

Abstract
The effects of consuming foods on the intestinal microbiome of obese individuals remain unclear. The objective of this study was to compare the effects of consuming low glycinin soymilk (LGS, 49.5% β-conglycinin/6% glycinin), conventional soymilk (S, 26.5% β-conglycinin/38.7% glycinin) or bovine milk (M, 0% β-conglycinin/0% glycinin) on the intestinal microbiome in overweight and obese men. In a randomized double-blind study, participants (64 men, BMI > 25, 20–45 y old), organized in three groups, consumed 500 mL of LGS, S or M daily for 3 mo. Three fecal samples were collected before (baseline) and after 3 mo of consumption. Dietary energy and macronutrient intake were monitored monthly and remained constant throughout the study (p > 0.05). Microbial composition was analyzed with qPCR and bTEFAP. Within groups, qPCR analysis showed that the total bacteria increased in all treatments over time (p < 0.001). Bacteroides-Prevotella (p = 0.001) and Lactobacillus (p < 0.001) increased in LGS and M, respectively. Bifidobacterium was significantly reduced in LGS (p = 0.003) and S (p < 0.001). Bacterial diversity decreased for LGS, S and M (p = 0.004, 0.005, 0.001; respectively). Unweighted UniFrac analysis revealed that the microbial communities were more similar within than between individuals. The Firmicutes to Bacteroidetes ratio decreased in both LGS and S groups and remained relatively unchanged in the M group (Time p = 0.012; Interaction p = 0.059). Indicator analysis revealed several genera that were indicative of each treatment including Lactobacillus and Prevotella. Consumption of the three beverages differentially altered the microbiota in overweight and obese men including a potentially beneficial alteration of the Firmicutes to Bacteroidetes ratio in both soymilk groups.
Keywords: obesity, soymilk, microbiota, low glycinin, firmicutes, bacteroidetes
Introduction
Researchers have begun to uncover links between the complex gut microbiome and its relationship with obesity.1 Turnbaugh and colleagues2 demonstrated a potential causal link between obesity and gut microbiota using a murine model. Around the same time, Ley et al.3 demonstrated that there were differences in the gut bacteria of obese and lean adults. These two foundational studies provide strong evidence of a link between the gut microbiome and obesity. Research published since have tried to elucidate mechanisms for how bacteria influence adiposity. Since the gut microbiota is directly involved in metabolism of food and its conversion to energy, it is logical to conclude that the gut microbiota can cause caloric excess contributing to the development of obesity.1,4,5 Gut microbiota have been shown to suppress expression of fasting-induced adipose factor (Fiaf) which increased storage of excess calories as fat.6 Cani et al.7 have shown that bacterial lipopolysaccharide (LPS) is linked to high-fat diet-induced inflammation in ob/ob mice. Additionally, antibiotics and bifidobacteria supplementation can lower plasma LPS levels in mice and can reduce the effects of high-fat diet-induced metabolic disorders. Therefore, the gut microbiota should be a focus of further research into the etiology of obesity.
Over the past 2 decades, a wealth of knowledge has been gained detailing the health benefits of soybeans.8 A great deal of this research has focused on soybean protein isolate (SPI) which is primarily composed of glycinin and β-conglycinin proteins (50–70% of total protein).9,10 SPI has been shown to contribute to a reduced risk of cardiovascular disease in humans possibly due to lowered blood triglycerides, decreased LDL and total cholesterol.9 Clinical trials have shown that a decrease in serum triglycerides, visceral fat, lipid accumulation and body fat ratio can be attributed to the consumption of β-conglycinin.10,11 Additionally, clinical trials have shown favorable weight loss with the general consumption of soymilk.12,13 New varieties of soybeans with different protein profiles are now available. The ratio of β-conglycinin and glycinin present in soy may produce different positive effects in human health. Thus, β-conglycinin has received particular attention as a potential agent to improve health of overweight individuals.14
In this study, the gut microbiome composition of overweight and obese men was analyzed following consumption of low glycinin soymilk (high in β-conglycinin), conventional soymilk and bovine milk for three months. Changes in the gut microbiome were measured by qPCR and high-throughput 16S rDNA sequencing.
Results
Subjects and treatments
Male subjects (n = 64) aged 32 ± 7 y with an average initial BMI of 29 ± 4 completed this study out of 81 recruited subjects. The subject’s age and BMI at baseline did not differ significantly among groups. The subjects were randomly distributed into one of three treatments: low glycinin soymilk (LGS, n = 19), conventional soymilk (S, n = 23) or bovine milk (M, n = 22). The nutrient profiles of the treatments are listed in Table 2. The composition of isoflavones was comparable between the low glycinin soymilk and conventional soymilk.
Table 2. Chemical composition of treatments; low glycinin soymilk (LGS), conventional soymilk (S) and bovine milk (M)1.
| Calories | Fat (g) | Carbohydrates (g) | Fiber (%) | Protein (g) | Calcium (mg) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| LGS |
206 |
± 0 |
7 |
± 0 |
23 |
± 0.5 |
< 1.0 |
14 |
± 0.1 |
699 |
± 3 |
| M |
186 |
± 0.1 |
7 |
± 0 |
17 |
± 0.2 |
< 1.0 |
14 |
± 0.1 |
446 |
± 2 |
| S | 209 | ± 0.5 | 7 | ± 0.2 | 23 | ± 0.5 | < 1.0 | 14 | ± 0.2 | 660 | ± 3 |
1Data expressed as means ± SD per 500 mL of milk. Each participant consumed 500 ml per day with a >95% compliance rate.
Participant compliance with consumption of test materials was high (95% of packages consumed per month on average) and there were no compliance differences among treatments. No adverse effects of the treatments were reported by the subjects.
Protein, carbohydrate, fat, fiber and energy intake of the participants were not different among groups during the study (p > 0.05, Table 3). Physical activity did not change during the period of study in any of the groups (data not shown). Specifically, mild to moderate physical activity (p = 0.209) and strenuous physical activity (p = 0.489) were not different among treatments and did not differ more than one hour per week within every group. Every month during the study, participants were asked to record their bowel movements for one week and note any changes in daily frequency. There were no significant changes in daily defecation frequency (p = 0.575) reported by the subjects. In addition, treatment (p = 0.612), time (p = 0.236) and treatment x time interaction (p = 0.998) did not impact the percent dry weight of feces.
