

Abstract
To define the complete sRNA population of the halophilic archaeon Haloferax volcanii, we employed high throughput sequencing. cDNAs were generated from RNA ranging in size from 17 to 500 nucleotides isolated from cells grown at three different conditions to exponential and stationary phase, respectively. Altogether, 145 intergenic and 45 antisense sRNAs were identified. Comparison of the expression profile showed different numbers of reads at the six different conditions for the majority of sRNAs. A striking difference in the number of sRNA reads was observed between cells grown under standard vs. low salt conditions. Furthermore, the six highest numbers of reads were found for low salt conditions. In contrast, only slight differences between sRNA reads at different growth temperatures were detected. Attempts to delete four sRNA genes revealed that one sRNA gene is essential. The three viable sRNA gene deletion mutants possessed distinct phenotypes. According to microarray analyses, the removal of the sRNA gene resulted in a profound change of the transcriptome when compared with the wild type. High throughput sequencing also showed the presence of high concentrations of tRNA derived fragments in H. volcanii. These tRF molecules were shown to have different amounts of reads at the six conditions analyzed. Northern analysis was used to confirm the presence of the tRNA-derived fragments.
Keywords: Haloferax volcanii, archaea, high throughput sequencing, sRNAs, tRFs
Introduction
Small RNAs are involved in regulation of gene expression in all three domains of life.1-3 The modes of function have been studied in detail for eukaryotic and bacterial small RNAs. In eukaryotes a variety of small RNA types are known nowadays. The micro RNAs (miRNAs), for example, are small RNAs of about 21 nucleotides and they function primarily by base pairing with their target mRNAs, mediating the inhibition of translation or triggering degradation.4-6 Recently, a novel class of small RNAs was identified in eukaryotes: RNA fragments derived from tRNAs and tRNA precursors.7,8 In human cells these fragments are second most abundant to miRNAs and are processed from mature tRNAs or precursor-tRNAs. tRNA 5′ fragments, tRNA 3′ fragments and tRNA 3′ trailer sequences have been detected as stable molecules. Processing of these molecules seems to involve Dicer9,10 as well as the tRNA 3′ processing endonuclease tRNase Z.11-13 They can be generated by different means and show specific expression patterns. Recent studies suggest that these tRNA fragments are not random by-products of tRNA biogenesis and degradation, but are an abundant and novel class of short RNAs.
In bacteria RNA regulators range in size from app. 50–300 nucleotides and the majority regulate their target RNA by short, imperfect base-pairing interactions.14 In contrast, very little is known about small regulatory RNAs in the third domain of life, the Archaea.15,16 The first archaeal small RNA populations were identified several years ago with RNomics and bioinformatics in Archaeoglobus fulgidus, Methanocaldococcus jannaschii, Pyrococcus furiosus and Sulfolobus solfataricus.17-20 At that time no efficient genetic systems were available for these Archaea to analyze the sRNA genes further, e.g., via generation of deletion mutants. Recently, high throughput sequencing was employed to identify the complete population of small RNAs in a given species. Using this method it was shown that in S. solfataricus 310 small RNAs are present21 and in Methanosarcina mazei 242 small RNAs were identified15,16,22,.
The goal of this study was to identify the complete sRNA population of the halophilic archaeon Haloferax volcanii. H. volcanii is a halophilic organism, which requires 2.1 M NaCl for optimal growth and contains similar concentrations of salt intracellularly to cope with high salt concentration in the medium. The genome has recently been sequenced and annotated.23 H. volcanii is studied as an archaeal model organism because it is easy to cultivate and easily genetically modified.24-26 Recently, RNomics approaches revealed that H. volcanii contains 21 intergenic sRNAs and 18 antisense RNAs.27-29 In this former study only RNAs in the size range of 130–460 nucleotides were analyzed from cells grown only under a single condition. Using bioinformatics analyses the sRNA population of H. volcanii was predicted to contain more than 100 sRNAs.30 To complement these former studies, we used here high throughput sequencing (HTS) of RNAs in the size range of 17–500 nucleotides isolated from cells grown at three different growth conditions to exponential and stationary phase, respectively, to elucidate the complete small RNA population of H. volcanii.
Results and Discussion
Identification of small RNAs using high throughput sequencing
H. volcanii cultures were either grown under optimal growth conditions (45°C; 18% salt), high temperature conditions (48.5°C; 18% salt) or low salt conditions (45°C; 15% salt). From each culture one sample was taken in exponential phase and a second one in stationary phase. Total RNA was size fractionated and RNA from 17 to 500 nucleotides was isolated and transcribed into cDNA. All together six cDNA libraries were constructed and investigated. The subsequent sequencing yielded 4.4 million reads, of which 42% were sequences of tRNA molecules, 5S rRNA and 16S and 23S rRNA fragments. rRNA sequences were removed from the data set and the remaining sequences were mapped to the H. volcanii genome. In total, 145 potential intergenic sRNAs and 45 antisense sRNAs were identified (Table S1 and data not shown). The 21 intergenic sRNAs and 18 antisense sRNAs previously identified by RNomics28 were also present in this data set. We also found RNAs, which mapped to open reading frames, representing potential sense sRNAs. Since it remains to be determined whether these RNAs are stable degradation fragments of the respective mRNAs, we did not investigate these RNAs further. The identified antisense RNAs were investigated separately (Heyer, et al. in preparation). High throughput sequencing (HTS) also showed the presence of high concentrations of tRNA derived fragments in H. volcanii (Tables S3 and S6). This novel class of small RNAs are RNA fragments derived from tRNAs and tRNA precursors as recently identified in humans.9,13 Here, we show that tRFs are also present in Archaea. From the 51 tRNA genes encoded in H. volcanii 11 generate tRFs, which show different numbers of reads at the different conditions (Table S3). Northern analyses confirmed the stable presence of the tRNA-derived fragments (Fig. 1).
