

Abstract
The tumor suppressor p53 is a sequence-specific transcription factor that activates the expression of genes involved in apoptosis, cell cycle arrest and senescence. p53 can also inhibit gene expression and this effect is partly mediated by inducing several microRNAs (miRNAs). MiRNAs have emerged as a new class of regulators of the expression and function of eukaryotic genomes. Tumor suppressive or oncogenic functions have been attributed to some miRNAs. Recent studies have shown that p53 can alter the transcription of several miRNAs, and in some cases, it can also influence miRNA maturation. Conversely, miRNAs can also modulate the abundance and activity of p53 by direct or indirect mechanisms. Moreover, mutant p53 can actively repress the expression of some miRNAs that are activated by wild-type p53. In this review, we discuss recent evidences of this crosstalk between miRNAs and the p53 network and also highlight its implications in cancer.
Keywords: cancer, miRNA, miRNA dysregulation, mutant p53, p53, wild-type p53
Introduction
RNA interference (RNAi) is a naturally occurring gene-silencing phenomenon that occurs in fungi, plants and animals. MiRNAs are the predominant and most widely studied class of endogenous small non-coding RNAs that post-transcriptionally silence gene expression through the RNAi pathway.1-6 MiRNAs control diverse biological processes from nematodes to mammals, predominantly by binding to the 3′ untranslated region (UTR) of target mRNAs.7-10 Many miRNAs are evolutionarily conserved in several organisms: almost 50% of C.elegans miRNAs have homologs in humans, suggesting important roles for these tiny molecules in essential biological processes.7,11 To date, 1527 distinct precursors and 1921 mature miRNAs have been identified in humans (miRBase version 18), and majority of protein-coding genes are miRNA targets.12-15 Each miRNA can regulate hundreds of genes and each gene can be regulated by multiple miRNAs, resulting in complex combinatorial post-transcriptional regulation of gene expression.16
Inappropriate repair of damaged DNA in a normal cell can lead to oncogene activation which in turn can drive cell proliferation and/or survival in the absence of physiological stimuli.17 In response to DNA damage, the tumor suppressor p53 is induced and causes growth arrest or apoptosis, depending upon the extent of DNA damage.18-21 In about 50% of human cancers, p53 function is compromised mainly due to deletion or point mutations in the TP53 gene.19,20,22 Therefore, the mechanisms by which p53 achieves tumor suppression have been subject to intense investigation.
Majority of the downstream effects of p53 activation are mediated through its intrinsic effects as a transcription factor that regulates the expression of a wide variety of genes.23-27 The cellular effects of p53 are partly mediated by its ability to upregulate anti-proliferative and proapoptotic genes such as p21 (G1 arrest), 14–3-3σ (G2 arrest) and PUMA (apoptosis). Interestingly, p53 also suppresses the expression of numerous genes23 including those involved in regulation of cell proliferation28,29 and apoptosis.30 In other words, the p53 transcriptional response involves both activation and repression of hundreds of genes. In addition to direct effects of p53 on the promoters of protein-coding genes, p53 activation has recently been shown to modulate the expression of miRNAs, which in turn, can dampen the expression of hundreds of proteins. The miRNAs upregulated by p53 could provide an attractive mechanism to explain post-transcriptional inhibition of gene expression upon p53 activation. Such a mechanism may be particularly important during the stress response since it does not require the translation of effector proteins and may facilitate regulation of numerous processes by p53. Furthermore, miRNAs can also regulate p53 itself by direct or indirect mechanisms, suggesting that miRNAs are key components of the p53 network. Here, we provide an overview of the expanding universe of the p53 master regulatory network and discuss the potential roles of the crosstalk between miRNAs and p53 in tumor suppression and cancer prevention.
miRNA Biogenesis and Mechanism of Action
Mature miRNAs are ~22 nucleotides (nt) long, but these functional, single stranded molecules are the products of a complex, multistep processing mechanism. Endogenous miRNA-coding loci may be embedded in the exons or introns of other genes or in intergenic regions. Early studies suggested that majority of miRNAs are intergenic but it is now clear that miRNAs are generally located in the intronic regions of protein-coding or non-coding genes.11,31,32 Cleavage of the intron during miRNA biogenesis does not impact splicing of the host gene because it occurs between the splicing commitment step and the intron excision step. Thus, excision of miRNA from an intron and mRNA splicing is a highly regulated process to ensure proper miRNA biogenesis and protein synthesis from a single primary transcript.
Nearly 50% of human miRNA loci are found clustered near other miRNAs, and these clusters are transcribed together as polycistronic transcriptional units.33 Transcription of miRNAs may be contextually dependent on the promoters of surrounding genes or by discrete, miRNA-specific promoters. In most cases, transcription of miRNAs is mediated by RNA-polymerase II, which generates a primary transcript several kilobases in length that contains stem-loop structures. This stem-loop structure is initially processed in the nucleus by the RNaseIII Drosha, its cofactor DGCR8 and several other proteins that together form the microprocessor complex.11,34 The product of Drosha cleavage is a precursor hairpin miRNA (pre-miRNA), which is exported to the cytoplasm by the nuclear transporter exportin5 in association with the GTP-bound form of Ran-GTPase. In the cytoplasm, the pre-miRNA undergoes its final cleavage step, with the RNase Dicer cutting the hairpin near its terminal loop, yielding a 22 nt miRNA duplex composed of a guide strand and a passenger strand.35 The strand with relatively less stable 5′ base pairs is selected as the guide strand and will go on to interact with the target mRNA while the passenger strand is generally degraded.36
To silence gene expression, miRNAs are assembled into a RNA-induced silencing complex (RISC) consisting of several components including Dicer, Argonaute proteins, TRBP and the dsRNA-binding protein PACT. Base-pairing of miRNAs to target mRNAs results in mRNA degradation and/or translational suppression. The ability of a miRNA to suppress gene expression largely depends on the complementarity between the target mRNA 3′UTR and positions 2–8 of the 5′ end of the miRNA, known as seed sequences,8,37,38 but some 3′UTRs can also be regulated by seed-independent mechanisms.39-41 As a consequence of imperfect base-pairing between the miRNA guide strand and its target mRNA, a single miRNA can suppress the 3′UTRs of hundreds of genes12,15,42,43 but this regulatory promiscuity also makes identifying miRNA targets challenging.44 Although miRNAs predominantly act by binding to the 3′UTR, there is evidence in support of a functional role for miRNA binding sites in the coding region, though the effects are weaker than for sites in the 3′UTR.45-49 Irrespective of the location of the miRNA-binding site within the target mRNA, most early studies showed that mammalian miRNAs inhibited gene expression by suppressing translation. However, recent studies suggest that mammalian miRNAs mainly modulate mRNA stability.12,42,50,51 Finally, greater than 50% of human genes are thought to be directly targeted by miRNAs, and the indirect effects extend even further, indicating that miRNAs have an important and well-conserved role in regulating global gene expression.
Dysregulation of miRNAs in Cancer
The expression of miRNAs is perturbed in cancer cells compared with corresponding normal tissues. MiRNAs can act as tumor suppressor genes or oncogenes and can have key functions in tumorigenesis and tumor progression.52-61 The first link between miRNAs and cancer was provided by genetic studies of chronic lymphocytic leukemia (CLL) in which the breakpoint of a CLL translocation was found to map to the miR-15a-16–1 locus at chromosome 13q14, a region typically deleted in CLL.62 Genetic deletion of this locus in mice causes CLL and other lymphoproliferative disorders.63 Several other tumor suppressor miRNAs, including members of the let-7 family map to fragile sites and are deleted in some breast, lung, ovarian, and cervical cancers. Oncogenic miRNA loci such as the miR-17:92 and miR-106b-25 clusters are frequently amplified in some cancers.64 Although these studies emphasize the importance of specific miRNAs acting as tumor suppressors or oncogenes, global downregulation of miRNAs is a frequent event in human cancers.65,66
Recent studies have identified tumor-specific genetic defects in the miRNA-processing machinery, such as the genes encoding TARBP2,67 Dicer 168 and exportin 5.69 Alterations in maturation of specific miRNAs can also cause changes in miRNA expression in cancer. For instance, in undifferentiated tumors maturation of the tumor suppressor let-7 is blocked by the RNA-binding proteins LIN28 and LIN28B.70 These findings underscore the relevance of the miRNA-processing machinery in cellular transformation, and defects in these mechanisms may contribute to miRNA dysregulation in cancer. In addition to genetic changes, dysregulation of miRNAs in cancer can also be mediated by epigenetic changes.54 For instance, in some cancers, miR-20071 and miR-12772 are aberrantly silenced by alterations in promoter methylation.
Finally, the same transcription factors that control mRNA expression can also regulate miRNA transcription. For instance, the proto-oncogene MYC activates the miR-17–92 cluster and causes widespread downregulation of miRNA genes including tumor suppressor miRNAs such as miR-15a/16–1, miR-34a, and let-7 family members.73,74 Interestingly, suppression of let-7 by MYC is not due to altered transcription of let-7 genes75 but instead, is mediated by transcriptional induction of Lin28B by MYC, which in turn, results in inhibition of maturation of let-7 family of miRNAs. It appears that the MYC-regulated miRNAs affect virtually all aspects of the MYC oncogenic program, including proliferation, survival, metabolism, angiogenesis, and metastasis.76 Besides MYC, other key proteins involved in transcriptional regulation, including E2F1,77,78 ZEB179,80 and p53 can also regulate the transcription of several miRNAs.81-84
Transcriptional Regulation of miRNAs by p53
In the p53 tumor suppressor network, many of the functions normally associated with p53 may also be executed by miRNAs. As a transcription factor, p53 directly regulates the transcription of a growing number of miRNAs (Fig. 1), acting both as a transactivator of tumor-suppressive miRNAs and a repressor of some oncogenic miRNAs. MiRNAs upregulated by p53 often target anti-apoptotic and pro-proliferative genes, thus reinforcing the function of p53,83 or they may even feedback to regulate p53 itself.85-87 It is not yet clear whether miRNAs are important for initiating the effects of p53 or for maintaining them. Further research will be needed to understand the role miRNAs play in mediating cell death and tumor suppression in the p53 response.88,89 It is already evident that our current understanding of p53 will continue to change as more interactions with miRNAs are discovered.
