondrial and cytoplasmic ribosomes (Fig. 8). The importance of correctly positioning the acceptor stem, which holds the nascent polypeptide chain, is evident from the rRNA sequence conservation for regions interacting with the tRNA. Furthermore, additional contacts are made between the tRNA and the mitoribosome at this position during mitochondrial translation by protein(s) in the central protuberance (the so-called P-site finger, PSF). The conserved protein models that we fit to the EM density are not in positions to participate in these new interactions, so the PSF interaction can be attributed to extention(s) of one of the 12 unmodeled bacterial homologs or, more likely, to a MRP unique to mitochondrial translation.
Interactions at the A-site are also highly conserved (Fig. 8), showing the importance of positioning the A- and P-site tRNAs during catalysis of the peptidytransferase reaction.18 Contacts are also maintained between the highly conserved rRNA helix 69 and the minor groove of the tRNA D-stem. But interactions with the A-site finger (ASF), which interacts with the D- and T-loops of the tRNA in cytoplasmic ribosomes,21 are lost because of sequence reduction in domain II of the LSU rRNA (Fig. 1b). These tRNA loops exhibit a large degree of variation in size and content in mitochondria,46,47 suggesting that the rRNA and tRNA sequences that normally interact have co-evolved to accommodate structural changes. D- and T-loop structures are not sensed by the ribosome at the P-site, and new interactions with the PSF are located at the T-stem. It has been suggested that reductions in the size of both D- and T-stems and loops in some mitochondrial tRNAs may cause a dramatic change in the preferred angle between the two arms of these tRNAs,46,48 and transient electric birefringence studies have supported this suggestion.49 Our model of the LSU mitoribosome would be more accommodating to these differences at the A-site, but it is not clear what, if any, affect this difference may play in translational fidelity.
The ribosomal E-site is markedly different in mitoribosomes, as most of the rRNA regions responsible for interacting with the E-site tRNA in the bacterial ribosome are absent in the mitoribosome.24 The regions of rRNA sequence that bind the E-site tRNA during translation are almost completely missing in the mito-rRNA (Fig. 8). The one interaction that might be structurally conserved occurs near the base of the L1 arm in the rRNA model. The sequence in this region is markedly different in the mito-rRNA compared with prokaryotes, so any interaction will be different, if not completely lost. Furthermore, the observation that the tRNA binds strongly in the P-site, but not in the E-site, suggests that the E-site is either very weak or non-existent in the mitoribosome.13
Many interactions between the prokaryotic 23S rRNA and E-site tRNA involve contacts near the L1-BD.21 These interactions and the flexibility of the L1 arm (Fig. 9b, L1) suggest that E-site occupancy on the ribosome may be coupled to dynamic motions in this structure.25-27 The mitochondrial L1 protein is readily identifiable in the EM density, interacting with the shortened mitochondrial rRNA stem loop in domain V (Figs. 4b), but the protein is positioned far from the putative E-site (Fig. 9a). While this orientation may in part be due to the inherent flexibility of the L1-BD, the truncation of the rRNA may limit the range of motion. The EM density corresponding to L1 makes contacts with neighboring, unassigned protein density (Fig. 4b, marked with *), which may further restrict its mobility. Since the L1 protein is conserved in the mitoribosome, it is probably important for translation, but it may not have a direct role in tRNA binding.
Figure 9.
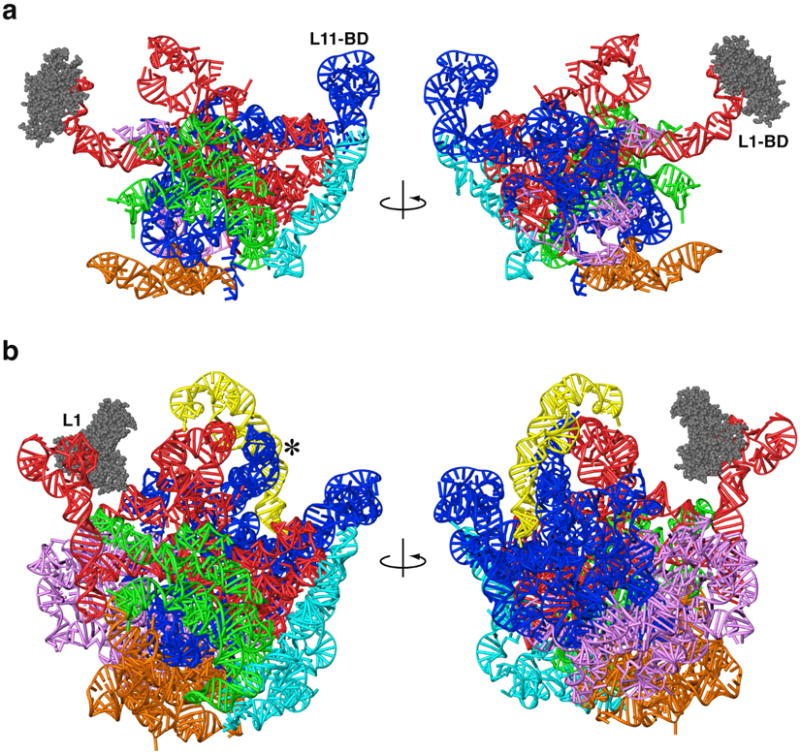
Three-dimensional models of mitochondrial and archaeal large ribosomal subunit rRNAs. Models are shown from the subunit-interface side (left) and from the solvent side (right). a. Mitochondrial rRNA, showing a dramatic reduction compared to archaea. b. H. marismortui rRNA from X-ray crystallography.30 The L1-arm (L1) was modeled using structural data from other crystallographic structures.21,38 The A-site finger (blue RNA helix adjacent to *) was modeled using sequence data and cryo-EM density from E. coli.67 Six domains in both models (a and b) are identified by different colors: I (purple), II (dark blue), III (orange), IV (green), V (red) and VI (light blue). The 5S rRNA in the archaeon (yellow) is absent in the mitoribosome. L1 proteins for both models are shown with space-filling representations (grey).
