

Abstract
Tumor protein (TP)-p53 family members (TP63, TP63 and TP73) are guardians of the genome and key players in orchestrating the cellular response to cisplatin treatment. Cisplatin-induced phosphorylation of ΔNp63α was shown to have a role in regulating intracellular ΔNp63α protein levels. We previously found that squamous cell carcinoma (SCC) cells exposed to cisplatin displayed the ATM-dependent phosphorylation of ΔNp63α (p-ΔNp63α), which is critical for the transcriptional regulation of specific downstream mRNAs and microRNAs and is likely to underlie the chemoresistance of SCC cells. However, SCC cells expressing non-p-ΔNp63α became more cisplatin-resistant. We also found that p-ΔNp63α forms complexes with a number of proteins involved in cell death response through regulation of cell cycle arrest, apoptosis, autophagy, RNA splicing and chromatin modifications. Here, we showed that p-ΔNp63α induced ARG1, GAPDH, and CPT2 gene transcription in cisplatin-sensitive SCC cells, while non-p-ΔNp63α increased a transcription of CAD, G6PD and FASN genes in cisplatin-resistant SCC cells. We report that the p-ΔNp63α-dependent regulatory mechanisms implicated in the modulation of plethora of pathways, including amino acid, carbohydrate, lipid and nucleotide metabolisms, thereby affect tumor cell response to cisplatin-induced cell death, suggesting that the ATM-dependent ΔNp63α pathway plays a role in the resistance of tumor cells to platinum therapy.
Keywords: P53, p63, cisplatin, squamous cell carcinomas, protein interactions, metabolomics
Introduction
Cisplatin used in cancer chemotherapy against different human tumors inhibits DNA replication, cell cycle arrest and cell death.1-3 Although cisplatin triggers cell death, its prolonged use often induces cell resistance against itself in a tumor cell population.1-4 Tumor protein (TP)-p53 family members (TP63, TP63 and TP73) are guardians of the genome and key players in orchestrating the cellular response to cisplatin treatment.5-7 The role of TP53 in cell death and metabolism is well defined by its downstream binding to specific gene promoters or interacting with extensive protein network as well as by upstream regulatory phosphorylation and acetylation of TP53 and intracellular translocations.8-12
TP53 is a transcription factor that mediates cell response to various stresses by promoting expression of specific genes that induce cell cycle arrest, DNA repair, senescence, cell death and alter various metabolic pathways.8,10,12,13 TP53 was found to regulate TP53-induced glycolysis regulator (TIGAR, encodes a protein with sequence similarity to the bisphosphate domain of the glycolytic enzyme that degrades fructose-2, 6-bisphosphate), which switches glucose from glycolysis to the pentose phosphate pathway.12,14 Downregulation of TP53-dependent SCO2 [SCO cytochrome oxidase deficient homolog 2, which acts as a copper chaperone, transporting copper to the Cu(A) site on the cytochrome c oxidase subunit II, COX2] causes a shifting of ATP production from oxidative phosphorylation to glycolysis.12
TP53 can also affect expression of other genes involved in carbohydrate metabolism (e.g., glyceraldehyde-3-phosphate dehydrogenase, GAPDH, glucose-6-phosphate dehydrogenase, G6PD, hexokinase-2, HK2, malate dehydrogenase-1, MDH1, phosphoglycerate mutase, PGM); amino acid metabolism (e.g., carbamoyl phosphate synthetase-2, CAD, ornithine carbamoyltransferase, OTC, glutaminase-2, GLS2); lipid metabolism [e.g., apolipoprotein D (APOD), carnitine palmitoyl-transferase (CPT), fatty acid synthase (FASN), ferredoxin reductase (FDXR), guanidine-acetate methyltransferase (GAMT)] and nucleotide metabolism [e.g., adenosine deaminase (ADA), dihydroorotate dehydrogenase (DHODH), hypoxanthine phosphoribosyl transferase (HPRT), ribonucleotide reductase (RRM2)].15-30 TP53 can interact with the mTOR pathway [e.g., AMP-activated protein kinase (AMPK)], which coordinates cell growth by sensing nutrient availability and balancing anabolic and catabolic processes.31,32 Genotoxic stress causes p53-dependent suppression of mTOR activity via transcriptional activation of AMPKβ, TSC2, IGF-BP3 and PTEN.31-33
The energy for malignant tumor growth is supplied by a non-oxidative pathway, glycolysis, in contrast to that of normal cells, which generate energy from oxidative breakdown of pyruvate within the mitochondria. The inactivation of TP53 normal function by mutations upregulates glycolysis and downregulates oxidative phosphorylation in cancer cells, suggesting that TP53 status plays a pivotal role in the underlying mechanism for the “Warburg effect.”34-36
Tp63 express two main classes of isoforms: isoforms with the transactivation (TA-) domain and dominant-negative isoforms, which are truncated at the NH2-terminus (ΔN-). TAp63 isoforms show pro-apoptotic activities, whereas ΔNp63 isoforms often show anti-apoptotic functions. ΔNp63α can directly interfere with the transcriptional function of the TA-isoforms of the p53 family over apoptotic target genes (CD-95, TNFR, TRAIL-R, BAX, PIG3, PERP, RAD9, APAF1, CASP-3, -8 and -9) induced by chemotherapeutic drugs, thereby contributing to chemoresistance.37,38
Phosphorylation of p63 (e.g., ΔNp63α) has become a matter of extensive investigation in our laboratory for the last decade and was shown to have a role in regulating intracellular ΔNp63α protein levels.39-44 We previously found that squamous cell carcinoma (SCC) cells exposed to cisplatin displayed the ATM-dependent phosphorylation of wild type (wt) ΔNp63α generating phosphorylated (p)-ΔNp63α, which is critical for the transcriptional regulation of specific downstream mRNAs and microRNAs, and is likely to underlie the chemosensitivity/resistance of SCC cells.39-44 However, SCC cells expressing non-p-ΔNp63α became more cisplatin-resistant.42-44 Our recent report showed that SCC cells spontaneously developed resistance to cisplatin treatment (SCC-25CP) in contrast to cisplatin-sensitive SCC-25 cells, which displayed the greater ratio between non-p-ΔNp63α and p-ΔNp63α levels, which might play a role in cisplatin resistance displayed by SCC-25CP cells.41
Understanding the multiple mechanisms by which p-ΔNp63α modulates the response of SCC cells to cisplatin, including those that effect metabolic pathways, will provide us with crucial information about a role for the ATM-dependent ΔNp63α pathway in the resistance of tumor cells to platinum therapy.
Results
Cisplatin induces multiple metabolic changes in SCC-11 and SCC-11M cells
Cisplatin is effective for induction of neoplastic cell death, especially for squamous cell carcinomas (SCC).2 However, a common limitation of this treatment is the eventual development of resistance to cisplatin chemotherapy.1-3 The transcription factor ΔNp63α is phosphorylated upon exposure to cisplatin and subsequently triggers a gene program controlling stress response and cell survival.39-44 Our previous studies showed that cisplatin-sensitive SCC cells display a greater ratio of phosphorylated (p)-ΔNp63α/non-p-ΔNp63α as compared with cells that are resistant to cisplatin treatment.39-44 Using a metabolomics approach,45-48 we compared global metabolic profiles of a SCC cell line expressing p-ΔNp63α (SCC-11) or non-p-ΔNp63α with an altered ability to be phosphorylated (SCC-11M, S385G mutation in the ATM kinase site of ΔNp63α). Cells were treated with control medium (Ctrl) or with 10 μg/ml (CisMn) for 16 h. A total of 103 metabolites were upregulated, and 98 metabolites were downregulated in SCC-11 cells compared with SCC-11M cells (both treated with cisplatin, Table S1 and Fig. S1). The highest changers were divided on the following groups: amino acids, peptides, carbohydrates, energy, lipids, nucleotides, vitamins and cofactors and xenobiotics (Fig. S2–S9).
Amino acid and peptide metabolism
As we compared values obtained from SCC-11 cells to those from SCC-11M cells (both under cisplatin exposure), we found that 19 metabolites increased, nine decreased, and 17 remained unchanged (Fig. S2). We observed that while urea, glutamine, leucine and dimethylarginine increased in SCC-11 cells, many metabolites (e.g., reduced glutathione, S-methylthioadenosine, glutamate, citrulline, ornithine, cysteine, cysteine-glutathione disulfide, N-acetylaspartate) dramatically decreased (Fig. S2). These data suggest that the urea and glutathione metabolic pathways are likely to be altered in SCC-11M cells compared with SCC-11M cells. We further found that among 49 peptides, seven increased, while others decreased (40) or unchanged (2) in SCC-11 cells compared with SCC-11M (both treated with cisplatin), as shown in Figure S3.
Carbohydrate and energy metabolism
We then found clear alterations in glucose metabolism observed in SCC cells under cisplatin pressure. Among 27 metabolites, three increased (e.g., fructose-2, 6-biphosphate), 14 decreased, and 10 remained unchanged (Fig. S4 and S5). Despite minimal changes in glucose, production of glycolytic intermediates (e.g., glucose 6-phosphate, fructose 6-phosphate, 3-phosphoglycerate, 2-phosphoglycerate and lactate) was reduced in SCC-11 cells compared with SCC-11M cells (Fig. S4). Together with decreased levels of the pentose phosphate pathway intermediates (e.g., 6-phosphogluconate, ribulose 5-phosphate/xylulose 5-phosphate, ribose 5-phosphate, and sedoheptulose 7-phosphate and ribose, these changes are suggestive of lesser glucose utilization in SCC-11 cells than in SCC-11M cells (Fig. S4). When comparing SCC-11 cells exposed to cisplatin and SCC-11 cells grown in control medium, there is an apparent decrease in glucose uptake and/or utilization, as slight reductions in glycolytic and pentose phosphate pathway intermediates were also observed. However, we observed reduction in lactate levels and elevation of most glycolytic and pentose phosphate pathway intermediates in SCC-11M cells exposed to cisplatin compared with untreated SCC-11M cells. These data suggest that p-ΔNp63α is not required for the cisplatin-induced dampening of glucose metabolism, but non-p-ΔNp63α may influence how glucose is utilized following treatment of SCC-11M cells with cisplatin.