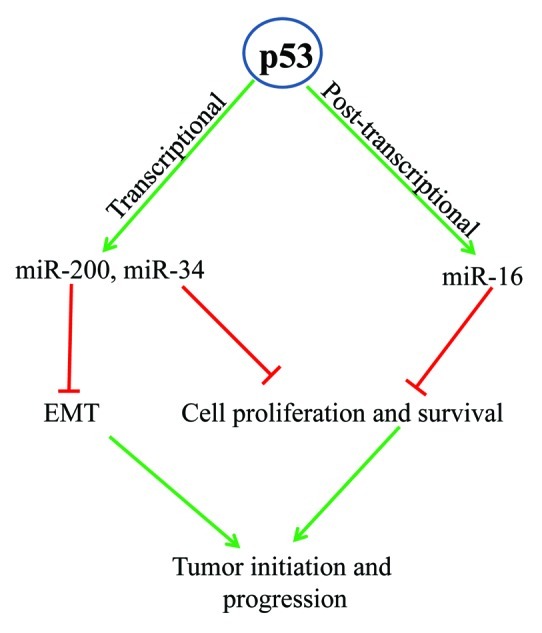
Figure 1. Regulation of miRNA biogenesis by p53. p53 enhances the expression of miRNAs including miR-200, miR-34 and miR-16. During DNA damage, p53 transcriptionally activates miR-34 family and post-transcriptionally upregulates miR-16. These miRNAs, inhibit the expression of several genes involved in cell proliferation and survival. Basal p53 levels can also regulate miRNAs such as the miR-200 family to inhibit EMT. By inducing miRNAs, p53 can exert its tumor suppressive function to inhibit tumor initiation and progression.
miRNAs Transcriptionally Upregulated by p53
The miR-34 family was among the first to be connected with p53 and has since been the subject of intense study in relation to cancer. In mammals, the miR-34 family consists of three miRNAs encoded by two loci: miR-34a is transcribed independently whereas miR-34b and miR-34c share a primary transcript. Both miR-34a and the miR-34b/c polycistron have p53-responsive elements in their promoter regions.81 Because the p53 protein binds to DNA in a sequence-specific manner and generally activates proximal genes in trans, the genes it transcriptionally regulates can often be found by chromatin immunoprecipitation (ChIP). Genome-wide ChIP for p53 initially revealed putative binding sites proximal to many miRNAs.90 The presence of p53 binding sites upstream of both miR-34 family loci suggested a conserved and functionally significant role for miR-34 in the p53 pathway. This miRNA family plays a key role in mediating the anti-proliferative and pro-apoptotic effects of p53 by targeting cell-cycle genes and proto-oncogenes.82,84 After forced expression of miR-34, upregulated genes were enriched for those classified in gene ontology as cell cycle, DNA repair, mitotic checkpoint, and DNA integrity checkpoint. Downregulated transcripts were enriched for cell proliferation and angiogenesis genes. Significantly, p53 is known to activate and repress these same gene categories.91 Using a novel biochemical approach to identify miRNA targets, we have recently uncovered the cellular transcripts bound by miR-34a.92 We found that miR-34a plays a critical role in growth factor signaling and regulates a network of genes involved in cellular proliferation and cell survival.
Although miR-34 was one of the first miRNA families shown to be regulated by p53, the downstream targets of miR-34 in response to DNA damage are less well known. Recently, the p53-miR-34 axis has been shown to regulate Snail-dependent epithelial-to-mesenchymal transition (EMT).93 EMT is a developmental program that may be reactivated in some cancers, allowing for epithelial cancer cells to adopt a more mobile mesenchymal phenotype and spread to distant sites. The miR-34 family members have been shown to directly target Snail1, a known regulator of cell motility and matrix metalloproteinase activity. It should also be noted that miR-34 can also be regulated by p53-independent mechanisms; miR-34a is dramatically induced during megakaryocyte differentiation of K562 cells (which do not express p53).94 Another example of p53-independent miR-34 function is the recent finding that MYC overexpression can override p53 regulation of miR-34. MYC and miR-34 are partners in a reciprocal negative feedback loop: miR-34 targets MYC, while MYC represses miR-34 transcription.95 miR-34 expression is often lost in many human cancers, either as a result of p53 mutation or deletion of the miR-34a locus at 1p36.87 Changes in miR-34 levels may cause dramatic reprogramming of gene expression and push the cell decisively toward apoptosis (increased miR-34) or survival (decreased miR-34).
While miR-34 has just recently been implicated in EMT via the Snail1 pathway, the miR-200 family consisting of 5 miRNAs (miR-200a,b,c, miR-141, and miR-429) has been known for some time to play an important role in regulating EMT by downregulating Zeb1 and Zeb2.96,97 Zeb1 and Zeb2 repress E-cadherin transcription and decreased E-cadherin expression is a hallmark of EMT. Not only do all the members of the miR-200 family target Zeb1 and Zeb2, but ectopic expression of miR-200s in mesenchymal cells can trigger a reverse EMT or MET (mesenchymal to epithelial transition) consistent with restored E-cadherin expression.96-98 This reversion from a motile mesenchymal phenotype back to an epithelial phenotype is thought to be necessary for the final colonization steps of the metastatic process. p53 binds to a sequence in the miR-200c promoter to transactivate miR-200c (Fig. 1).99 By knocking down p53 in human mammary carcinoma cells (MCF12A), this study showed that p53 expression correlates directly with an epithelial phenotype. Expression of miR-200c in p53 knockdown cells mitigated the loss of p53 and suppressed EMT and stem cell phenotypes. Additionally, miR-200c was seen to directly target the 3′UTR of the polycomb gene BMI1, suggesting that p53 can act through miR-200 to regulate both EMT and stem cell phenotypes. Another recent study expanded on this finding to show that p53 can also regulate EMT through miR-200a, miR-200b, and miR-192.100 Shortly after these reports, p53 and miR-200 were also found to control EMT and invasion by a mechanism independent of Zeb1/2 and E-cadherin. miR-200 family expression correlates positively with metastasis in human breast cancers, which seems counterintuitive given its previously reported role as a suppressor of EMT. However, Korpal et al. found that by targeting the mRNA of the membrane secretory protein sec23a, miR-200 inhibits the secretion of anti-metastatic factors such as Igfbp4 and Tinagl1.101 Therefore, miR-200 seems to have a biphasic role in the metastatic cascade: in the initial stages of invasion and intravasation, low miR-200 expression favors tumor progression by decreasing E-cadherin and increased motility; in later stages, high miR-200 levels promotes metastatic colonization. These data support and help explain an earlier study that showed miR-200 expression to be elevated in metastatic breast cancer cell lines relative to isogenic non-metastatic cells102 and that enforced miR-200 expression conferred metastatic capability to a non-metastatic cell line. The case of miR-200 illustrates the difficulty in classifying some miRNAs as tumor suppressive or oncogenic simply by looking at the changes in its expression.
Although several groups have demonstrated changes in miRNA expression following the induction of p53, it is important to consider the method of p53 induction that was employed. Studies using doxorubicin or other genotoxic agents to induce p53 expression also cause non-specific effects associated with DNA damage. An alternate method of modulating p53 levels entails the use of the pharmacologic agent nutlin-3, which inhibits the interaction of p53 with its antagonist MDM2. Several recent reports have established that a different set of miRNAs are upregulated by p53 in response to nutlin-3 as compared with those elevated in the context of DNA damage. The miRNA clusters consisting of miR-192, miR-194, and miR-215 were upregulated after p53 activation by nutlin-3 in the sarcoma line SJSA.103 In this system, the change in miR-194 cluster expression in response to p53 activation was even more profound than that of miR-34—the poster-child of p53-inducible miRNAs. Pichiorri and Croce, et al. conducted a similar study in cell lines and samples derived from multiple myeloma and found strong upregulation of miR-192, miR-194, and miR-215 after treatment with nutlin-3. However, of these miRNAs, only miR-194 was differentially expressed between multiple myeloma cell lines with wild-type or mutant TP53. Significantly, the core promoter region for the miR-194–192 cluster is hypermethylated in multiple myeloma, suggesting that clonal selection favors silencing of these miRNAs. Functionally, miR-192, miR-194, and miR-215 target MDM2 (Fig. 2) and may be involved in a positive feedback loop with p53. Methylation of the miR-194–192 promoter may tip the balance in multiple myeloma toward p53 acetylation and contribute to tumorigenesis.85

Figure 2. Post-transcriptional regulation of p53 by miRNAs. p53 is regulated by several miRNAs. miR-125b, miR-504, miR-25 and miR-30d directly bind to the 3′UTR of p53 mRNA and inhibit its expression. In fibroblasts, miR-122, inhibits CPEB leading to decreased p53 mRNA translation. In livercells, miR-122 inhibits cyclin G1 3′UTR and upregulates p53 levels via decreased MDM2. miR-34, miR-192, miR-194, miR-215, miR-605 and miR-29 regulate the regulators of p53 to indirectly activate p53.