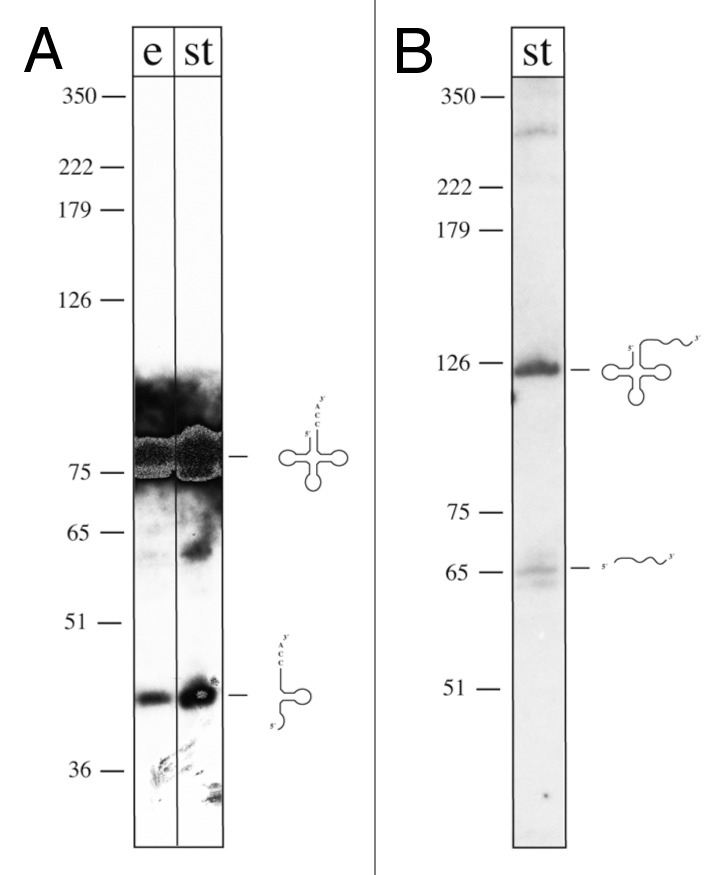
Figure 1. Northern analyses of tRFs. (A) Analysis of fragments derived from tRNAGln (CTG). RNA was extracted from cells grown under high temperature conditions [48.5°C; at exponential (lane e) and stationary phase (lane st)], separated on denaturing PAGE, transferred to membranes and hybridized with a probe against the 3′ half of tRNAGln (CTG) (including the terminal CCA triplet). The mature tRNA is detected with this probe as well as a tRNA derived fragment of about 40 nucleotides. A DNA size marker is given at the left in nucleotides (note that DNA runs approx. 10% faster than RNA on denaturing PAGE). (B) Analysis of fragments derived from tRNAHis (GTG). RNA was extracted from cells grown under low salt conditions (15% salt concentration in the medium; stationary phase), separated on denaturing PAGE, transferred to membranes, and hybridized with a probe against the 3′ trailer of the tRNAHis precursor. The tRNA precursor (including the tRNA and the 3′ trailer) is detected with this probe (RNA of about 120 nucleotides) as well as the 3′ trailer of about 60 nucleotides. A DNA size marker is given on the left in nucleotides (note that DNA runs approx. 10% faster than RNA on denaturing PAGE).
The study presented here was restricted to RNA populations from cells grown under three different conditions; the analysis of more growth conditions would probably identify additional sRNAs. Similar HTS approaches were performed for only two other archaeal species, S. solfataricus and M. mazei. For S. sulfolobus 125 intergenic sRNAs and 185 cis-antisense sRNAs were identified; here likewise RNA from three different growth conditions (three different carbon sources) was analyzed.21 In M. mazei 199 intergenic sRNAs and 43 cis-antisense sRNAs were found in cDNA libraries derived from two different conditions (different nitrogen sources).15,22 Altogether, these data suggest that the number of sRNAs in Archaea will be around 200–300 similar to the number of sRNAs expected in bacteria.3
Table S1 summarizes the expression profiles of the 145 identified intergenic sRNAs. Forty four of these sRNAs were present at all three growth conditions (standard, high temperature and low salt) with varying degrees of expression. Differential transcript levels under the different conditions are discussed below. To be on the save side, only sRNAs with a statistical meaningful number of reads (> 100 reads) were analyzed, and a more than two-fold difference in read numbers was taken as an indication of differential sRNA levels.
Changes in sRNA expression between exponential and stationary phase
Only three sRNAs had consistent growth rate-dependent differential levels under all three conditions (Hts39, Hts43 and Hts70), all of them were unregulated in stationary phase. Interestingly, a variety of sRNAs were highly growth phase regulated under only one or two conditions, e.g., Hts1 and Hts2 showed 3,810 and 7,277 reads, respectively, at low salt during exponential phase, whereas under none of the other five conditions the number of reads exceeded 42. Furthermore, several sRNAs exhibited a different direction of growth phase-dependent control under different conditions. Striking examples are Hts5, Hts71 and Hts107, which were downregulated toward stationary phase under standard conditions, but in contrast, were upregulated toward stationary phase under low salt conditions, while Hts144 behaved the opposite way. These and many more examples seem to indicate that the sRNA profiles of stationary phase cells depend on the conditions under which the cells entered stationary phase.
Changes in sRNA expression upon high temperature
Twenty-three sRNAs had higher levels during growth at the standard temperature compared with the elevated temperature, while not a single sRNA had a higher level at the elevated temperature at both growth phases. Seven of these sRNAs could not be detected at all at the elevated temperature (Hts5, Hts34, Hts45, Hts66, Hts127, Hts131 and Hts142), the most prominent example is Hts5 that had 3,074 reads at normal temperature during exponential phase. Six sRNAs had higher levels at the elevated temperature, but only at one of the two growth phases (Hts27, Hts71, Hts96, Hts99, Hts101 and Hts107), while they were not regulated or regulated in the opposite direction in the other growth phase. Taken together, sRNAs do not seem to play a prominent role in the adaptation of H. volcanii to high temperature stress.