On the opposing side of the LSU of the mitoribosome, the spatial orientations of conserved structural domains near the periphery of the mitoribosome are conserved. Specifically, the L11-BD is positioned by a unique structure (when compared to Archaea, Fig. 9), because of sequence reduction in the region that connects the conserved cofactor binding domain and the core of the particle (Fig. 3, orange region in domain II). The crystal structure of mitochondrial EF-Tu in complex with GDP50 is similar to homologous bacterial structures from Escherichia coli51 and Thermus aquaticus52, which would suggest that the overall binding of cofactor with the ribosome in mitochondria is comparable to that in bacteria.
Unique mito-rRNA structures (ie. the sequence leading to the L11-BD) may also be stabilized by additional protein interactions found in the mitoribosome. The placement of 16 MRP models begins the assignment of structure to the increased protein mass in the large subunit of the mitoribosome. The unique MRP sequences suggest that their role in translation is likely specific to mitochondria. The unidentified protein densities are mostly localized to the peripheral regions of the large subunit, and all of the proteins synthesized by mitoribosomes are inserted into the inner mitochondrial membrane. Therefore, unique MRPs appear to be associated with positioning the mitoribosome during cotranslational insertion of nascent polypeptides into the membrane53 and/or stabilizing extended rRNA structures created by the large reductions in sequence.
The conservation of rRNA structure in bovine mitochondria does not directly correlate with sequence conservation. The overall base content is very G-poor and A-rich when compared to Archaeal (H. marismortui) and Bacterial (E. coli) species (Fig. 10a). While guanine is the most frequent base in these prokaryotic rRNAs (30%), it is sharply reduced in bovine mitochondria (18%). In contrast, adenine content (25% in prokaryotes) jumps to 38% in the mitochondrion. The fraction of nucleotides that are base paired drops from ∼60% in prokaryotes to 45% in mitochondria (Fig. 10b). This is partly due to the reduction in guanine content without a corresponding reduction in cytosines, since the latter have fewer prospective base pairing partners. The increase in adenine content also contributes to the decreased base pairing in the mitochondrial rRNA secondary structure (Fig. 10b), because adenines pair less frequently than other bases. For example, 62% of adenines are unpaired in the E. coli 16S rRNA, while only 30% of G, C and U bases are unpaired.35,54 An even larger fraction of adenines are unpaired in the E. coli 23S rRNA (Fig. 10b). The increased adenine content in the mitoribosome results in reduced base pairing and facilitates formation of two unique structural features: (1) “stretched” regions that reach across large distances to connect functional regions whose positions are not changed (e.g., the L11-BD, whose position is maintained at the periphery to interact with translation cofactors); and (2) large, unpaired loops at helix ends that may assume globular structures and serve as recognition elements for some of the MRP binding, e.g., the A-rich loops in domains II and III (grey in Fig. 3).
Figure 10.

Sequence composition of LSU rRNAs from Bos taurus mitochondrion (red), H. marismortui (blue) and E. coli (green). a. The mitochondrial rRNA exhibits a significant reduction in guanine (G) content and increase in adenine (A) content relative to H. marismortui (an archaeon) and E. coli (a eubacterium). b. The base paired fractions for each species. The frequency of base pairing is substantially lower in the mitochondrial ribosome, especially for cytosine.
It is not clear why evolutionary pressures lead to such a remarkable decrease in the size of the LSU mito-rRNA and a corresponding increase in protein content, but two structural principles have emerged from the present study. First, the key functional sites occupy essentially the same positions as in prokaryotic ribosomes. Second, the significant decrease in G content and increase in A content leads to a marked reduction in the fraction of base-paired nucleotides, yielding unique structures that can span large distances to maintain the three-dimensional organization of structures essential for translation.