A recent study showed that the gene Panthothenate kinase 1 (PANK1) and its embedded intronic miRNA miR-107 are co-regulated by p53.104 This study also showed that miR-107 targets the cell cycle genes CDK6 and p130, which are involved in G1/S progression.104 A contemporaneous study by Yamakuchi et al. confirmed upregulation of miR-107 by p53 and suggested a role for miR-107 in suppressing the cell’s response to hypoxia by targeting HIF1-2. Additionally, forced expression of miR-107 in mice reduced tumor growth, angiogenesis, and VEGF expression.105
Alterations in the non-protein coding genomic region PVT1 are associated with many human cancers: the locus is frequently amplified or overexpressed in some cancers. No long non-coding transcripts have been found within PVT1, but the locus does express 6 annotated miRNAs: miR-1204, miR-1205, miR-1206, miR-1207–5p, miR-1207–3p, and miR-1208.106 Barsotti et al. have shown that this cluster may be involved in a positive-feedback loop with p53. p53 binds to a region proximal to miR-1204 and increases its expression. Upregulation of miR-1204, in turn, results in an increase in p53 expression and potentiates cell death, but the mechanism for this feedback remains unclear.107
miRNAs Transcriptionally Repressed by p53
Although p53 acts primarily to activate the transcription of tumor suppressive miRNAs, recent evidence suggests that p53 can also directly inhibit the expression of oncogenic miRNAs, including the miR-17–92 cluster. The miR-17–92 cluster on chromosome 13 comprises 7 miRNAs that are transcribed as a polycistron. Expression of this cluster is induced by MYC 74 and is increased in breast, colon, pancreas, lung and prostate carcinomas.108 miR-17–92 is known to exert an anti-apoptotic influence in response to MYC by targeting the transcript of the pro-apoptotic protein Bim.109 Consistent with its putative role as an oncogenic miRNA cluster, miR-17-92 is transcriptionally repressed by p53 in hypoxic conditions, resulting in increased apoptosis.110 Yan et al. showed an inverse correlation between p53 status and miR-17-92 expression in a paired comparison of colorectal carcinoma and normal colonic tissue. A similar repression of miR-17-92 was also seen in response to DNA damage. This study also used ChIP to demonstrate direct binding of p53 to a binding site overlapping a crucial non-consensus TATA box in the miR-17-92 promoter, thus interfering with binding of the TATA box binding protein (TBP) and preventing the recruitment of transcriptional machinery. In addition to the miR-17-92 cluster, some members of the let-7 family are also repressed by p53. The let-7 family are well-documented tumor suppressors that target growth related genes such as RAS, CDC25A and CYCLIN D1.111,112 Previously, expression of let-7a and let-7b had been shown to decrease under ionizing radiation or cytotoxic chemotherapy, but p53 was not implicated.113 Recently, however, Saleh et al. have shown that under genotoxic stress, p53 binds to an enhancer upstream of let-7a3 and let-7b, which presumably competitively prevents binding of other transcriptional activators (perhaps by a mechanism similar to that outlined above for miR-17-92) leading to downregulation of these let-7 family members. Because tissues with higher let-7 expression have been shown to be more sensitive to radiation and genotoxic chemotherapy, it has been suggested that let-7 mimics could be used to potentiate adjuvant cancer therapy.114
Post-Transcriptional Regulation of miRNAs by p53
Most research on p53 and miRNA focuses on the transcriptional effects of p53 on miRNA expression. However, p53 also has transcription-independent functions.115,116 Indeed, a recent study117 suggests that p53’s influence on miRNA might extend beyond transcriptional regulation. In HCT116 cells and human fibroblasts, p53 immunoprecipitated with the miRNA-processing protein Drosha through the DEAD-box RNA helicase DDX5 (also known as p68). Drosha and its associated DEAD-box proteins are components of the microprocessor complex, which processes and cleaves primary miRNA transcripts to yield pre-miRNA hairpins. The observed association of p53 with DDX5 correlated with an increase in the levels of several different mature and precursor miRNAs after DNA damage, including miR-16–1 (Fig. 1), miR-143, miR-145 and miR-206; primary transcript levels were unchanged. These miRNAs modulate the expression of cell proliferation and stemness associated genes,118,119 and their regulation by p53 suggests a role for p53 controlling global gene expression and cell fate. A similar post-transcriptional regulation of miRNA biogenesis involving p68/p78 has also been shown to be mediated by other nuclear proteins such as SMAD4, Nanog and estrogen receptor- ±.120-122 In sum, these findings suggest a novel function of p53 in miRNA maturation and suggest that several transcription factors interact with the Drosha processing machinery to regulate miRNA expression.
Regulation of p53 by miRNAs
The crosstalk between the p53 network and miRNAs is further substantiated by recent studies on regulation of p53 by miRNAs. To date, several miRNAs including miR-125b, miR-504, miR-25, miR-30d, miR-34a, miR-122, miR-29, miR-192, miR-194 and miR-215 have been shown to regulate p53 abundance and/or activity. Among these miR-125b, miR-504, miR-25 and miR-30d negatively regulate p53 by binding to its 3′UTR (Fig. 2) whereas the others indirectly influence p53 abundance and/or activity by regulating the regulators of p53 (Fig. 2).
p53-Suppressing miRNAs
Bioinformatic tools predict potential targeting of the p53 3′UTR by several miRNAs. Indeed, miR-125b, a miRNA highly abundant in the brain, downregulates p53 in both zebrafish and humans123 by binding to the 3′UTR of p53 mRNA (Fig. 2). Overexpression of miR-125b decreased endogenous p53 levels and inhibited apoptosis in human neuroblastoma cells and human lung fibroblast cells. Conversely, knockdown of miR-125b increased the abundance of p53 protein and induced apoptosis in human lung fibroblasts and in the zebrafish brain. Importantly, this phenotype could be rescued by either an ablation of endogenous p53 function or ectopic expression of miR-125b in zebrafish. miR-125b was downregulated when zebrafish embryos were treated with gamma-radiation or camptothecin, and introduction of miR-125b suppressed the increase of p53 and inhibited apoptosis. These findings suggested that miR-125b is an important negative regulator of p53 and p53-induced apoptosis during development and also during the stress response. In a subsequent study,124 the same group recently showed that in addition to p53, miR-125b down-modulates the expression of 20 novel targets in the p53 network. These targets include both apoptosis regulators like BAK1, IGFBP3, ITCH, PUMA, PRKRA, TP53INP1, TP53, ZAC1, and cell-cycle genes such as CYCLIN C, CDC25C, CDKN2C, EDN1, PPP1CA, SEL1L, in the p53 network. Although each miRNA-target pair was rarely conserved between human, mouse and zebrafish, regulation of the p53 pathway was conserved at the network level. These findings imply that miR-125b is an important regulator of the p53 pathway and plays a key role in cellular homeostasis and tumorigenesis.
In addition to these studies on miR-125b, a recent study employed a bioinformatic approach to identify candidate p53-regulating miRNAs. Among the top 5 putative p53-targeting miRNAs, miR-504 was found to downregulate p53 (Fig. 2) by binding to 2 sites in the 3′UTR of p53 mRNA.125 In response to stress, overexpression of miR-504 reduced p53 abundance, impaired p53-mediated apoptosis and cell cycle arrest. Furthermore, miR-504 overexpression promoted tumor growth in mice and this effect was not observed in an isogenic p53-deficient cell line, suggesting that the oncogenic function of miR-504 is mediated through downregulation of p53. In another recent study, a luciferase reporter screening approach was used to identify miRNAs directly targeting the 3′UTR of p53 mRNA.126 Of the 65 miRNAs that inhibited luciferase expression, miR-25 and miR-30d directly downregulated p53 mRNA and protein levels (Fig. 2) and also reduced the expression of genes that are transactivated by p53. Overexpression of these miRNAs inhibited p53-mediated apoptosis, cell cycle arrest and senescence. Conversely, antagonizing miR-25 or miR-30d resulted in increased endogenous p53 expression and elevated apoptosis in several cell lines.
Taken together, the above-mentioned studies demonstrate that miR-125b, miR-504, miR-25 and miR-30d negatively regulate the basal levels of p53 by direct association with the 3′UTR of p53 mRNA. miR-125b and miR-504 did not repress the p53 3′UTR in the study by Kumar et al., suggesting that their screening approach may have false negatives. Another possibility is that the effect of these miRNAs on p53 may depend on the cellular context. Of note, other family members of miR-25 and miR-30d (miR-92a,b and miR-30a,b,c,e) were not identified as p53 regulators126 in the luciferase reporter screen even though they have the same seed sequence. These findings suggest that in addition to the seed sequence, regulation of p53 by miR-25 and miR-30d requires additional base-pairing between the 3′UTR of p53 and 3′ends of these miRNAs. It is noteworthy that of these four p53-inhibiting miRNAs, miR-25, miR-125b and miR-30d belong to gene families that are conserved in fly, zebrafish, mouse and humans127 while miR-504 is mammalian-specific. Finally, the 3′UTR of p53 is not well conserved between human and mouse except for a ~100-nt region, meaning that these miRNAs cannot directly regulate p53 in mouse cells. It would be interesting to see whether similar to miR-125b, the other p53-repressing miRNAs may still inhibit the p53 pathway in mouse cells by targeting a distinct set of genes in the p53 network.
p53-Activating miRNAs
In addition to direct suppression of p53 levels, several miRNAs including miR-34a, miR-605, miR-122, miR-29, miR-192, miR-194 and miR-215 can indirectly activate p53 (Fig. 2). miR-34a, a miRNA transcriptionally regulated by p53, was found to regulate SIRT1 87 and YY1,128 two proteins that negatively regulate p53. SIRT1 regulates p53 dependent apoptosis by deacteylating and destabilizing p53129 whereas YY1 downregulates p53 by stimulating p53 ubiquitination and degradation.130 p53 induces miR-34a expression, which in turn increases the abundance and activity of p53 by post-transcriptionally suppressing SIRT1 and YY1 through direct binding to their 3′UTRs. It is plausible that the positive feedback loop involving p53, miR-34a and its target genes, SIRT1 and YY1 may enable p53 to maintain miR-34a expression. A similar positive feedback loop has been identified for miR-605, miR-192, miR-194 and miR-215 and p53.85,86 These miRNAs are transcriptional targets of p53 and also upregulate p53 by downregulating MDM2 (Fig. 2), a gene that controls the half-life of p53 protein.