Table 3. Average daily dietary macronutrient intake during three months of consumption of low glycinin soymilk (LGS), conventional soymilk (S) and bovine milk (M)1.
| Protein (g) | Carbohydrates (g) | Fat (g) | Fiber (g) | Energy (Kcal) | ||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Baseline |
LGS |
108 |
± 9 |
377 |
± 51 |
109 |
± 14 |
30 |
± 3 |
2904 |
± 329 |
|||||||||||||||||||||||||
| M |
76 |
± 8 |
243 |
± 24 |
69 |
± 8 |
14 |
± 1 |
1930 |
± 176 |
||||||||||||||||||||||||||
| S |
89 |
± 7 |
268 |
± 19 |
94 |
± 10 |
15 |
± 1 |
2690 |
± 435 |
||||||||||||||||||||||||||
| |
p = 0.04 |
p = 0.03 |
p = 0.07 |
p = 0.09 |
p = 0.20 |
|||||||||||||||||||||||||||||||
| |
||||||||||||||||||||||||||||||||||||
| |
|
Protein (g) |
Carbohydrates (g) |
Fat (g) |
Fiber (g) |
Energy (Kcal) |
||||||||||||||||||||||||||||||
| 1 Month |
LGS |
95 |
± 11 |
320 |
± 36 |
99 |
± 11 |
21 |
± 3 |
2615 |
± 267 |
|||||||||||||||||||||||||
| M |
77 |
± 9 |
237 |
± 26 |
81 |
± 12 |
17 |
± 2 |
2023 |
± 235 |
||||||||||||||||||||||||||
| S |
91 |
± 5 |
330 |
± 48 |
110 |
± 21 |
22 |
± 4 |
2695 |
± 389 |
||||||||||||||||||||||||||
| |
p = 0.39 |
p = 0.21 |
p = 0.43 |
p = 0.40 |
p = 0.29 |
|||||||||||||||||||||||||||||||
| |
||||||||||||||||||||||||||||||||||||
| |
|
Protein (g) |
Carbohydrates (g) |
Fat (g) |
Fiber (g) |
Energy (Kcal) |
||||||||||||||||||||||||||||||
| 2 Month |
LGS |
89 |
± 10 |
267 |
± 38 |
86 |
± 15 |
20 |
± 3 |
2244 |
± 295 |
|||||||||||||||||||||||||
| M |
79 |
± 9 |
266 |
± 25 |
91 |
± 11 |
15 |
± 2 |
2135 |
± 124 |
||||||||||||||||||||||||||
| S |
79 |
± 4 |
250 |
± 18 |
88 |
± 6 |
16 |
± 1 |
2248 |
± 197 |
||||||||||||||||||||||||||
| |
p = 0.49 |
p = 0.92 |
p = 0.99 |
p = 0.20 |
p = 0.96 |
|||||||||||||||||||||||||||||||
| |
||||||||||||||||||||||||||||||||||||
| |
|
Protein (g) |
Carbohydrates (g) |
Fat (g) |
Fiber (g) |
Energy (Kcal) |
||||||||||||||||||||||||||||||
| 3 Month |
LGS |
97 |
± 12 |
307 |
± 36 |
85 |
± 9 |
23 |
± 2 |
2395 |
± 244 |
|||||||||||||||||||||||||
| M |
69 |
± 7 |
234 |
± 28 |
69 |
± 8 |
14 |
± 2 |
1892 |
± 212 |
||||||||||||||||||||||||||
| S |
94 |
± 6 |
299 |
± 27 |
102 |
± 9 |
18 |
± 2 |
2542 |
± 193 |
||||||||||||||||||||||||||
| |
p = 0.02 |
p = 0.24 |
p = 0.02 |
p = 0.17 |
p = 0.10 |
|||||||||||||||||||||||||||||||
| Interaction2 | p = 0.17 | p = 0.05 | p = 0.09 | p = 0.62 | p = 0.13 | |||||||||||||||||||||||||||||||
1Data expressed as means ± SD. 2Interaction p-values determined by repeated measures ANOVA.
Characterization of gut microbiota by quantitative PCR
Quantitative PCR (qPCR) was used to determine the fecal bacterial composition via primers specific for the 16s rRNA gene to compare the change in total bacterial abundance, Bacteroides-Prevotella, Bifidobacterium, and Lactobacillus following 3 mo of consumption. The qPCR values are reported as log10 copy number per g dry feces (Fig. 1). The change in copy number was not significantly different among treatments for the total bacteria, Bacteroides-Prevotella, Bifidobacterium, and Lactobacillus (p = 0.213, p = 0.374, p = 0.592, p = 0.142; respectively). Within treatments, total bacterial copy numbers increased significantly after the consumption for LGS, S and M (p < 0.001, p < 0.001, p = 0.002; respectively). The copy number of Bacteroides-Prevotella increased significantly in LGS (p = 0.001) but not S or M (p > 0.05). The copy number of Lactobacillus increased significantly within M (p < 0.001) but did not significantly change within LGS and S (p > 0.05). The Bifidobacterium copy numbers decreased significantly within LGS and S (p = 0.003, p < 0.001; respectively) and increased within the M group though not significantly (p = 0.216).

Figure 1. Change in copy number for the total microbiota (A), Bacteroides-Prevotella (B), Bifidobacterium (C) and Lactobacillus (D) after three months. Values reported as Log10 of the #copies/g dry matter. *P-value within treatments. **P-value among treatments
Characterization of gut microbiota by bacterial Tag-encoded FLX amplicon pyrosequencing
384 samples were analyzed (6 samples per subject) resulting in 2,815,029 total raw sequences of the V1-V3 hypervariable region of the 16S rRNA gene. After trimming protocols were applied, 1,787,001 high quality bacterial 16S rRNA sequences were produced with an average amplicon length of 491bp. We obtained an average of 4604–4730 sequences per sample which yielded an average of 1064 (LGS), 1019 (S) and 1043 (M) unique operational taxonomic units (OTUs; 97% cutoff) at baseline and 938 (LGS), 915 (S) and 923 (M) OTUs per sample (97% cutoff) after three months of treatment (Table 4). This reduction in bacterial diversity following the consumption of the treatments was statistically significant for LGS, S and M (p = 0.004, 0.005, 0.001; respectively, Table 4). Bacterial richness, estimated using ACE and Chao1 indices, also showed a decline over the three months with each treatment (Table 4). Combined with the ACE and Chao1 indices, the rarefaction curves showed sufficient leveling off to conclude that our data included good coverage of the estimated OTUs present in our samples (Fig. 2). Lastly, bacterial communities in the samples from the same individual were more similar than communities in samples from different subjects at both baseline and 3 mo (Fig. 3).
Table 4. Microbial analysis from participant fecal samples by bacterial Tag-encoded FLX amplicon pyrosequencing of the hypervariable V1-V3 region of the 16S rRNA gene1.
| Sequenced Used | OTUs 97%2 | ACE 97%3 | Chao1 97%3 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| |
Baseline |
3 Months |
Baseline |
3 Months |
p-value* |
Baseline |
3 Months |
Baseline |
3 Months |
| LGS |
4687 ± 259 |
4730 ± 224 |
1064 ± 207 |
938 ± 147 |
0.004 |
2191 ± 783 |
1606 ± 375 |
1749 ± 473 |
1396 ± 271 |
| M |
4604 ± 490 |
4625 ± 341 |
1043 ± 207 |
923 ± 142 |
0.001 |
2150 ± 685 |
1539 ± 337 |
1722 ± 414 |
1365 ± 244 |
| S | 4689 ± 336 | 4671 ± 175 | 1019 ± 173 | 915 ± 80 | 0.005 | 2065 ± 586 | 1653 ± 268 | 1663 ± 373 | 1386 ± 148 |
1Data expressed as means ± SD. 2OTUs = Operational taxonomic units. 3ACE and Chao1 measure estimated bacterial richness. *p-value represents significant changes from baseline. Differences in OTUs 3% among treatments were not significant (p = 0.227).

Figure 2. Rarefaction curves of the OTUs at 97% distance. A single line represents one participant, each participant represented at baseline (A, C, E) and 3 mo (B, D, F). LGS (A,B), M (C,D) and S (E, F).