Changes in sRNA expression upon low salt concentrations
In stark contrast to the two conditions discussed above, a striking difference in the sRNA profiles of cells grown under standard vs. low salt conditions was observed. Nineteen sRNAs had higher levels at the low salt conditions, while 24 had higher levels during growth at the optimal salt concentration. The six highest numbers of reads were found for low salt conditions, five of them for exponentially growing cells (Hts3: 13,189 reads, Hts144: 7,775 reads, Hts2: 7,277 reads, Hts51: 4,888 reads and Hts1: 3,810 reads), one for stationary phase cells (Hts43: 4,634 reads). But also in the other direction the differences were sometimes high, e.g., there were five sRNAs with more than 1,000 reads at the standard salt concentration and a considerably lower number of reads under low salt (Hts10, Hts12, Hts25, Hts71 and Hts145), and six sRNAs that could only be detected under standard salt concentration and were not at all detectable in cells grown at low salt (Hts34, Hts66, Hts97, Hts127, Hts131 and Hts142). Taken together, these results indicate that many sRNAs play important roles for H. volcanii in the adaptation to specific salt concentrations, and further experiments will unravel their specific functions.
Expression of candidate intergenic sRNAs
To confirm the expression of the sRNAs and to determine their size, we used northern analyses for selected sRNAs (Hts 1–21)(Fig. 2 and data not shown). From the 21 sRNAs tested 19 were shown to be expressed. For the remaining two (Hts7 and Hts19), showing no signal in the northern, the HTS data indicate a very low expression level, probably below the detection limit of the northern (Table S1).
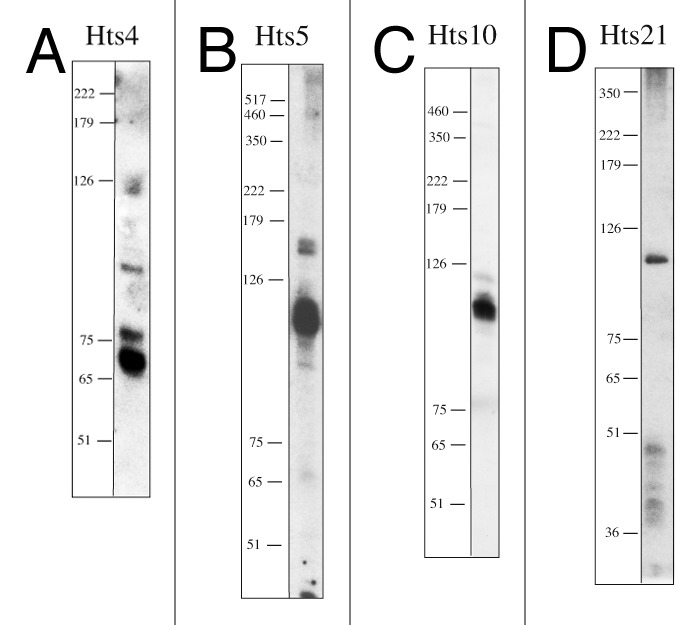
Figure 2. Northern analyses of Hts sRNAs. RNA was separated on denaturing PAGE and transferred to membranes. Membranes were probed with oligonucleotides complementary to the sequences of Hts4, Hts5, Hts10 and Hts21 (Table S2). A DNA size marker is given at the left in nucleotides (note that DNA runs approximately 10% faster than RNA on denaturing PAGE). (A) For the analysis of Hts4, RNA was extracted from cells grown under low salt conditions (15% salt concentration; 45°C; at exponential phase). Hts4 is efficiently expressed and probably processed since several RNA molecules are detectable. (B) For the analysis of Hts5, RNA was extracted from cells grown under standard conditions (18% salt concentration; 45°C; at exponential phase), Hts5 is efficiently expressed, two RNA fragments are detected in the northern with sizes of app. 110 nt and 150 nt. (C) For the analysis of Hts10, RNA was extracted from cells grown under standard conditions (18% salt concentration; 45°C; at stationary phase), Hts10 is expressed with a size of approximately 110 nt. (D) For the analysis of Hts21, RNA was extracted from cells grown under low salt conditions (15% salt concentration; 45°C; at exponential phase), Hts21 is expressed with a size of approximately 120 nt, in addition some signals of about 36–50 nts are also detected.
Generation of sRNA gene deletion strains
To investigate the biological function of the candidate sRNAs, we aimed to generate deletion strains for four sRNA genes. We selected the genes for the sRNAs Hts4, Hts5, Hts10 and Hts21, because of the following reasons: Hts4, Hts5 and Hts10 were chosen since they showed different amounts of reads at different conditions. Further genes flanking the respective hts gene have a different orientation, therefore the respective hts gene must be expressed independently. All three sRNAs have a high number of reads at a specific condition (Hts4: 13,189 reads at low salt concentrations in exponential phase; Hts5: 3,074 reads at standard conditions in exponential phase and Hts10: 1,959 reads at standard conditions in stationary phase, Table S1). Furthermore, northern analyses confirmed for all three Hts sRNAs expressions of RNA molecules in the size range of 65–110 nucleotides. Hts21 was chosen as example for an sRNA which was only expressed at one condition (low salt concentrations: 257 reads, Table S1).
Using the pop in-pop out method,31 the sRNA genes were replaced by the trpA gene.32 The genomic location and deletion strategy for one sRNA gene, hts10, is shown exemplarily in Figure 3. Deletion strains were verified using PCR and Southern blots (Fig. 3 and data not shown). Except for sRNA Hts5 all sRNA genes could be successfully deleted. The pop in step for the deletion of the hts5 gene was successful, but subsequent removal of the hts5 gene did not work. The gene for Hts5 is located on the main chromosome between genes for a hypothetical protein (HVO_2394) and a putative ATP:cob(I)alamin adenosyltransferase (HVO_2395). Northern analysis shows two fragments, with the major fragment being about 110 nucleotides long (Fig. 2B). According to the HTS analysis, Hts5 is strongly expressed at exponential phase under normal conditions (3,074 reads) (Table S1). The high concentration at normal conditions in exponential phase together with the failure to generate an hts5 gene deletion strain might indicate that the Hts5 sRNA is essential. Likewise, it is possible, that the hts5 deletion strain had a severe growth defect and grew considerably slower on the selection plates therefore remaining undetected.