Lipid metabolism
When comparing the cisplatin-treated SCC-11and SCC-11M cells, we observed that 41 metabolites increased, 32 decreased, and 31 remained unchanged (Fig. S6). We then found that SCC-11 cells exhibited greater production of prostaglandin E2, linoleate (18: 2n6), caprylate, carnitine and arachidonate (20: 4n6), cholesterol, laurate, palmitate (16: 0), oleic ethanolamide, palmitoylethanolamide, 1-palmitoleoylglycerophosphoinositol, 1-oleoylglycero- phosphoinositol and glycerophosphoetanolamine than SCC-11M cells (both under cisplatin pressure, Fig. S6). While treatment of SCC-11 cells with cisplatin increased production of prostaglandin E2 compared with untreated SCC-11 cells, exposure of SCC-11M cells to cisplatin resulted in decreased levels of fatty acid precursors and prostaglandin E2, with a concomitant increase in the prostaglandin E2 degradation product 13, 14-dihydro-15-keto-prostaglandin A2. We further found that membrane degradation products glycerophosphoryl-choline, 1-myristoylglycerophosphorylcholine, 1-stearoyl- glycerophosphoetanolamine and glycerol 3-phosphate decreased in SCC-11 cells compared with SCC-11M cells (both treated with cisplatin). We also showed that the levels of arachidonate (20: 4n6), 2-arachidonoyl-glycerol and 1-arachidonoylglycerolphosphocholine decreased in SCC-11 cells compared with SCC-11M cells (Fig. S6). Upon cisplatin exposure, SCC-11 cells (compared with SCC-11M cells) showed a slight decrease of cholesterol precursors, lanosterol and lathosterol as well as sphinganine, sphingosine and palmitoylsphingomyelin (Fig. S6).
Nucleotide metabolism
We then showed that five metabolites increased, 15 decreased, eight remained unchanged in SCC-11 cells compared with SCC-11M cells (Fig. S7).
Cofactor/vitamin metabolism
We further showed that two metabolites increased, eight decreased, and two remained unchanged in SCC-11 cells (compared with SCC-11M cells) exposed to cisplatin (Fig. S8). The level of acetyl-CoA dramatically increased, while 3′-dephospho-CoA and nicotine derivatives (nicotinate, nicotinamide, nicotinamide ribonucleotide and nicotinamide adenine dinucleotide) substantially decreased in SCC-11 cells compared with SCC-11M cells.
Cisplatin induces the metabolic gene expression in SCC-11 and SCC-11M cells
To further examine the molecular mechanism underlying the metabolic changes induced by the transcriptional factor, ΔNp63α, in the phosphorylated and non-phosphorylated states. Using SCC-11 and SCC-11M cells exposed to control medium and 10 μg/ml cisplatin for 16 h, we performed global cDNA expression chip array, ChIP-on-chip array and DNA methylation chip array analyses (Supplemental Methods). As a result of this systemic global investigation, we found that many metabolic genes are induced by p-ΔNp63α in SCC-11 cells upon cisplatin exposure (e.g., SREBF1, NQO1, RRM2B, ALDH7A1, TYMS, DHFR, TK1, ACAD9, GCLM, DCK, GSR, ATP11B, LNPEP, PARP1, ARG1, ATOX1, ALDH3, CA2, CEPT1, GAPDH, ETNK1, CPT2, PFKP, PNP, PKM2, COASY, NMRK2 and ATP7B, see Table S2). On the other hand, numerous metabolic genes are also induced by non-p-ΔNp63α in SCC-11M cells under same conditions (e.g., GYS1, XDH1, DIO2, TALDO1, TKT, CBS, LOX, ASNA1, IDO1, ACLY, G6PD, CAD, ENO1, FASN, PTGS1, ALOX5, FADS1, CTPS1, DAGLB, HK3, NAPRT1, DEGS1, NMNAT1, PLA2G4, PGK1, NADSYN1 and ADA, see Table S2).
After the ChIP-on-chip array analysis, we further found that the cisplatin exposure induced the ΔNp63α protein binding to the following gene promoter sequences in SCC-11 cells (CPT2, ETNK1, GAPDH, ATP7B, PNP, SREBF1, COASY, NMRK2, DCK, ATOX1, ARG1, see Table S2) or in SCC-11M cells (e.g., PLA2G4, NMNAT1, ENO1, PFKFB3, NADSYN1, ACACA, FASN, ACLY, CAD, XDH, ADA, TKT, ACAD9, HPGD, CD38, ALDH7A1, HK3, DAGLB, CYP51A1, ASL, NAPRT1, PTGS1, PGK1, G6PD, see Table S4).
Knowing that the DNA methylation of specific promoter sequences leads to transcriptional inactivation of expression,49,50 we performed the DNA methylation chip array analysis and observed that the cisplatin exposure induced the methylation of 91 gene promoters in SCC-11 cells (Table S5), and 156 gene promoters in SCC-11M cells (Table S6). Among those promoters methylated in SCC-11 cells, we found the following genes: ACACA, ACO1, CD38, DGKB, DIO2, CAD, FADS1, FASN, OTC, PFKFB3, PLA2G4, G6PD, NMNAT1 and NAPRT1, see Table S5). We further found that the following gene promoters (e.g., ADCY1, 3 and 8, AK1, ARG1, COASY, GAPDH, CPT2, ETNK1, NMRK2 and PKM2, Table S6) were methylated in SCC-11M cells.
Upon inspection of specific gene promoter sequences (ARG1, GAPDH, PKM2 and CPT2), we found that these sequences contain potential consensus binding elements for TP63 and SREBF1 (Figs. S10–13), while promoter sequences for CAD, G6PD, PFKFB3 and FASN contain potential binding elements for TP53, TP63 and E2F (Figs. S14–17) as defined elsewhere.51-58 We thus investigated the role of these transcription factors in regulation of gene expression in SCC-11 and SCC-11M cells upon cisplatin exposure.
P-ΔNp63α induces expression of specific metabolic genes in SCC-11 cells upon cisplatin exposure
To further examine the selected gene targets upregulated by p-ΔNp63α in response to SCC-11 cells to cisplatin treatment, we have undertaken the study of the following gene expression and regulation: ARG1, GAPDH, PKM2 and CPT2 (Figs. 1–4). As arginase 1 (ARG1) catalyzes the critical step of urea metabolism (l-arginine + H2O to L-ornintine + Urea, Fig. 1A), ARG1 mRNA and protein levels dramatically increased (5-fold for mRNA) in SCC-11 cells upon cisplatin exposure, while no such increase observed in SCC-11M cells (Fig. 2B). We then showed that the increasing binding of p-ΔNp63α and SREBF1 to the specific region (while no binding was found to the non-specific region) of the ARG1 promoter in SCC-11 cells after cisplatin treatment, but not in SCC-11M cells (Fig. 1C). We then found that while the scrambled siRNA had no effect on p-ΔNp63α binding (enrichment) to the ARG1 promoter, SREBF1 siRNA substantially inhibited such binding to the ARG1 promoter in SCC-11 cells under cisplatin treatment (Fig. 1D). We next demonstrated an additive effect of p-ΔNp63α and SREBF1 on the luciferase reporter activity driven by the ARG1 promoter in SCC-11 cells upon cisplatin exposure (Fig. 1E).

Figure 1. P-ΔNp63α induces the expression of ARG1 in SCC-11 cells upon cisplatin exposure. (A) Metabolite reaction of arginase 1 (ARG1). Grey arrow indicates the increase in SCC-11 cells, while black arrow indicates the decrease in SCC-11M cells. (B, upper panel) qPCR expression analysis of ARG1 mRNA obtained from SCC-11 and SCC-11M cells treated with control medium (Con) and 10μg/ml cisplatin (CIS). The values from SCC-11 cells treated with control medium designated as 1. The qPCR values presented as relative units (RU) (B, lower panel) Immunoblot analysis of ARG1 in SCC-11 and SCC-11M cells treated with control medium (Con) or cisplatin (CIS). Loading was normalized by β-actin expression. (C) ChIP assay of the binding of p-ΔNp63α and SREBF1 to the ARG1 promoter (enrichment) in SCC-11 cells exposed to control medium (Con) and cisplatin (CIS). (D) Quantitative analysis of enrichment (ChIP/Input ratio) of the ARG1 promoter with the p-ΔNp63α and SREBF1 transcription factors in SCC-11 cells treated with cisplatin. ChiP/Input ratio values presented as relative units (RU). SCC-11 cells were transfected with the scrambled and SREBF1 siRNA and analyzed for ChIP/Input enrichment. (E) Luciferase reporter assay of the ARG1 promoter in SCC-11 and SCC-11M cells treated with control medium (Con) and cisplatin (CIS). SCC-11 cells were transfected with the scrambled and SREBF1 siRNA and analyzed for the ARG1 promoter-driven luciferase activity presented as relative units (RU). The values from SCC-11 cells treated with control medium designated as 1. All experiments were performed in triplicates. p < 0.05.