Methods
Comparative Sequence Analysis
The rRNA sequences were aligned manually with the alignment editor AE2 (developed by T.Macke55). This program runs on SUN Microsystems computers on the Solaris operating system. Ribosomal RNA sequences are aligned by juxtapositioning the nucleotides that map to the same elements in the secondary and tertiary stucture models to the same columns in the alignment. The rRNA structure models were predicted with covariation analysis,35 a method that identifies a conserved set of base pairings and helices in a group of aligned sequences. The secondary structure diagrams for the B. taurus mitochondrial LSU rRNA were templated from previously predicted mammalian mitochondrial LSU rRNA structure models,56 and modified for unique features in the B. taurus structure. The structure diagrams were drawn with the interactive secondary structure program XRNA (B.Weiser and H.Noller, University of California, Santa Cruz).
RNA Homology Modeling
The model for the mito-rRNA is largely based on the crystal structure of the large subunit from Haloarcula marismortui30 (PDB accession code: 1JJ2). To begin, conserved RNA helices were identified based on the comparative sequence analysis. Having identified homologous regions in the structure, simple changes could be made in the cases where a base pair or an individual base (in a loop or bulge) could be changed to the corresponding nucleotide(s) found in the mitochondrial sequence (i.e. A-U pair changed to a C-G) using the Biopolymer module of the Insight-II software package (Molecular Simulations, Inc., San Diego, CA). Other changes involve mutations that result in non-canonical base pairing where normal base pairs are found in the H. marismortui structure. In still other cases, a canonical base pair may be found in the mitochondrial structure where a non-canonical pair exists in the Archaeal ribosome. For these cases, the pair found in the mitochondrial structure is superposed on the pair found in the H. marismortui structure. A round of energy minimization was used to satisfy the backbone geometry while preserving the hydrogen bond interactions between the base pairs.
For regions where a greater difference is found between the sequences, the mitochondrial rRNA were modeled based on previously characterized structures with similar or identical sequences.29 These include common motifs found in RNA tertiary structures;57 such as tetraloops,58 U-turns,59 and so on. For structures that are not as common, RNA structures previously determined by X-ray crystallography are used as a library, or database, for generating a three-dimensional model. Sometimes sequence comparisons suggested more than one possible structure. In such cases, fits to the cryo-EM density were used to determine which candidate was more feasible.
Additional/Alternate Secondary Structure Predictions
The Bos taurus mitochondrial rRNA sequence was taken from the genomic sequence NC_001567 using the Entrez genome database at NCBI.60 Potential secondary structures for regions of interest were predicted by mfold36 and the Alifold34 program in the Vienna RNA package. Default settings were used to predict secondary structure with mfold. Elements of secondary structure that were consistent across several thermodynamic predictions, levels of lineage, or both, were selected as potential secondary structures. These were used to predict three-dimensional structures that were validated or rejected by fitting to the cryo-EM density.
Multiple sequence alignments were created with the mitochondrial LSU rRNAs from Bos taurus and related organisms (Bovinae, Bovidae, Pecora, Ruminantia, and Cetarteriodactyla), as defined the NCBI Taxonomy Browser.60 The sequences were obtained using Entrez (nucleotide or genome) and aligned using ClustalW.61 Regions of interest were cut from each alignment and submitted to Alifold twice, once with a default covariance weight of 1, and again with a covariance weight of 10. In both cases isolated base pairs were allowed. Alternative secondary structures were modeled in three dimensions and then examined in the cryo-EM density map.
Protein Homology Modeling
The sequences of all 48 proteins of the mitoribosome6-8 were identified and searched against the non-redundant sequence database (nr) and the protein database (PDB) using the program BLASTP.62 MRPs homologous to ribosomal proteins in the same family were modeled for those cases where the crystal structure has been determined. ClustalW61 was used to create sequence alignments. We used the aligned sequences in the modeling program MODELLER663 to create structural models. Most of the templates selected for the modeling contained only the Cα carbon of each amino acid, due to limited resolution in the crystal structures. Therefore, the remaining atomic positions were extrapolated using probability density functions.63 This process can sometimes lead to positioning that is unfavorable, so some manual adjustments were made with the protein using Insight-II (Molecular Simulations, Inc., San Diego, CA). The models were optimized by steepest decent energy minimization to remove any unfavorable bonds, angles and steric conflicts. The structural characteristics of the models were then examined with PROCHECK.38 Regions that could not be modeled were further checked against the nr and PDB database to determine if homologous sequences or structures could be determined using just those short, unmodeled regions.