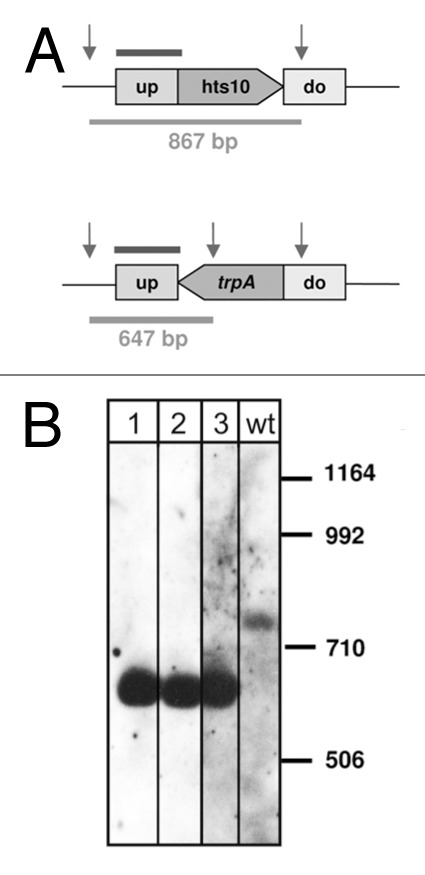
Figure 3. Deletion of hts10 gene. (A) The genomic localizations of the wild type gene and the deletion for the sRNA gene are shown schematically. The probe used for Southern blot analysis is indicated by a bar above the genome. Relevant SalI restriction sites are indicated by vertical arrows, and sizes of resulting SalI fragments are indicated below in base pairs. (B) Southern blot for deletion strain Δhts10. Lane wt: wild type, lanes 1, 2, 3: deletion strains, sizes of the DNA marker are given in base pairs at the right.
Characterization of selected sRNA genes: genomic localization, sRNA length and analysis of deletion strains
The sRNA gene deletion strains for sRNA Hts4, Hts10 and Hts21 and the parent strain H119 were grown in synthetic medium in microtiter plates and compared under ten different conditions. Growth was monitored on four different sole carbon and energy sources (glucose, xylose, acetate, casamino acids) and under low salt and high salt conditions (1.2 and 4 M NaCl). In addition, the effect of four different stress conditions was characterized [shift from 2.1 M to 1.3 M NaCl, temperature downshift, 1% (v/v) EtOH and 4% mM paraquat]. Average OD600 values of three biological replicates were determined and the length of the lag phase and the growth yield of mutants and parent strain were compared. The growth yields are summarized in Table S4, and selected results are mentioned in the following paragraphs. In summary, these and nearly 30 additional sRNA gene deletion mutants (Jaschinski et al. in preparation) and parent strain typically grew indistinguishably under the majority of conditions and the mutants exhibited a phenotype under one or a few conditions. Interestingly, the phenotypes included—depending on mutant and condition—a longer or a shorter lag phase and a lower and a higher growth yield.
Analysis of strain Δhts4
sRNA Hts4 is encoded on the main chromosome between genes HVO_2019 and HVO_2020, which encode a putative PRC-barrel domain and a protein of unknown function (DUF502). The sRNA is expressed at all conditions with lower expression levels at standard and high temperature conditions, but extremely strong expression at low salt concentrations (exponential phase, 13,189 reads, which is the highest number of reads found and 1,771 reads at stationary phase) (Table S1). According to the northern, three major fragments are visible of 65 nucleotides, 75 nucleotides and 110 nucleotides length, indicating that processing of a precursor is happening (Fig. 2A). The mutant strain Δhts4 had a slight growth defect on the carbon sources glucose and acetate and growth yields between 76% and 87% at the high NaCl concentration of 4 M (Fig. 4; Table S4). For analysis of ethanol and oxidative stress glucose was used as carbon source. The lower growth yields after application of ethanol and oxidative stress (79% and 80%) could, therefore, be due to the carbon source and not be caused by the application of stress. The mutant showed no significant differences to the parent strain under optimal and low NaCl concentrations and after the application of other stress conditions (Table S4).
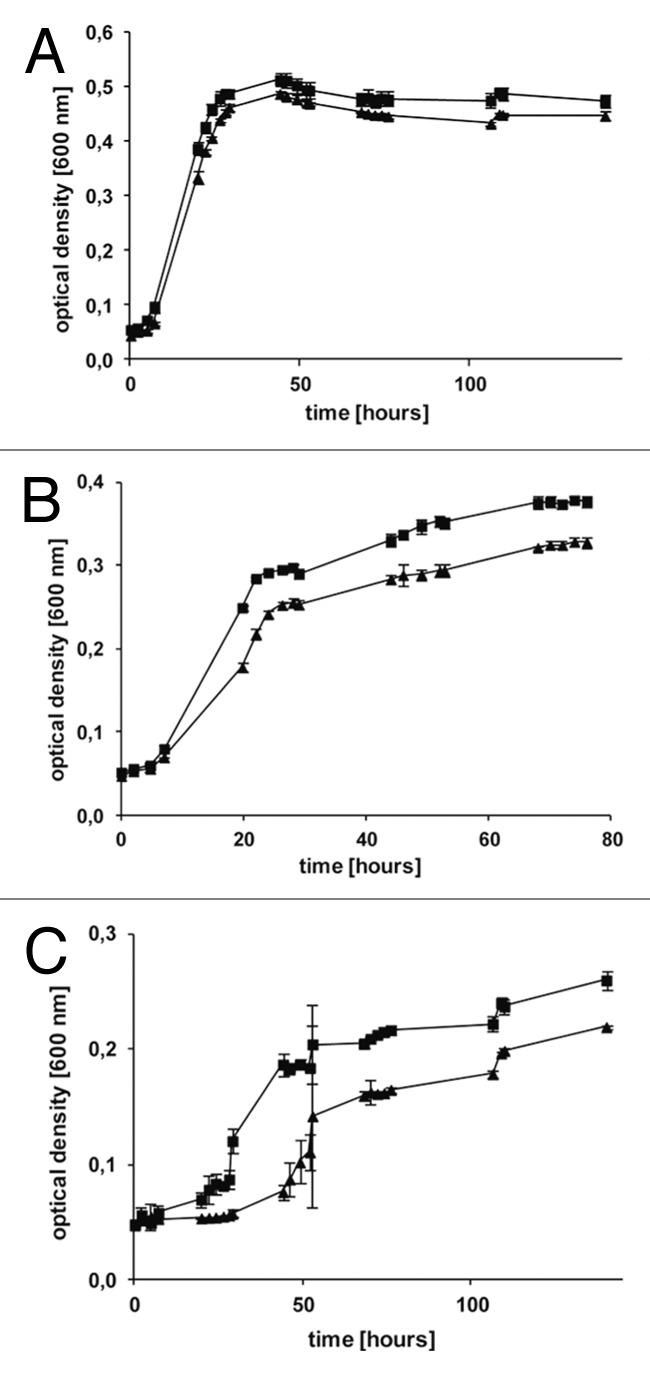
Figure 4. Selected growth curves of mutant Δhts4 and the parent strain H119. Growth of the deletion mutant of the sRNA Hts4 (triangles) and parent strain H119 (squares) was measured at 600 nm, and the average values of three independent cultures and their standard deviations are shown. Growth experiments were performed in microtiterplates with synthetic media supplemented with three different carbon sources casamino acids (A), glucose (B) and acetate (C).
