

Conspectus

The DNA duplex is an exquisite macromolecular array that stores genetic information to encode proteins and regulate pathways, but its unique structure imparts chemical function that allows it also to mediate charge transport (CT). We have utilized diverse platforms to probe DNA CT, using spectroscopic, electrochemical, and even genetic methods. These studies have established powerful features of DNA CT chemistry. DNA CT can occur over long molecular distances as long as the bases are well stacked; perturbations in base stacking as arise with single base mismatches, DNA lesions, and the binding of some proteins that kink the DNA, all serve to inhibit DNA CT. Significantly, single molecule studies of DNA CT show that ground state CT can occur over 34 nm as long as the duplex is well stacked; one single base mismatch inhibits CT. The DNA duplex is an effective sensor for the integrity of the base pair stack. Moreover the efficiency of DNA CT is what one would expect for a stack of graphite sheets, equivalent to the stack of DNA base pairs, and independent of the sugar-phosphate backbone.
Since DNA CT offers a means to carry out redox chemistry from a distance, we have considered how this chemistry might be used for long range signaling in a biological context. We have taken advantage of our chemical probes and platforms to characterize DNA CT also in the context of the cell. CT can occur over long distances, perhaps funneling damage to particular sites and insulating others from oxidative stress. Significantly, transcription factors that activate the genome to respond to oxidative stress can also be activated from a distance through DNA CT.
Numerous proteins work to maintain the integrity of the genome and increasingly they have been found to contain [4Fe-4S] clusters that do not appear to carry out either structural or enzymatic roles. Using electrochemical methods, we find that DNA binding shifts the redox potentials of the clusters, activating them towards oxidation at physiological potentials. We have proposed a model describing how repair proteins may utilize DNA CT to efficiently search the genome for lesions. Importantly, many of these proteins are in low copy number, and thus a processive mechanism is insufficient to explain how they find and repair lesions before the cell divides. Using atomic force microscopy and genetic assays, we find that repair proteins proficient at DNA CT are able to relocalize in the vicinity of DNA lesions and, within the cell, they cooperate in their repair of lesions. Conversely, proteins defective in DNA CT cannot relocalize in the vicinity of lesions and do not provide help to other proteins involved in repair within the cell; moreover these genetic defects are associated with disease in human protein analogues. As we continue to unravel this chemistry and discover more proteins with redox cofactors involved in genome maintenance, we are learning more regarding opportunities for long range signaling and sensing, and more examples of DNA CT chemistry that may play critical roles within the cell.
Introduction
The structure of double helical DNA, with two dynamic strands of complementary bases that encode a meaningful sequence, has long been considered as an elegant construct for the storage, expression, and transmission of the genetic instructions of life. Beyond sequence, however, we are just beginning to understand how the electronic properties of this beautiful macromolecular array may serve also an important role in directing biological processes. Since the initial suggestions of DNA conductivity, 1numerous studies have confirmed and expanded our understanding of this chemistry, termed DNA-mediated charge transport (DNA CT). We have observed that DNA is a highly effective conductor of charge and it is the overlapping π orbitals of the stacked bases that function as the path for this conductivity. DNA CT requires electronic coupling of the donor and acceptor to the π-stack, and the conductivity from a donor to an acceptor is highly sensitive to the structural integrity of the stacked path of intervening bases. Indeed, DNA CT reports upon the integrity of the base pair stack. The rate of DNA CT can be very fast, occurring on a picosecond timescale, but this rate is gated by the dynamical motions of the bases and those of the donor and acceptor, as they move in and out of CT-active conformations.2 Significantly, as long as the array is well stacked, the distance dependence of this rate is extremely shallow, allowing DNA CT to occur efficiently over hundreds of base pairs, and likely much farther.
We and others have now thoroughly characterized DNA CT through photooxidation studies, spectroscopy, and electrochemistry.2–4 DNA CT has been probed in solution, on surfaces, and with single molecule techniques, using a variety of donors and acceptors. Here we describe some of these key studies. Importantly, from this solid foundation, we are now also in a position to ask the question that puts this fundamental characteristic of DNA into context: How might Nature utilize DNA CT in biology?
Diverse Platforms Reveal the Characteristics of DNA CT
Just like the characteristics of gene expression and inheritance, the characteristics of DNA CT originate from the unique structure of DNA, the overlapping π orbitals of the stacked nucleotide bases. Early on, it was noted that the aromatic bases of DNA, the very bases that encode genetic information, stack next to each other with an interplanar spacing similar to graphite and thus, like sheets of graphite, could form a conductive path of overlapping π orbitals that extends down the helical axis (Figure 1).1 However, a critical feature that distinguishes the DNA duplex from solid π-stacked materials is that DNA is a macromolecular array in solution, with dynamical changes in π-stacking that occur on the picosecond-millisecond time scales.
Figure 1.
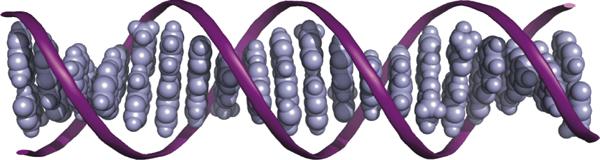
The structure of DNA facilitates charge transport. The aromatic bases (blue) of DNA stack with each other like a pile of coins and are wrapped by a sugar phosphate backbone (purple ribbon). The overlapping π orbitals of these stacked bases form a conductive core down the helical axis that mediates the flow of charge. This structure resembles that of stacked graphite sheets and, indeed, undamaged, well stacked DNA is found to have the same conductivity as that measured perpendicular to graphite.6 Importantly, the conformation of this structure is not static; dynamic motions within this macromolecular array contribute to the mechanistic complexity of DNA CT.