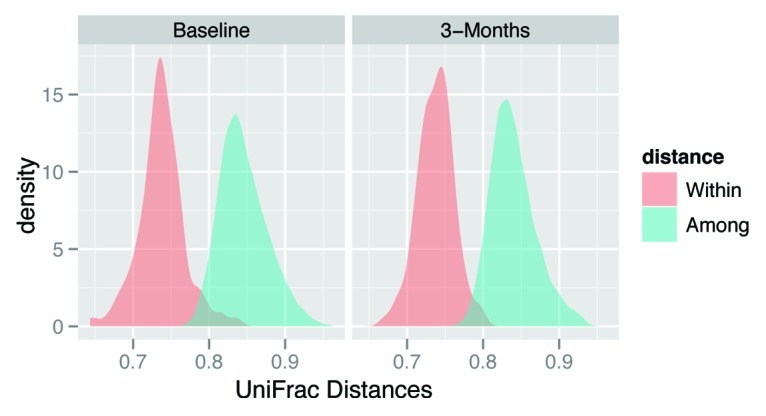
Figure 3. Variability in bacterial community structure within and among subjects. Unweighted UniFrac distance quantifies differences in bacterial community composition, and the observed probability distribution of unweighted UniFrac distances within and among subjects is shown at baseline and at 3 mo.
Eleven different bacterial phyla were detected in our study. The relative abundance of Firmicutes decreased for both soymilk groups (LGS and S) while staying unchanged in the M group (Table 5, p = 0.03). In contrast, the relative abundance of Bacteroidetes increased in both soymilk groups (LGS and S) and decreased in the M group, although this interaction only approached statistical significance (Table 5, p = 0.07). The Firmicutes to Bacteroidetes ratio decreased over the course of the experiment in both the LGS and S treatment groups and remained relatively unchanged in the M treatment group (Time p = 0.012; treatment x time interaction p = 0.059). The relative abundance of members in the phylum Proteobacteria significantly increased in all groups (Time p < 0.01) following the consumption of the milks.
Table 5. Phylogenic abundance profile related with low glycinin soymilk (LGS), conventional soymilk (S) and bovine milk (M)1.
| LGS | M | S | ANOVA2 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Phylum |
Baseline |
3 Months |
Baseline |
3 Months |
Baseline |
3 Months |
Treatment |
Time |
Interaction |
|
Actinobacteria |
0.10 |
0.32 |
0.29 |
0.43 |
0.13 |
0.24 |
0.14 |
< 0.01 |
0.27 |
|
Bacteroidetes |
15.93 |
18.45 |
19.44 |
18.12 |
16.53 |
20.25 |
0.95 |
0.04 |
0.07 |
|
Chloroflexi |
0.00 |
0.01 |
0.00 |
0.00 |
0.00 |
0.01 |
0.05 |
< 0.01 |
0.18 |
|
Cyanobacteria |
0.00 |
0.00 |
0.00 |
0.01 |
0.00 |
0.01 |
0.76 |
< 0.01 |
0.67 |
|
Firmicutes |
83.43 |
79.59 |
77.02 |
77.64 |
82.93 |
78.08 |
0.49 |
0.01 |
0.03 |
|
Fusobacteria |
1.18 |
1.10 |
4.30 |
3.74 |
1.09 |
0.96 |
0.42 |
0.65 |
0.96 |
|
Proteobacteria |
0.65 |
2.24 |
0.87 |
1.87 |
0.55 |
1.22 |
0.42 |
< 0.01 |
0.93 |
|
Tenericutes |
0.01 |
0.00 |
0.01 |
0.01 |
0.01 |
0.00 |
0.94 |
0.34 |
0.64 |
| TM7 |
0.00 |
0.01 |
0.00 |
0.01 |
0.00 |
0.01 |
0.58 |
< 0.01 |
0.40 |
|
Synergistetes |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.92 |
0.49 |
0.26 |
|
Verrucomicrobia |
0.25 |
0.07 |
0.27 |
0.09 |
0.06 |
0.17 |
0.94 |
0.28 |
0.10 |
| F / B ratio3 | 5.23 | 4.31 | 3.96 | 4.28 | 5.01 | 3.86 | > 0.05 | 0.012 | 0.059 |
1Data expressed as average relative abundance. 2ANOVA: Prior to statistical analysis, relative abundances were transformed using an arcsin transformation. Mixed effects ANOVA, p < 0.05. 3F /B ratio was calculated by the relative abundance of Firmicutes divided by the relative abundance of Bacteroidetes.
When viewed from a multivariate perspective, the change in the bacterial community over time was the most distinct between M and S (Fig. 4), although the treatment effect only approached statistical significance (p = 0.057). For subjects who consumed bovine milk, Roseburia tended to increase whereas Prevotella tended to decrease. The opposite was true for subjects who consumed conventional soymilk. Subjects who consumed low glycinin soymilk, however, tended to experience increases in Faecalibacterium.

Figure 4. Biplot illustrating the results of RDA. Here, each point represents a subject, and distances among points represent differences among subjects’ microbiomes. As a result, points close together represent subjects who responded similarly to treatment. Ellipses represent the 95% CI for each treatment group centroid (i.e., multivariate mean), and arrows reflect the contribution of genera to each axis. Prior to analysis, relative abundance of each genus within each subject was averaged across three replicates and then adjusted for baseline conditions. Differences among treatment groups approached significance (p = 0.057).
Indicator analysis38 was used to identify individual genera that were indicative of a particular treatment group (Fig. 5). For many of the genera such as Lactobacillus, the change in indicator score was similar for LGS and S yet different from M. In fact, the indicator score for Lactobacillus increased for the M group yet decreased for both the LGS and S groups, which is consistent with the qPCR results (Fig. 1). For other genera, LGS and S do not have similar indicator scores. For example, the indicator score from Prevotella increased for the S group yet was relatively unchanged for the other groups which are consistent with our multivariate analysis (Fig. 4). Particularly, the indicator analysis demonstrated that the genus Pseudomonas was more abundant in LGS (1.63% ± 5.59) whereas M and S were less than 1% (0.16% ± 0.34, 0.13% ± 0.36; respectively) after 3 mo of consumption.