Analysis of strain Δhts10
sRNA Hts10 is encoded on the main chromosome between genes HVO_2213 and HVO_2214, which encode a hypothetical protein and a transducer protein Htr36. The sRNA is expressed at all conditions with the highest expression in stationary phase under standard conditions (1,959 reads) (Table S1). According to the northern, the most prominent fragment is about 110 nucleotides in length (Fig. 2C). The Δhts10 mutant grew indistinguishable from the parent strain under nearly all conditions tested, except for a small reduction of the growth yield on acetate as carbon source (87%) and upon growth in 4 M NaCl (89% compared with the parent strain) (Table S4). In contrast to the reduced growth yield at high salt, the growth yield is slightly greater than that of the parent strain in low salt (113%).
Analysis of strain Δhts21
sRNA Hts21 is encoded on the main chromosome between genes HVO_2583 and HVO_2584, which encode a hydroxymethylglutaryl-CoA reductase (NADPH-dependent) and a protein with similarities to the replication protein of pNRC100. According to the HTS data, the sRNA is expressed only at low salt concentrations during exponential phase (Table S1). Northern analysis revealed a prominent RNA with about 120 nucleotides and some smaller fragments of about 36–50 nucleotides (Fig. 2D). The deletion strain Δhts21 grew indistinguishable from the parent strain under most conditions, but has a slightly higher growth yield upon growth on glucose (120%) or acetate (134%) as carbon source (Table S4). The growth yield after an osmotic down-shock is considerably higher than that of the parent strain (264%).
To unravel the biological function of sRNAs the generation of sRNA gene deletion mutants and the analysis of their phenotype is an effective approach, which is confirmed by observations made with the sRNA deletion mutants generated in this study. While one sRNA gene proved to be essential, deletion mutants could be obtained for the other three sRNA genes and all three showed very distinct phenotypes, suggesting a role in metabolic regulation and stress adaptation, respectively.
Comparison of the transcriptomes of mutant Δhts10 and the parent strain
The deletion strain Δhts10 behaved in most aspects indistinguishably from the parent strain (see above). Therefore, its transcriptome was compared with that of the parent strain to unravel whether phenotypically silent differences in transcript levels could be observed. Both strains were grown to mid-exponential growth phase (4 x 108 cells/ml) and the transcriptomes were compared using a self-constructed DNA microarray. Three independent biological replicates were performed, which included a dye swap. A scatter plot of the average signals is shown in Figure 5. Most signals were very close to the diagonal underscoring that the expression of most genes was unchanged. A few spots have a lower intensity in the deletion mutant, but the transcript level ratio was close to two-fold and thus the difference was not very large. However, one genomic region was represented by three independent spots, indicating that the difference was real (Table 1). The region encodes an extremely large protein of 2257 amino acids annotated as “Muc19 precursor” (HVO_2160), which is highly enriched in threonines (14%) and serines (10%) indicating that it has the typical high water-binding capacity of mucines.

Figure 5. Comparison of the transcriptomes of mutant Δhts10 and parent strain H119. Both strains were grown to mid-exponential growth phase. RNA was isolated from three independent cultures each and compared using a self-constructed DNA microarray (compare Materials and Methods). Average values of the signals for the transcripts of the mutant (y-axis) and the parent strain (x-axis) are shown. The solid line represents the diagonal and the two broken lines a 2-fold difference between mutant and parent strain.
Table 1. Genes with different transcript levels in the Δhts10 deletion mutant and the parent strain H119, as revealed with DNA microarray analysis.
|
A |
| HVO number | gene name / function | no. of spots |
regulation level |
|---|---|---|---|
| HVO_0259 |
hypothetic protein |
1 |
55.28 |
| HVO_0255 - 0258 |
spermine/spermidine synthase family protein; conserved protein; XerC/D-like integrase |
1 |
16.61 |
| HVO_0160 |
CAAX N-terminal protease family, transmembrane |
1 |
14.27 |
| HVO_2079 |
conserved protein |
1 |
12.82 |
| HVO_0266 - 0271 |
all hypothetic proteins |
5 |
11.15 |
| HVO_B0128 |
glucose 1-dehydrogenase related protein |
1 |
5.26 |
| HVO_0260 - 0261 |
PhiH1 repressor-like; putative repressor phrH2 |
1 |
5.07 |
| HVO_2447 - 2448 |
probable integral membrane protein; transcription initiation factor TFB |
1 |
4.60 |
| HVO_0299 |
membrane protein, putative |
1 |
4.20 |
|
B |
| HVO number | gene name / function | no. of spots |
regulation level |
|---|---|---|---|
| HVO_2160 |
Muc19 precursor, putative |
3 |
0.44 |
| HVO_2401 | glycine cleavage system P-protein | 1 | 0.49 |
Genes upregulated in the Δhts10 deletion mutant and (B) genes downregulated in the Δhts10 deletion mutant. Column 1 (HVO number) shows the gene number (see www.halolex.mpg.de), column 2 (gene name/ function) lists the gene name and annotated function, column 3 (no. of spots) shows the number of times the gene is represented on the microarray and column 4 (regulation level (mean value)) lists the fold induction of repression. Average values of three independent biological replicates are shown, which included one dye swap.