Several of the platforms we have developed to probe DNA CT are illustrated in Figure 2. We have examined DNA CT using photophysical methods, appending donors and acceptors to either end of the DNA duplex. We have examined long-range oxidative damage to DNA using DNA-binding photooxidants to promote oxidation of guanines in the duplex from a distance. Additionally we have probed DNA CT in the ground state, both in electrochemical studies and in single molecule nanoscale devices. Remarkably, despite the diversity of techniques, we have observed the same features: DNA CT can occur over long molecular distances, but DNA CT is extremely sensitive to perturbations in stacking with and across the duplex.
Figure 2.
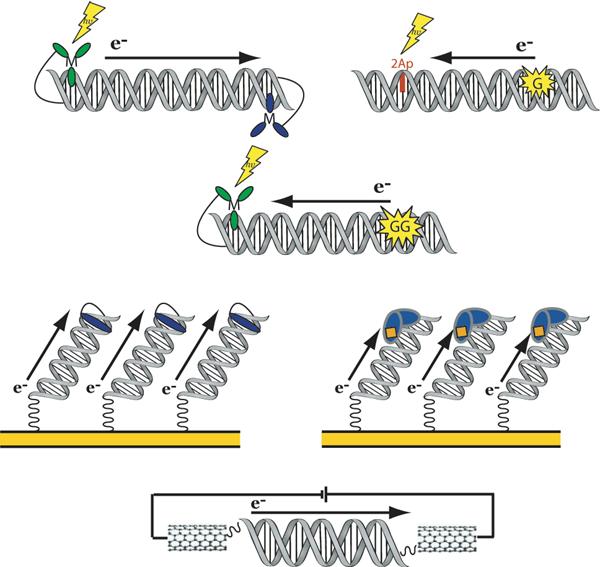
Platforms for the study of DNA CT. Top row, spectroscopic solution platforms: (left) photoactivated luminescence and quenching between a covalent metallointercalator and acceptor pair and (right) photoactivated fluorescence of base analog 2-aminopurine and quenching by guanine. Second row, a biochemical solution platform: photoactivated oxidation of guanine doublets by a covalently bound metallointercalator. Third row, electrochemical surface platforms: (left) DNA-modified electrodes with a covalent redox probe and (right) DNA-modified electrodes with a bound protein that contains a redox-active cofactor. Fourth row, a single molecule platform: covalent attachment of DNA across a gap in a carbon nanotube device.
Coupling to the DNA π-stack
In order to build effective platforms for study, we quickly identified the first, critical characteristic of this process: DNA CT requires effective electronic coupling of the donor and acceptor to the base pair π-stack. Our first platform to measure DNA CT utilized metallointercalators that were covalently attached to 15-mer DNA duplexes in solution.2 The donor and acceptor, Ru(phen)2dppz2+ and Rh(phi)2phen3+, both have aromatic ligands that allow them to intercalate and interact directly with the DNA π-stack. When the donor is photoexcited, complete quenching occurs due to rapid CT to the acceptor. This quenching occurs only when the donor and acceptor are bound to the same duplex and does not occur when Ru(phen)2(phen′)2+, a poor intercalator, is substituted for the acceptor. This result illustrates that DNA CT is necessarily an intraduplex process, that donors and acceptors can access through effective coupling into the base pair π-stack. In a later variation on this platform, we used modified bases to examine photoinduced CT. Here too it was evident that coupling with the base stack is key.5
Sensitivity to Perturbations of the DNA π-stack
Studies with our solution-based platforms also revealed a second important characteristic: DNA CT is highly sensitive to the integrity of the π-stack of the bases between the donor and the acceptor. We observed that not only is the quenching of a covalent, intercalating donor and acceptor pair lost when the double helix is melted, even the introduction of a single base mismatch between the donor and acceptor severely decreases the quenching yield. This sensitivity to perturbations of the π-stack was perhaps best illustrated recently in single-molecule studies of DNA CT in a carbon nanotube device (Figure 3).6 In these experiments, a DNA oligonucleotide functionalized at its 3′ and 5′ ends with alkyl amines is made to covalently bridge an etched gap in a carbon nanotube circuit using amide chemistry6, and a well matched complementary strand is floated into the device to form a duplex. The current flow in the device may be compared to that in the carbon nanotube before etching the gap. With the well matched duplex, significant current is obtained. We can however, then, wash away the non-covalently associated complementary strand and replace it with a strand containing a single base change to yield a C:A mismatch; then the current decreases ~100-fold. We can then replace the strand with another to generate a G:T mismatch. In this case, the duplex is thermodynamically as stable as if there were an AT base pair at that position, yet the decreased current flow through the device remains. However, when we wash off the mismatched strand and then float in the well matched complement, the full current is restored. The attenuation in ground state CT is associated with the presence of the single base mismatch. Using this platform to measure the conductivity of properly stacked DNA, we were also able to validate that the base pair stack is very similar to that for layered sheets of graphite over the same gap.
Figure 3.