Figure 4. P-ΔNp63α induces the expression of CPT2 in SCC-11 cells upon cisplatin exposure. (A) Metabolite reaction of carnitine palmitoyltransferase 2 (CPT2). Grey arrow indicates the increase in SCC-11 cells, while black arrow indicates the decrease in SCC-11M cells. (B, upper panel) qPCR expression analysis of CPT2 mRNA obtained from SCC-11 and SCC-11M cells treated with control medium (Con) and 10 μg/ml cisplatin (CIS). The values from SCC-11 cells treated with control medium designated as 1. The qPCR values presented as relative units (RU). (B, lower panel) Immunoblot analysis of CPT2 in SCC-11 and SCC-11M cells treated with control medium (Con) or cisplatin (CIS). Loading was normalized by β-actin expression. (C) ChIP assay of the binding of p-ΔNp63α and SREBF1 to the CPT2 promoter (enrichment) in SCC-11 cells exposed to control medium (Con) and cisplatin (CIS). (D) Quantitative analysis of enrichment (ChIP/Input ratio) of the CPT2 promoter with the p-ΔNp63α and SREBF1 transcription factors in SCC-11 cells treated with cisplatin. ChiP/Input ratio values presented as relative units (RU). SCC-11 cells were transfected with the scrambled and SREBF1 siRNA and analyzed for ChIP/Input enrichment. (E) Luciferase reporter assay of the CPT2 promoter in SCC-11 and SCC-11M cells treated with control medium (Con) and cisplatin (CIS). SCC-11 cells were transfected with the scrambled and SREBF1 siRNA and analyzed for the CPT2 promoter-driven luciferase activity presented as relative units (RU). The values from SCC-11 cells treated with control medium designated as 1. All experiments were performed in triplicates. p < 0.05.

Figure 2. P-ΔNp63α induces the expression of GAPDH in SCC-11 cells upon cisplatin exposure. (A) Metabolite reaction of glyceraldehyde-3-phosphate dehydrogenase. Grey arrow indicates the increase in SCC-11 cells, while black arrow indicates the decrease in SCC-11M cells. (B, upper panel) qPCR expression analysis of GAPDH mRNA obtained from SCC-11 and SCC-11M cells treated with control medium (Con) and 10 μg/ml cisplatin (CIS). The values from SCC-11 cells treated with control medium designated as 1. The qPCR values presented as relative units (RU). (B, lower panel) Immunoblot analysis of GAPDH in SCC-11 and SCC-11M cells treated with control medium (Con) or cisplatin (CIS). Loading was normalized by β-actin expression. (C) ChIP assay of the binding of p-ΔNp63α and SREBF1 to the GAPDH promoter (enrichment) in SCC-11 cells exposed to control medium (Con) and cisplatin (CIS). (D) Quantitative analysis of enrichment (ChIP/Input ratio) of the GAPDH promoter with the p-ΔNp63α and SREBF1 transcription factors in SCC-11 cells treated with cisplatin. ChiP/Input ratio values presented as relative units (RU). SCC-11 cells were transfected with the scrambled and SREBF1 siRNA and analyzed for ChIP/Input enrichment. (E) Luciferase reporter assay of the GAPDH promoter in SCC-11 and SCC-11M cells treated with control medium (Con) and cisplatin (CIS). SCC-11 cells were transfected with the scrambled and SREBF1 siRNA and analyzed for the GAPDH promoter-driven luciferase activity presented as relative units (RU). The values from SCC-11 cells treated with control medium designated as 1. All experiments were performed in triplicates. p < 0.05.
We next showed that mRNA and protein expression for glyceraldehyde-3-phosphate dehydrogenase (GAPDH), which catalyzes the reaction (glyceraldehyde-3-phosphate + NAD+ + Pi to D-glycerate-1, 3-biphosphate + NADH + H+, Fig. 2A), was greatly increased by cisplatin in SCC-11 cells but not in SCC-11M cells (Fig. 2B). We observed that the increasing binding of p-ΔNp63α and SREBF1 to the specific region (while no binding was found to the non-specific region) of the GAPDH promoter in SCC-11 cells after cisplatin treatment, but not in SCC-11M cells (Fig. 2C). We found that while the scrambled siRNA had no effect on p-ΔNp63α binding (enrichment) to the GAPDH promoter, SREBF1 siRNA greatly inhibited such binding in SCC-11 cells under cisplatin treatment (Fig. 2D). We then showed a cumulative effect of p-ΔNp63α and SREBF1 on the GAPDH promoter-luciferase reporter in SCC-11 cells upon cisplatin exposure (Fig. 2E).
We then showed that the mRNA/protein expression for pyruvate kinase M2 (PKM2), which catalyzes the reaction (Phosphoenolpyrivate + ADP to Pyruvate + ATP, Figure 3A) very much increased by cisplatin in SCC-11 cells but not in SCC-11M cells (Fig. 3B). We then showed that the increasing binding of p-ΔNp63α and SREBF1 to the specific region (while no binding was found to the non-specific region) of the PKM2 promoter in SCC-11 cells after cisplatin treatment, but not in SCC-11M cells (Fig. 3C). We also showed that while the scrambled siRNA had no effect on p-ΔNp63α binding (enrichment) to the PKM2 promoter, SREBF1 siRNA greatly inhibited such binding in SCC-11 cells under cisplatin treatment (Fig. 3D). We next showed a supplementary effect of p-ΔNp63α and SREBF1 on the PKM2 promoter-driven luciferase reporter in SCC-11 cells upon cisplatin exposure (Fig. 3E).

Figure 3. P-ΔNp63α induces the expression of PKM2 in SCC-11 cells upon cisplatin exposure. (A) Metabolite reaction of pyruvate kinase M2 (PKM2). Grey arrow indicates the increase in SCC-11 cells, while black arrow indicates the decrease in SCC-11M cells. (B, upper panel) qPCR expression analysis of PKM2 mRNA obtained from SCC-11 and SCC-11M cells treated with control medium (Con) and 10 μg/ml cisplatin (CIS). The values from SCC-11 cells treated with control medium designated as 1. The qPCR values presented as relative units (RU). (B, lower panel) Immunoblot analysis of PKM2 in SCC-11 and SCC-11M cells treated with control medium (Con) or cisplatin (CIS). Loading was normalized by β-actin expression. (C) ChIP assay of the binding of p-ΔNp63α and SREBF1 to the PKM2 promoter (enrichment) in SCC-11 cells exposed to control medium (Con) and cisplatin (CIS). (D) Quantitative analysis of enrichment (ChIP/Input ratio) of the PKM2 promoter with the p-ΔNp63α and SREBF1 transcription factors in SCC-11 cells treated with cisplatin. ChiP/Input ratio values presented as relative units (RU). SCC-11 cells were also transfected with the scrambled and SREBF1 siRNA and analyzed for ChIP/Input enrichment. (E) Luciferase reporter assay of the PKM2 promoter in SCC-11 and SCC-11M cells treated with control medium (Con) and cisplatin (CIS). SCC-11 cells were transfected with the scrambled and SREBF1 siRNA and analyzed for the PKM2 promoter-driven luciferase activity presented as relative units (RU). The values from SCC-11 cells treated with control medium designated as 1. All experiments were performed in triplicates. p < 0.05.
We finally showed that the mRNA/protein expression for carnitine palmitoyltransferase 2 (CPT2), which catalyzes the reaction (Acetyl-Carnitine to Acetyl-CoA + Carnitinei, Fig. 4A) dramatically increased by cisplatin in SCC-11 cells but not in SCC-11M cells (Fig. 4B). We then showed the increasing binding of p-ΔNp63α and SREBF1 to the specific region (while no binding was found to the non-specific region) of the CPT2 promoter in SCC-11 cells after cisplatin treatment, but not in SCC-11M cells (Fig. 4C). We also showed that while the scrambled siRNA had no effect on p-ΔNp63α binding (enrichment) to the CPT2 promoter, SREBF1 siRNA greatly inhibited such binding in SCC-11 cells under cisplatin treatment (Fig. 4D). We next demonstrated an accompanying effect of p-ΔNp63α and SREBF1 on the CPT2 promoter-driven luciferase reporter activity in SCC-11 cells upon cisplatin exposure (Fig. 4E).
Non-p-ΔNp63α induces expression of specific metabolic genes in SCC-11M cells upon cisplatin exposure
To further examine the selected gene targets upregulated by non-p-ΔNp63α in response of SCC-11M cells to cisplatin treatment, we have undertaken the study of the following genes expression and regulation: CAD, G6PD, PFKFB3 and FASN (Figs. 5–8).