In contrast, many more transcripts had a higher level in the deletion mutant and the regulation level was considerably higher (Table 1; Table S5). The transcript levels of about 30 genomic regions were at least two-fold higher in the mutant than in the parent strain, and the highest induction level was nearly sixtyfold (Table 1; Table S5). A high fraction of genes is annotated to encode conserved proteins without known function. Nine microarray spots including those with the highest difference in signal ratios represent a single genomic region comprised of the genes HVO_0255 to HVO_0270. These 16 genes encode 12 hypothetical proteins or conserved hypothetical proteins, a spermine/spermidine synthase like protein, a XerC/D-like integrase and two putative repressor proteins. The microarray spots represent different parts of this gene cluster, therefore, at least several of these genes had higher transcript levels in the Δhts10 mutant.
Target analysis using the program IntaRNA33 revealed that the Hts10 RNA is able to bind to all upregulated mRNAs (Table S8). Four target groups can be defined which bind to similar regions of the Hts10 sRNA (Table S8). Further analyses will have to show if the Hts sRNA10 indeed interacts with the predicted target RNA regions.
Taken together the in-depth analysis of one deletion mutant with microarrays revealed that although the phenotype was not very pronounced the transcriptome change caused by the removal of the sRNA gene was profound.
Materials and Methods
Strains and culture conditions
H. volcanii strain H119 (ΔpyrE2, ΔleuB, ΔtrpA)32 and the sRNA gene deletion mutants based on H119 (Δhts4, Δhts10, Δhts21) (mutant construction is described below) were grown aerobically at 45°C in Hv-YPC medium or in Hv-Ca medium.41E. coli strains DH5α (Invitrogen) and GM12134 were grown aerobically at 37°C in 2YT medium.
High throughput sequencing
Total RNA was prepared from H. volcanii cells35 which were grown with different salt concentrations (15% and 18% salt) and at different temperatures (45°C and 48,5°C). From each RNA preparation 10 µg were sent to GATC Biotech AG for high throughput sequencing. Shortly, RNA was size fractionated on 12% PAGE and RNA from 17–500 nucleotides length was eluted. The eluted RNA fraction was treated with tobacco acid pyrophosphatase to convert 5′ triphosphate groups to 5′-monophosphate. Then, the RNA was polyadenylated using poly(A)-polymerase and an RNA oligo was ligated to the 5′ ends. RNA was reverse transcribed using an oligo(dT)-adaptor primer and M-MLV-RNase H- reverse transcriptase. The resulting cDNAs were amplified by PCR and sequenced using the Illumina/ Solexa sequencer yielding 36 nucleotide long reads. Sequences from rRNAs and tRNAs were removed from the output, and the remaining sequences were mapped against the genome.
Northern blot analysis
Total RNA was extracted as described above. To analyze expression of sRNAs, cells were grown with different salt concentrations: 15% salt, 18% salt and 23% salt. In addition, cultures were grown at different temperatures (30°C, 45°C, 48,5°C). For all growth conditions, cells were harvested at exponential phase (OD650 = 0.5) and at stationary phase (OD650 = 1.1–1.4). From these different RNA preparations, 10 µg each were separated on 8% denaturing gels, which were subsequently transferred to nylon membranes. DNA-oligonucleotide probes complementary to the sRNAs were used as probes for hybridization (primers used are listed in Table S7).
Construction of sRNA deletion mutants
Deletion mutants of Hts sRNA genes were constructed using the pop in-pop out method described previously.31,32,34 The sRNA genes were amplified with 500 additional base pairs upstream and downstream and then cloned into the integration vector pTA131 (primers used are listed in Table S7). An inverse PCR reaction was performed on the resulting plasmid to delete the entire sRNA gene and to introduce an SnaBI restriction site. The PCR product was ligated and subsequently digested with SnaBI. The trpA marker gene was then cloned into the plasmid, yielding plasmids which contain the up- and downstream regions of the sRNA genes and the trpA marker gene instead of the respective sRNA gene. H. volcanii strain H11932 was transformed with the plasmids to yield pop in clones. To generate the pop out strains, cells were plated on medium containing 5-fluoro-orotic acid (5-FOA). Chromosomal DNA was isolated from wild type and potential deletion mutants. Southern blot hybridization was performed as described with the following modifications: 10 µg of SalI digested DNA were separated on a 0.8% agarose gel and transferred to a nylon membrane (HybondTM-N, GE Healthcare).36,37 As hybridization probe the hts10 gene upstream region was generated using PCR with primers hts10up and hts10IP1, which was subsequently labeled using a DIG random prime according to the manufacturers protocol (Roche). Deletion strains for sRNA genes hts4, hts5 and hts21 were likewise confirmed by Southern blot analyses (data not shown) (probes and primers used are listed in Table S7).
Phenotyping of sRNA gene deletion mutants
Growth experiments were performed in 96-well microtiter plates (round-bottom plates; Sarstedt) as described previously.38 In short, H. volcanii precultures were grown under standard conditions to the early exponential growth phase (OD600 = 0.3 ± 0.1). To inhibit evaporation of water during the time of incubation all outer wells were filled with 200 µl of 1 M NaCl, which generated an “evaporation barrier.” The cells of the preculture were collected by centrifugation, washed once in basal salts (medium without carbon source) and resuspended in basal salts to yield cell suspensions with an OD600 of 0.375. Following this, 130 µl of prewarmed medium in 96-well microtiter plates were inoculated with 20 µl of the respective precultures. Thereby, all cultures had an identical optical density of OD600 = 0.05. The cultures were incubated on a Titramax 1000 rotary shaker (Heidolph) with 1100 rpm at 42°C. At selected time points the OD600 was measured using the microtiter plate photometer Spectramax 340 (Molecular Devices). The data were exported and Microsoft Excel was used to calculate average values with their variance and to generate growth curves. Growth of mutants and the parent strain was characterized under ten different conditions. Standard conditions were defined as aerobic growth in synthetic medium with 0.5% (w/v) glucose as sole carbon and energy source with a NaCl concentration of 2.1 M.38 Three other carbon and energy sources were tested, i.e., xylose (0.25% w/v), acetate (40 mM) and casamino acids [0.25% (w/v)]. In addition to the optimal salt concentration of 2.1 M NaCl also a very low concentration of 1.2 M and a high concentration of 4 M NaCl were used. Furthermore, four different stress conditions were applied to cultures of mid-exponential growth phase: oxidative stress was induced by the addition of 4 mM paraquat, organic solvent stress was induced by the addition of 1% (v/v) ethanol, a temperature down-shift was induced by removing half of the culture and replacing it with ice-cold medium, and an osmotic down-shift to 1.3 M NaCl was induced by removing half of the culture and replacing it with synthetic medium with an NaCl concentration of 0.5 M.