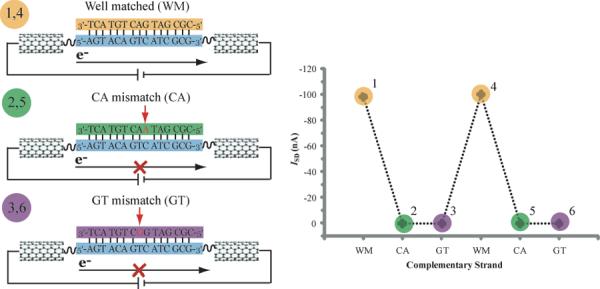
Single molecule experiments illustrate the sensitivity of DNA CT to mismatches. Left illustration: DNA CT is measured in single molecules of DNA that covalently bridge a gap in a carbon nanotube device. One strand (blue) is covalently attached by its 3′ and 5′ ends while the other strand can be freely exchanged between well matched complements (orange) and strands that introduce a single base mismatch (green, purple). Right plot: the device was connected with a series of well matched and mismatched strands and the source-drain current (ISD) measured at the gating voltage VG = −3 V is shown for each. The colors and numbers of the points in the series correspond to the different strands in the left illustration. This result clearly shows that current through the device is cut off in duplexes that contain a single base mismatch and restored when the DNA in the gap is re-hybridized with its well matched complement. Adapted with permission from reference 6.
This exquisite sensitivity in DNA CT to perturbations in stacking provided the basis for our development of ground state DNA CT-based sensors.7 In this platform, DNA duplexes are attached to gold electrodes by an alkane-thiol linker and are modified on the distal end with a redox-active probe molecule.7–9 Importantly, as in solution studies, the redox probe must be associated with the DNA in a way that allows for effective electronic coupling to the π-stack in order to report a signal that is DNA-mediated. Thus, this platform provides sensitive feedback about the integrity of the DNA π-stack between the electrode surface and the redox probe; even small structural perturbations are detected by the resulting attenuation in the redox signal. We have used this platform to detect a variety of distortions to the structure of the DNA π-stack including all base mismatches, a variety of base lesions, and protein binding.7–10 Importantly, it is specifically perturbations to the stacked bases that inhibit the flow of charge; we can even introduce nicks in the intervening DNA, breaking the sugar-phosphate backbone, as long as the base pair stack is unperturbed.
Shallow Distance Dependence
In these studies was revealed also another critical characteristic of this chemistry: the distance dependence is very shallow. To probe the distance dependence of DNA CT, we developed a platform that allows for the measurement in solution of long-range guanine oxidation from a distal, covalent metallointercalator photooxidant; after irradiation, the location of DNA-mediated photooxidation in the duplex is determined by biochemical sequencing.11, 12 Irrespective of the DNA-bound photooxidant, we observed significant oxidative damage at the 5′-G of guanine doublets, the site of lowest oxidation potential in the DNA. Indeed, in an oligonucleotide duplex with the Rh intercalator tethered to one end, we observed oxidation not only at the 5′-G of the guanine doublet in the center of the duplex but also at the 5′-G of the guanine doublet located near the distal end of the duplex. With this platform we observed photooxidation that was insensitive to the separation distance up to the longest distance measured, 20 nm (60 bp).12 Moreover, what was key to generating oxidative damage to DNA from a distance was the integrity of the intervening stack, independent of the DNA-bound photooxidant employed.
More recently, we examined ground state DNA CT electrochemically in a multiplexed chip platform. Using this platform we measured DNA CT to a distal redox probe over 34 nm (100 bp).8 We found that, remarkably, the redox signal size and the degree of signal attenuation from a mismatched base was the same as what is observed for much smaller (17 bp) duplexes. Also, like the shorter duplexes, we determined that the rate of DNA CT through the 100-mer is still limited by the tunneling rate through the alkane-thiol linker. Interestingly, this 100-mer DNA was among the longest documented molecular wires (34 nm).
DNA CT in a Biological Context: Long-range signaling
Given that CT chemistry can occur over long molecular distances and is so sensitive to intervening mismatches and lesions that perturb the base pair stack, we have begun to explore how DNA CT might be exploited within the cell. We have considered several ways in which DNA-mediated CT could act as a conduit to funnel damage to distant sites, trigger transcription of genes to activate cellular defense pathways, or even provide a platform for DNA-bound repair proteins to signal one another in their search for lesions.
In early studies, we addressed these topics by probing hole migration in the nucleus of the cell, in functioning mitochondria, and in a nucleosome core particle.4,13 Our results were consistent with the idea that long-range CT, mediated via π-stacking of the base pairs, can proceed across regions of DNA in the cell that would otherwise be inaccessible, generating damage at remote points from the site of the tethered oxidant. We also saw that proteins may modulate DNA CT. TATA-binding proteins, for example, kink DNA duplexes at their recognition sites, attenuating CT,9 while other proteins do not perturb stacking when they bind; and facilitate CT by stabilizing the duplex. In fact, even in a nucleosome containing a tethered photooxidant, we observed damage at distant sites in a pattern equivalent to that in the DNA in the absence of bound histones.13 Certainly, then, while we may consider that DNA packaged in the nucleosome is protected from some kinds of damage, it is not the case that it is protected from long-range oxidative damage through DNA CT.
Activating Transcription (SoxR)
A variety of signaling cascades are triggered by reactive oxygen species. In enteric bacteria, SoxR regulates this response by activating transcription of soxS, which then enhances the expression of genes required for defense. Each monomer of SoxR contains a [2Fe-2S] cluster that is not required for protein folding.14 Though SoxR (dimer) has a similar affinity for its promoter in the apo, oxidized [2Fe-2S]2+, and reduced [2Fe-2S]1+ forms, only the oxidized protein activates transcription. When the bacterium is undergoing oxidative stress, guanine radicals are generated, and we asked if these radicals might be considered a first signal to activate the genome to respond.