Determining protein interactions
A simple Python script was written to determine the interactions between RNA and proteins using various cutoff distances, based on the resolution of the crystal structure. This procedure works well for the H. marismortui structure, because the all-atom detail allows the use of a 3Å cutoff to determine specific hydrogen bonding pairs between RNA and protein atoms. Not all mammalian MRPs have a high level of homology to proteins in the Archaeal structure, so specific interactions cannot be determined for all proteins. But homologous proteins are also found in the crystal structures from Thermus thermophilus21 (PDB accession code: 1GIY), and Deinococcus radiodurans25 (PDB accession code: 1NKW). These structures contain only Cα coordinates for the proteins, and 1GIY provides only the phosphate positions for each nucleotide. Therefore, P – Cα distances were measured using a 6Å cutoff for homologous protein structures in these eubacterial complexes, providing a list of neighboring residues between the RNA and protein. MRP-L11 is homologous to Escherichia coli L11, so the all-atom crystal structure of the L11-RNA fragment64 (PDB accession code: 1KC8) was used to determine specific interactions (3Å cutoff) between the RNA and protein. Protein/protein interactions were analyzed using the same approach.
Once a list of interactions was determined for each protein model, restraints in the rigid-body Monte Carlo refinement were applied to pairs of pseudoatoms defining interacting residues in both the proteins and nucleic acid. Thirteen of the 16 proteins have interactions with the rRNA that could be expressed in such restraints, but L17, L19 and L24 did not have such interactions, due to deletions in the mitochondrial rRNA and MRP sequences.
Structure refinement
Cryo-EM density is incorporated as a structural restraint for refinement of the model in YAMMP, our in-house molecular modeling package.32 The vector lattice (VLAT) force field term defines the cryo-EM density as a three-dimensional potential, providing a score for the fit of the model to the density (documentation available at http://rumour.biology.gatech.edu). Refinement is done using the rigid body Monte Carlo module in YAMMP.
The mitochondrial rRNA was modeled using a reduced representation with one pseudoatom per nucleotide.33 The initial refinement was performed by treating the entire RNA homology model as a rigid unit, and it was subjected to 2,000,000 steps of Monte Carlo refinement with simulated annealing as a rigid body, starting at 1,000K with cooling to 10K.
A second round of refinement was used to optimize the local fit of each helix to the corresponding density. Each helix was treated as a separate rigid unit (52 units total, Fig. 2), and refinement was used to improve the total VLAT score. To maintain connectivity between helices along the RNA chain, harmonic bonds were included in the energy calculation as “tethers” between connected helices:
where Eβi denotes the energy of the ith bond; kβi is the force constants for the ith bond; βi is the ith bond; and βio is the corresponding equilibrium or ideal value. The ideal bond length is determined for each bond as the crystallographically determined distance between consecutive nucleotides in two rigid units. The force constant was set at 100 kcal/mol• Å2 for “normal” distances between consecutive homologous nucleotides (usually 6-7Å). In some cases gaps were present due to sequences that could not be modeled (grey regions, Fig. 2). A smaller force constant (1 kcal/mol•Å2) was used for these cases to allow more conformational freedom. A non-bond term was also added to the energy calculation to prevent interpenetration of the helices:
where Eij is the non-bond interaction energy between atoms i and j; kij is the non-bond force constant (set to 100 kcal/mol•Å2) for the atom pair ij; rij is the distance between atoms i and j; and rijo is the minimum distance allowed between the two atoms. The goal of the non-bond term is to provide volume exclusion so that double helices do not interpenetrate, and a value of rijo = 7.5Å was used to achieve this.
This second round of refinement was performed using multiple rigid-body Monte Carlo with simulated annealing. The starting temperature was set at 100K, with a final temperature of 10K reached after 2,000,000 steps. The final position of each rigid unit was accepted or rejected based on the energy score and by visual inspection with “O”.65 The original all-atom structures were superimposed onto the refined phosphate positions and covalently linked, except for the regions where gaps occur. A final round of energy minimization resolved small structural discrepancies caused by structural rearrangements during the refinement protocol.
Having placed the modeled rRNA structure in the density from EM, the proteins were then placed using a similar protocol, with the RNA structure held in a fixed position. Each protein was treated as an independent rigid body (17 rigid bodies total, 1 RNA and 16 proteins). The overall placement of the L7/12 dimer was based on an earlier X-ray study21 and was fit both as a dimer and as two monomers, to determine the structural organization that would best agree with the density and the conserved interactions between the two proteins. A consensus structure was determined from both methods. Bonds were included in the calculations using the previously determined P – Cα distances for conserved residues between the RNA and protein as the ideal bond length (βio) and a bond force constant (kβ) of 10 kcal/mol•Å2. The proteins were manually placed in positions consistent with previous ribosomal complexes. 1,000,000 steps of rigid body Monte Carlo with simulated annealing was performed over a range of starting temperatures (1000K, 100K, and 10K) because of the varied restraints associated with each protein. Those with more restraints could sample conformations at higher temperatures, while those with fewer restraints required lower temperatures to prevent extensive motions. All simulations were annealed to a final temperature of 1K. The final position for each protein structure was evaluated both visually using “O”65 and quantitatively from the final energy score.