Figure 5. Non-p-ΔNp63α induces the expression of CAD in SCC-11M cells upon cisplatin exposure. (A) Metabolite reaction of carbamoyl-phosphate synthetase 2 (CAD). Grey arrow indicates the decrease in SCC-11 cells, while black arrow indicates the increase in SCC-11M cells. (B, upper panel) qPCR expression analysis of CAD mRNA obtained from SCC-11 and SCC-11M cells treated with control medium (Con) and 10 μg/ml cisplatin (CIS). The values from SCC-11 cells treated with control medium designated as 1. The qPCR values presented as relative units (RU). (B, lower panel) Immunoblot analysis of CAD in SCC-11 and SCC-11M cells treated with control medium (Con) or cisplatin (CIS). Loading was normalized by β-actin expression. (C) ChIP assay of the binding of p-ΔNp63α and E2F1 to the CAD promoter (enrichment) in SCC-11M cells exposed to control medium (Con) and cisplatin (CIS). (D) Quantitative analysis of enrichment (ChIP/Input ratio) of the CAD promoter with the p-ΔNp63α and E2F1 transcription factors in SCC-11M cells treated with cisplatin. ChiP/Input ratio values presented as relative units (RU). SCC-11M cells were transfected with the scrambled and E2F1 siRNA and analyzed for ChIP/Input enrichment. (E) Luciferase reporter assay of the CAD promoter in SCC-11 and SCC-11M cells treated with control medium (Con) and cisplatin (CIS). SCC-11M cells were transfected with the scrambled and E2F1 siRNA and analyzed for the CAD promoter-driven luciferase activity presented as relative units (RU). The values from SCC-11 cells treated with control medium designated as 1. All experiments were performed in triplicates. p < 0.05.

Figure 8. Non-p-ΔNp63α induces the expression of FASN in SCC-11M cells upon cisplatin exposure. (A) Metabolite reaction of fatty acid synthase (FASN). Grey arrow indicates the decrease in SCC-11 cells, while black arrow indicates the increase in SCC-11M cells. (B, upper panel) qPCR expression analysis of FASN mRNA obtained from SCC-11 and SCC-11M cells treated with control medium (Con) and 10 μg/ml cisplatin (CIS). The values from SCC-11 cells treated with control medium designated as 1. The qPCR values presented as relative units (RU). (B, lower panel) Immunoblot analysis of FASN in SCC-11 and SCC-11M cells treated with control medium (Con) or cisplatin (CIS). Loading was normalized by β-actin expression. (C) ChIP assay of the binding of p-ΔNp63α and E2F1 to the FASN promoter (enrichment) in SCC-11M cells exposed to control medium (Con) and cisplatin (CIS). (D) Quantitative analysis of enrichment (ChIP/Input ratio) of the FASN promoter with the p-ΔNp63α and E2F1 transcription factors in SCC-11M cells treated with cisplatin. ChiP/Input ratio values presented as relative units (RU). SCC-11M cells were transfected with the scrambled and E2F1 siRNA and analyzed for ChIP/Input enrichment. (E) Luciferase reporter assay of the FASN promoter in SCC-11 and SCC-11M cells treated with control medium (Con) and cisplatin (CIS). SCC-11M cells were transfected with the scrambled and E2F1 siRNA and analyzed for the FASN promoter-driven luciferase activity presented as relative units (RU). The values from SCC-11 cells treated with control medium designated as 1. All experiments were performed in triplicates. p < 0.05.
First, we found that the mRNA/protein expression for carbamoyl-phosphate synthetase 2 (CAD), which catalyzes the reaction (ATP + HCO3 + L-glutamine + H2O to ADP + Pi + L-glutamate + Carbamoyl-phosphate, Fig. 5A), was significantly increased by cisplatin in SCC-11M cells, while it is decreased in SCC-11 cells (Fig. 5B). We then showed that the increasing binding of non-p-ΔNp63α and E2F1 to the specific region (while no binding was found to the non-specific region) of the CAD promoter in SCC-11M cells after cisplatin treatment, but not in SCC-11 cells (Fig. 5C). We also showed that while the scrambled siRNA had no effect on non-p-ΔNp63α binding (enrichment) to the CAD promoter, E2F1 siRNA greatly inhibited such binding in SCC-11M cells under cisplatin treatment (Fig. 5D). We next showed a cumulative effect of non-p-ΔNp63α and E2F1 on the CAD promoter-driven luciferase reporter activity in SCC-11M cells upon cisplatin exposure (Fig. 5E).
Second, we found that the mRNA/protein expression for glucose-6-phosphate dehydrogenase (G6PD), which catalyzes the reaction (glucose-6-phosphate to 6-phospho-δ-lactone, Fig. 6A), was significantly increased by cisplatin in SCC-11M cells, while it is decreased in SCC-11 cells (Fig. 6B). We then showed that the increasing binding of non-p-ΔNp63α and E2F1 to the specific region (while no binding was found to the non-specific region) of the G6PD promoter in SCC-11M cells after cisplatin treatment, but not in SCC-11 cells (Fig. 6C). We also showed that while the scrambled siRNA had no effect on non-p-ΔNp63α binding (enrichment) to the G6PD promoter, E2F1 siRNA greatly inhibited such binding in SCC-11M cells under cisplatin treatment (Fig. 6D). We next demonstrated a complementary effect of non-p-ΔNp63α and E2F1 on the G6PD promoter-driven luciferase reporter activity in SCC-11M cells upon cisplatin exposure (Fig. 6E).

Figure 6. Non-p-ΔNp63α induces the expression of G6PD in SCC-11M cells upon cisplatin exposure. (A) Metabolite reaction of glucose-6-phosphate dehydrogenase (G6PD). Grey arrow indicates the decrease in SCC-11 cells, while black arrow indicates the increase in SCC-11M cells. (B, upper panel) qPCR expression analysis of G6PD mRNA obtained from SCC-11 and SCC-11M cells treated with control medium (Con) and 10μg/ml cisplatin (CIS). The values from SCC-11 cells treated with control medium designated as 1. The qPCR values presented as relative units (RU). (B, lower panel) Immunoblot analysis of G6PD in SCC-11 and SCC-11M cells treated with control medium (Con) or cisplatin (CIS). Loading was normalized by β-actin expression. (C) ChIP assay of the binding of p-ΔNp63α and E2F1 to the G6PD promoter (enrichment) in SCC-11M cells exposed to control medium (Con) and cisplatin (CIS). (D) Quantitative analysis of enrichment (ChIP/Input ratio) of the G6PD promoter with the p-ΔNp63α and E2F1 transcription factors in SCC-11M cells treated with cisplatin. ChiP/Input ratio values presented as relative units (RU). SCC-11M cells were transfected with the scrambled and E2F1 siRNA and analyzed for ChIP/Input enrichment. (E) Luciferase reporter assay of the G6PD promoter in SCC-11 and SCC-11M cells treated with control medium (Con) and cisplatin (CIS). SCC-11M cells were transfected with the scrambled and E2F1 siRNA and analyzed for the G6PD promoter-driven luciferase activity presented as relative units (RU). The values from SCC-11 cells treated with control medium designated as 1. All experiments were performed in triplicates. p < 0.05.
Third, we found that the mRNA/protein expression for fructose-2, 6-biphosphatase-3 (PFKFB3), which catalyzes the reaction (β-D-fructose-2, 6-biphosphate to D-Fructose-6-phosphate, Fig. 7A), was significantly increased by cisplatin in SCC-11M cells, while it is decreased in SCC-11 cells (Fig. 7B). We then showed increasing binding of non-p-ΔNp63α and E2F1 to the specific region (while no binding was found to the non-specific region) of the PFKFB3 promoter in SCC-11M cells after cisplatin treatment, but not in SCC-11 cells (Fig. 7C). We also showed that while the scrambled siRNA had no effect on non-p-ΔNp63α binding (enrichment) to the PFKFB3 promoter, E2F1 siRNA greatly inhibited such binding in SCC-11M cells under cisplatin treatment (Fig. 7D). We next demonstrated a cumulative effect of non-p-ΔNp63α and E2F1 on the PFKFB3 promoter-driven luciferase activity in SCC-11M cells upon cisplatin exposure (Fig. 7E).

Figure 7. Non-p-ΔNp63α induces the expression of PFKFB3 in SCC-11M cells upon cisplatin exposure. (A) Metabolite reaction of fructose-2, 6-biphosphatase (PFKFB3). Grey arrow indicates the decrease in SCC-11 cells, while black arrow indicates the increase in SCC-11M cells. (B, upper panel) qPCR expression analysis of PFKFB3 mRNA obtained from SCC-11 and SCC-11M cells treated with control medium (Con) and 10 μg/ml cisplatin (CIS). The values from SCC-11 cells treated with control medium designated as 1. The qPCR values presented as relative units (RU). (B, lower panel) Immunoblot analysis of PFKFB3 in SCC-11 and SCC-11M cells treated with control medium (Con) or cisplatin (CIS). Loading was normalized by β-actin expression. (C) ChIP assay of the binding of p-ΔNp63α and E2F1 to the PFKFB3 promoter (enrichment) in SCC-11M cells exposed to control medium (Con) and cisplatin (CIS). (D) Quantitative analysis of enrichment (ChIP/Input ratio) of the PFKFB3 promoter with the p-ΔNp63α and E2F1 transcription factors in SCC-11M cells treated with cisplatin. ChiP/Input ratio values presented as relative units (RU). SCC-11M cells were transfected with the scrambled and E2F1 siRNA and analyzed for ChIP/Input enrichment. (E) Luciferase reporter assay of the PFKFB3 promoter in SCC-11 and SCC-11M cells treated with control medium (Con) and cisplatin (CIS). SCC-11M cells were transfected with the scrambled and E2F1 siRNA and analyzed for the PFKFB3 promoter-driven luciferase activity presented as relative units (RU). The values from SCC-11 cells treated with control medium designated as 1. All experiments were performed in triplicates. p < 0.05.