We first used DNA electrochemical studies to see whether the redox cofactors of SoxR could be accessed in a DNA-mediated reaction and to determine the DNA-bound potential.15 We found using our DNA electrodes that there is ~500 mV positive shift in potential when SoxR binds DNA, so that DNA binding actually turns SoxR into the oxidation sensor for the genome. But can guanine radicals serve as the oxidant? We showed first that reduced SoxR inhibits guanine damage by filling the radical hole with its own electron, resulting in the oxidized form of the protein; there is, however, no attenuation in damage with oxidized SoxR. We then examined the SoxR response to DNA damage in E. coli cell cultures. When cells were treated with Rh(phi)2(bpy)3+, our intercalating photooxidant, we found enhanced expression levels of the soxS RNA product. We then tethered the photooxidant to a 180-mer duplex of DNA containing the SoxR binding site and the SoxS promoter to see if we could activate transcription from a distance anaerobically by DNA CT.16 Notably, the transcription product was only observed upon irradiation of samples containing reduced SoxR and Rh-tethered DNA, which were positioned 270 Å from each other (Figure 4). While these data do not prove that guanine radicals are the oxidant for SoxR within the cell, these data do establish that guanine radicals are capable of oxidizing SoxR from a distance, activating the genome to respond.
Figure 4.
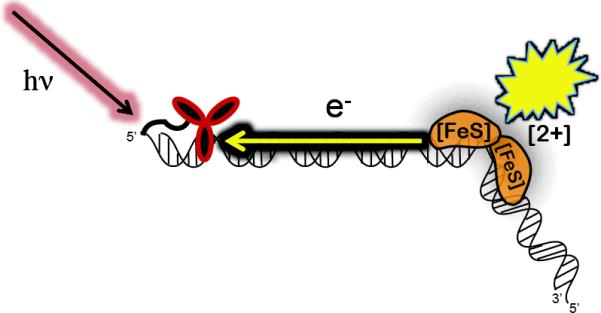
DNA CT triggers transcription. We have utilized a tethered metal photooxidant to oxidize bound SoxR protein (orange) from a distance.16
Thus, CT over long ranges may play many roles across the genome, funneling damage to some locations, insulating other sites from damage through stacking perturbations, even activating transcription from a distance to quickly defend the cell from oxidative stress.4 It is clear that in a biological context, DNA CT may “wear many hats.”
DNA Charge Transport in Repair
A powerful characteristic of DNA CT that we observed through several platforms was the ability of DNA CT to report on the integrity of the DNA. DNA CT chemistry can easily distinguish base pair mismatches and lesions from well-matched bases. It seemed therefore important to consider whether DNA CT might play some role in the cell in the search for DNA damage by repair proteins. Could repair proteins utilize DNA CT in signaling damage across the genome? This question became even more intriguing to consider given the fact that a subset of DNA repair proteins had been found to contain [4Fe-4S] clusters, a common redox cofactor in bioinorganic chemistry, and these cofactors were conserved in archaea, bacteria, and man.
We thus began to examine metalloproteins from the base excision repair (BER) pathway. The proteins from the BER network recognize and repair DNA bases that have been damaged by oxidative stress.17 Left unrepaired, these lesions threaten the integrity of the genome. We first employed electrochemical techniques to examine whether DNA damage products found in nature influence DNA-mediated signaling.4, 7 Just as we established that single base mismatches inhibit DNA CT, as they disrupt base pair stacking, common base lesions have this effect as well. How might BER proteins exploit this unique property in the context of the cell? Notably, a subset of BER proteins contain [4Fe-4S]2+ clusters that are not required for the protein to fold into a native conformation.17 Having no clear structural role, we wondered: why are the iron-sulfur cofactors present? Crystal structures (with DNA) of two BER proteins from E. coli, MutY and Endonuclease III (EndoIII), have revealed the proximity of the [4Fe-4S] cluster to the DNA backbone.18 Importantly, mutations at residues near the cluster in the human homologues of these proteins have been shown to lead to a predisposition toward colorectal cancer. When we examined EndoIII and MutY on DNA electrodes, we observed midpoint potentials of ~80 mV vs. NHE.4, 19 Not only are these DNA-bound potentials characteristic of high-potential iron proteins, they are also just in the range required for a biological redox switch. Furthermore, we observed a large negative shift in potential (>−200 mV) as the protein bound DNA. The shift in potential for the 2+/ 3+ couple for the [4Fe-4S] cluster necessitates that these proteins have a much higher affinity (Kd:1000 fold) for DNA when oxidized.
Thus, we proposed a model for how these proteins could cooperate in the first step of repair4, 19: when a BER protein is in the vicinity of DNA, it is activated toward oxidation (i.e. by a guanine radical). As the protein binds to DNA, with a shift in potential, it then releases an electron that travels through an intact base pair stack to a distally bound protein that gets reduced, loses its affinity for DNA, and dissociates. This process persists so long as the intervening region of DNA is free of lesions. In fact, this process corresponds to a scan of this region of the genome; as long as the intervening DNA is intact, electron transfer through the DNA can proceed, promoting the dissociation of the repair proteins away from the well matched strands. When a lesion is present, however, CT is attenuated, interrupting the signal; the repair proteins can no longer be reduced with concomitant dissociation. Thus, the second protein never receives the signal and remains bound, slowly proceeding toward the site of the lesion (Figure 5). Importantly, the two proteins must have similar DNA-bound potentials to cooperate in this search. The driving force for the redistribution of repair proteins near lesions and away from intact DNA is the concentration of oxidized protein, generated as a result of oxidative stress.