Microarray analysis
Cultures of the control strain H119 and the sRNA gene deletion mutant Δhts10 were grown to mid exponential growth phase. Three independent biological replicates were performed. Cells were harvested by centrifugation and RNA was isolated with the RNA isolation midi kit (Qiagen) according to the manufacturer’s instructions, but omitting the RW1 buffer to enable the isolation small RNAs. A DNase treatment was included, while the RNA was bound to the ion exchange column to exclude DNA contamination. The RNA concentration was determined photometrically and the integrity was controlled using an analytical agarose gel. The RNAs isolated from the control culture and the Δhts10 culture were reverse transcribed using random hexamer oligonucleotides and M-MLV reverse transcriptase RNase H minus (Promega). Cy3-dUTP or Cy5-dUTP were added to label the control sample and the Δhts10 sample, respectively, and in one replicate the addition was reversed (dye swap) to exclude systematic errors due to different properties of the two dyes. The microarray analysis was performed using a self-constructed DNA microarray comprised of PCR fragments39 to which 60mer oligonucleotides were later added for the detection of sRNAs.30 Slide preparation, competitive hybridization with the labeled cDNAs, washing and microarray scanning using a GenePix 4200A scanner were performed as described.39 The data were exported and Microsoft Excel was used for data filtering, background subtraction, calculation of average signals and their variances and for data sorting as described.40 The program Acuity (Molecular Devices) was used for data normalization.
Supplementary Material
Acknowledgments
We are grateful to Elli Bruckbauer for her expert technical help. Work was funded by the German Research Council (Deutsche Forschungsgemeinschaft) through the priority program SPP 1258 “Sensory and regulatory RNAs in Prokaryotes” (grants DFG Ma1538/11–2 and So264/14–2). We thank members of the priority program for helpful discussions.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/rnabiology/article/20826
References
- 1.Storz G. An expanding universe of noncoding RNAs. Science. 2002;296:1260–3. doi: 10.1126/science.1072249. [DOI] [PubMed] [Google Scholar]
- 2.Hüttenhofer A, Schattner P, Polacek N. Non-coding RNAs: hope or hype? Trends Genet. 2005;21:289–97. doi: 10.1016/j.tig.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 3.Brantl S. Bacterial chromosome-encoded small regulatory RNAs. Future Microbiol. 2009;4:85–103. doi: 10.2217/17460913.4.1.85. [DOI] [PubMed] [Google Scholar]
- 4.Meister G. miRNAs get an early start on translational silencing. Cell. 2007;131:25–8. doi: 10.1016/j.cell.2007.09.021. [DOI] [PubMed] [Google Scholar]
- 5.Guo H, Ingolia NT, Weissman JS, Bartel DP. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature. 2010;466:835–40. doi: 10.1038/nature09267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet. 2010;11:597–610. doi: 10.1038/nrg2843. [DOI] [PubMed] [Google Scholar]
- 7.Pederson T. Regulatory RNAs derived from transfer RNA? RNA. 2010;16:1865–9. doi: 10.1261/rna.2266510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sobala A, Hutvagner G. Transfer RNA-derived fragments: origins, processing, and functions. Wiley Interdiscip Rev RNA. 2011;2:853–62. doi: 10.1002/wrna.96. [DOI] [PubMed] [Google Scholar]
- 9.Cole C, Sobala A, Lu C, Thatcher SR, Bowman A, Brown JW, et al. Filtering of deep sequencing data reveals the existence of abundant Dicer-dependent small RNAs derived from tRNAs. RNA. 2009;15:2147–60. doi: 10.1261/rna.1738409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jinek M, Doudna JA. A three-dimensional view of the molecular machinery of RNA interference. Nature. 2009;457:405–12. doi: 10.1038/nature07755. [DOI] [PubMed] [Google Scholar]
- 11.Schiffer S, Rösch S, Marchfelder A. Assigning a function to a conserved group of proteins: the tRNA 3′-processing enzymes. EMBO J. 2002;21:2769–77. doi: 10.1093/emboj/21.11.2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hartmann RK, Gössringer M, Späth B, Fischer S, Marchfelder A. The making of tRNAs and more - RNase P and tRNase Z. Prog Mol Biol Transl Sci. 2009;85:319–68. doi: 10.1016/S0079-6603(08)00808-8. [DOI] [PubMed] [Google Scholar]
- 13.Lee YS, Shibata Y, Malhotra A, Dutta A. A novel class of small RNAs: tRNA-derived RNA fragments (tRFs) Genes Dev. 2009;23:2639–49. doi: 10.1101/gad.1837609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Storz G, Vogel J, Wassarman KM. Regulation by small RNAs in bacteria: expanding frontiers. Mol Cell. 2011;43:880–91. doi: 10.1016/j.molcel.2011.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schmitz R, Jäger D, Jellen-Ritter A. Babski. J, Soppa J, Marchfelder A. 2011. Archaea employ small RNAs as regulators. In: Hess WR, Marchfelder A, eds. Regulatory RNAs in Prokaryotes Wien: Springer. pp 131-45. [Google Scholar]