The stability of p53 is also regulated by the oncogenic phosphatase Wip1 (PPM1D), which is itself induced by p53 following DNA damage. Wip1 dephosphorylates p38/MAPK and MDM2, resulting in decreased phosphorylation and acetylation of p53 and ultimately suppressing p53-mediated apoptosis and cell-cycle arrest.131 Zhang et al. have reported that the p53-inducible miRNA, miR-16,117 directly targets Wip1. miR-16 expression increases sharply after treatment with the DNA damaging agent neocarzinostatin (NCZ), and although WIP1 transcription increased at a similar rate, there was a significant lag between WIP1 mRNA accumulation and detection of Wip1 protein due to translational inhibition by miR-16. The delayed expression of Wip1 allows the damaged cell to complete a DNA-damage responsive signaling cascade, and the subsequent gradual de-repression of Wip1 terminates the p53-mediated damage response. miR-16 was also seen to be decreased in mammospheres cultured from breast cancer stem and progenitor cells, indicating a significant permissive role for miR-16 in the DNA damage response as well as a role for p53 in restricting the development of cancer stem cells.132
Another study identified miRNAs that activate p53 by investigating the effects of 91 cancer-associated miRNAs on the luciferase expression of a reporter plasmid containing 13 p53 binding sites upstream of a luciferase gene. Of the 10 miRNAs (miR-186, miR-187, miR-95, miR-191, miR-181b, miR-155, miR-29a, miR-183, miR-125a and miR-302b) that significantly enhanced luciferase activity, miR-29a133 was chosen for further analysis since it also upregulated endogenous p53 protein levels, induced cell cycle arrest and decreased viability. The authors went on to show that miR-29 (Fig. 2) repressed the 3′UTR of p85 ± (a regulatory subunit of PI3 kinase (PI3K)) and CDC42 (which regulates cell migration and cell-cycle progression). Importantly, the authors showed that inhibition of miR-29 inhibited p53 function when the cells were treated with DNA-damaging agents that activate p53 activation and cause apoptosis. These findings suggest that miR-29 contributes to the DNA-damage response.
Upregulation and activation of p53 has also been shown to be mediated by miR-122,134 a liver-specific miRNA which is commonly downregulated in human and mouse liver cancer. miR-122 directly inhibits the expression of cyclin G1, a protein that forms a complex with the phosphatase, PP2A. Because PP2A is known to regulate phosphorylation of MDM2 (phosphorylated MDM2 is less active), downregulation of cyclin G1 inhibits PP2A, which in turn results in increased levels of phosphorylated MDM2. By directly repressing the 3′UTR of cyclin G1, miR-122 decreases MDM2 activity and enhances p53 levels and activity (Fig. 2). Interestingly, even though miR-122 accounts for almost 70% of the total miRNAs in liver and is thought to be liver-specific, a recent study135 showed that it is expressed in human skin fibroblasts where it inhibits the translation of p53 mRNA by directly suppressing the 3′UTR of CPEB, a protein that regulates polyadenylation of p53 mRNA. The authors proposed a model by which Gld2 (a non-canonical poly(A) polymerase) stabilizes miR-122 by catalyzing the addition of a single adenylate residue to its 3′end. In the absence of Gld2, miR-122 is destabilized and downregulated. Downregulation of miR-122 de-represses the 3′UTR of CPEB and upregulates CPEB. CPEB binds to the 3′UTR of p53 mRNA and recruits Gld4 (another non-canonical poly(A) polymerase) to increase p53 mRNA polyadenylation and translation, resulting in increased p53 protein levels and induction of senescence. Thus, translation of p53 mRNA and senescence is coordinately regulated by the Gld2/miR-122/CPEB/Gld4 axis. Interestingly, these studies show that miR-122 can either upregulate p53 by increasing the half-life of p53 protein through cyclin G1-PP2A-MDM2 or downregulate p53 by regulating p53 mRNA translation through CPEB-Gld4 in different cell types suggesting that the function of miR-122 is context-dependent. Although p53 is indirectly regulated by miR-122, it may be a key downstream effector of miR-122 (Fig. 2). It would be important to identify the targets and function of miR-122 in p53-deficient cells. These studies suggest that miRNAs not only are downstream effectors of p53, but they also regulate the regulators of p53 to indirectly upregulate p53 protein levels and p53 activity.
MiRNAs and Mutant p53
Unlike most tumor suppressor genes, the vast majority of cancer-associated mutations in p53 lead to the production of a full-length protein. In most cases, these mutations involve a single amino acid change that abrogates the tumor suppressor function of p53.26,136,137 Mutant p53 also possesses activities of its own, distinct from those of wild-type p53, and actively contributes to several aspects of tumor progression including genomic instability, cell survival, cell migration and invasion.27,136,138,139 Importantly, some of these gain-of-functions of mutant p53 have also been validated in animal models.140 Many groups have extensively investigated the mechanisms underlying the gain-of-function of mutant p53, and currently 3 mechanisms are well-established. First, mutant p53 can associate with transcription factors such as E2F1,141 VDR142 and NF-Y143 and is recruited to core promoters to regulate gene transcription.136,139 Second, mutant p53 can antagonize the activity of its family members, p63 and p73, and impair their transcriptional functions.144,145 Inhibition of p63 or p73 by mutant p53 is perhaps the most accepted mechanism of gene regulation by mutant p53. Third, even though many missense mutations in p53 occur in the DNA binding domain and thereby prevent p53 from activating its canonical target genes, mutant p53 can still directly bind to DNA via its N-terminal transactivation domain and drive the expression of its own set of target genes.146,147 Furthermore, in contrast to sequence-specific and DNA-structure dependent binding of wild-type p53, mutant p53 has been shown to associate with specific DNA elements such as matrix attachment regions (MAR) to regulate gene transcription.148 Taken together, these findings provide mechanistic insights on mutant p53 gain-of-function and suggest that mutant p53 can transactivate a number of wild-type p53-independent genes. If mutant p53 was associated with these transcription factors on each of their target promoters across the entire genome, one would expect mutant p53 to drive the expression of hundreds of genes. Corecruitment of these transcription factors and mutant p53 to specific promoters may involve interactions with coactivators or corepressors to assist in specific targeting of a smaller number of genes. For instance, mutant p53 is tethered to DNA by the transcription factor NF-Y and a corepressor to inhibit NF-Y target genes.143
Two recent studies have expanded the biochemical function of mutant p53 and shown that mutant p53 can also regulate miRNAs. In the first study,117 the tumor derived p53 mutants C135Y, R175H, and R273H regulate miRNA biogenesis post-transcriptionally by interfering with Drosha’s ability to bind primary miRNA transcripts. Overexpression of these p53 mutants decreased mature and precursor miRNA levels of miR-16–1, miR-143, and miR-205.117 The authors hypothesized that loss or mutation of p53 interferes with the assembly of the microprocessor complex, consisting of Drosha, DGCR8, and several DEAD-box RNA helicases including p72 and p68. In another study, qRT-PCR analysis of 11 miRNAs identified miR-128 as a miRNA upregulated by mutant p53R175H. Upregulation of miR-128 was achieved by transactivating the expression of its host gene, ARPP21.149 The authors went on to show that miR-128 directly suppressed the expression of the transcriptional repressor, E2F5 by binding to its 3′UTR. Downregulation of E2F5 by miR-128 derepressed p21 (CDKN1A) expression and cytoplasmic localization. Nuclear localization of p21 promotes growth arrest whereas cytoplasmic p21 prevents apoptosis by suppression of pro-caspase 3. The authors showed that the anti-apoptotic effect of miR-128 is mediated by its ability to suppress E2F5, which in turn induces p21. The induced p21 is predominantly cytoplasmic, possibly due to phosphorylation by AKT, and inhibits apoptosis by inhibiting pro-caspase 3. Taken together, their results provide evidence that miR-128 induction by mutant p53 contributes to the well-established chemoresistance activity of mutant p53. Interestingly, in mice, the p53 family member, p63, has been shown to regulate the transcription of Dicer.150 Because some p53 mutants can inhibit p63, it remains to be seen whether a global decrease in miRNAs occurs in mutant p53 expressing cells. Furthermore, because miRNAs and mutant p53 are known to modulate cell migration, invasion and metastasis, it would be worthwhile to investigate the role of miRNAs in these specific aspects of tumor progression regulated by mutant p53.