Acknowledgments
We thank Jamie Cannone for help with generating the secondary structure diagrams. This work was funded by grants from the National Institutes of Health to S.C.H. (GM53827), R.R.G. (GM067317) and R.K.A. (GM61576), and from the Human Frontier Science Program to R.K.A. (RGY232003).
References
- 1.Attardi G. Animal mitochondrial DNA: an extreme example of genetic economy. Int Rev Cytol. 1985;93:93–145. doi: 10.1016/s0074-7696(08)61373-x. [DOI] [PubMed] [Google Scholar]
- 2.Chomyn A, Cleeter MW, Ragan CI, Riley M, Doolittle RF, Attardi G. URF6, last unidentified reading frame of human mtDNA, codes for an NADH dehydrogenase subunit. Science. 1986;234:614–8. doi: 10.1126/science.3764430. [DOI] [PubMed] [Google Scholar]
- 3.Brenner C, Kroemer G. Apoptosis. Mitochondria--the death signal integrators. Science. 2000;289:1150–1. doi: 10.1126/science.289.5482.1150. [DOI] [PubMed] [Google Scholar]
- 4.Wallace DC. Mitochondrial diseases in man and mouse. Science. 1999;283:1482–8. doi: 10.1126/science.283.5407.1482. [DOI] [PubMed] [Google Scholar]
- 5.Kurland CG. Evolution of mitochondrial genomes and the genetic code. Bioessays. 1992;14:709–14. doi: 10.1002/bies.950141013. [DOI] [PubMed] [Google Scholar]
- 6.O'Brien TW, Liu J, Sylvester JE, Mougey EB, Fischel-Ghodsian N, Thiede B, Wittmann-Liebold B, Graack HR. Mammalian mitochondrial ribosomal proteins (4). Amino acid sequencing, characterization, and identification of corresponding gene sequences. J Biol Chem. 2000;275:18153–9. doi: 10.1074/jbc.M909762199. [DOI] [PubMed] [Google Scholar]
- 7.Suzuki T, Terasaki M, Takemoto-Hori C, Hanada T, Ueda T, Wada A, Watanabe K. Structural compensation for the deficit of rRNA with proteins in the mammalian mitochondrial ribosome. Systematic analysis of protein components of the large ribosomal subunit from mammalian mitochondria. J Biol Chem. 2001;276:21724–36. doi: 10.1074/jbc.M100432200. [DOI] [PubMed] [Google Scholar]
- 8.Koc EC, Burkhart W, Blackburn K, Moyer MB, Schlatzer DM, Moseley A, Spremulli LL. The large subunit of the mammalian mitochondrial ribosome. Analysis of the complement of ribosomal proteins present. J Biol Chem. 2001;276:43958–69. doi: 10.1074/jbc.M106510200. [DOI] [PubMed] [Google Scholar]
- 9.Kenmochi N, Suzuki T, Uechi T, Magoori M, Kuniba M, Higa S, Watanabe K, Tanaka T. The human mitochondrial ribosomal protein genes: mapping of 54 genes to the chromosomes and implications for human disorders. Genomics. 2001;77:65–70. doi: 10.1006/geno.2001.6622. [DOI] [PubMed] [Google Scholar]
- 10.Yang D, Oyaizu Y, Oyaizu H, Olsen GJ, Woese CR. Mitochondrial origins. Proc Natl Acad Sci U S A. 1985;82:4443–7. doi: 10.1073/pnas.82.13.4443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gray MW, Burger G, Lang BF. The origin and early evolution of mitochondria. Genome Biol. 2001;2:REVIEWS1018. doi: 10.1186/gb-2001-2-6-reviews1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Patel VB, Cunningham CC, Hantgan RR. Physiochemical properties of rat liver mitochondrial ribosomes. J Biol Chem. 2001;276:6739–46. doi: 10.1074/jbc.M005781200. [DOI] [PubMed] [Google Scholar]
- 13.Sharma MR, Koc EC, Datta PP, Booth TM, Spremulli LL, Agrawal RK. Structure of the mammalian mitochondrial ribosome reveals an expanded functional role for its component proteins. Cell. 2003;115:97–108. doi: 10.1016/s0092-8674(03)00762-1. [DOI] [PubMed] [Google Scholar]
- 14.Koc EC, Burkhart W, Blackburn K, Moseley A, Koc H, Spremulli LL. A proteomics approach to the identification of mammalian mitochondrial small subunit ribosomal proteins. J Biol Chem. 2000;275:32585–91. doi: 10.1074/jbc.M003596200. [DOI] [PubMed] [Google Scholar]
- 15.Suzuki T, Terasaki M, Takemoto-Hori C, Hanada T, Ueda T, Wada A, Watanabe K. Proteomic Analysis of the Mammalian Mitochondrial Ribosome. Identification of protein components in the 28 S small subunit. J Biol Chem. 2001;276:33181–33195. doi: 10.1074/jbc.M103236200. [DOI] [PubMed] [Google Scholar]