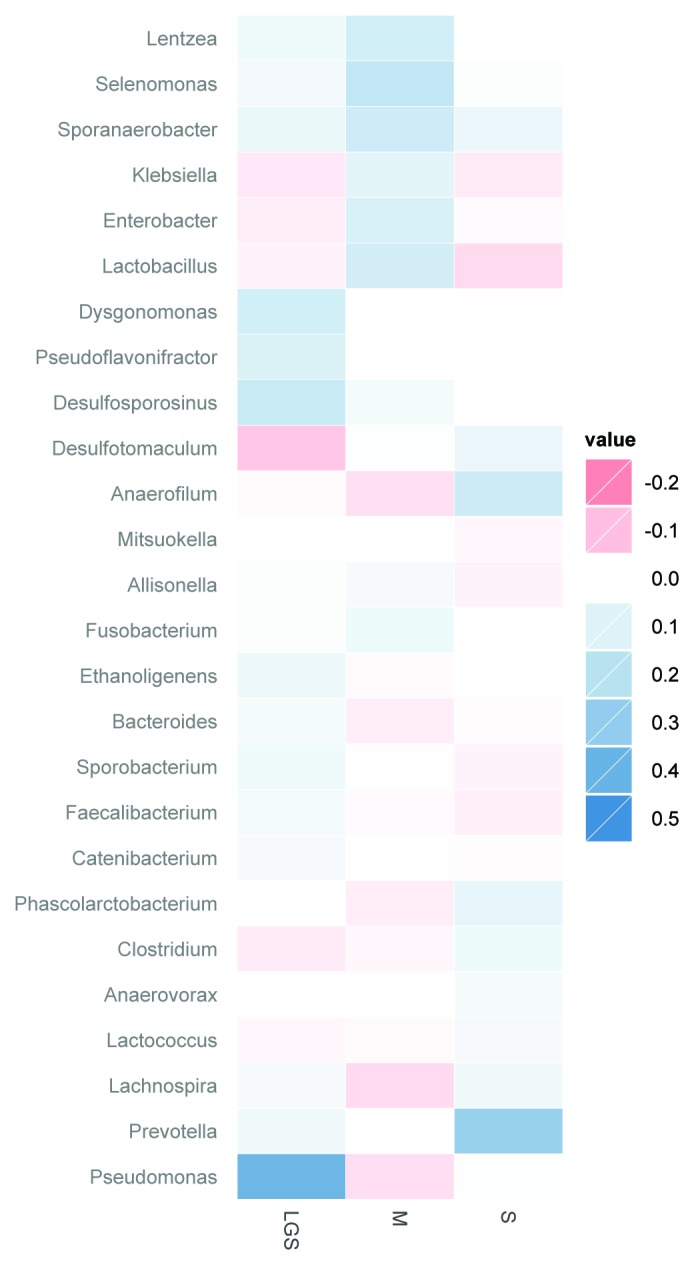
Figure 5. Change in indicator value scores (3 mo – baseline) after 3 mo of treatment. Indicator value scores reflect the fidelity and relative abundance of each genera (e.g., a score of 1 reflects a genus that is ubiquitous and highly abundant in all samples of a particular type), and were calculated at baseline and at 3 mo. Positive values are indicative of indicator scores that increased after treatment. All genera with indicator scores that had p < 0.10 at 3 mo are shown.
Discussion
While β-conglycinin has previously been shown to positively lessen the risk factors of developing obesity,9,45 the potential impact of β-conglycinin on the adult human gut microbiota is unknown. This study analyzed the changes in gut microbiota due to the consumption of a low glycinin soymilk, conventional soymilk or bovine milk over three months in overweight and obese men. The gut microbiota was analyzed using two distinct molecular based approaches, quantitative PCR (qPCR) and bacterial tag-encoded FLX amplicon pyrosequencing (bTEFAP).
The total bacterial 16S rDNA copy number was measured with universal primers by qPCR and increased significantly in all treatment groups. Gut microbiota composition has been demonstrated to be diet-responsive where minor shifts in dietary intake can cause a shift in bacterial numbers.46,47 While our participants were asked to maintain their normal diets throughout the course of this study (3 mo), it is difficult to ignore the changes in food consumption of free living human subjects. In addition, the use of dietary recall is problematic as indicated by the wide variation seen in the study.
The overall diversity of species (OTUs at 97% cutoff) measured by bTEFAP decreased at the end of this study for all three treatment groups. While reduced diversity has been shown in obese people as compared with lean people this does not explain the reduction in diversity seen in this study.48 All of our participants were either overweight or obese at baseline and their BMI did not change over the course of the study.
The collection of three fecal samples from each individual at both time points allowed us to determine the variability in bacterial community structure within and among subjects. This analysis showed that the microbial composition was more similar when comparing an individual to themselves than to other individuals which is consistent with previously reported data.48,49 Importantly, this result indicates that increasing the total number of participants may be a better use of resources than collecting multiple samples per person.
The Firmicutes and Bacteroidetes phyla have been shown to directly correlate with obesity in several studies.3,48,50 We demonstrated here that the relative abundance of Firmicutes significantly decreased whereas the relative abundance of Bacteroidetes significantly increased following consumption of LGS and S. Correspondingly, the Firmicutes to Bacteroidetes ratio decreased significantly in the LGS and S groups. There was no change in relative abundance of Firmicutes, Bacteroidetes or their ratio with respect to the M group.
The significant increase in relative abundance of Proteobacteria did not result from consumption of solely soymilk. The Proteobacteria phylum contains gram-negative bacteria with an outer membrane rich in lipopolysaccharides (LPS) and includes important genera like Pseudomonas, Klebsiella, Enterobacter, Salmonella, and Escherichia, among others. LPS has pro-inflammatory activity and high levels have been related with obesity.51 Since the subjects were already overweight and obese, the increase in the relative abundance of Proteobacteria may be a concern. In all three groups, the proportion of participants with detectable levels of Pseudomonas, a member of the phylum Proteobacteria, were greater following consumption and was not unique to the LGS group (LGS increased from 32% to 100%; M increased from 35% to 65%; S increased from 13% to 91%). While Pseudomonas spp are a normal part of the gut microbiota,52 the significance of the increase in relative abundance in LGS and the increase in individuals with detectable Pseudomonas spp in all three groups is unknown.
We analyzed three genera by qPCR: Lactobacillus, Bacteroides-Prevotella and Bifidobacterium (Fig. 1). The Bacteroides-Prevotella group significantly increased in total abundance in LGS after 3 mo of consumption (Fig. 1). Bacteroides was shown to be indicative of LGS by the multivariate and indicator analysis (Fig. 4 and Fig. 5). Additionally, Prevotella was indicative of S by multivariate and indicator analysis. Bacteroides and Prevotella are both members of the phylum Bacteroidetes. For both LGS and S the Firmicutes to Bacteroidetes ratio decreased which is consistent with an increase in abundance (or indicator score) of these two genera.
The total abundance (qPCR) of the genus Lactobacillus significantly increased in M after 3 mo (Fig. 1). Lactobacillus was also shown to have a higher indicator score in M (Fig. 5). Lactobacillus spp have been shown to have health promoting capabilities and many are defined as probiotics. The total abundance of Lactobacillus was low at baseline in the M group and increased to similar amounts seen in the LGS and S group. The increase in Lactobacillus was only seen in the M group. Since Lactobacillus spp are effective lactose fermenters, it is reasonable to suspect the increase in lactobacilli in the M group could be related to the lactose in the milk.
Interestingly, the multivariate analysis (Fig. 4) identified other members of the Firmicutes that were predominant in one treatment or another. The genus Faecalibacterium was more abundant in the LGS group. In addition, the genera Eubacterium and Clostridium were more abundant in the S group. The M group had a higher abundance of Roseburia. Of these four genera, only Faecalibacterium and Clostridium had a higher indicator score for LGS and S, respectively. These four genera contain species which have been classified as butyrate producers which might have obesity implications.2
Subjects were distributed into groups using a stratified randomization. However, the gut microbiota of the three groups varied at baseline. Analyses of the gut microbiota composition by DNA based molecular methods have inherent biases due to extraction methods, primers and sequencing method. In this study, care was taken to reduce the bias associated with the microbiota analysis. Fecal extraction was randomly performed to ensure that participant samples were not extracted together. Primer sets (qPCR and bTEFAP) were consistent between all samples and did not change during this study. Extracted DNA were pooled and analyzed on a single plate for qPCR analysis. Additionally, extracted DNA samples were run on a single bTEFAP plate. Therefore, the cause of the differences between the groups at baseline is unknown. We chose to focus our analysis on the change over time due to the treatments within groups rather than between groups. Since our sample to sample variation was larger between individuals than within individuals (Fig. 3) we believe this analysis was appropriate.