Figure 5.
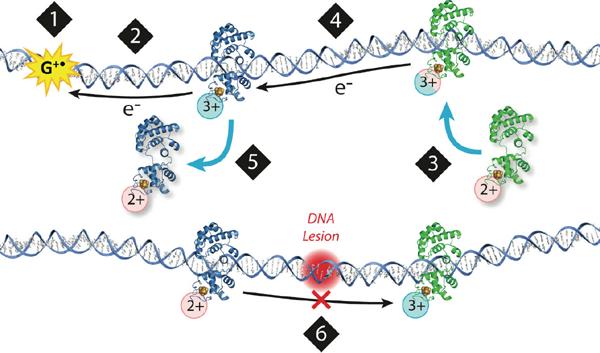
Model for a DNA-mediated search by repair proteins. 1) When the cell undergoes oxidative stress, guanine radicals are formed, triggering a repair protein to bind DNA. 2) DNA-binding protein is oxidized, releasing an electron that repairs the guanine radical. 3) Another repair protein binds to a distant site. As it binds to DNA, there is a shift in the redox potential of the protein, making it more easily oxidized. 4) The protein could then send an electron through the DNA base pair stack that travels to a distally bound protein, scanning the intervening region for damage. 5) If the base pair stack is intact, charge transport occurs between proteins. The repair protein that receives the electron is reduced and dissociates. 6) If a lesion is present (red), charge transport is attenuated, and the repair proteins will remain bound in the oxidized form and slowly proceed to the site of damage.
We have carried out calculations to determine the distances over which these CT's must occur for this to be beneficial and lead to a more efficient search process.19 Without invoking DNA CT, simple calculations, using facilitated protein diffusion, the copy numbers measured for these proteins, and ignoring protein traffic on the strand, indicate that the genome search time in E. coli is significantly longer than the lifetime of the bacterium! But including DNA CT over even a few hundred base pairs is sufficient to rapidly improve the efficiency of the search, and these distances for DNA CT have been well documented.
Signaling Leads to Redistribution
Based on our model for the search process, the repair proteins redistribute in the vicinity of lesions, and the search is reduced to the neighborhood of the lesion, eliminating vast populations of intact DNA. We have utilized single-molecule atomic force microscopy (AFM) to assay for the redistribution of EndoIII onto strands containing a single C:A mismatch.19 DNA strands for the AFM assay were prepared via PCR with primers that were designed to generate two short duplexes (~1.9 kbp), that are then ligated to yield a long strand (~3.8 kbp) with a C:A mismatch in the middle of the sequence. Importantly, the C:A mismatch attenuates CT, but it is not a specific substrate for EndoIII. Based on our search model, we would expect that the proteins redistribute onto the mismatched strands. In fact, we find a greater density of EndoIII on the mismatched long strands of DNA. Also consistent with the model, when both long and short strands are matched, there is no accumulation of EndoIII on the longer strands. As the model predicts, DNA-mediated CT thus drives proteins from intact regions to cluster in the vicinity of mismatched sites or lesions. Additionally, we found the redistribution of EndoIII was more pronounced with oxidative stress, suggesting that oxidized protein can further expedite the search for lesions.
Helper Function Assay to Probe DNA CT in the Cell
Since our model requires that charge travels between two redox-active proteins that are bound to DNA, it is possible that proteins that exhibit similar midpoint potentials, even if they are from distinct pathways and perform different functions, could signal one another. In this regard, the two proteins help one another in their search. We can test this genetically by assaying whether the in vivo activity of one repair protein is aided by the presence of another.19 This assay for “helper function” employed a strain, CC104, that contains a single cytosine to adenine substitution in the lacZ Glu 461 codon (lac−). Since MutY can prevent GC to TA transversions by excising adenine mispaired with 8-oxo-G, reversion of the CC104 strain from lac− to lac+ is indicative of a decrease in the ability of MutY to find and repair its substrate. Consistent with our model, we find that when EndoIII is knocked out of the CC104 strain (nth−) MutY activity decreases.19 We then complemented the nth− strain with a plasmid encoding a glycolytically inactive mutant of EndoIII (D138A), and in this case MutY activity was restored; though this mutant could not perform the excision reaction, it still maintained a [4Fe-4S] cluster and thus could carry out DNA CT signaling. To establish a link with DNA CT we also complemented the nth− strain with a plasmid encoding the Y82A mutant that we had shown was defective in CT. In this case there was no restoration of MutY activity (Figure 6). Thus, our results from the assay for helper function indicate that EndoIII and MutY help one another in the search for damage as long as the proteins can perform DNA-mediated CT. Significantly, we also tested Y82A and other EndoIII mutants in our AFM assay and here we established the clear correlation between the ability to redistribute on the mismatched strand and the ability to carry out DNA CT electrochemically. Thus, through the Y82A mutant, we actually established a link between the ability to carry out DNA CT on an electrode, in the cell, and also to find the mismatched lesion by AFM.
Figure 6.