- 16.O'Brien TW. Evolution of a protein-rich mitochondrial ribosome: implications for human genetic disease. Gene. 2002;286:73–9. doi: 10.1016/s0378-1119(01)00808-3. [DOI] [PubMed] [Google Scholar]
- 17.Youngman EM, Brunelle JL, Kochaniak AB, Green R. The active site of the ribosome is composed of two layers of conserved nucleotides with distinct roles in peptide bond formation and peptide release. Cell. 2004;117:589–99. doi: 10.1016/s0092-8674(04)00411-8. [DOI] [PubMed] [Google Scholar]
- 18.Sievers A, Beringer M, Rodnina MV, Wolfenden R. The ribosome as an entropy trap. Proc Natl Acad Sci U S A. 2004;101:7897–901. doi: 10.1073/pnas.0402488101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weinger JS, Parnell KM, Dorner S, Green R, Strobel SA. Substrate-assisted catalysis of peptide bond formation by the ribosome. Nat Struct Mol Biol. 2004;11:1101–6. doi: 10.1038/nsmb841. [DOI] [PubMed] [Google Scholar]
- 20.Cannone JJ, Subramanian S, Schnare MN, Collett JR, D'Souza LM, Du Y, Feng B, Lin N, Madabusi LV, uumlller KM, Pande N, Shang Z, Yu N, Gutell RR. The Comparative RNA Web (CRW) Site: an online database of comparative sequence and structure information for ribosomal, intron, and other RNAs. BMC Bioinformatics. 2002;3:2. doi: 10.1186/1471-2105-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yusupov MM, Yusupova GZ, Baucom A, Lieberman K, Earnest TN, Cate JH, Noller HF. Crystal structure of the ribosome at 5.5 A resolution. Science. 2001;292:883–96. doi: 10.1126/science.1060089. [DOI] [PubMed] [Google Scholar]
- 22.Agrawal RK, Penczek P, Grassucci RA, Frank J. Visualization of elongation factor G on the Escherichia coli 70S ribosome: the mechanism of translocation. Proc Natl Acad Sci U S A. 1998;95:6134–8. doi: 10.1073/pnas.95.11.6134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stark H, Rodnina MV, Rinke-Appel J, Brimacombe R, Wintermeyer W, van Heel M. Visualization of elongation factor Tu on the Escherichia coli ribosome. Nature. 1997;389:403–6. doi: 10.1038/38770. [DOI] [PubMed] [Google Scholar]
- 24.Mears JA, Cannone JJ, Stagg SM, Gutell RR, Agrawal RK, Harvey SC. Modeling a minimal ribosome based on comparative sequence analysis. J Mol Biol. 2002;321:215–34. doi: 10.1016/s0022-2836(02)00568-5. [DOI] [PubMed] [Google Scholar]
- 25.Harms J, Schluenzen F, Zarivach R, Bashan A, Gat S, Agmon I, Bartels H, Franceschi F, Yonath A. High resolution structure of the large ribosomal subunit from a mesophilic eubacterium. Cell. 2001;107:679–88. doi: 10.1016/s0092-8674(01)00546-3. [DOI] [PubMed] [Google Scholar]
- 26.Agrawal RK, Spahn CM, Penczek P, Grassucci RA, Nierhaus KH, Frank J. Visualization of tRNA movements on the Escherichia coli 70S ribosome during the elongation cycle. J Cell Biol. 2000;150:447–60. doi: 10.1083/jcb.150.3.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tama F, Valle M, Frank J, Brooks CL., 3rd Dynamic reorganization of the functionally active ribosome explored by normal mode analysis and cryo-electron microscopy. Proc Natl Acad Sci U S A. 2003;100:9319–23. doi: 10.1073/pnas.1632476100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gutell RR, Lee JC, Cannone JJ. The accuracy of ribosomal RNA comparative structure models. Curr Opin Struct Biol. 2002;12:301–10. doi: 10.1016/s0959-440x(02)00339-1. [DOI] [PubMed] [Google Scholar]
- 29.Tung CS, Joseph S, Sanbonmatsu KY. All-atom homology model of the Escherichia coli 30S ribosomal subunit. Nat Struct Biol. 2002;9:750–5. doi: 10.1038/nsb841. [DOI] [PubMed] [Google Scholar]
- 30.Ban N, Nissen P, Hansen J, Moore PB, Steitz TA. The complete atomic structure of the large ribosomal subunit at 2.4 Å resolution. Science. 2000;289:905–20. doi: 10.1126/science.289.5481.905. [DOI] [PubMed] [Google Scholar]