In conclusion, consumption of LGS, S and M resulted in changes in the gut microbiota in overweight and obese men as assessed by qPCR and bTEFAP. These molecular based approaches generated similar conclusions. Additionally, the Firmicutes to Bacteroidetes ratio decreased in the LGS and S groups yet was unchanged in the M group. We also identified several genera that changed differentially due to the consumption of the soymilks or bovine milk, such as Pseudomonas, Bacteroides, Prevotella, Lactobacillus, Clostridium, Eubacterium, Roseburia, and Faecalibacterium. The identification of specific bacterial genera allows for a better understanding of the microbial response to dietary interventions. Future research could provide evidence to link the beneficial health effects of soymilk to the development and treatment of obesity, possibly due to a change in the gut microbiome.
Methods
Ethics statement
This study was a randomized, double-blind trial. It was conducted in accordance with good clinical practice guidelines in compliance with the declaration of Helsinki and approval of the Institutional Review Board of the University of Illinois at Urbana Champaign (IRB #09454). Participants voluntarily participated in the study and signed an informed consent form.
Subjects
Male participants (20–45 y of age) with a body mass index (BMI) ranging from 25–44 were recruited voluntarily from the campus of the University of Illinois at Urbana Champaign. Participants were excluded from the study if they were taking antibiotics, a smoker, an athlete, a vegetarian and / or lactose intolerant. Only male participants were recruited to avoid the variability in gut microbiota produced by monthly hormonal changes in women. Participants (n = 81), were staggered from May to December 2009 and 64 subjects completed the study. Age and BMI, were determined at baseline to organize subjects within groups using a stratified randomization. Subjects were asked to avoid the consumption of dietary supplements, antibiotics and to inform investigators if they became ill or were prescribed any new medication. Participants were asked to avoid the consumption of fluid milk (other than the provided milk) during the course of the study; however solid dairy products such as cheese were permitted. Participants also agreed to maintain their regular diet and physical activity during the study.
Treatments
The low glycinin/high β-conglycinin soybean seeds were provided by The Monsanto Co. (Saint Louis, MO). The corresponding whole soybean milk powders, including the milk powder produced from traditional soybean seeds were manufactured by Archer Daniels Midland (Decatur, IL). The liquid preparation and packaging of the soymilk and bovine milk were conducted using an Ultra High Temperature procedure (Tetra Pak Co., Danton, TX). Final beverage products were stored for 2 weeks and microbiological analyses were then performed by Tetra Pak to ensure the safety of the beverages prior to the shipment to the University of Illinois at Urbana Champaign. The soymilk or bovine milks were provided to participants in 250 ml Tetra Pak containers, there were no differences in appearance between the three milks. Compliance of product consumption was determined by counting the number of clipped cardboard codes returned monthly to investigators and by a signed declaration of the number of beverage packages consumed.
Study design
Eighty one volunteers who met the inclusion criteria were randomly distributed into three groups and asked to consume 500 mL per day of low glycinin soymilk (LGS, 49.5% β-conglycinin/6% glycinin), conventional soymilk (S, 26.5% β-conglycinin/38.7% glycinin) or bovine milk (M, 0% β-conglycinin/0% glycinin). The study comprised of a one week washout period in which subjects were requested to avoid the consumption of any food product containing soy; a list of common products to avoid was provided. The washout period was followed by three months of soymilk or bovine milk consumption according to the experimental protocol. Subjects also participated in a monthly one-week bowel movement recall.
Sample collection
Participants donated 3 fecal samples over the course of 5–6 d during the wash-out week (baseline) and during the final week of the 3 mo study. Fecal material was deposited in a sealed container by the participant (Fisher Scientific, Fair Lawn, NJ) and placed in a styrofoam box containing ice packs (U-line, Pleasant Prairie, WI). Fecal material was delivered and flash frozen within two hours of defecation. Four total samples (3 - 200 mg tubes and 1 - 1 g tube, Sarstedt, Newton, NC) were collected from each sample. Fecal samples were stored at -80°C until subsequent analysis. One 200 mg tube of fecal material was weighed, placed in an aluminum weigh boat and dried at 105°C for 24h to calculate % dry matter.15
Dietary and physical activity assessment
A five-day dietary record was used to assess monthly nutrient intake. Serving sizes were reported according to USDA guidelines for the US population (USDA, 1996). Dietary records were analyzed with the Food Processor SQL, Nutrition Analysis and Fitness Software (version 10.5.0, ESHA Research). A seven-day physical activity recall (SDPAR) was conducted by trained interviewers every month.16 Subjects were asked to list the frequency, length and intensity of physical activity seven days prior to the interview. Changes in hours per day of mild-moderate and strenuous physical activity were analyzed throughout the study.
DNA extraction
DNA was isolated from 200–300 mg of feces using a modified bead beating method.17,18 Briefly, samples were lysed using a three minute homogenization step with beads using a vortexer. Extracted DNA was re-suspended in TE buffer overnight at 4°C instead of mechanical pipetting. These modifications were used to minimize DNA shearing for downstream applications. The DNA was purified using QIAmpDNA Stool Kits (Qiagen, Valencia, CA). Extracted DNA was quantified using a NanoDrop ND-1000 Spectrophotometer (Nano-Drop, Wilmington, DE). The DNA samples were normalized to a final concentration of 100 ng/µl.
General quantification of microbiota using quantitative PCR
The stock (100 ng/µl) DNA was further diluted to a final concentration of 100 pg/µl for the Universal and Bacteroides-Prevotella primers and 10 ng/µl for the Lactobacillus and Bifidobacterium primers. The primers used for qPCR are listed in Table 1 based on published sequences.7,19-21 Primer coverage and specificity were checked with the Ribosomal Database Project’s Probe Match search tool. Coverage is reported as the percentage of the database’s total number of organism sequences from the target groups that the primers match. Specificity is reported as the percentage of amplicons matched across the entire database that correspond to organisms of the target group. Unclassified organisms were not considered as targets, but were included in the totals that the primers matched (Date accessed 4/26/2012).22
Table 1. Primers used for quantification of Total bacteria, Bacteroides-Prevotella, Bifidobacteria and Lactobacillus.1.
| Target group | Primer Name | Sequence (5’-3’) | Coverage2 | Specificity3 | Reference |
|---|---|---|---|---|---|
| Total bacteria |
Uni331F |
TCCTACGGGAGGCAGCAGT |
ND4 |
ND4 |
Jernberg et al. 2007 |
| Uni797R |
GGACTACCAGGGTATCTATCCTGTT |
||||
| Bacteroides-Prevotella |
AllBac296F |
GAG AGG AAG GTC CCCCAC |
73.3 |
86.0 |
Cani et al. 2008 |
| AllBac2112R |
CGCTACTTGGCTGGTTCAG |
||||
| Bifidobacterium |
Bif164F |
GGGTGGTAATGCCGGATG |
24.6 |
99.3 |
Kok et al. 1996 |
| Bif662R |
CCACCGTTACACCGGGAA |
||||
| Lactobacillus | LacF |
AGCAGTAGGGAATCTTCCA |
68 | 89.4 | Rinttila et al. 2004 |
| LacR | CACCGCTACACATGGAG |
1Coverage was determined by the probe match function on the Ribosomal Database Project. Accessed 4/26/2012. 2Percentage of the database’s total number of organism sequences from the target groups that the primers match. 3Percentage of amplicons matched across the entire database that correspond to organisms of the target group. 4ND = not determined.