Conclusions and View to the Future
Since the discovery of let-7 in C. elegans, miRNAs have come to the fore as key regulators of gene expression. Although it was believed that most miRNAs may act more as fine-tuners than on/off switches, a recent study showed that miRNAs can act both as a switch and a fine-tuner of target gene expression.151 The authors show that although the average extent of target gene repression by miRNAs for a population of cells is generally modest, repression among individual cells varies dramatically and depends on the abundance of the target mRNA. A miRNA can act as a switch if the target mRNA levels in a specific cell is below a threshold, and as a fine-tuner in cells expressing higher target mRNA levels. This phenomenon may play a key role in cancer and could provide a proliferative and survival advantage to a pool of cells in a tissue due to the greatly increased potency of a miRNA in cells where the miRNA is capable of acting as a switch. The importance of miRNAs is also reflected by the fact that some miRNAs are very abundant (some miRNAs are expressed up to 50,000 copies/cell)152 and each miRNA can regulate a wide variety of cellular processes by suppressing the expression of hundreds of genes. As our understanding of the tumor suppressive and oncogenic roles of miRNAs has increased, we are attempting to understand this regulatory scenario: miRNAs regulate gene expression, but what regulates miRNA expression? Here we have reviewed recent and illustrative studies demonstrating that miRNAs are involved in a crosstalk with one of the most well-studied cellular networks, that of p53-the guardian of the genome. The crosstalk between p53 and miRNAs is particularly important given the multiple lines of evidence that indicate a major role for miRNAs153-155 and p53 during the stress response, and this is further exemplified by the fact that the stress phenotype is a hallmark of cancer.156
In this review we have discussed how (a) p53 regulates the abundance of specific miRNAs and enables p53 to post-transcriptionally inhibit the expression of large number of proteins, and (b) miRNAs can directly or indirectly modulate p53 abundance and p53 activity to inhibit or promote p53-associated functions including proliferation, apoptosis, cell migration and invasion. While miRNAs act at the post-transcriptional level, p53, as a transcription factor works to regulate miRNA expression by altering the activity of upstream promoters. p53 can act conventionally as a transcriptional transactivator or may have a repressive effect by blocking the binding of essential transcriptional machinery to the promoter.23,28,30 Furthermore, p53 interacts with proteins involved in the biogenesis of miRNAs to modulate miRNA maturation at a post-transcriptional level. More work will be needed to establish a clear picture of a physiological role of p53 in regulating miRNA expression and activity. Future directions should include further investigation of reciprocal feedback between p53 and miRNAs. Many of the above relationships between the p53 network and miRNAs have only been considered in the context of a single mode of p53 induction, e.g., DNA damage or hypoxia. It will be necessary for future work to integrate and reconcile the different responses seen in different systems.
While these studies enhance our knowledge of the crosstalk between the key tumor suppressor gene p53 and miRNAs, some issues should be kept in mind before establishing a biological relationship between a miRNA and target gene. In particular, several studies on miRNAs show that specific miRNAs regulate p53 in the context of overexpressing a given miRNA in cells that did not normally express that miRNA. These results may not apply to endogenous levels of miRNAs expressed. Given the fact that each miRNA can regulate hundreds of distinct mRNAs, the abundance of the miRNA in the cell type being used for investigating its function should be considered in any functional analyses. Finally, given the well-established links between mutant p53 and tumor progression and response to chemotherapy, future studies on miRNAs and mutant p53 may provide novel mechanistic insights about the gain-of-function phenotypes of mutant p53 in human cancers.
Acknowledgments
We thank Walter Bodmer (Oxford University) and members of the Lal lab for comments and suggestions. We apologize for being unable to cite all related publications due to space limitations. This work was supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research.
Footnotes
Previously published online: www.landesbioscience.com/journals/rnabiology/article/20146
References
- 1.Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–11. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 2.Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843–54. doi: 10.1016/0092-8674(93)90529-Y. [DOI] [PubMed] [Google Scholar]
- 3.Ghildiyal M, Zamore PD. Small silencing RNAs: an expanding universe. Nat Rev Genet. 2009;10:94–108. doi: 10.1038/nrg2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet. 2004;5:522–31. doi: 10.1038/nrg1379. [DOI] [PubMed] [Google Scholar]
- 5.Mendell JT. MicroRNAs: critical regulators of development, cellular physiology and malignancy. Cell Cycle. 2005;4:1179–84. doi: 10.4161/cc.4.9.2032. [DOI] [PubMed] [Google Scholar]
- 6.Bushati N, Cohen SM. microRNA functions. Annu Rev Cell Dev Biol. 2007;23:175–205. doi: 10.1146/annurev.cellbio.23.090506.123406. [DOI] [PubMed] [Google Scholar]
- 7.Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–5. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 8.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–97. doi: 10.1016/S0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 9.Berezikov E. Evolution of microRNA diversity and regulation in animals. Nat Rev Genet. 2011;12:846–60. doi: 10.1038/nrg3079. [DOI] [PubMed] [Google Scholar]
- 10.Rigoutsos I. Short RNAs: how big is this iceberg? Current biology: CB 2010; 20:R110-3. [DOI] [PubMed]
- 11.Kim VN, Han J, Siomi MC. Biogenesis of small RNAs in animals. Nat Rev Mol Cell Biol. 2009;10:126–39. doi: 10.1038/nrm2632. [DOI] [PubMed] [Google Scholar]
- 12.Baek D, Villén J, Shin C, Camargo FD, Gygi SP, Bartel DP. The impact of microRNAs on protein output. Nature. 2008;455:64–71. doi: 10.1038/nature07242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Friedman RC, Farh KK, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19:92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 15.Selbach M, Schwanhäusser B, Thierfelder N, Fang Z, Khanin R, Rajewsky N. Widespread changes in protein synthesis induced by microRNAs. Nature. 2008;455:58–63. doi: 10.1038/nature07228. [DOI] [PubMed] [Google Scholar]
- 16.Krek A, Grün D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, et al. Combinatorial microRNA target predictions. Nat Genet. 2005;37:495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- 17.Lowe SW, Cepero E, Evan G. Intrinsic tumour suppression. Nature. 2004;432:307–15. doi: 10.1038/nature03098. [DOI] [PubMed] [Google Scholar]
- 18.Bode AM, Dong Z. Post-translational modification of p53 in tumorigenesis. Nat Rev Cancer. 2004;4:793–805. doi: 10.1038/nrc1455. [DOI] [PubMed] [Google Scholar]
- 19.Braithwaite AW, Prives CL. p53: more research and more questions. Cell Death Differ. 2006;13:877–80. doi: 10.1038/sj.cdd.4401938. [DOI] [PubMed] [Google Scholar]
- 20.Junttila MR, Evan GI. p53--a Jack of all trades but master of none. Nat Rev Cancer. 2009;9:821–9. doi: 10.1038/nrc2728. [DOI] [PubMed] [Google Scholar]
- 21.Meek DW. Tumour suppression by p53: a role for the DNA damage response? Nat Rev Cancer. 2009;9:714–23. doi: 10.1038/nrc2716. [DOI] [PubMed] [Google Scholar]
- 22.Vousden KH, Lu X. Live or let die: the cell’s response to p53. Nat Rev Cancer. 2002;2:594–604. doi: 10.1038/nrc864. [DOI] [PubMed] [Google Scholar]
- 23.Menendez D, Inga A, Resnick MA. The expanding universe of p53 targets. Nat Rev Cancer. 2009;9:724–37. doi: 10.1038/nrc2730. [DOI] [PubMed] [Google Scholar]
- 24.Riley T, Sontag E, Chen P, Levine A. Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol. 2008;9:402–12. doi: 10.1038/nrm2395. [DOI] [PubMed] [Google Scholar]
- 25.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–10. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 26.Vousden KH, Lane DP. p53 in health and disease. Nat Rev Mol Cell Biol. 2007;8:275–83. doi: 10.1038/nrm2147. [DOI] [PubMed] [Google Scholar]
- 27.Vousden KH, Prives C. Blinded by the Light: The Growing Complexity of p53. Cell. 2009;137:413–31. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- 28.McKenzie L, King S, Marcar L, Nicol S, Dias SS, Schumm K, et al. p53-dependent repression of polo-like kinase-1 (PLK1) Cell Cycle. 2010;9:4200–12. doi: 10.4161/cc.9.20.13532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Spurgers KB, Gold DL, Coombes KR, Bohnenstiehl NL, Mullins B, Meyn RE, et al. Identification of cell cycle regulatory genes as principal targets of p53-mediated transcriptional repression. J Biol Chem. 2006;281:25134–42. doi: 10.1074/jbc.M513901200. [DOI] [PubMed] [Google Scholar]
- 30.Murphy M, Ahn J, Walker KK, Hoffman WH, Evans RM, Levine AJ, et al. Transcriptional repression by wild-type p53 utilizes histone deacetylases, mediated by interaction with mSin3a. Genes Dev. 1999;13:2490–501. doi: 10.1101/gad.13.19.2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim VN. MicroRNA biogenesis: coordinated cropping and dicing. Nat Rev Mol Cell Biol. 2005;6:376–85. doi: 10.1038/nrm1644. [DOI] [PubMed] [Google Scholar]
