

Surmounting multiple tumor suppressive barriers is fundamental to cancer onset, but also predicts that their restoration may pose a powerful anticancer strategy. The proof of this principle has been elegantly demonstrated by the restoration of p53-induced tumor suppression (reviewed in ref. 1). The promyelocytic leukemia protein (PML) is emerging as a key tumor suppressor that is inactivated in multiple cancer types and is thus a candidate for therapeutic restoration. PML is the indispensable component of PML nuclear bodies, which are structures that coalesce in response to cellular stress and are implicated in growth inhibitory functions.2 Three important criteria distinguish PML as an attractive candidate for tumor suppression. First, PML inhibits the growth of tumor cells by regulating multiple pathways, including those activating the p53 family and inhibiting the PI3K/AKT pathway.2 Further, PML has been substantiated to inhibit tumor development in various mouse models.2 Second, PML expression is frequently downregulated or lost in a wide spectrum of human cancers, including prostate adenocarcinoma, B-lymphoma, lung, colon and breast adenocarcinomas.3,4 Third, PML is rarely mutated, and its downregulation occurs predominantly at the post-translational level. These criteria render PML as a promising candidate for restoration of tumor suppression, and intense research has been directed toward delineating its regulation at the post-translational level. These studies revealed that the stability of PML is regulated by multiple mechanisms (Fig. 1) including phosphorylation (by Caseine Kinase 2, CK2, Big MAP kinase 1, BMK1), isomerization (by PIN1), acetylation (by p300 histone acetyl-transferase and SIRT1), SUMOylation (by PIAS1) and ubiquitination of SUMOylated PML by RNF4 (the RING finger protein 4) (refs. 5–8 and references therein). The stability of PML and formation of PML nuclear bodies are regulated under physiological conditions by the E3 ligase E6AP (E6-associated protein, E6AP9).
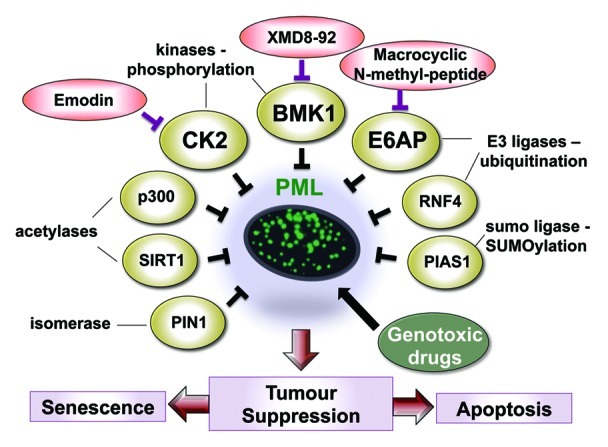
Figure 1. Restoration of tumor suppression by targeting negative regulators of PML using small-molecule inhibitors. Diagramatic presentation of the major inhibitors (yellow ovals) of PML by post-translational modifications, including phosphorylation, SUMOylation, isomerization, acetylation and ubiquitination. Specific inhibitors targeting several of these proteins (red ovals), two of which (XMD8–92 and emodin) have been shown to restore PML-induced tumor suppression. Combinations of small molecule inhibitors with genotoxic DNA damaging drugs (green oval) such as etoposide may prove to have a synergistic anticancer effect.
Identification of these modes of PML regulation has led to the proposition of novel approaches for the restoration of tumor suppression by PML. Intriguingly, recent studies identified several small-molecule inhibitors capable of targeting key negative regulators of PML, thereby restoring PML levels/functions. The efficacy of these inhibitors as anticancer drugs warrants further exploration (Fig. 1). Pandolfi and his team found that CK2-mediated phosphorylation of PML promoted its proteasomal degradation.7 Application of a pharmacological inhibitor of CK2, emodin, to established lung cancer xenografts, provided the first evidence for tumor suppression by reactivation of PML in vivo.7 Importantly, elevated levels of CK2 are frequently found in NSCLC and inversely correlate with PML abundance. Similarly, relieving PML from BMK1-mediated inhibition using a small-molecule inhibitor of BMK1, XMD8–92, suppressed cancer cell proliferation in vitro and xenografts growth in vivo.8 Recently, we demonstrated elevated levels of E6AP in 60% of human Burkitt lymphomas and identified it as a critical player responsible for the loss of PML in these B lymphomas.4 Importantly, downregulation of E6AP in a Myc-induced mouse model as well as in human lymphoma cells elicited PML-mediated senescence, which efficiently suppressed cancer growth. It will be interesting to test whether an E6AP inhibitor, such as the recently discovered anti-E6AP N-methyl peptide,10 could efficiently restore tumor suppression by PML in B lymphoma.
Intriguingly, PML has also been identified to lie at the heart of molecular machinery, driving anticancer response to genotoxic drugs that are commonly used in the clinic (reviewed in ref. 2). This provides a rationale for combinatorial treatment of cancer cells with small-molecule inhibitors, such as those described here, together with genotoxic drugs. This approach is likely to result in the stabilization and activation of PML, leading to an efficient growth suppression of cancer cells with low or no PML expression. This exciting progress in the PML field also raises new fundamental questions. For example, PML was shown to promote self-renewal of CML initiating cells, and a loss of PML exhausts the leukemic stem cell pool.11 This needs to be considered when PML is targeted, as well as the potential impact on other types of stem cells.6 Importantly, the stage during tumorigenesis at which tumor suppression needs to be restored is critical, as elegantly shown for p53 (reviewed in ref. 1). Genetically engineered mouse models in which PML expression can be turned on and off reversibly in a tissue-dependent manner would provide powerful tools to address these questions and test novel approaches to restore tumor suppression by PML.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/22043
References
- 1.Junttila MR, et al. Nat Rev Cancer. 2009;9:821–9. doi: 10.1038/nrc2728. [DOI] [PubMed] [Google Scholar]
- 2.Bernardi R, et al. Nat Rev Mol Cell Biol. 2007;8:1006–16. doi: 10.1038/nrm2277. [DOI] [PubMed] [Google Scholar]
- 3.Gurrieri C, et al. J Natl Cancer Inst. 2004;96:269–79. doi: 10.1093/jnci/djh043. [DOI] [PubMed] [Google Scholar]
- 4.Wolyniec K, et al. Blood. 2012;120:822–32. doi: 10.1182/blood-2011-10-387647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Petrie K, et al. Nat Cell Biol. 2008;10:507–9. doi: 10.1038/ncb0508-507. [DOI] [PubMed] [Google Scholar]
- 6.Salomoni P, et al. Cell Death Dis. 2012;3:e247. doi: 10.1038/cddis.2011.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scaglioni PP, et al. Cell. 2006;126:269–83. doi: 10.1016/j.cell.2006.05.041. [DOI] [PubMed] [Google Scholar]
- 8.Yang Q, et al. Cancer Cell. 2010;18:258–67. doi: 10.1016/j.ccr.2010.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Louria-Hayon I, et al. Cell Death Differ. 2009;16:1156–66. doi: 10.1038/cdd.2009.31. [DOI] [PubMed] [Google Scholar]
- 10.Yamagishi Y, et al. Chem Biol. 2011;18:1562–70. doi: 10.1016/j.chembiol.2011.09.013. [DOI] [PubMed] [Google Scholar]
- 11.Ito K, et al. Nature. 2008;453:1072–8. doi: 10.1038/nature07016. [DOI] [PMC free article] [PubMed] [Google Scholar]