Primers were purchased from Integrated DNA Technologies (Coralville, IA). Quantitative PCR (qPCR) was performed using the Applied Biosystems 7900HT Fast Real-Time PCR System (Applied Biosystems, Carlsbad, CA). Each 10 µl qPCR reaction included 5 µl of 2Χ Power SYBR Green PCR Master mix (Applied Biosystems), bovine serum albumin at a final concentration of 1 μg BSA /μL (New England Bio Labs), 0.5 µM of each primer and 2 µl of the DNA sample. The PCR conditions were 50°C for 2 min, 95°C for 10 min, followed by 40 cycles of 95°C for 15 sec, and 60°C for 1 min. Following amplification, a dissociation step was included to analyze the melting profile of the amplified products and ensure that no extraneous products were generated. Standard curves (102 to 107 16S rRNA gene copies per reaction) were generated using purified 2.1TOPO-TA plasmids (Invitrogen, Carlsbad, CA), according to manufacturer’s instructions. The plasmids contained the 16S rRNA gene of Lactobacillus rhamnosus (used for Lactobacillus and universal quantization), Bifidobacterium longum, and Bacteroides fragilis. The qPCR values are reported as log10 copy number per g dry feces.
Massively parallel bacterial tag-encoded FLX amplicon pyrosequencing
Bacterial tag-encoded FLX amplicon pyrosequencing (bTEFAP) was performed using Gray28F (5′GAGTTTGATCNTGGCTCAG) and Gray519R (5′GTNTTACNGCGGCKGCTG) as described in previous studies.23-26 Initial generation of the sequencing library used a one-step PCR with a total of 30 cycles, a mixture of Hot Start and HotStar high fidelity taq polymerases, and amplicons originating and extending from the 28F for bacterial diversity. bTEFAP analyses was performed using the Roche 454 FLX instrument (Roche, Indianapolis, IN) with titanium reagents, titanium procedures were performed at the Research and Testing Laboratory (RTL, Lubbock, TX) based upon RTL protocols.27
Bacterial diversity
Following sequencing, all failed sequence reads, low quality sequence ends and tags and primers were removed and sequence collections were depleted of any non-bacterial ribosome sequences and chimeras using B2C228 as has been described previously.25,26,29,30 To determine the identity of bacteria in the remaining sequences, sequences were de-noised, assembled into clusters and queried using a distributed BLASTn .NET algorithm31 against a custom database of high quality 16S bacterial sequences derived from NCBI (01–01–11 or current version). Database sequences were characterized as high quality based upon similar criteria utilized by RDP version 9.32 Using a .NET and C# analysis pipeline the resulting BLASTn outputs were compiled and validated using taxonomic distance methods, and data reduction analysis were performed as described in the standard method.28,31,33-35
Bacterial identification
Based upon the above BLASTn derived sequence identity (percent of total length query sequence which aligns with a given database sequence) and using taxonomic distance methods, the bacteria were classified at the appropriate taxonomic levels based upon the following criteria. Sequences with identity scores, to known or well characterized 16S sequences, greater than 97% identity (< 3% divergence) were resolved at the species level, between 95% and 97% at the genus level, between 90% and 95% at the family and between 85% and 90% at the order level, 80% and 85% at the class and 77% and 80% at phyla. After resolving based upon these parameters, the percentage of each bacterium was individually analyzed for each sample providing relative abundance information within and among the individual samples based upon relative numbers of reads within each. Evaluations presented at each taxonomic level, including percentage compilations represent all sequences resolved to their primary identification or their closest relative.23,36,37
Statistical analysis
Subject’s characteristics at baseline and physical activity were analyzed with one-way ANOVA. Dietary intake data was analyzed by repeated measures ANOVA. Bacterial diversity, change in OTUs from baseline, was analyzed with a student t-test. These analyses were performed using SAS version 9.3 (SAS Institute, Cary, NC).
The relative abundances (transformed via the arcsin transformation) of phyla and several genera, were analyzed individually using a mixed-effects ANOVA. Treatment (LGS, M, and S) and time (baseline vs. 3 mo) were considered fixed effects. Subject was considered a random effect. This mixed-effects model was also used to analyze qPCR based abundance of total bacteria, Bacteroides-Prevotella, Bifidobacterium, and Lactobacillus, as well as species richness (ACE and Chao1) and the Firmicutes to Bacteroidetes ratio. This ratio was log transformed prior to analysis.
For each time period, indicator analysis38 was used to identify individual genera that were indicative of low glycinin soymilk, conventional soymilk or bovine milk samples. Indicator analysis provides an indicator score that synthesizes information about occurrence and abundance of individual taxa, and provides a randomization test of the degree to which taxa are indicative of a particular state (low glycinin soymilk vs. conventional soymilk vs. bovine milk). We then examined the change in indicator scores from baseline to three months as a measure of treatment effect.
Multivariate differences among treatment groups at baseline and at 3 mo were evaluated using distance based redundancy analysis (dbRDA).39 For this analysis, distances among samples were first calculated using the unweighted UniFrac distance measure,40 which measures the phylogenetic distances among samples. Because differences among treatment groups existed at baseline, another dbRDA also was performed to account for baseline differences (i.e., standardizing relative abundances at 3 mo to baseline conditions). In this analysis, the relative abundance of each genus was averaged for each subject at baseline and at 3 mo (n = 3 per time period). Then, the change in relative abundance (3 mo – baseline) was used in the analysis. UniFrac was calculated using Qiime,41 and all other analyses were conducted in R,42 using the vegan43 and labdsv 44 packages.
Acknowledgments
E.G.M., M.M. and N.B. designed research; D.F.R., J.H. and S.D. conducted research; D.F.R. and S.C. performed statistical analysis; D.F.R., J.H., and M.M. wrote the paper; E.G.M. and M.M. made revisions and additions to the manuscript. E.G.M. and M.M. had primary responsibility for final manuscript content. This study was supported by the Monsanto Company to E.G.M., USDA Cooperative State Research, Education and Extension Service (CSREES), AG548 2005–34505–15767 Future Foods IL to E.G.M., and Illinois Soybean Association, IL to E.G.M.. Authors acknowledge the assistance of Archer Daniels Midland, Company, Decatur, IL, in particular of Tom Gottemoller for the production of soymilk powders and Russ Egbert and Joe Richardson for the processing of the soymilks. We would also like to extend acknowledgment to all participants in the study.