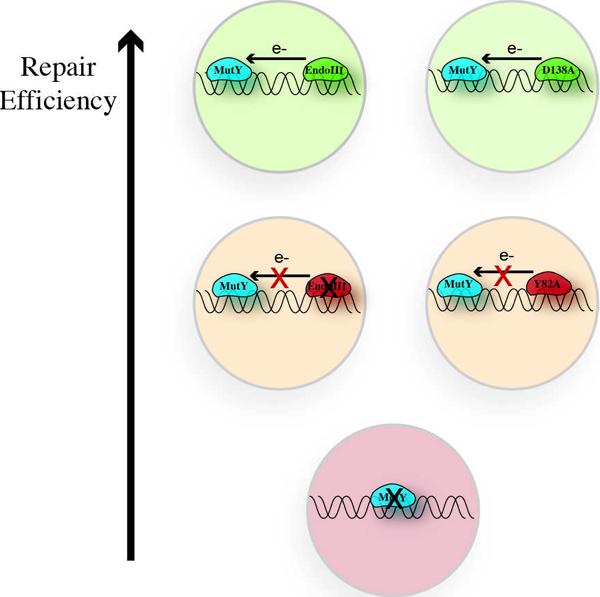
Schematic illustrating the helper function assay to monitor MutY activity. When EndoIII or D138A, an EndoIII mutant that is CT proficient but glycolytically active, is available to help in the search for damage, MutY repairs lesions efficiently (green). If EndoIII is knocked out or Y82A, a mutant deficient in CT, is present, MutY efficiency decreases (yellow). When MutY is knocked out, repair is not observed (red).
Repair Proteins from Different Organisms are Proficient at DNA CT
The search for damage in the genome is not a process limited to BER glycosylases. And curiously, neither is the presence of [4Fe-4S] clusters in these DNA-binding proteins. Proteins from a wide variety of organisms such as E. coli and archaea are being identified as DNA-binding proteins that contain [4Fe-4S] clusters with no obvious enzymatic role.20 For example, XPD, an archaeal protein, is a 5′–3′ helicase that is part of the TFIIH machinery and required for nucleotide excision repair. Mutations in XPD in humans are moreover associated with a host of diseases associated with poor DNA repair: trichothiodystrophy, xeroderma pigmentosum, premature aging, and early onset of cancers.20–22 Interestingly, recently three crystal structures were determined for XPD's from archaea, and while the structures were quite close for the three helicases, two contained the [4Fe-4S] clusters and one did not. Clearly the clusters are not necessary to maintain the protein structure and are relatively labile.
We then tested XPD from Sulfolobus acidocaldarious electrochemically to determine its DNA-bound potential.10 Remarkably, we obtained the same value, ~80 mV versus NHE, that we had seen with the BER enzymes. But XPD is an ATP-dependent helicase, and when we added ATP, we observed an increase in the XPD redox signal dependent upon the ATP concentration. It was apparent that we could monitor helicase function electrically. This electronic effect became more clear upon examination of mutants prepared based upon their association with human disease. The G34R mutation, which blocks ATP binding and thus shows poor ATPase activity, shows a slow increase in XPD signal with ATP when monitored electrically (Figure 7).10, 20 Thus the electrochemistry provides a facile way to monitor helicase function. The result is more remarkable still to consider given that the [4Fe-4S] cluster is 30 Å from the ATP binding site. Whether this is a property that is exploited within the cell is something we need still to determine.
Figure 7.
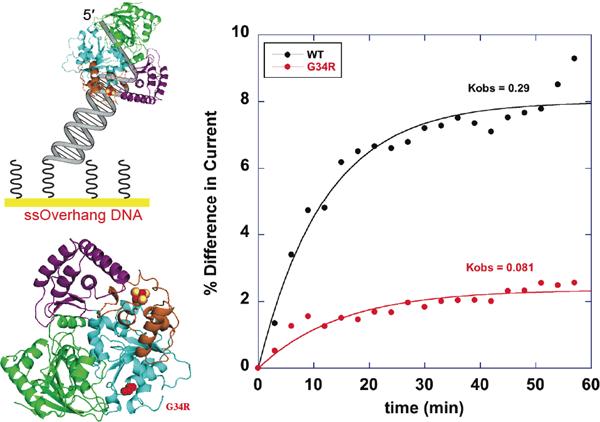
(Top Left) Schematic displaying well matched DNA that contains a single-strand overhang, tethered to a gold electrode. The electrochemical setup is used to monitor the DNA-bound redox potential of SaXPD. (Bottom Left) Crystal structure of SaXPD with G34R mutation and the [4Fe4S] cluster shown in space filling models. (Right) ATP-dependent CT of WT (black) and G34R (red). The initial rate constant observed for G34R is lower than that for WT SaXPD. Reprinted and adapted with permission from reference 10. Copyright 2011 American Chemical Society.
We next asked whether we could observe signaling between XPD and EndoIII, given that these proteins have similar redox potentials and DNA-binding affinities. We thus tested their abilities to redistribute onto mismatched strands in our AFM assay.23 Would they cooperate in finding DNA lesions? Significantly, our results were consistent with this signaling. In mixtures, with protein loadings of ~2 proteins/ 3 kb strand, we found that the XPD/EndoIII pair would redistribute onto the mismatched strand as well or better than in samples containing solely either EndoIII or XPD. If instead we mixed the protein with mutants defective in protein/DNA CT, for example, WT XPD with equimolar Y82A EndoIII, or WT EndoIII with L325V XPD (an XPD mutant with low redox signal), we would not see a redistribution onto the mismatched strand. Significantly, these results establish DNA CT signaling between two different DNA repair proteins. The results are more remarkable to consider, given that here the proteins are from two completely different organisms. What they have in common is the presence of the [4Fe-4S] cluster at a similar DNA-bound redox potential.