- 32.Rodriguez A, Griffiths-Jones S, Ashurst JL, Bradley A. Identification of mammalian microRNA host genes and transcription units. Genome Res. 2004;14(10A):1902–10. doi: 10.1101/gr.2722704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee Y, Jeon K, Lee JT, Kim S, Kim VN. MicroRNA maturation: stepwise processing and subcellular localization. EMBO J. 2002;21:4663–70. doi: 10.1093/emboj/cdf476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Han J, Lee Y, Yeom KH, Nam JW, Heo I, Rhee JK, et al. Molecular basis for the recognition of primary microRNAs by the Drosha-DGCR8 complex. Cell. 2006;125:887–901. doi: 10.1016/j.cell.2006.03.043. [DOI] [PubMed] [Google Scholar]
- 35.Czech B, Hannon GJ. Small RNA sorting: matchmaking for Argonautes. Nat Rev Genet. 2011;12:19–31. doi: 10.1038/nrg2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Khvorova A, Reynolds A, Jayasena SD. Functional siRNAs and miRNAs exhibit strand bias. Cell. 2003;115:209–16. doi: 10.1016/S0092-8674(03)00801-8. [DOI] [PubMed] [Google Scholar]
- 37.Brodersen P, Voinnet O. Revisiting the principles of microRNA target recognition and mode of action. Nat Rev Mol Cell Biol. 2009;10:141–8. doi: 10.1038/nrm2619. [DOI] [PubMed] [Google Scholar]
- 38.Djuranovic S, Nahvi A, Green R. A parsimonious model for gene regulation by miRNAs. Science. 2011;331:550–3. doi: 10.1126/science.1191138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lal A, Navarro F, Maher CA, Maliszewski LE, Yan N, O’Day E, et al. miR-24 Inhibits cell proliferation by targeting E2F2, MYC, and other cell-cycle genes via binding to “seedless” 3’UTR microRNA recognition elements. Mol Cell. 2009;35:610–25. doi: 10.1016/j.molcel.2009.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shin C, Nam JW, Farh KK, Chiang HR, Shkumatava A, Bartel DP. Expanding the microRNA targeting code: functional sites with centered pairing. Mol Cell. 2010;38:789–802. doi: 10.1016/j.molcel.2010.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chi SW, Hannon GJ, Darnell RB. An alternative mode of microRNA target recognition. Nat Struct Mol Biol. 2012;19:321–7. doi: 10.1038/nsmb.2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–33. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carthew RW, Sontheimer EJ. Origins and Mechanisms of miRNAs and siRNAs. Cell. 2009;136:642–55. doi: 10.1016/j.cell.2009.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thomas M, Lieberman J, Lal A. Desperately seeking microRNA targets. Nat Struct Mol Biol. 2010;17:1169–74. doi: 10.1038/nsmb.1921. [DOI] [PubMed] [Google Scholar]
- 45.Chi SW, Zang JB, Mele A, Darnell RB. Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature. 2009;460:479–86. doi: 10.1038/nature08170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Forman JJ, Coller HA. The code within the code: microRNAs target coding regions. Cell Cycle. 2010;9:1533–41. doi: 10.4161/cc.9.8.11202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Forman JJ, Legesse-Miller A, Coller HA. A search for conserved sequences in coding regions reveals that the let-7 microRNA targets Dicer within its coding sequence. Proc Natl Acad Sci U S A. 2008;105:14879–84. doi: 10.1073/pnas.0803230105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee EK, Gorospe M. Coding region: the neglected post-transcriptional code. RNA Biol. 2011;8:44–8. doi: 10.4161/rna.8.1.13863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tay Y, Zhang J, Thomson AM, Lim B, Rigoutsos I. MicroRNAs to Nanog, Oct4 and Sox2 coding regions modulate embryonic stem cell differentiation. Nature. 2008;455:1124–8. doi: 10.1038/nature07299. [DOI] [PubMed] [Google Scholar]
- 50.Guo H, Ingolia NT, Weissman JS, Bartel DP. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature. 2010;466:835–40. doi: 10.1038/nature09267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fabian MR, Sonenberg N, Filipowicz W. Regulation of mRNA translation and stability by microRNAs. Annu Rev Biochem. 2010;79:351–79. doi: 10.1146/annurev-biochem-060308-103103. [DOI] [PubMed] [Google Scholar]
- 52.Croce CM. Causes and consequences of microRNA dysregulation in cancer. Nat Rev Genet. 2009;10:704–14. doi: 10.1038/nrg2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Esquela-Kerscher A, Slack FJ. Oncomirs - microRNAs with a role in cancer. Nat Rev Cancer. 2006;6:259–69. doi: 10.1038/nrc1840. [DOI] [PubMed] [Google Scholar]
- 54.Esteller M. Non-coding RNAs in human disease. Nat Rev Genet. 2011;12:861–74. doi: 10.1038/nrg3074. [DOI] [PubMed] [Google Scholar]
- 55.Hammond SM. MicroRNAs as tumor suppressors. Nat Genet. 2007;39:582–3. doi: 10.1038/ng0507-582. [DOI] [PubMed] [Google Scholar]
- 56.Ma L, Teruya-Feldstein J, Weinberg RA. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature. 2007;449:682–8. doi: 10.1038/nature06174. [DOI] [PubMed] [Google Scholar]
- 57.Ma L, Young J, Prabhala H, Pan E, Mestdagh P, Muth D, et al. miR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin and cancer metastasis. Nat Cell Biol. 2010;12:247–56. doi: 10.1038/ncb2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nicoloso MS, Spizzo R, Shimizu M, Rossi S, Calin GA. MicroRNAs--the micro steering wheel of tumour metastases. Nat Rev Cancer. 2009;9:293–302. doi: 10.1038/nrc2619. [DOI] [PubMed] [Google Scholar]
- 59.Tavazoie SF, Alarcón C, Oskarsson T, Padua D, Wang Q, Bos PD, et al. Endogenous human microRNAs that suppress breast cancer metastasis. Nature. 2008;451:147–52. doi: 10.1038/nature06487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Valastyan S, Reinhardt F, Benaich N, Calogrias D, Szász AM, Wang ZC, et al. A pleiotropically acting microRNA, miR-31, inhibits breast cancer metastasis. Cell. 2009;137:1032–46. doi: 10.1016/j.cell.2009.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 61.Ventura A, Jacks T. MicroRNAs and cancer: short RNAs go a long way. Cell. 2009;136:586–91. doi: 10.1016/j.cell.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, Noch E, et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A. 2002;99:15524–9. doi: 10.1073/pnas.242606799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Klein U, Lia M, Crespo M, Siegel R, Shen Q, Mo T, et al. The DLEU2/miR-15a/16-1 cluster controls B cell proliferation and its deletion leads to chronic lymphocytic leukemia. Cancer Cell. 2010;17:28–40. doi: 10.1016/j.ccr.2009.11.019. [DOI] [PubMed] [Google Scholar]
- 64.Calin GA, Liu CG, Sevignani C, Ferracin M, Felli N, Dumitru CD, et al. MicroRNA profiling reveals distinct signatures in B cell chronic lymphocytic leukemias. Proc Natl Acad Sci U S A. 2004;101:11755–60. doi: 10.1073/pnas.0404432101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6:857–66. doi: 10.1038/nrc1997. [DOI] [PubMed] [Google Scholar]
- 66.Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–8. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- 67.Melo SA, Ropero S, Moutinho C, Aaltonen LA, Yamamoto H, Calin GA, et al. A TARBP2 mutation in human cancer impairs microRNA processing and DICER1 function. Nat Genet. 2009;41:365–70. doi: 10.1038/ng.317. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 68.Hill DA, Ivanovich J, Priest JR, Gurnett CA, Dehner LP, Desruisseau D, et al. DICER1 mutations in familial pleuropulmonary blastoma. Science. 2009;325:965. doi: 10.1126/science.1174334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Melo SA, Moutinho C, Ropero S, Calin GA, Rossi S, Spizzo R, et al. A genetic defect in exportin-5 traps precursor microRNAs in the nucleus of cancer cells. Cancer Cell. 2010;18:303–15. doi: 10.1016/j.ccr.2010.09.007. [DOI] [PubMed] [Google Scholar]
- 70.West JA, Viswanathan SR, Yabuuchi A, Cunniff K, Takeuchi A, Park IH, et al. A role for Lin28 in primordial germ-cell development and germ-cell malignancy. Nature. 2009;460:909–13. doi: 10.1038/nature08210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Davalos V, Moutinho C, Villanueva A, Boque R, Silva P, Carneiro F, et al. Dynamic epigenetic regulation of the microRNA-200 family mediates epithelial and mesenchymal transitions in human tumorigenesis. Oncogene. 2012;31:2062–74. doi: 10.1038/onc.2011.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Saito Y, Liang G, Egger G, Friedman JM, Chuang JC, Coetzee GA, et al. Specific activation of microRNA-127 with downregulation of the proto-oncogene BCL6 by chromatin-modifying drugs in human cancer cells. Cancer Cell. 2006;9:435–43. doi: 10.1016/j.ccr.2006.04.020. [DOI] [PubMed] [Google Scholar]
- 73.Chang TC, Yu D, Lee YS, Wentzel EA, Arking DE, West KM, et al. Widespread microRNA repression by Myc contributes to tumorigenesis. Nat Genet. 2008;40:43–50. doi: 10.1038/ng.2007.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.O’Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT. c-Myc-regulated microRNAs modulate E2F1 expression. Nature. 2005;435:839–43. doi: 10.1038/nature03677. [DOI] [PubMed] [Google Scholar]
- 75.Chang TC, Zeitels LR, Hwang HW, Chivukula RR, Wentzel EA, Dews M, et al. Lin-28B transactivation is necessary for Myc-mediated let-7 repression and proliferation. Proc Natl Acad Sci U S A. 2009;106:3384–9. doi: 10.1073/pnas.0808300106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bui TV, Mendell JT. Myc: Maestro of MicroRNAs. Genes & cancer 2010; 1:568-75. [DOI] [PMC free article] [PubMed]