Glossary
Abbreviations:
- LGS
low glycinin soymilk
- S
conventional soymilk
- M
bovine milk. BMI, body mass index
- SPI
soy protein isolate
- qPCR
real-time quantitative PCR
- bTEFAP
bacterial tag-encoded FLX amplicon pyrosequencing
- OTU
operational taxonomic units
Disclosure of Potential Conflicts of Interest
N.B. was an employee at Monsanto Co. at the time of the study. S.C. is an employee of Research and Testing Laboratory. S.D. was an employee of Research and Testing Laboratory at the time of this study. S.D. is currently employed by Molecular Research LP, Shallowater, TX. E.G.M. receives grant support from Monsanto Co., Archer Daniels Midland Co., and the Illinois Soybean Association.
Footnotes
Previously published online: www.landesbioscience.com/journals/gutmicrobes/article/21578
References
- 1.DiBaise JK, Zhang H, Crowell MD, Krajmalnik-Brown R, Decker GA, Rittmann BE. Gut microbiota and its possible relationship with obesity. Mayo Clin Proc. 2008;83:460–9. doi: 10.4065/83.4.460. [DOI] [PubMed] [Google Scholar]
- 2.Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–31. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- 3.Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444:1022–3. doi: 10.1038/4441022a. [DOI] [PubMed] [Google Scholar]
- 4.Raoult D. Obesity pandemics and the modification of digestive bacterial flora. Eur J Clin Microbiol Infect Dis. 2008;27:631–4. doi: 10.1007/s10096-008-0490-x. [DOI] [PubMed] [Google Scholar]
- 5.Armougom F, Henry M, Vialettes B, Raccah D, Raoult D. Monitoring bacterial community of human gut microbiota reveals an increase in Lactobacillus in obese patients and Methanogens in anorexic patients. PLoS One. 2009;4:e7125. doi: 10.1371/journal.pone.0007125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bäckhed F, Manchester JK, Semenkovich CF, Gordon JI. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc Natl Acad Sci U S A. 2007;104:979–84. doi: 10.1073/pnas.0605374104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, et al. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes. 2008;57:1470–81. doi: 10.2337/db07-1403. [DOI] [PubMed] [Google Scholar]
- 8.Messina M. Insights gained from 20 years of soy research. J Nutr. 2010;140:2289S–95S. doi: 10.3945/jn.110.124107. [DOI] [PubMed] [Google Scholar]
- 9.Xiao CW. Health effects of soy protein and isoflavones in humans. J Nutr. 2008;138:1244S–9S. doi: 10.1093/jn/138.6.1244S. [DOI] [PubMed] [Google Scholar]
- 10.Martinez-Villaluenga C, Bringe NA, Berhow MA, Gonzalez de Mejia E. Beta-conglycinin embeds active peptides that inhibit lipid accumulation in 3T3-L1 adipocytes in vitro. J Agric Food Chem. 2008;56:10533–43. doi: 10.1021/jf802216b. [DOI] [PubMed] [Google Scholar]
- 11.Kohno M, Hirotsuka M, Kito M, Matsuzawa Y. Decreases in serum triacylglycerol and visceral fat mediated by dietary soybean beta-conglycinin. J Atheroscler Thromb. 2006;13:247–55. doi: 10.5551/jat.13.247. [DOI] [PubMed] [Google Scholar]
- 12.Faghih Sh, Abadi AR, Hedayati M, Kimiagar SM. Comparison of the effects of cows’ milk, fortified soy milk, and calcium supplement on weight and fat loss in premenopausal overweight and obese women. Nutr Metab Cardiovasc Dis. 2011;21:499–503. doi: 10.1016/j.numecd.2009.11.013. [DOI] [PubMed] [Google Scholar]
- 13.Lukaszuk JM, Luebbers P, Gordon BA. Preliminary study: soy milk as effective as skim milk in promoting weight loss. J Am Diet Assoc. 2007;107:1811–4. doi: 10.1016/j.jada.2007.07.008. [DOI] [PubMed] [Google Scholar]
- 14.Martinez-Villaluenga C, Dia VP, Berhow M, Bringe NA, Gonzalez de Mejia E. Protein hydrolysates from beta-conglycinin enriched soybean genotypes inhibit lipid accumulation and inflammation in vitro. Mol Nutr Food Res. 2009;53:1007–18. doi: 10.1002/mnfr.200800473. [DOI] [PubMed] [Google Scholar]
- 15.Hayward G, Pavlicik V. A corrected method for dry matter determination for use in anaerobic digester control. Biological Wastes. 1990;34:101–11. doi: 10.1016/0269-7483(90)90011-G. [DOI] [Google Scholar]
- 16.Sallis JF. Seven-day physical activity recall. 1997;29(suppl 6):89–103. [Google Scholar]
- 17.Barry KA, Hernot DC, Middelbos IS, Francis C, Dunsford B, Swanson KS, et al. Low-level fructan supplementation of dogs enhances nutrient digestion and modifies stool metabolite concentrations, but does not alter fecal microbiota populations. J Anim Sci. 2009;87:3244–52. doi: 10.2527/jas.2008-1659. [DOI] [PubMed] [Google Scholar]
- 18.Yu Z, Morrison M. Improved extraction of PCR-quality community DNA from digesta and fecal samples. Biotechniques. 2004;36:808–12. doi: 10.2144/04365ST04. [DOI] [PubMed] [Google Scholar]
- 19.Jernberg C, Löfmark S, Edlund C, Jansson JK. Long-term ecological impacts of antibiotic administration on the human intestinal microbiota. ISME J. 2007;1:56–66. doi: 10.1038/ismej.2007.3. [DOI] [PubMed] [Google Scholar]
- 20.Kok RG, de Waal A, Schut F, Welling GW, Weenk G, Hellingwerf KJ. Specific detection and analysis of a probiotic Bifidobacterium strain in infant feces. Appl Environ Microbiol. 1996;62:3668–72. doi: 10.1128/aem.62.10.3668-3672.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rinttilä T, Kassinen A, Malinen E, Krogius L, Palva A. Development of an extensive set of 16S rDNA-targeted primers for quantification of pathogenic and indigenous bacteria in faecal samples by real-time PCR. J Appl Microbiol. 2004;97:1166–77. doi: 10.1111/j.1365-2672.2004.02409.x. [DOI] [PubMed] [Google Scholar]
- 22.Cole JR, Chai B, Farris RJ, Wang Q, Kulam-Syed-Mohideen AS, McGarrell DM, et al. The ribosomal database project (RDP-II): introducing myRDP space and quality controlled public data. Nucleic Acids Res. 2007;35(Database issue):D169–72. doi: 10.1093/nar/gkl889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dowd SE, Callaway TR, Wolcott RD, Sun Y, McKeehan T, Hagevoort RG, et al. Evaluation of the bacterial diversity in the feces of cattle using 16S rDNA bacterial tag-encoded FLX amplicon pyrosequencing (bTEFAP) BMC Microbiol. 2008;8:125. doi: 10.1186/1471-2180-8-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Callaway TR, Dowd SE, Wolcott RD, Sun Y, McReynolds JL, Edrington TS, et al. Evaluation of the bacterial diversity in cecal contents of laying hens fed various molting diets by using bacterial tag-encoded FLX amplicon pyrosequencing. Poult Sci. 2009;88:298–302. doi: 10.3382/ps.2008-00222. [DOI] [PubMed] [Google Scholar]