- 16.Marchfelder A, Fischer S, Brendel J, Stoll B, Maier LK, Jäger D, et al. Small RNAs for Defence and Regulation in Archaea. Extremophiles. 2012 doi: 10.1007/s00792-012-0469-5. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Klein RJ, Misulovin Z, Eddy SR. Noncoding RNA genes identified in AT-rich hyperthermophiles. Proc Natl Acad Sci U S A. 2002;99:7542–7. doi: 10.1073/pnas.112063799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schattner P. Searching for RNA genes using base-composition statistics. Nucleic Acids Res. 2002;30:2076–82. doi: 10.1093/nar/30.9.2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tang TH, Bachellerie JP, Rozhdestvensky T, Bortolin ML, Huber H, Drungowski M, et al. Identification of 86 candidates for small non-messenger RNAs from the archaeon Archaeoglobus fulgidus. Proc Natl Acad Sci U S A. 2002;99:7536–41. doi: 10.1073/pnas.112047299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tang TH, Rozhdestvensky TS, d’Orval BC, Bortolin ML, Huber H, Charpentier B, et al. RNomics in Archaea reveals a further link between splicing of archaeal introns and rRNA processing. Nucleic Acids Res. 2002;30:921–30. doi: 10.1093/nar/30.4.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wurtzel O, Sapra R, Chen F, Zhu Y, Simmons BA, Sorek R. A single-base resolution map of an archaeal transcriptome. Genome Res. 2010;20:133–41. doi: 10.1101/gr.100396.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jäger D, Sharma CM, Thomsen J, Ehlers C, Vogel J, Schmitz RA. Deep sequencing analysis of the Methanosarcina mazei Gö1 transcriptome in response to nitrogen availability. Proc Natl Acad Sci U S A. 2009;106:21878–82. doi: 10.1073/pnas.0909051106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hartman AL, Norais C, Badger JH, Delmas S, Haldenby S, Madupu R, et al. The complete genome sequence of Haloferax volcanii DS2, a model archaeon. PLoS One. 2010;5:e9605. doi: 10.1371/journal.pone.0009605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Allers T, Mevarech M. Archaeal genetics - the third way. Nat Rev Genet. 2005;6:58–73. doi: 10.1038/nrg1504. [DOI] [PubMed] [Google Scholar]
- 25.Leigh JA, Albers SV, Atomi H, Allers T. Model organisms for genetics in the domain Archaea: methanogens, halophiles, Thermococcales and Sulfolobales. FEMS Microbiol Rev. 2011;35:577–608. doi: 10.1111/j.1574-6976.2011.00265.x. [DOI] [PubMed] [Google Scholar]
- 26.Soppa J. Functional genomic and advanced genetic studies reveal novel insights into the metabolism, regulation, and biology of Haloferax volcanii. Archaea. 2011;2011:602408. doi: 10.1155/2011/602408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Soppa J, Straub J, Brenneis M, Jellen-Ritter A, Heyer R, Fischer S, et al. Small RNAs of the halophilic archaeon Haloferax volcanii. Biochem Soc Trans. 2009;37:133–6. doi: 10.1042/BST0370133. [DOI] [PubMed] [Google Scholar]
- 28.Straub J, Brenneis M, Jellen-Ritter A, Heyer R, Soppa J, Marchfelder A. Small RNAs in haloarchaea: identification, differential expression and biological function. RNA Biol. 2009;6:281–92. doi: 10.4161/rna.6.3.8357. [DOI] [PubMed] [Google Scholar]
- 29.Fischer S, Benz J, Späth B, Jellen-Ritter A, Heyer R, Dörr M, et al. Regulatory RNAs in Haloferax volcanii. Biochem Soc Trans. 2011;39:159–62. doi: 10.1042/BST0390159. [DOI] [PubMed] [Google Scholar]
- 30.Babski J, Tjaden B, Voss B, Jellen-Ritter A, Marchfelder A, Hess WR, et al. Bioinformatic prediction and experimental verification of sRNAs in the haloarchaeon Haloferax volcanii. RNA Biol. 2011;8 doi: 10.4161/rna.8.5.16039. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 31.Bitan-Banin G, Ortenberg R, Mevarech M. Development of a gene knockout system for the halophilic archaeon Haloferax volcanii by use of the pyrE gene. J Bacteriol. 2003;185:772–8. doi: 10.1128/JB.185.3.772-778.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Allers T, Ngo HP, Mevarech M, Lloyd RG. Development of additional selectable markers for the halophilic archaeon Haloferax volcanii based on the leuB and trpA genes. Appl Environ Microbiol. 2004;70:943–53. doi: 10.1128/AEM.70.2.943-953.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith C, Heyne S, Richter AS, Will S, Backofen R. Freiburg RNA Tools: a web server integrating INTARNA, EXPARNA and LOCARNA. Nucleic Acids Res. 2010;38(Web Server issue):W373-7. doi: 10.1093/nar/gkq316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Allers T, Barak S, Liddell S, Wardell K, Mevarech M. Improved strains and plasmid vectors for conditional overexpression of His-tagged proteins in Haloferax volcanii. Appl Environ Microbiol. 2010;76:1759–69. doi: 10.1128/AEM.02670-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nieuwlandt DT, Palmer JR, Armbruster DT, Kuo Y-P, Oda W, Daniels CJ. 1995. A rapid procedure for the isolation of RNA from Haloferax volcanii. In: Robb FT, Place AR, Sowers KR, Schreier HJ, DasSarma S, Fleischmann EM, eds. Archaea: A laboratory manual New York: Cold Spring Harbour Press. pp 161. [Google Scholar]
- 36.Sambrook J, Russell D. 2001. Molecular Cloning: A Laboratory Manual New York: Cold Spring Harbour Press. [Google Scholar]
- 37.Hölzle A, Fischer S, Heyer R, Schütz S, Zacharias M, Walther P, et al. Maturation of the 5S rRNA 5′ end is catalyzed in vitro by the endonuclease tRNase Z in the archaeon H. volcanii. RNA. 2008;14:928–37. doi: 10.1261/rna.933208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jantzer K, Zerulla K, Soppa J. Phenotyping in the archaea: optimization of growth parameters and analysis of mutants of Haloferax volcanii. FEMS Microbiol Lett. 2011;322:123–30. doi: 10.1111/j.1574-6968.2011.02341.x. [DOI] [PubMed] [Google Scholar]
- 39.Zaigler A, Schuster SC, Soppa J. Construction and usage of a onefold-coverage shotgun DNA microarray to characterize the metabolism of the archaeon Haloferax volcanii. Mol Microbiol. 2003;48:1089–105. doi: 10.1046/j.1365-2958.2003.03497.x. [DOI] [PubMed] [Google Scholar]
- 40.Lange C, Zaigler A, Hammelmann M, Twellmeyer J, Raddatz G, Schuster SC, et al. Genome-wide analysis of growth phase-dependent translational and transcriptional regulation in halophilic archaea. BMC Genomics. 2007;8:415. doi: 10.1186/1471-2164-8-415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. www.haloarchaea.com/resources/halohandbook/ Halohandbook_2008_v7.pdf
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.