Forth, we found that the mRNA/protein expression for fatty acid synthase (FASN), which catalyzes the reaction (acetyl-CoA + malonyl-CoA + NADPH + H+ to palmitate + CO2 + NADP++ CoASH + H2O, Fig. 8A), was significantly increased by cisplatin in SCC-11M cells, while it is decreased in SCC-11 cells (Fig. 8B). We then showed that the increasing binding of non-p-ΔNp63α and E2F1 to the specific region (while no binding was found to the non-specific region) of the FASN promoter in SCC-11M cells after cisplatin treatment, but not in SCC-11 cells (Fig. 8C). We also showed that while the scrambled siRNA had no effect on non-p-ΔNp63α binding (enrichment) to the FASN promoter, E2F1 siRNA greatly inhibited such binding in SCC-11M cells under cisplatin treatment (Fig. 8D). We next demonstrated an additive effect of non-p-ΔNp63α and E2F1 on the FASN promoter-driven luciferase activity in SCC-11M cells upon cisplatin exposure (Fig. 8E).
Silencing of specific ΔNp63α gene targets attenuates the cisplatin sensitivity of SCC-11 cells and cisplatin resistance of SCC-11M cells
Our previous reports showed that alteration of the critical step of ATM-dependent phosphorylation of ΔNp63α triggers a distinct gene expression program leading to a pro-apoptotic cell response in the case of p-ΔNp63α in SCC-11 cells sensitive to cisplatin-induced apoptosis, while non-p-ΔNp63α expression in SCC-11M cells renders them more resistant to cisplatin-induced cell death. In this report, we showed the specific metabolic genes upregulated by either p-ΔNp63α or non-p-ΔNp63α implicated in cell response to cisplatin treatment. We thus further examine whether the siRNA silencing of these metabolic proteins could potentially affect the response of SCC-11cells or SCC-11M cells to cisplatin exposure. We transfected SCC-11 cells with the scrambled siRNA and ARG1 siRNA or CPT2 siRNA for 32 h to inhibit expression of ARG1 or CPT2, while SCC-11M cells were transfected with the scrambled siRNA and G6PD siRNA or FASN siRNA to inhibit expression of G6PD or FASN. Resulting cells were treated with control medium (Con) and 10 μg/ml cisplatin (CIS) for 1–5 d and then tested cell viability under cisplatin pressure using MTT assay (Fig. 9). SCC-11 cells showed a greater viability under cisplatin treatment, when they expressed less ARG1 or CPT2 inhibited by the corresponding siRNAs (Fig. 9A). However, SCC-11M cells transfected with G6PD or FASN siRNAs showed a lesser chance for survival under cisplatin than SCC-11M cells transfected with the scrambled siRNA (Fig. 9B).

Figure 9. siRNA-dependent attenuation of sensitivity of SCC-11 cells and resistance of SCC-11M cells to cisplatin-induced cell death. SCC-11 cells were transfected with the scrambled and ARG1 siRNA (A), or the scrambled and CPT2 siRNA (B), while SCC-11M cells were transfected with the scrambled or G6PD siRNA (C) or the scrambled and FASN siRNA (D) for 32 h. Resulting cells were exposed to control medium (Con) or 10 μg/ml cisplatin (CIS) for 5 d, as indicated. Cell viability (MTT) assay was repeated three times. The bars are the mean ± SD of triplicate; p < 0.05.
Differential protein interactions of ΔNp63α with SREBF1 and E2F1 n SCC-11and SCC-11M cells upon cisplatin exposure
Our previous global quantitative study (iTRAQ) showed that cisplatin induced the p-ΔNp63α protein interactions enriched with SREBF1 in SCC-11 cells and the non-p-ΔNp63α interactions enriched with E2F1 in SCC-11M cells.59 Using immunoprecipitation analysis, we showed here that p-ΔNp63α, indeed, associated with SREBF1 and NF-YA in nuclear lysates of SCC-11 cells upon cisplatin exposure, while it failed to do so in SCC-11M cells (Fig. 10A and B) suggesting importance of the phosphorylated state for ΔNp63α to bind SREBF1 and NF-YA proteins. However, cisplatin treatment greatly enhanced the non-p-ΔNp63α nuclear protein complexes with TP53, NF-YA and E2F1 in SCC-11M cells (Fig. 10A and C).
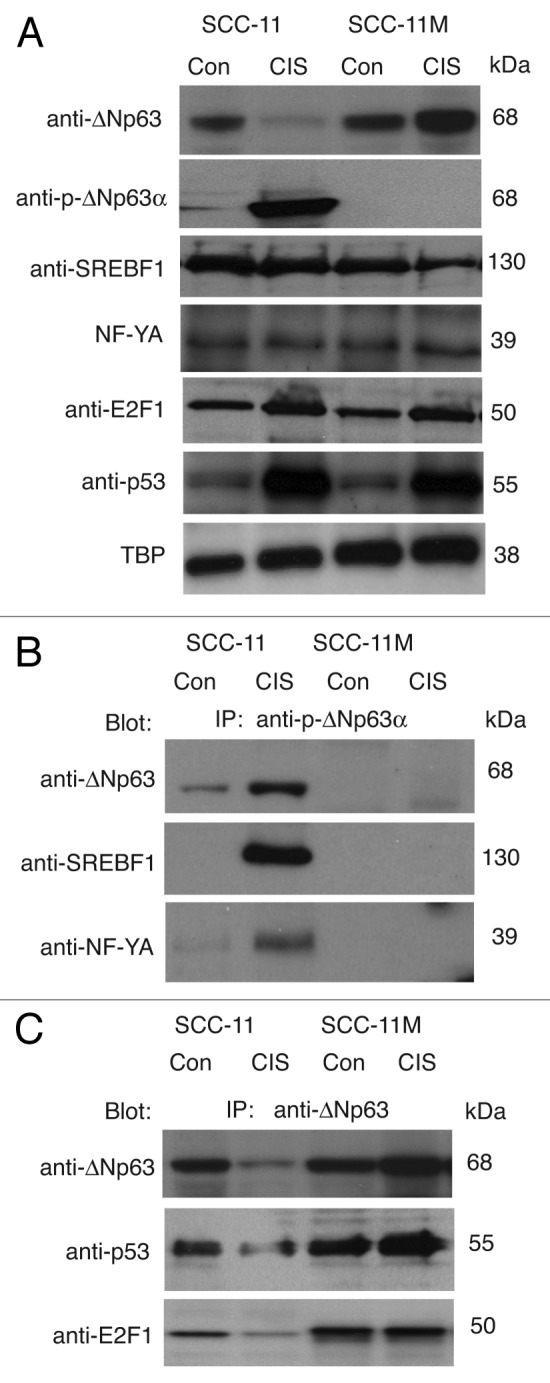
Figure 10. P-ΔNp63α/SREBF1 and non-p-ΔNp63α/E2F1 protein complexes formed in SCC cells upon cisplatin exposure. (A) Immunoblotting analysis of SCC-11 and SCC-11M cells exposed to control medium (Con) and 10 μg/ml cisplatin (CIS). Nuclear lysates were probed for expression of specific transcription factors with indicated antibodies. Levels of TBP served as loading controls. (B) Immunoprecipitation (IP) with anti-p-ΔNp63α antibody followed by immunoblotting with anti-SREBF1 and NF-Y antibodies. (C) IP with anti-ΔNp63 antibody followed by immunoblotting with anti-E2F1 and anti-p53 antibodies.
Discussion
Platinum-based drugs (e.g., cisplatin) are often used to treat solid malignancies, including testicular, ovarian, head and neck, colorectal, bladder and lung cancers. Prolonged cisplatin treatment often results in the development of chemoresistance. Since cisplatin constitutes the major therapeutic option, the development of chemosensitization strategies constitutes a goal with important clinical implications.4
Among molecular mechanisms implicated in chemoresistance to anticancer drugs are DNA and histone methylation processes that control gene expression, RNA processing, microRNA functions and multiple protein-protein interactions that also involve regulation of many metabolic pathways.4,6,7,10,39-45,49,50,52,59 Cisplatin exerts anticancer effects mainly via DNA damage response and the induction of mitochondrial apoptosis. Coordinated activation of the pro-apoptotic Bcl-2 family and the caspase family during apoptosis often leads to permeabilization of the mitochondrial outer membrane and release of multiple enzymes functioning as regulators of energy production and metabolism.60 A recent study shows that since acetyl-Coenzyme A (CoA) is a key cofactor for the protein N-α-acetyltransferase complex, protein acetylation is under metabolic regulation.61 The protein N-α-acetylation is regulated by Bcl-xL, a major anti-apoptotic mitochondrial protein, and thus supports a mechanism by which metabolism can regulate the activation of apoptotic program contributing to tumor cell apoptotic sensitivity or resistance.61 Interestingly, acetyl-L-carnitine regulates the acetyl-CoA levels that provide a source of acetyl groups for metabolic and acetylation-regulated processes.62,63 Acetyl-L-carnitine/cisplatin combination caused the activation of p53, associated with protein acetylation and induction of p53 target genes, leading to apoptosis and cell cycle arrest.62-64
In this paper, we found that the SCC cells sensitive (SCC-11) and resistant (SCC-11M) to cisplatin treatment showed differential increase or decrease in metabolite levels involved in amino acid metabolism, carbohydrate metabolism, lipid metabolism, and nucleotide metabolism. We found apparent increases in oxidative stress, inflammation, membrane remodeling, glucose utilization and mitochondrial energy production, which are likely to support a more anti-apoptotic phenotype for SCC-11M cells (expressing non-p-ΔNp63α) than SCC-11 cells (expressing p-ΔNp63α) upon cisplatin exposure. We found that cisplatin treatment upregulated a few amino acids, (dimethylarginine, glutamine, leucine), urea and dipeptides, while it downregulated several amino acids (citrulline, cysteine, glutamate, 5-methylthioadenosine, N-acetylaspartate, ornithine, reduced glutathione) in SCC-11 cells compared with SCC-11M cells. Intriguingly, the metabolites involved in the urea cycle were affected the most. We further found that a few carbohydrate/energy metabolites (e.g., fructose-2, 6-biphosphate, glucose-2, 6-biphosphate and phosphate) and lipid metabolites (caprylate, carnitine, cholesterol, lactate, oleates, palmitate, palmitoylates and prostaglandin E2) were upregulated in SCC-11 cells upon cisplatin exposure, while other components of glycolysis and penthose phosphate pathways and many arachidonates were downregulated. We then found that while a few members of nucleotide biosynthesis or degradation (e.g., xanthine, 2’-deoxyinosine) were upregulated in SCC-11 cells, the majority of them were downregulated by cisplatin treatment.