- 25.Wolcott RD, Gontcharova V, Sun Y, Dowd SE. Evaluation of the bacterial diversity among and within individual venous leg ulcers using bacterial tag-encoded FLX and titanium amplicon pyrosequencing and metagenomic approaches. BMC Microbiol. 2009;9:226. doi: 10.1186/1471-2180-9-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smith DM, Snow DE, Rees E, Zischkau AM, Hanson JD, Wolcott RD, et al. Evaluation of the bacterial diversity of pressure ulcers using bTEFAP pyrosequencing. BMC Med Genomics. 2010;3:41. doi: 10.1186/1755-8794-3-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dowd SE, Sun Y, Wolcott RD, Domingo A, Carroll JA. Bacterial tag-encoded FLX amplicon pyrosequencing (bTEFAP) for microbiome studies: bacterial diversity in the ileum of newly weaned Salmonella-infected pigs. Foodborne Pathog Dis. 2008;5:459–72. doi: 10.1089/fpd.2008.0107. [DOI] [PubMed] [Google Scholar]
- 28.Gontcharova V, Youn E, Sun Y, Wolcott RD, Dowd SE. A comparison of bacterial composition in diabetic ulcers and contralateral intact skin. Open Microbiol J. 2010;4:8–19. doi: 10.2174/1874285801004010008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ishak HD, Plowes R, Sen R, Kellner K, Meyer E, Estrada DA, et al. Bacterial diversity in Solenopsis invicta and Solenopsis geminata ant colonies characterized by 16S amplicon 454 pyrosequencing. Microb Ecol. 2011;61:821–31. doi: 10.1007/s00248-010-9793-4. [DOI] [PubMed] [Google Scholar]
- 30.Dowd SE, Sun Y, Secor PR, Rhoads DD, Wolcott BM, James GA, et al. Survey of bacterial diversity in chronic wounds using pyrosequencing, DGGE, and full ribosome shotgun sequencing. BMC Microbiol. 2008;8:43. doi: 10.1186/1471-2180-8-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dowd SE, Zaragoza J, Rodriguez JR, Oliver MJ, Payton PR. Windows. NET Network Distributed Basic Local Alignment Search Toolkit (W.ND-BLAST) BMC Bioinformatics. 2005;6:93. doi: 10.1186/1471-2105-6-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, et al. The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 2009;37(Database issue):D141–5. doi: 10.1093/nar/gkn879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Handl S, Dowd SE, Garcia-Mazcorro JF, Steiner JM, Suchodolski JS. Massive parallel 16S rRNA gene pyrosequencing reveals highly diverse fecal bacterial and fungal communities in healthy dogs and cats. FEMS Microbiol Ecol. 2011;76:301–10. doi: 10.1111/j.1574-6941.2011.01058.x. [DOI] [PubMed] [Google Scholar]
- 34.Guerrero FD, Dowd SE, Djikeng A, Wiley G, Macmil S, Saldivar L, et al. A database of expressed genes from Cochliomyia hominivorax (Diptera: Calliphoridae) J Med Entomol. 2009;46:1109–16. doi: 10.1603/033.046.0518. [DOI] [PubMed] [Google Scholar]
- 35.Sen R, Ishak HD, Estrada D, Dowd SE, Hong E, Mueller UG. Generalized antifungal activity and 454-screening of Pseudonocardia and Amycolatopsis bacteria in nests of fungus-growing ants. Proc Natl Acad Sci U S A. 2009;106:17805–10. doi: 10.1073/pnas.0904827106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stephenson MF, Mfuna L, Dowd SE, Wolcott RD, Barbeau J, Poisson M, et al. Molecular characterization of the polymicrobial flora in chronic rhinosinusitis. J Otolaryngol Head Neck Surg. 2010;39:182–7. [PubMed] [Google Scholar]
- 37.Andreotti R, Pérez de León AA, Dowd SE, Guerrero FD, Bendele KG, Scoles GA. Assessment of bacterial diversity in the cattle tick Rhipicephalus (Boophilus) microplus through tag-encoded pyrosequencing. BMC Microbiol. 2011;11:6. doi: 10.1186/1471-2180-11-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dufrene M, Legendre P. Species assemblages and indicator species: The need for a flexible asymmetrical approach. Ecol Monogr. 1997;67:345–66. [Google Scholar]
- 39.Anderson MJ, Willis TJ. Canonical analysis of principal coordinates: A useful method of constrained ordination for ecology. Ecology. 2003;84:511–25. doi: 10.1890/0012-9658(2003)084[0511:CAOPCA]2.0.CO;2. [DOI] [Google Scholar]
- 40.Lozupone C, Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol. 2005;71:8228–35. doi: 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–6. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.R Development Core Team. R: A language and environment for statistical computing. 2011; ISBN 3-900051-07-0. [Google Scholar]
- 43.Oksanen J, Blanchet FG, Kindt R, Legendre PO. Vegan: Community ecology package. R package version. 2011;1:17-–8. [Google Scholar]
- 44.Roberts DW. Labdsv: Ordination and multivariate analysis for ecology. 2010:1.4–1. [Google Scholar]
- 45.Baba T, Ueda A, Kohno M, Fukui K, Miyazaki C, Hirotsuka M, et al. Effects of soybean beta-conglycinin on body fat ratio and serum lipid levels in healthy volunteers of female university students. J Nutr Sci Vitaminol (Tokyo) 2004;50:26–31. doi: 10.3177/jnsv.50.26. [DOI] [PubMed] [Google Scholar]
- 46.Duncan SH, Lobley GE, Holtrop G, Ince J, Johnstone AM, Louis P, et al. Human colonic microbiota associated with diet, obesity and weight loss. Int J Obes (Lond) 2008;32:1720–4. doi: 10.1038/ijo.2008.155. [DOI] [PubMed] [Google Scholar]
- 47.Walker AW, Ince J, Duncan SH, Webster LM, Holtrop G, Ze X, et al. Dominant and diet-responsive groups of bacteria within the human colonic microbiota. ISME J. 2011;5:220–30. doi: 10.1038/ismej.2010.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457:480–4. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Costello EK, Lauber CL, Hamady M, Fierer N, Gordon JI, Knight R. Bacterial community variation in human body habitats across space and time. Science. 2009;326:1694–7. doi: 10.1126/science.1177486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schwiertz A, Taras D, Schäfer K, Beijer S, Bos NA, Donus C, et al. Microbiota and SCFA in lean and overweight healthy subjects. Obesity (Silver Spring) 2010;18:190–5. doi: 10.1038/oby.2009.167. [DOI] [PubMed] [Google Scholar]
- 51.Amar J, Burcelin R, Ruidavets JB, Cani PD, Fauvel J, Alessi MC, et al. Energy intake is associated with endotoxemia in apparently healthy men. Am J Clin Nutr. 2008;87:1219–23. doi: 10.1093/ajcn/87.5.1219. [DOI] [PubMed] [Google Scholar]
- 52.Wang M, Ahrné S, Jeppsson B, Molin G. Comparison of bacterial diversity along the human intestinal tract by direct cloning and sequencing of 16S rRNA genes. FEMS Microbiol Ecol. 2005;54:219–31. doi: 10.1016/j.femsec.2005.03.012. [DOI] [PubMed] [Google Scholar]