- 31.Bourne PE, Addess KJ, Bluhm WF, Chen L, Deshpande N, Feng Z, Fleri W, Green R, Merino-Ott JC, Townsend-Merino W, Weissig H, Westbrook J, Berman HM. The distribution and query systems of the RCSB Protein Data Bank. Nucleic Acids Res. 2004;32(Database issue):D223–5. doi: 10.1093/nar/gkh096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tan RKZ, Harvey SC. Yammp: Development of a molecular mechanics program using the modular programming method. J Comput Chem. 1993;14:455–70. [Google Scholar]
- 33.Malhotra A, Tan RK, Harvey SC. Modeling large RNAs and ribonucleoprotein particles using molecular mechanics techniques. Biophys J. 1994;66:1777–95. doi: 10.1016/S0006-3495(94)80972-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hofacker IL. Vienna RNA secondary structure server. Nucleic Acids Res. 2003;31:3429–31. doi: 10.1093/nar/gkg599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gutell RR, Weiser B, Woese CR, Noller HF. Comparative anatomy of 16-S-like ribosomal RNA. Prog Nucleic Acid Res Mol Biol. 1985;32:155–216. doi: 10.1016/s0079-6603(08)60348-7. [DOI] [PubMed] [Google Scholar]
- 36.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–15. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Suzuki T, Terasaki M, Takemoto-Hori C, Hanada T, Ueda T, Wada A, Watanabe K. Structural compensation for the deficit of rRNA with proteins in the mammalian mitochondrial ribosome. J Biol Chem. 2001;276:21724–36. doi: 10.1074/jbc.M100432200. [DOI] [PubMed] [Google Scholar]
- 38.Nikulin A, Eliseikina I, Tishchenko S, Nevskaya N, Davydova N, Platonova O, Piendl W, Selmer M, Liljas A, Drygin D, Zimmermann R, Garber M, Nikonov S. Structure of the L1 protuberance in the ribosome. Nat Struct Biol. 2003;10:104–8. doi: 10.1038/nsb886. [DOI] [PubMed] [Google Scholar]
- 39.Ogle JM, Carter AP, Ramakrishnan V. Insights into the decoding mechanism from recent ribosome structures. Trends Biochem Sci. 2003;28:259–66. doi: 10.1016/S0968-0004(03)00066-5. [DOI] [PubMed] [Google Scholar]
- 40.Matthews DE, Hessler RA, Denslow ND, Edwards JS, O'Brien TW. Protein composition of the bovine mitochondrial ribosome. J Biol Chem. 1982;257:8788–94. [PubMed] [Google Scholar]
- 41.Graack HR, Wittmann-Liebold B. Mitochondrial ribosomal proteins (MRPs) of yeast. Biochem J. 1998;329:433–48. doi: 10.1042/bj3290433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vila-Sanjurjo A, Ridgeway WK, Seymaner V, Zhang W, Santoso S, Yu K, Cate JH. X-ray crystal structures of the WT and a hyper-accurate ribosome from Escherichia coli. Proc Natl Acad Sci U S A. 2003;100:8682–7. doi: 10.1073/pnas.1133380100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Agrawal RK, Heagle AB, Penczek P, Grassucci RA, Frank J. EF-G-dependent GTP hydrolysis induces translocation accompanied by large conformational changes in the 70S ribosome. Nat Struct Biol. 1999;6:643–7. doi: 10.1038/10695. [DOI] [PubMed] [Google Scholar]
- 44.Agrawal RK, Lata RK, Frank J. Conformational variability in Escherichia coli 70S ribosome as revealed by 3D cryo-electron microscopy. Int J Biochem Cell Biol. 1999;31:243–54. doi: 10.1016/s1357-2725(98)00149-6. [DOI] [PubMed] [Google Scholar]
- 45.Diaconu M, Kothe U, Schlunzen F, Fischer N, Harms JM, Tonevitsky AG, Stark H, Rodnina MV, Wahl MC. Structural basis for the function of the ribosomal L7/12 stalk in factor binding and GTPase activation. Cell. 2005;121:991–1004. doi: 10.1016/j.cell.2005.04.015. [DOI] [PubMed] [Google Scholar]
- 46.Steinberg S, Cedergren R. Structural compensation in atypical mitochondrial tRNAs. Nat Struct Biol. 1994;1:507–10. doi: 10.1038/nsb0894-507. [DOI] [PubMed] [Google Scholar]
- 47.Hanada T, Suzuki T, Yokogawa T, Takemoto-Hori C, Sprinzl M, Watanabe K. Translation ability of mitochondrial tRNAsSer with unusual secondary structures in an in vitro translation system of bovine mitochondria. Genes Cells. 2001;6:1019–30. doi: 10.1046/j.1365-2443.2001.00491.x. [DOI] [PubMed] [Google Scholar]
- 48.Steinberg S, Leclerc F, Cedergren R. Structural rules and conformational compensations in the tRNA L-form. J Mol Biol. 1997;266:269–82. doi: 10.1006/jmbi.1996.0803. [DOI] [PubMed] [Google Scholar]