Upon treatment with cisplatin, cellular metabolites involved in energy production, oxidative stress, membrane turnover and, ultimately, in cell death were differentially affected in SCC-11/SCC-11M cells (e.g., increase of CoA and decrease of 3′-dephospho-CoA, nicotinate, nicotinamide, nicotinamide mono- and dinucleotides), suggesting that phosphorylation of the ΔNp63α transcription factor may be necessary for mediating several cytotoxic metabolic effects associated with cisplatin treatment. Our findings support the idea that phosphorylation of ΔNp63α is a key regulator of metabolic response of SCC cells to cisplatin exposure. Metabolic profiling studies involving genetic manipulation of other proteins associated with ΔNp63α signaling, including the Ataxia-Telangiectasia mutated (ATM) kinase that is responsible for phosphorylation of ΔNp63α, may further clarify the role of this key transcription factor in modulating sensitivity to cisplatin therapy.28
Since cisplatin was shown to induce the ATM-dependent phosphorylation of ΔNp63α in SCC-11 cells, while SCC-11M cells were designed to abolish this process, we suggested that in contrast to non-p-ΔNp63α (ΔNp63α-S385G), the p-ΔNp63α transcription factor plays a critical role in regulation of tested metabolic pathways through a regulation of specific metabolic gene promoters.
Our global expression chip, ChiP-on-chip and methylation chip array analyses narrowed down a list of potential target gene promoters that are likely to be affected by p-ΔNp63α or non-p-ΔNp63α in SCC cells treated with cisplatin. We thus found that several metabolic genes (ARG1, GADPH, PKM2 and CPT2) were upregulated in SCC-11 cells (expressing p-ΔNp63α) upon cisplatin exposure through a direct binding and functional activation of the corresponding promoters shown by qPCR, ChIP-PCR and promoter-reporter luciferase assays. All of the tested promoters were negatively affected by SREBF1 silencing, suggesting the critical involvement of apoptosis-inducible SREBF1 in transcriptional control of these genes.55 On the other hand, a few metabolic genes (CAD, G6PD, PFKFB3, and FASN) were upregulated in SCC-11M cells (expressing non-p-ΔNp63α) upon cisplatin exposure tested by qPCR, ChIP-PCR and promoter-reporter luciferase assays. And their activities were attenuated by E2F1 silencing, suggesting the role for these promoters in E2F1-mediated induction of cell cycle, cell proliferation and cell survival.56
To ascertain the effect of tested p-ΔNp63α and non-p-ΔNp63α targets on a survival of SCC-11 cells and SCC-11M cells, we further examined whether silencing of the specific metabolic mRNA would lead to alterations in cell viability upon cisplatin treatment.
Previous studies showed that l-arginine deprivation enhances apoptosis.65 Mitochondrial membrane permeabilization is a rate-limiting step of cell death.61 Proteomics/mass spectrometry analysis of proteins released from purified mitochondria shows a total of 79 known proteins.66 Among known proteins, several may have indirect or direct pro-apoptotic properties (e.g., arginase 1 (enzyme that turns arginine into ornithine and urea).66 Similarly, ARG1 mRNA and protein were upregulated during cisplatin-induced hepatotoxicity shown by an integrative expression array/mass spectrometry analysis.67 While the cytosolic CPT1 produces acetyl-CoA carnitine (from acetyl-CoA and L-carnitine), the mitochondrial inner membranal CPT2 degrades acetyl-CoA carnitine back to acetyl-CoA and L-carnitine.20,62,63 The fatty acid palmitate can induce apoptosis, which could be inhibited by overexpression of anti-apoptotic Bcl-2 and exacerbated by carnitine suggesting the pro-apoptotic role for CPT2 (refs. 60 and 61).
On the other hand, the doxorubicin-resistant cancer cells exhibited a decreased doxorubicin accumulation, increased reduced glutathione, increased activity of the pentose phosphate pathway and G6PD.68 G6PD is the principal source of the NADPH, which is required by many enzymes of the antioxidant pathway.69,70 Inactivation of both G6PD and FASN with siRNA or inhibitors led to increased reactive oxygen species and apoptosis and decreased proliferation.69-72 FASN forced expression protected cells from apoptosis induced by camptothecin and caused drug resistance, while reducing the FASN expression increased drug sensitivity of cancer cells.71-73 Both G6PD and FASN are direct targets of TP53 family members, including TP63 and TP73.18,30,64,74,75 Knockdown of ΔNp63 decreased cell viability by inducing apoptosis via reduction of FASN expression.75 The FASN forced expression partially rescues cells from cell death induced by ΔNp63 silencing, while inhibition of FASN triggers apoptosis in human cancer cells.74,75
We showed that siRNA silencing of ARG1 and CPT2 led to a partial reduction of the cell death phenotype of cisplatin-sensitive SCC-11 cells, while siRNA knockdown of G6PD and FASN partially increased the sensitivity of resistant SCC-11M cells to cisplatin-induced cell death.
We further evaluated the role for p-ΔNp63α and SREBF1 in transcriptional regulation of gene expression in SCC-11 cells or non-p-ΔNp63α, TP53 and E2F1 in transcriptional regulation of gene expression in SCC-11M cells and found that the cisplatin exposure induced protein complexes between p-ΔNp63α, SREBF1 and NF-YA proteins in nuclear lysates of SCC-11 cells. However, cisplatin treatment greatly enhanced the non-p-ΔNp63α nuclear protein complexes with TP53, NF-YA and E2F1 in SCC-11M cells.
SREBF-1 and -2 were found to serve as regulatory hubs that control lipid metabolism, cell growth or cell death.76,77 Casp2 and Casp7 were found to be the SREBF-responsive gene, raising the possibility for caspase cascade to participate in the control of cholesterol/ triacylglycerol levels.78,79 Activated caspases were shown to release SREBF from the endoplasmic reticulum membrane, leading to induction of sterol-regulated genes at an early stage in the apoptotic cascade, suggesting that they may play a role in the proper execution of the apoptotic program.80
Specific cellular metabolic phenotypes were robustly associated with platinum chemosensitivity, while several of the metabolic perturbations have been associated with the acquisition of drug resistance (e.g., “Warburg effect,” an increase in glucose uptake and glycolysis to lactate even in normal oxygen conditions).81-83 However, higher rates of glutaminolysis, fatty acid and lipid metabolism and nucleotide synthesis also correlate with degree of platinum sensitivity/resistance.81
As previously reported by others, the higher levels of citrate, phosphoenolpyruvate, glutamate and 2-oxoglutarate shown in platinum-sensitive tumor cells are consistent with decreased Krebs cycle rate and increased diversion of glycolytic intermediates into anabolic pentose phosphate pathway, which, in turn, feeds nucleotide synthesis.81 To replace Krebs cycle intermediates and NADPH, tumor cells increase the uptake of glutamine and its conversion to oxaloacetate via glutamate and 2-oxoglutarate.81 Coordination between gene transcript and metabolite levels was observed in nucleotide metabolism, revealing a robust association between increased nucleotide synthesis and tumor cell platinum chemosensitivity.81
Increased expression of nucleotide pathway enzymes (e.g., uracil phosphoribosyl transferase, hypoxanthine-guanine phosphoribosyl transferase) in platinum-sensitive cells was accompanied by decrease in purine and pyrimidines (guanine, guanosine, hypoxanthine, inosine, uracil and uridine) and increase in cytidine monophosphate.81 Adding the extracellular purines to cells abolished cisplatin cytotoxicity, suggesting that the metabolome may have a causal influence on platinum sensitivity.81 Apolipoprotein E and low-density lipoprotein receptor as well as methionine derivatives may contribute to platinum resistance, while β-alanine derivatives were associated with platinum sensitivity.80 Since chemotherapeutic sensitivity is in part determined by the metabolic phenotype that suggests metabolic enzymes may be potential targets for both drug naive and chemoresistant patients.45,62,65,81-84
Materials and Methods
Antibodies
We used a rabbit polyclonal antibody Ab-1 directed against human ΔNp63 (EMD Chemicals), rabbit anti-human p63 (p73L) monoclonal antibody (clone Y289, #NB110–57309) obtained from Novus Biologicals. A mouse monoclonal antibody to p63 (4A4, sc-8431, Santa Cruz Biotechnology) and a monoclonal antibody against human β-actin (Sigma) were also used. Anti-wild type p53 (clone DO-1, #554293), anti-sterol responsive element binding factor (SREBF1, clone IgG-2A4, #557036) and anti-E2F1 (#554213, clone KH95/E2F) antibodies were obtained from BD PharMingen. We also used the rabbit polyclonal antibody against human NF-YA (NBP1–19146, Novus Biologicals) and a mouse monoclonal antibody against TATA-binding protein (TBP, 1TBP18, ab818, Abcam). Custom rabbit polyclonal antibody against phosphorylated peptide encompassing the ΔNp63α protein sequence (ATM motif, NKLPSV-pS-QLINPQQ, residues 379–392) was prepared and purified against the phosphorylated peptide vs. non-phosphorylated peptide with the aid of Sigma Genosys.39 We then used the following antibodies: anti-arginase 1 (ARG1, sc-365547) from Santa Cruz Biotechnology; anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH, NB300–322), anti-pyruvate kinase M2 (PKM2, AT4M3, NBP1–48531); anti-carnitine palmitoyltransfecrase 2 (CPT2, H00001376-B01P), anti-carbamoyl-phosphate synthetase 2 (CAD, NB100–61614), anti-glucoso-6-phosphate dehydrogenase (G6PD, NB100–236), anti-fructose-2, 6-biphosphatase (PFKFB3, H00005209-M08) and anti-fatty acid synthase (FASN, NBP1–84733), all from Novus Biologicals.