Considerations for the future
These studies, a mix of photophysics, electrochemistry, bioinorganic chemistry and nucleic acid chemistry, carried out over more than twenty years, have helped to elucidate a unique and powerful chemistry, DNA charge transport. DNA CT can indeed occur over long molecular distances in a reaction similar to that in π-stacked solids but different in that the chemistry is gated by the dynamics of the DNA base pair stack in solution. In that respect, DNA CT chemistry reports on the characteristics and integrity of the DNA base pair stack and, as long as the stack is intact, the DNA duplex can be utilized as a medium for long-range signaling between redox partners.
As we have carried out these studies, more and more DNA-binding proteins involved in genome maintenance, proteins such as fancJ, primase, Dna2, and even DNA polymerases22, 24, have been discovered to contain [4Fe-4S] clusters. It remains now for us to determine whether these proteins too are involved in long-range signaling across the genome. Is DNA CT signaling a means not only to activate the genome to respond to stress but also to protect the genome through signaling among proteins where repair is needed before transcription and replication occur? There is much research that still needs to be done and many complex questions still to be addressed. What is clear, however, is that DNA CT chemistry offers a powerful means not only for chemists to probe these functions but also for proteins within the cell to carry out these important biological functions, and Nature, as the best chemist of all, tends to take advantage of the chemistry available.
Acknowledgements
We are grateful to the NIH for their financial support. We thank Dr. Eric D. Olmon for his help designing and constructing figures. We thank also our many coworkers involved in these experiments who had the courage and imagination to explore this chemistry.
Biography
Pamela Sontz received her BS in biochemistry with departmental honors and high distinction from Indiana University in 2006. While at Indiana, she worked on the synthesis and characterization of metalloenediyne compounds to probe Bergman cyclization in the laboratory of Jeffrey M. Zaleski. Currently, Pam is a Ralph M. Parson's graduate fellow in Professor Barton's laboratory investigating the role of DNA charge transport in a biological context. She has utilized single-molecule atomic force microscopy to probe the first step in lesion detection by repair metalloproteins.
Natalie Muren received her B.A. in chemistry from Willamette University in 2006. There she worked on the synthesis of novel aminoglycoside antibiotics and studied RNA-binding by this class of drugs in the laboratory of Professor Sarah R. Kirk. Natalie's graduate work in Professor Jacqueline K. Barton's group involves both fundamental studies of DNA charge transport and the use of this sensitive process for the electrochemical detection of clinically relevant DNA-binding proteins.
Jacqueline K. Barton is the Arthur and Marian Hanisch Memorial Professor of Chemistry and Chair of the Division of Chemistry and Chemical Engineering at the California Institute of Technology. She received her B.A. at Barnard College and Ph.D. at Columbia University. After faculty positions at City University of New York and Columbia, she joined the faculty at Caltech in 1989. Professor Barton's work is focused on the chemistry of DNA.
References
- 1.Eley DD, Spivey DI. Semiconductivity of organic substances. Part 9. Nucleic acid in the dry state. Trans. Faraday Soc. 1962;58:411–415. [Google Scholar]
- 2.Genereux JG, Barton JK. Mechanisms for DNA Charge Transport. Chem. Rev. 2010;110:1642–1662. doi: 10.1021/cr900228f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wagenknecht HA, editor. Charge Transfer in DNA: From Mechanism to Application. Wiley-VCH, Weinheim; Germany: 2005. [Google Scholar]
- 4.Genereux JC, Boal AK, Barton JK. DNA-Mediated Charge Transport in Redox Sensing and Signaling. J. Am. Chem. Soc. 2010;132:891–905. doi: 10.1021/ja907669c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kelley SO, Barton JK. Electron Transfer Between Bases in Double Helical DNA. Science. 1999;283:375–381. doi: 10.1126/science.283.5400.375. [DOI] [PubMed] [Google Scholar]
- 6.Guo X, Gorodetsky AA, Hone J, Barton JK, Nuckolls C. Conductivity of a single DNA duplex bridging a carbon nanotube gap. Nature Nanotech. 2008;3:163–167. doi: 10.1038/nnano.2008.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Boon EM, Ceres DM, Drummond TG, Hill MG, Barton JK. Mutation detection by electrocatalysis at DNA-modified electrodes. Nat. Biotechnol. 2000;18:1096–1100. doi: 10.1038/80301. [DOI] [PubMed] [Google Scholar]; (b) Boal AK, Yavin E, Lukianova OA, O'Shea VL, David SS, Barton JK. DNA-bound activity of DNA repair glycosylases containing [4Fe-4S] clusters. Biochemistry. 2005;44:8397–8407. doi: 10.1021/bi047494n. [DOI] [PubMed] [Google Scholar]