- 77.Emmrich S, Pützer BM. Checks and balances: E2F-microRNA crosstalk in cancer control. Cell Cycle. 2010;9:2555–67. doi: 10.4161/cc.9.13.12061. [DOI] [PubMed] [Google Scholar]
- 78.Woods K, Thomson JM, Hammond SM. Direct regulation of an oncogenic micro-RNA cluster by E2F transcription factors. J Biol Chem. 2007;282:2130–4. doi: 10.1074/jbc.C600252200. [DOI] [PubMed] [Google Scholar]
- 79.Brabletz S, Brabletz T. The ZEB/miR-200 feedback loop--a motor of cellular plasticity in development and cancer? EMBO Rep. 2010;11:670–7. doi: 10.1038/embor.2010.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wellner U, Schubert J, Burk UC, Schmalhofer O, Zhu F, Sonntag A, et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat Cell Biol. 2009;11:1487–95. doi: 10.1038/ncb1998. [DOI] [PubMed] [Google Scholar]
- 81.Bommer GT, Gerin I, Feng Y, Kaczorowski AJ, Kuick R, Love RE, et al. p53-mediated activation of miRNA34 candidate tumor-suppressor genes. Current biology. 2007;17:1298–307. doi: 10.1016/j.cub.2007.06.068. [DOI] [PubMed] [Google Scholar]
- 82.Chang TC, Wentzel EA, Kent OA, Ramachandran K, Mullendore M, Lee KH, et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell. 2007;26:745–52. doi: 10.1016/j.molcel.2007.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.He L, He X, Lowe SW, Hannon GJ. microRNAs join the p53 network--another piece in the tumour-suppression puzzle. Nat Rev Cancer. 2007;7:819–22. doi: 10.1038/nrc2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Raver-Shapira N, Marciano E, Meiri E, Spector Y, Rosenfeld N, Moskovits N, et al. Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol Cell. 2007;26:731–43. doi: 10.1016/j.molcel.2007.05.017. [DOI] [PubMed] [Google Scholar]
- 85.Pichiorri F, Suh SS, Rocci A, De Luca L, Taccioli C, Santhanam R, et al. Downregulation of p53-inducible microRNAs 192, 194, and 215 impairs the p53/MDM2 autoregulatory loop in multiple myeloma development. Cancer Cell. 2010;18:367–81. doi: 10.1016/j.ccr.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 86.Xiao J, Lin H, Luo X, Luo X, Wang Z. miR-605 joins p53 network to form a p53:miR-605:Mdm2 positive feedback loop in response to stress. EMBO J. 2011;30:524–32. doi: 10.1038/emboj.2010.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yamakuchi M, Ferlito M, Lowenstein CJ. miR-34a repression of SIRT1 regulates apoptosis. Proc Natl Acad Sci U S A. 2008;105:13421–6. doi: 10.1073/pnas.0801613105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hermeking H. p53 enters the microRNA world. Cancer Cell. 2007;12:414–8. doi: 10.1016/j.ccr.2007.10.028. [DOI] [PubMed] [Google Scholar]
- 89.Hermeking H. MiR-34a and p53. Cell Cycle. 2009;8:1308. doi: 10.4161/cc.8.9.8511. [DOI] [PubMed] [Google Scholar]
- 90.Wei CL, Wu Q, Vega VB, Chiu KP, Ng P, Zhang T, et al. A global map of p53 transcription-factor binding sites in the human genome. Cell. 2006;124:207–19. doi: 10.1016/j.cell.2005.10.043. [DOI] [PubMed] [Google Scholar]
- 91.Giono LE, Manfredi JJ. The p53 tumor suppressor participates in multiple cell cycle checkpoints. J Cell Physiol. 2006;209:13–20. doi: 10.1002/jcp.20689. [DOI] [PubMed] [Google Scholar]
- 92.Lal A, Thomas MP, Altschuler G, Navarro F, O’Day E, Li XL, et al. Capture of microRNA-bound mRNAs identifies the tumor suppressor miR-34a as a regulator of growth factor signaling. PLoS Genet. 2011;7:e1002363. doi: 10.1371/journal.pgen.1002363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kim NH, Kim HS, Li XY, Lee I, Choi HS, Kang SE, et al. A p53/miRNA-34 axis regulates Snail1-dependent cancer cell epithelial-mesenchymal transition. J Cell Biol. 2011;195:417–33. doi: 10.1083/jcb.201103097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Navarro F, Gutman D, Meire E, Cáceres M, Rigoutsos I, Bentwich Z, et al. miR-34a contributes to megakaryocytic differentiation of K562 cells independently of p53. Blood. 2009;114:2181–92. doi: 10.1182/blood-2009-02-205062. [DOI] [PubMed] [Google Scholar]
- 95.Sotillo E, Laver T, Mellert H, Schelter JM, Cleary MA, McMahon S, et al. Myc overexpression brings out unexpected antiapoptotic effects of miR-34a. Oncogene. 2011;30:2587–94. doi: 10.1038/onc.2010.634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Park SM, Gaur AB, Lengyel E, Peter ME. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008;22:894–907. doi: 10.1101/gad.1640608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, et al. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol. 2008;10:593–601. doi: 10.1038/ncb1722. [DOI] [PubMed] [Google Scholar]
- 98.Korpal M, Kang Y. The emerging role of miR-200 family of microRNAs in epithelial-mesenchymal transition and cancer metastasis. RNA Biol. 2008;5:115–9. doi: 10.4161/rna.5.3.6558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chang CJ, Chao CH, Xia W, Yang JY, Xiong Y, Li CW, et al. p53 regulates epithelial-mesenchymal transition and stem cell properties through modulating miRNAs. Nat Cell Biol. 2011;13:317–23. doi: 10.1038/ncb2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kim T, Veronese A, Pichiorri F, Lee TJ, Jeon YJ, Volinia S, et al. p53 regulates epithelial-mesenchymal transition through microRNAs targeting ZEB1 and ZEB2. J Exp Med. 2011;208:875–83. doi: 10.1084/jem.20110235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Korpal M, Ell BJ, Buffa FM, Ibrahim T, Blanco MA, Celià-Terrassa T, et al. Direct targeting of Sec23a by miR-200s influences cancer cell secretome and promotes metastatic colonization. Nat Med. 2011;17:1101–8. doi: 10.1038/nm.2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Dykxhoorn DM, Wu Y, Xie H, Yu F, Lal A, Petrocca F, et al. miR-200 enhances mouse breast cancer cell colonization to form distant metastases. PLoS One. 2009;4:e7181. doi: 10.1371/journal.pone.0007181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Braun CJ, Zhang X, Savelyeva I, Wolff S, Moll UM, Schepeler T, et al. p53-Responsive micrornas 192 and 215 are capable of inducing cell cycle arrest. Cancer Res. 2008;68:10094–104. doi: 10.1158/0008-5472.CAN-08-1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Böhlig L, Friedrich M, Engeland K. p53 activates the PANK1/miRNA-107 gene leading to downregulation of CDK6 and p130 cell cycle proteins. Nucleic Acids Res. 2011;39:440–53. doi: 10.1093/nar/gkq796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Yamakuchi M, Lotterman CD, Bao C, Hruban RH, Karim B, Mendell JT, et al. P53-induced microRNA-107 inhibits HIF-1 and tumor angiogenesis. Proc Natl Acad Sci U S A. 2010;107:6334–9. doi: 10.1073/pnas.0911082107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Huppi K, Volfovsky N, Runfola T, Jones TL, Mackiewicz M, Martin SE, et al. The identification of microRNAs in a genomically unstable region of human chromosome 8q24. Mol Cancer Res. 2008;6:212–21. doi: 10.1158/1541-7786.MCR-07-0105. [DOI] [PubMed] [Google Scholar]
- 107.Barsotti AM, Beckerman R, Laptenko O, Huppi K, Caplen NJ, Prives C. p53-Dependent induction of PVT1 and miR-1204. J Biol Chem. 2012;287:2509–19. doi: 10.1074/jbc.M111.322875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A. 2006;103:2257–61. doi: 10.1073/pnas.0510565103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chen L, Li C, Zhang R, Gao X, Qu X, Zhao M, et al. miR-17-92 cluster microRNAs confers tumorigenicity in multiple myeloma. Cancer Lett. 2011;309:62–70. doi: 10.1016/j.canlet.2011.05.017. [DOI] [PubMed] [Google Scholar]
- 110.Yan HL, Xue G, Mei Q, Wang YZ, Ding FX, Liu MF, et al. Repression of the miR-17-92 cluster by p53 has an important function in hypoxia-induced apoptosis. EMBO J. 2009;28:2719–32. doi: 10.1038/emboj.2009.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Johnson CD, Esquela-Kerscher A, Stefani G, Byrom M, Kelnar K, Ovcharenko D, et al. The let-7 microRNA represses cell proliferation pathways in human cells. Cancer Res. 2007;67:7713–22. doi: 10.1158/0008-5472.CAN-07-1083. [DOI] [PubMed] [Google Scholar]
- 112.Yu F, Yao H, Zhu P, Zhang X, Pan Q, Gong C, et al. let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell. 2007;131:1109–23. doi: 10.1016/j.cell.2007.10.054. [DOI] [PubMed] [Google Scholar]
- 113.Weidhaas JB, Babar I, Nallur SM, Trang P, Roush S, Boehm M, et al. MicroRNAs as potential agents to alter resistance to cytotoxic anticancer therapy. Cancer Res. 2007;67:11111–6. doi: 10.1158/0008-5472.CAN-07-2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Saleh AD, Savage JE, Cao L, Soule BP, Ly D, DeGraff W, et al. Cellular stress induced alterations in microRNA let-7a and let-7b expression are dependent on p53. PLoS One. 2011;6:e24429. doi: 10.1371/journal.pone.0024429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sengupta S, Harris CC. p53: traffic cop at the crossroads of DNA repair and recombination. Nat Rev Mol Cell Biol. 2005;6:44–55. doi: 10.1038/nrm1546. [DOI] [PubMed] [Google Scholar]
- 116.Riley KJ, Maher LJ., 3rd p53 RNA interactions: new clues in an old mystery. RNA. 2007;13:1825–33. doi: 10.1261/rna.673407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Suzuki HI, Yamagata K, Sugimoto K, Iwamoto T, Kato S, Miyazono K. Modulation of microRNA processing by p53. Nature. 2009;460:529–33. doi: 10.1038/nature08199. [DOI] [PubMed] [Google Scholar]