- 49.Vacano E, Hagerman PJ. Analysis of birefringence decay profiles for nucleic acid helices possessing bends: the tau-ratio approach. Biophys J. 1997;73:306–17. doi: 10.1016/S0006-3495(97)78071-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Andersen GR, Thirup S, Spremulli LL, Nyborg J. High resolution crystal structure of bovine mitochondrial EF-Tu in complex with GDP. J Mol Biol. 2000;297:421–36. doi: 10.1006/jmbi.2000.3564. [DOI] [PubMed] [Google Scholar]
- 51.Song H, Parsons MR, Rowsell S, Leonard G, Phillips SE. Crystal structure of intact elongation factor EF-Tu from Escherichia coli in GDP conformation at 2.05 A resolution. J Mol Biol. 1999;285:1245–56. doi: 10.1006/jmbi.1998.2387. [DOI] [PubMed] [Google Scholar]
- 52.Kjeldgaard M, Nissen P, Thirup S, Nyborg J. The crystal structure of elongation factor EF-Tu from Thermus aquaticus in the GTP conformation. Structure. 1993;1:35–50. doi: 10.1016/0969-2126(93)90007-4. [DOI] [PubMed] [Google Scholar]
- 53.Stuart R. Insertion of proteins into the inner membrane of mitochondria: the role of the Oxa1 complex. Biochim Biophys Acta. 2002;1592:79–87. doi: 10.1016/s0167-4889(02)00266-5. [DOI] [PubMed] [Google Scholar]
- 54.Gutell RR, Cannone JJ, Shang Z, Du Y, Serra MJ. A story: unpaired adenosine bases in ribosomal RNAs. J Mol Biol. 2000;304:335–54. doi: 10.1006/jmbi.2000.4172. [DOI] [PubMed] [Google Scholar]
- 55.Larsen N, Olsen GJ, Maidak BL, McCaughey MJ, Overbeek R, Macke TJ, Marsh TL, Woese CR. The ribosomal database project. Nucleic Acids Res. 1993;21:3021–3. doi: 10.1093/nar/21.13.3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cannone JJ, Subramanian S, Schnare MN, Collett JR, D'Souza LM, Du Y, Feng B, Lin N, Madabusi LV, Muller KM, Pande N, Shang Z, Yu N, Gutell RR. The Comparative RNA Web (CRW) Site: an online database of comparative sequence and structure information for ribosomal, intron, and other RNAs: Correction. BMC Bioinformatics. 2002;3:15. doi: 10.1186/1471-2105-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hermann T, Patel DJ. Stitching together RNA tertiary architectures. J Mol Biol. 1999;294:829–49. doi: 10.1006/jmbi.1999.3312. [DOI] [PubMed] [Google Scholar]
- 58.Woese CR, Winker S, Gutell RR. Architecture of ribosomal RNA: constraints on the sequence of “tetra- loops”. Proc Natl Acad Sci U S A. 1990;87:8467–71. doi: 10.1073/pnas.87.21.8467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gutell RR, Cannone JJ, Konings D, Gautheret D. Predicting U-turns in ribosomal RNA with comparative sequence analysis. J Mol Biol. 2000;300:791–803. doi: 10.1006/jmbi.2000.3900. [DOI] [PubMed] [Google Scholar]
- 60.Wheeler DL, Church DM, Edgar R, Federhen S, Helmberg W, Madden TL, Pontius JU, Schuler GD, Schriml LM, Sequeira E, Suzek TO, Tatusova TA, Wagner L. Database resources of the National Center for Biotechnology Information: update. Nucleic Acids Res. 2004;32(Database issue):D35–40. doi: 10.1093/nar/gkh073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–80. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 63.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 64.Conn GL, Draper DE, Lattman EE, Gittis AG. Crystal structure of a conserved ribosomal protein-RNA complex. Science. 1999;284:1171–4. doi: 10.1126/science.284.5417.1171. [DOI] [PubMed] [Google Scholar]
- 65.Jones TA, Zou JY, Cowan SW, Kjeldgaard Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A. 1991;47(Pt 2):110–9. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 66.Kim SH, Quigley GJ, Suddath FL, McPherson A, Sneden D, Kim JJ, Weinzierl J, Rich A. Three-dimensional structure of yeast phenylalanine transfer RNA: folding of the polynucleotide chain. Science. 1973;179:285–8. doi: 10.1126/science.179.4070.285. [DOI] [PubMed] [Google Scholar]
- 67.Gabashvili IS, Agrawal RK, Spahn CM, Grassucci RA, Svergun DI, Frank J, Penczek P. Solution structure of the E. coli 70S ribosome at 11.5 Å resolution. Cell. 2000;100:537–49. doi: 10.1016/s0092-8674(00)80690-x. [DOI] [PubMed] [Google Scholar]