- 8.Slinker JD, Muren NB, Renfrew SE, Barton JK. DNA charge transport over 34 nm. Nat. Chem. 2011;3:230–235. doi: 10.1038/nchem.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boon EM, Salas JE, Barton JK. An electrical probe of protein-DNA interactions on DNA-modified surfaces. Nat. Biotechnol. 2002;20:282–286. doi: 10.1038/nbt0302-282. [DOI] [PubMed] [Google Scholar]
- 10.Mui TP, Fuss JO, Ishida JP, Tainer AJ, Barton JK. ATP-stimulated, DNA-mediated redox signaling by XPD, a DNA repair and transcription helicase. J. Am. Chem. Soc. 2011;133:16378–16381. doi: 10.1021/ja207222t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hall DB, Holmlin ER, Barton JK. Oxidative DNA damage through long-range electron transfer. Nature. 1996;382:731–735. doi: 10.1038/382731a0. [DOI] [PubMed] [Google Scholar]
- 12.Núñez ME, Hall DB, Barton JK. Long-range oxidative damage to DNA: effects of distance and sequence. Chem. Biol. 1999;6:85–97. doi: 10.1016/S1074-5521(99)80005-2. [DOI] [PubMed] [Google Scholar]
- 13.(a) Núñez ME, Noyes KT, Barton JK. Oxidative Charge Transport through DNA in Nucleosome Core Particles. Chem. Biol. 2002;9:403–415. doi: 10.1016/s1074-5521(02)00121-7. [DOI] [PubMed] [Google Scholar]; (b) Núñez ME, Holmquist GP, Barton JK. Evidence for DNA Charge Transport in the Nucleus. Biochemistry. 2001;40:12465–12471. doi: 10.1021/bi011560t. [DOI] [PubMed] [Google Scholar]; (c) Merino EJ, Davis ML, Barton JK. Common mitochondrial DNA mutations generated through DNA-mediated charge transport. Biochemistry. 2009;48:660–666. doi: 10.1021/bi801570j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ding H, Hidalgo E, Demple B. The redox state of the [2Fe-2S] clusters in SoxR protein regulates its activity as a transcription factor. J. Biol. Chem. 1996;271:33173–33175. doi: 10.1074/jbc.271.52.33173. [DOI] [PubMed] [Google Scholar]
- 15.Gorodetsky AA, Dietrich LEP, Lee PE, Demple B, Newman DK, Barton JK. DNA binding shifts the redox potential of the transcription factor SoxR. Proc. Natl. Acad. Sci. USA. 2008;105:3684–3689. doi: 10.1073/pnas.0800093105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee P, Demple B, Barton JK. DNA-mediated redox signaling for transcriptional activation of SoxR. Proc. Natl. Acad. Sci. USA. 2009;106:13164–13168. doi: 10.1073/pnas.0906429106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.David SS, Williams SD. Chemistry of Glycosylases and Endonucleases Involved in Base-Excision Repair. Chem. Rev. 1998;98:1221–1262. doi: 10.1021/cr980321h. [DOI] [PubMed] [Google Scholar]
- 18.(a) Thayer MM, Ahern H, Xing D, Cunningham RP, Tainer JA. Novel DNA binding motifs in the DNA repair enzyme endonuclease III crystal structure. EMBO J. 1995;14:4108–4120. doi: 10.1002/j.1460-2075.1995.tb00083.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Fromme JC, Banerjee A, Huang SJ, Verdine GL. Structural basis for removal of adenine mispaired with 8-oxoguanine by MutY adenine DNA glycosylase. Nature. 2004;427:652–656. doi: 10.1038/nature02306. [DOI] [PubMed] [Google Scholar]; (c) Porello SL, Williams SD, Kuhn H, Michaels ML, David SS. Specific Recognition of Substrate Analogs by the DNA Mismatch Repair Enzyme MutY. J. Am. Chem. Soc. 1996;118:10684–10692. [Google Scholar]; (d) Sampson JR, Jones S, Dolwani S, Cheadle JP. MutYH (MYH) and colorectal cancer. Biochem. Soc. Trans. 2005;33:679–683. doi: 10.1042/BST0330679. [DOI] [PubMed] [Google Scholar]
- 19.Boal AK, Genereux JC, Sontz PA, Gralnick JA, Newman DK, Barton JK. Redox signaling between DNA repair proteins for efficient lesion detection. Proc. Natl. Acad. Sci. USA. 2009;106:15237–15242. doi: 10.1073/pnas.0908059106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fan L, Fuss JO, Cheng QJ, Arvai AA, Hammel M, Roberts VA, Cooper PK, Tainer JA. XPD Helicase Structures and Activities: Insights into the Dancer and Aging Phenotypes from XPD Mutations. Cell. 2008;133:789–800. doi: 10.1016/j.cell.2008.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wolski SC, Kuper J, Hänzelmann P, Truglio JJ, Croteau DL, Van Houten V, Kisker C. Crystal structures of the FeS cluster-containing nucleotide excision repair helicase XPD. PLos Biol. 2008;6:1332–1342. doi: 10.1371/journal.pbio.0060149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.White MF, Dillingham MS. Iron-sulphur clusters in nucleic acid processing enzymes. Curr. Opin. Struct. Biol. 2012;22:94–100. doi: 10.1016/j.sbi.2011.11.004. [DOI] [PubMed] [Google Scholar]
- 23.Sontz PA, Mui TP, Fuss JO, Tainer JA, Barton JK. DNA charge transport as a first step in coordinating the detection of lesions by repair proteins. Proc. Natl. Acad. Sci. USA. 2012;109:1856–1861. doi: 10.1073/pnas.1120063109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Netz DJA, Stith CM, Stümpfig M, Köpf G, Vogel D, Genau HM, Stodola JL, Lill R, Burgers PMJ, Pierik AJ. Eukaryotic DNA polymerases require an iron-sulfur cluster for the formation of active complexes. Nature Chem. Bio. 2012;8:125–132. doi: 10.1038/nchembio.721. [DOI] [PMC free article] [PubMed] [Google Scholar]