- 118.Xu N, Papagiannakopoulos T, Pan G, Thomson JA, Kosik KS. MicroRNA-145 regulates OCT4, SOX2, and KLF4 and represses pluripotency in human embryonic stem cells. Cell. 2009;137:647–58. doi: 10.1016/j.cell.2009.02.038. [DOI] [PubMed] [Google Scholar]
- 119.Borralho PM, Kren BT, Castro RE, da Silva IB, Steer CJ, Rodrigues CM. MicroRNA-143 reduces viability and increases sensitivity to 5-fluorouracil in HCT116 human colorectal cancer cells. FEBS J. 2009;276:6689–700. doi: 10.1111/j.1742-4658.2009.07383.x. [DOI] [PubMed] [Google Scholar]
- 120.Bourguignon LY, Spevak CC, Wong G, Xia W, Gilad E. Hyaluronan-CD44 interaction with protein kinase C(epsilon) promotes oncogenic signaling by the stem cell marker Nanog and the Production of microRNA-21, leading to down-regulation of the tumor suppressor protein PDCD4, anti-apoptosis, and chemotherapy resistance in breast tumor cells. J Biol Chem. 2009;284:26533–46. doi: 10.1074/jbc.M109.027466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Davis BN, Hilyard AC, Lagna G, Hata A. SMAD proteins control DROSHA-mediated microRNA maturation. Nature. 2008;454:56–61. doi: 10.1038/nature07086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Yamagata K, Fujiyama S, Ito S, Ueda T, Murata T, Naitou M, et al. Maturation of microRNA is hormonally regulated by a nuclear receptor. Mol Cell. 2009;36:340–7. doi: 10.1016/j.molcel.2009.08.017. [DOI] [PubMed] [Google Scholar]
- 123.Le MT, Teh C, Shyh-Chang N, Xie H, Zhou B, Korzh V, et al. MicroRNA-125b is a novel negative regulator of p53. Genes Dev. 2009;23:862–76. doi: 10.1101/gad.1767609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Le MT, Shyh-Chang N, Khaw SL, Chin L, Teh C, Tay J, et al. Conserved regulation of p53 network dosage by microRNA-125b occurs through evolving miRNA-target gene pairs. PLoS Genet. 2011;7:e1002242. doi: 10.1371/journal.pgen.1002242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hu W, Chan CS, Wu R, Zhang C, Sun Y, Song JS, et al. Negative regulation of tumor suppressor p53 by microRNA miR-504. Mol Cell. 2010;38:689–99. doi: 10.1016/j.molcel.2010.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kumar M, Lu Z, Takwi AA, Chen W, Callander NS, Ramos KS, et al. Negative regulation of the tumor suppressor p53 gene by microRNAs. Oncogene. 2011;30:843–53. doi: 10.1038/onc.2010.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Griffiths-Jones S, Grocock RJ, van Dongen S, Bateman A, Enright AJ. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006;34(Database issue):D140–4. doi: 10.1093/nar/gkj112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Chen QR, Yu LR, Tsang P, Wei JS, Song YK, Cheuk A, et al. Systematic proteome analysis identifies transcription factor YY1 as a direct target of miR-34a. J Proteome Res. 2011;10:479–87. doi: 10.1021/pr1006697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A, et al. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell. 2001;107:137–48. doi: 10.1016/S0092-8674(01)00524-4. [DOI] [PubMed] [Google Scholar]
- 130.Sui G, Affar el B, Shi Y, Brignone C, Wall NR, Yin P, et al. Yin Yang 1 is a negative regulator of p53. Cell. 2004;117:859–72. doi: 10.1016/j.cell.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 131.Choi J, Nannenga B, Demidov ON, Bulavin DV, Cooney A, Brayton C, et al. Mice deficient for the wild-type p53-induced phosphatase gene (Wip1) exhibit defects in reproductive organs, immune function, and cell cycle control. Mol Cell Biol. 2002;22:1094–105. doi: 10.1128/MCB.22.4.1094-1105.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zhang X, Wan G, Mlotshwa S, Vance V, Berger FG, Chen H, et al. Oncogenic Wip1 phosphatase is inhibited by miR-16 in the DNA damage signaling pathway. Cancer Res. 2010;70:7176–86. doi: 10.1158/0008-5472.CAN-10-0697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Park SY, Lee JH, Ha M, Nam JW, Kim VN. miR-29 miRNAs activate p53 by targeting p85 alpha and CDC42. Nat Struct Mol Biol. 2009;16:23–9. doi: 10.1038/nsmb.1533. [DOI] [PubMed] [Google Scholar]
- 134.Fornari F, Gramantieri L, Giovannini C, Veronese A, Ferracin M, Sabbioni S, et al. MiR-122/cyclin G1 interaction modulates p53 activity and affects doxorubicin sensitivity of human hepatocarcinoma cells. Cancer Res. 2009;69:5761–7. doi: 10.1158/0008-5472.CAN-08-4797. [DOI] [PubMed] [Google Scholar]
- 135.Burns DM, D’Ambrogio A, Nottrott S, Richter JD. CPEB and two poly(A) polymerases control miR-122 stability and p53 mRNA translation. Nature. 2011;473:105–8. doi: 10.1038/nature09908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Muller PA, Vousden KH, Norman JC. p53 and its mutants in tumor cell migration and invasion. J Cell Biol. 2011;192:209–18. doi: 10.1083/jcb.201009059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Liu Y, Bodmer WF. Analysis of P53 mutations and their expression in 56 colorectal cancer cell lines. Proc Natl Acad Sci U S A. 2006;103:976–81. doi: 10.1073/pnas.0510146103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Brosh R, Rotter V. When mutants gain new powers: news from the mutant p53 field. Nat Rev Cancer. 2009;9:701–13. doi: 10.1038/nrc2693. [DOI] [PubMed] [Google Scholar]
- 139.Oren M, Rotter V. Mutant p53 gain-of-function in cancer. Cold Spring Harb Perspect Biol. 2010;2:a001107. doi: 10.1101/cshperspect.a001107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Derksen PW, Liu X, Saridin F, van der Gulden H, Zevenhoven J, Evers B, et al. Somatic inactivation of E-cadherin and p53 in mice leads to metastatic lobular mammary carcinoma through induction of anoikis resistance and angiogenesis. Cancer Cell. 2006;10:437–49. doi: 10.1016/j.ccr.2006.09.013. [DOI] [PubMed] [Google Scholar]
- 141.Fontemaggi G, Dell’Orso S, Trisciuoglio D, Shay T, Melucci E, Fazi F, et al. The execution of the transcriptional axis mutant p53, E2F1 and ID4 promotes tumor neo-angiogenesis. Nat Struct Mol Biol. 2009;16:1086–93. doi: 10.1038/nsmb.1669. [DOI] [PubMed] [Google Scholar]
- 142.Stambolsky P, Tabach Y, Fontemaggi G, Weisz L, Maor-Aloni R, Siegfried Z, et al. Modulation of the vitamin D3 response by cancer-associated mutant p53. Cancer Cell. 2010;17:273–85. doi: 10.1016/j.ccr.2009.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Di Agostino S, Strano S, Emiliozzi V, Zerbini V, Mottolese M, Sacchi A, et al. Gain of function of mutant p53: the mutant p53/NF-Y protein complex reveals an aberrant transcriptional mechanism of cell cycle regulation. Cancer Cell. 2006;10:191–202. doi: 10.1016/j.ccr.2006.08.013. [DOI] [PubMed] [Google Scholar]
- 144.Gaiddon C, Lokshin M, Ahn J, Zhang T, Prives C. A subset of tumor-derived mutant forms of p53 down-regulate p63 and p73 through a direct interaction with the p53 core domain. Mol Cell Biol. 2001;21:1874–87. doi: 10.1128/MCB.21.5.1874-1887.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Li Y, Prives C. Are interactions with p63 and p73 involved in mutant p53 gain of oncogenic function? Oncogene. 2007;26:2220–5. doi: 10.1038/sj.onc.1210311. [DOI] [PubMed] [Google Scholar]
- 146.Matas D, Sigal A, Stambolsky P, Milyavsky M, Weisz L, Schwartz D, et al. Integrity of the N-terminal transcription domain of p53 is required for mutant p53 interference with drug-induced apoptosis. EMBO J. 2001;20:4163–72. doi: 10.1093/emboj/20.15.4163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Weisz L, Zalcenstein A, Stambolsky P, Cohen Y, Goldfinger N, Oren M, et al. Transactivation of the EGR1 gene contributes to mutant p53 gain of function. Cancer Res. 2004;64:8318–27. doi: 10.1158/0008-5472.CAN-04-1145. [DOI] [PubMed] [Google Scholar]
- 148.Göhler T, Jäger S, Warnecke G, Yasuda H, Kim E, Deppert W. Mutant p53 proteins bind DNA in a DNA structure-selective mode. Nucleic Acids Res. 2005;33:1087–100. doi: 10.1093/nar/gki252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Donzelli S, Fontemaggi G, Fazi F, Di Agostino S, Padula F, Biagioni F, et al. MicroRNA-128-2 targets the transcriptional repressor E2F5 enhancing mutant p53 gain of function. Cell Death Differ. 2012;19:1038–48. doi: 10.1038/cdd.2011.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Su X, Chakravarti D, Cho MS, Liu L, Gi YJ, Lin YL, et al. TAp63 suppresses metastasis through coordinate regulation of Dicer and miRNAs. Nature. 2010;467:986–90. doi: 10.1038/nature09459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Mukherji S, Ebert MS, Zheng GX, Tsang JS, Sharp PA, van Oudenaarden A. MicroRNAs can generate thresholds in target gene expression. Nat Genet. 2011;43:854–9. doi: 10.1038/ng.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Lim LP, Lau NC, Weinstein EG, Abdelhakim A, Yekta S, Rhoades MW, et al. The microRNAs of Caenorhabditis elegans. Genes Dev. 2003;17:991–1008. doi: 10.1101/gad.1074403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Leung AK, Sharp PA. MicroRNA functions in stress responses. Mol Cell. 2010;40:205–15. doi: 10.1016/j.molcel.2010.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Li X, Cassidy JJ, Reinke CA, Fischboeck S, Carthew RW. A microRNA imparts robustness against environmental fluctuation during development. Cell. 2009;137:273–82. doi: 10.1016/j.cell.2009.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Xu P, Vernooy SY, Guo M, Hay BA. The Drosophila microRNA Mir-14 suppresses cell death and is required for normal fat metabolism. Current biology: CB 2003; 13:790-5. [DOI] [PubMed]
- 156.Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009;136:823–37. doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]