Cells and reagents
The squamous cell carcinoma (SCC) cell line JHU-011 (formerly known as JHU-029, expressing wt-TP53, wt-TP63 is amplified and ΔNp63α is overexpressed) was isolated from primary tissue at the Department of Otolaryngology/Head and Neck Surgery (JHMI) then obtained from the Head and Neck Cancer Research Division Tissue Bank, as described elsewhere.44,59 The original cell line was authenticated by a short tandem repeat profiling analysis using the AmpFISTR Identifiler PCR Amplification Lit (Applied Biosystems) with the following markers: amelogenin, CSF1PO, D12S317, D16S539, D18S51, D19S433, D21S11, D2S1338, D3S1358, D5S818, D7S820, D8S1179, FGA, THO1, TPOX and VWA at the JHMI Fragment Analysis Facility. The stable SCC cell lines expressing ΔNp63α-wt or ΔNp63α-S385G were generated using Flp-In technology.39,40 Cells were maintained in RPMI medium 1640 and 10% fetal bovine serum and incubated with control medium without cis-diamminedichloroplatinum (cisplatin, CIS, Sigma) or medium with10 μg/ml cisplatin (Sigma) for the indicated time periods. Cells were lysed with 50mM Tris, pH7.5, 100mM NaCl, 2 mM EDTA, 0.5% Triton X-100, 0.5% Brij-50, 1 mM PMSF, 0.5 mM NaF, 0.1 mM Na3VO4, 2X complete protease inhibitor cocktail, sonicated for 10 sec intervals and spun for 30 min at 15,000 × g. Total and nuclear supernatants were analyzed by immunoblotting, and the levels of tested proteins were normalized against β-actin or TATA-binding protein (TBP) levels, respectively.
Isolation of nuclear fractions
1–2 × 106 cells were resuspended in hypotonic lysis buffer (10 mM HEPES pH 7.9, 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA) added with protease inhibitors (Sigma). After resuspension, 0.6% Triton X-100 (final concentration) was added, and the nuclei were pelleted by centrifugation at 2,500–3,000 × g for 10 min at 4°C. Nuclear pellets were resuspended in the extract buffer (20 mM HEPES pH 7.9, 25% glycerol, 0.4 M NaCl, 0.1 mM EDTA, 0.1 mM EGTA), rocked for 15 min at 4°C and nuclear lysate (supernatant) was recovered by centrifugation at 10,000 × g for 5 min at 4°C.
qPCR assay
We used the High Capacity cDNA Reverse Transcription Kit (#4374966, Applied Biosystems) to produce single-stranded cDNA probes. Next, we performed a qPCR using the TaqMan® PreAmp Master Mix Kit (includes Gene Expression Master Mix) (#4387406, Applied Biosystems). The following mRNA assays were used: ARG1 (Hs00968979_m1), GAPDH (Hs02758991_g1), PKM2 (Hs00761782_s1), CPT2 (Hs00988962_m1), CAD (Hs00983188_m1), G6PD (Hs00166169_m1), PFKFB3 (Hs00998700_m1) and FASN (Hs01005622_m1). The reaction (20 μl) was performed at 50°C for 2 min, 95°C for 10 min, 40 cycles of 95°C for 15 sec and 60°C for 1 min. The independent biological experiments were performed twice and in triplicate. Each RNA sample was amplified in triplicate. Expression was normalized to the 18S RNA TaqMan probe (4319413E, Applied Biosystems) and expression levels were be determined as the average Ct of this control. This averaged value was then used to normalize the sample’s Ct. The average mRNA expression was be determined using the Mann-Whitney U test. Values obtained from the control samples (untreated SCC-11 cells) were designated as 1 (refs. 42 and 51).
Chromatin immunoprecipitation (ChIP)
Five × 106 cell equivalents of chromatin (2–2.5 kbp in size) were immunoprecipitated (IP) with 10 μg of anti-ΔNp63 antibody (or anti-p-ΔNp63α antibody as described elsewhere).42,51 After reversal of formaldehyde cross-linking, RNA-ase A and proteinase K treatments, IP-enriched DNAs were used for PCR amplification. PCR consisted of 40 cycles of 94°C for 30 s, 60°C for 30 s, and 72°C for 30 s using Taq DNA polymerase (Invitrogen). Although the tested promoters contain multiple potential TP63 binding sites, the regions for PCR were selected based on the efficiency of amplification, choosing the highest PCR outcome. The specific regions (containing tested binding sites defined by the web browser: http://www.cbrc.jp/research/db/TFSEARCH) and non-specific regions (containing no tested binding sites) of selected gene promoters (ARG1, GAPDH, PKM2, CPT2, CAD, G6PD, PFKFB3 and FASN) were amplified for ChIP-PCR assay (primers are underlined in Figs. S10–17). To quantify the binding of the specific transcription factors (e.g., p-ΔNp63α, non-p-ΔNp63α, SREBF1 and E2F1) to the selected gene promoter sequences (enrichment), we used qPCR. ChIP-PCR values were obtained from the ChIP, and input samples and were normalized to GAPDH PCR values. For each transcription factor, values obtained from the Input samples were designated as 1. ChIP/input ratio was plotted from all biological experiments using the Microsoft Excel software. Experiments were performed in triplicate.
Luciferase reporter assay
We used the following promoter-reporter plasmids for: ARG1 (S711640), GAPDH (S721624), PKM2 (S721648), CPT2 (S714498), FASN (S721853), PFKFB3 (S722433), G6PD (S720234), CAD (S704691) purchased from SwitchGear Genomics. For the promoter-mediated luciferase activity assay, a total of 5 × 104 cells/well in a 24-well plate were transfected with the control (empty) pLightSwitch_Prom vector [#S707592, a fully optimized reporter system that includes an improved luminescent reporter gene (RenSP)] using Fugene HD reagent (Roche) as previously described.51 The LightSwitch Luciferase Assay Reagent (SwitchGear Genomics) enables the monitoring of luciferase reporter signal. At 36 h, cells were treated without or with 10 μg/ml cisplatin for an additional 12 h. The RenSP Renilla luciferase activity was measured at 480 nm using a luminometer.
Transfection
Cells were transiently transfected with the scrambled siRNA and the following siRNAs (SREBF1 siRNA (h), sc-36557; E2F-1 siRNA (h), sc-29297; arginase I siRNA (h), sc-45206; glucose-6-phosphate dehydrogenase siRNA (h): sc-60667; and fatty acid synthase siRNA (h): sc-43758 obtained from Santa Cruz Biotechnology. Human glyceraldehyde-3-phosphate dehydrogenase siRNA (H00002597-R01), carnitine palmitoyltarnsferase-2 siRNA (H00001376-R01), and pyruvate kinase M2 siRNA (H00005315-R09) were purchased from Novus Biologicals. Transfection with 20 nM of siRNA was carried-out using Lipofectamine SiRNAMAX (Invitrogen) for 32 h post-transfection.52 Resulting cells were treated with control medium or 10 μg/ml cisplatin for 16 h.
Cell viability assay
104 cells/well in 96-well plates were incubated in serum-free medium with 5 μg/ml of the 3-(4,5-dimethyl thiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT assay, American Tissue Culture Collection) in the dark for 4 h at 37°C. Cells were lysed and incubated for 2 h at 37°C, and the measurements (A570 nm to A650 nm) were obtained on a Spectra Max 250 plate reader (Molecular Devices) as described elsewhere.52 Each assay was repeated three times in triplicate. Diagrams indicated the extent of cell viability expressed as a portion of control cells without cisplatin represented as 1.
Statistical analysis
Results were expressed as means ± SD from three independent experiments in triplicate. Differences in variables between experimental and control group were assessed by using the Student’s t test. Statistically significant difference was accepted at p < 0.05.
Concluding remarks
In the series of reports, we showed the critical importance of cisplatin-induced ATM-mediated phoshorylation of ΔNp63α, member of p53 family, to function as a transcriptional regulator of gene expression for mRNAs and microRNAs. We established that the knock-in mutation S385G in ΔNp63α that altered the ΔNp63α ability to be phosphorylated by ATM kinase ultimately affects cellular response of SCC cells to cisplatin exposure rendering cells more resistant to cell death consequences. We found that p-ΔNp63α forms complexes with a number of proteins that involved in cell death response through regulation of cell cycle arrest, apoptosis, autophagy, RNA splicing, chromatin modifications. Here, we observed that the p-ΔNp63α-dependent regulatory mechanisms implicated in modulation of plethora of pathways, including amino acid, carbohydrate, lipid and nucleotide metabolisms, thereby affecting tumor cell response to cisplatin-induced cell death.
Understanding the multiple mechanisms by which p-ΔNp63α modulates the response of SCC cells to cisplatin, including those that affect metabolic pathways, will support a notion that the ATM-dependent ΔNp63α pathway plays a role in the resistance of tumor cells to platinum therapy. (AUTHOR: Please cite refs. 85 and 86)
Supplementary Material
Acknowledgments
This work is dedicated to the memory of the late Dr. Marina Nikolayevna Kiprianova (Institute of Experimental Medicine, Academy of Medical Sciences), whose kind friendship and unparalleled dedication to science inspired the senior author for years after her untimely death. This study was supported in part by the Flight Attendant Research Institutions grant (#082469 to E.A.R.), and by National Cancer Institute grants K01-CA164092 and U01-CA84986 (R.G-P).
We are grateful to Malcolm Jefferson (formerly of Metabolon) for his dedication and critical statistical support.
Glossary
Abbreviations:
- CIS
cisplatin
- Con
control
- p
phosphorylated
- quantitative PCR
qPCR
- SCC
squamous cell carcinoma
- TP
tumor protein
- SREBF
sterol regulatory element binding factor
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- G6PD
glucose-6-phosphate dehydrogenase
- HK2
hexokinase-2
- MDH1
malate dehydrogenase-1
- PGM
phosphoglycerate mutase
- CAD
carbamoyl phosphate synthetase-2
- OTC
ornithine carbamoyltransferase
- GLS2
glutaminase-2
- APO
apolipoprotein
- CPT
carnitine palmitoyl-transferase
- FASN
fatty acid synthase
- FDXR
ferredoxin reductase
- GAMT
guanidine-acetate methyltransferase
- ADA
adenosine deaminase
- DHODH
dihydroorotate dehydrogenase
- HPRT
hypoxanthine phosphoribosyl transferase
- RRM2
ribonucleotide reductase
- AMPK
AMP-activated protein kinase
- TA
transactivation domain
- ATM
Ataxia-Telangiectasia Mutated
- siRNA
small interference RNA
- ChIP
chromatin immunoprecipitation
- qPCR
quantitative PCR
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental material may be found here: www.landesbioscience.com/journals/cc/article/22022
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/22022
References
- 1.Helmbach H, Kern MA, Rossmann E, Renz K, Kissel C, Gschwendt B, et al. Drug resistance towards etoposide and cisplatin in human melanoma cells is associated with drug-dependent apoptosis deficiency. J Invest Dermatol. 2002;118:923–32. doi: 10.1046/j.1523-1747.2002.01786.x. [DOI] [PubMed] [Google Scholar]
- 2.Zangen R, Ratovitski EA, Sidransky D. DeltaNp63α levels correlate with clinical tumor response to cisplatin. Cell Cycle. 2005;4:1313–5. doi: 10.4161/cc.4.10.2066. [DOI] [PubMed] [Google Scholar]
- 3.Kelland L. The resurgence of platinum-based cancer chemotherapy. Nat Rev Cancer. 2007;7:573–84. doi: 10.1038/nrc2167. [DOI] [PubMed] [Google Scholar]
- 4.Galluzzi L, Senovilla L, Vitale I, Michels J, Martins I, Kepp O, et al. Molecular mechanisms of cisplatin resistance. Oncogene. 2012;31:1869–83. doi: 10.1038/onc.2011.384. [DOI] [PubMed] [Google Scholar]
- 5.Flores ER, Tsai KY, Crowley D, Sengupta S, Yang A, McKeon F, et al. Tsai K, Crowley D, Sengupta S, Yang A, McKeon F, Jacks T. P63 and p73 are required for TP53-dependent apoptosis in response to DNA damage. Nature. 2002;416:560–4. doi: 10.1038/416560a. [DOI] [PubMed] [Google Scholar]
- 6.Rocco JW, Leong CO, Kuperwasser N, DeYoung MP, Ellisen LW. p63 mediates survival in squamous cell carcinoma by suppression of p73-dependent apoptosis. Cancer Cell. 2006;9:45–56. doi: 10.1016/j.ccr.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 7.Seitz SJ, Schleithoff ES, Koch A, Schuster A, Teufel A, Staib F, et al. Chemotherapy-induced apoptosis in hepatocellular carcinoma involves the p53 family and is mediated via the extrinsic and the intrinsic pathway. Int J Cancer. 2010;126:2049–66. doi: 10.1002/ijc.24861. [DOI] [PubMed] [Google Scholar]
- 8.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–10. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 9.Collavin L, Lunardi A, Del Sal G. p53-family proteins and their regulators: hubs and spokes in tumor suppression. Cell Death Differ. 2010;17:901–11. doi: 10.1038/cdd.2010.35. [DOI] [PubMed] [Google Scholar]
- 10.Ory B, Ellisen LW. A microRNA-dependent circuit controlling p63/p73 homeostasis: p53 family cross-talk meets therapeutic opportunity. Oncotarget. 2011;2:259–64. doi: 10.18632/oncotarget.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Trinh DL, Elwi AN, Kim SW. Direct interaction between p53 and Tid1 proteins affects p53 mitochondrial localization and apoptosis. Oncotarget. 2010;1:396–404. doi: 10.18632/oncotarget.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gottlieb E, Vousden KH. p53 regulation of metabolic pathways. Cold Spring Harb Perspect Biol. 2010;2:a001040. doi: 10.1101/cshperspect.a001040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Madan E, Gogna R, Bhatt M, Pati U, Kuppusamy P, Mahdi AA. Regulation of glucose metabolism by p53: emerging new roles for the tumor suppressor. Oncotarget. 2011;2:948–57. doi: 10.18632/oncotarget.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, et al. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006;126:107–20. doi: 10.1016/j.cell.2006.05.036. [DOI] [PubMed] [Google Scholar]
- 15.Ruiz-Lozano P, Hixon ML, Wagner MW, Flores AI, Ikawa S, Baldwin AS, Jr., et al. p53 is a transcriptional activator of the muscle-specific phosphoglycerate mutase gene and contributes in vivo to the control of its cardiac expression. Cell Growth Differ. 1999;10:295–306. [PubMed] [Google Scholar]
- 16.Hu W, Zhang C, Wu R, Sun Y, Levine A, Feng Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc Natl Acad Sci USA. 2010;107:7455–60. doi: 10.1073/pnas.1001006107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Erickson JW, Cerione RA. Glutaminase: a hot spot for regulation of cancer cell metabolism? Oncotarget. 2010;1:734–40. doi: 10.18632/oncotarget.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li JN, Gorospe M, Chrest FJ, Kumaravel TS, Evans MK, Han WF, et al. Pharmacological inhibition of fatty acid synthase activity produces both cytostatic and cytotoxic effects modulated by p53. Cancer Res. 2001;61:1493–9. [PubMed] [Google Scholar]
- 19.Buzzai M, Bauer DE, Jones RG, Deberardinis RJ, Hatzivassiliou G, Elstrom RL, et al. The glucose dependence of Akt-transformed cells can be reversed by pharmacologic activation of fatty acid β-oxidation. Oncogene. 2005;24:4165–73. doi: 10.1038/sj.onc.1208622. [DOI] [PubMed] [Google Scholar]
- 20.Deberardinis RJ, Lum JJ, Thompson CB. Phosphatidylinositol 3-kinase-dependent modulation of carnitine palmitoyltransferase 1A expression regulates lipid metabolism during hematopoietic cell growth. J Biol Chem. 2006;281:37372–80. doi: 10.1074/jbc.M608372200. [DOI] [PubMed] [Google Scholar]
- 21.Nakano K, Bálint E, Ashcroft M, Vousden KH. A ribonucleotide reductase gene is a transcriptional target of p53 and p73. Oncogene. 2000;19:4283–9. doi: 10.1038/sj.onc.1203774. [DOI] [PubMed] [Google Scholar]
- 22.Mathupala SP, Heese C, Pedersen PL. Glucose catabolism in cancer cells. The type II hexokinase promoter contains functionally active response elements for the tumor suppressor p53. J Biol Chem. 1997;272:22776–80. doi: 10.1074/jbc.272.36.22776. [DOI] [PubMed] [Google Scholar]
- 23.Lee SM, Kim JH, Cho EJ, Youn HD. A nucleocytoplasmic malate dehydrogenase regulates p53 transcriptional activity in response to metabolic stress. Cell Death Differ. 2009;16:738–48. doi: 10.1038/cdd.2009.5. [DOI] [PubMed] [Google Scholar]
- 24.Suzuki S, Tanaka T, Poyurovsky MV, Nagano H, Mayama T, Ohkubo S, et al. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc Natl Acad Sci USA. 2010;107:7461–6. doi: 10.1073/pnas.1002459107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Khutornenko AA, Roudko VV, Chernyak BV, Vartapetian AB, Chumakov PM, Evstafieva AG. Pyrimidine biosynthesis links mitochondrial respiration to the p53 pathway. Proc Natl Acad Sci USA. 2010;107:12828–33. doi: 10.1073/pnas.0910885107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ashur-Fabian O, Har-Zahav A, Shaish A, Wiener Amram H, Margalit O, Weizer-Stern O, et al. apoB and apobec1, two genes key to lipid metabolism, are transcriptionally regulated by p53. Cell Cycle. 2010;9:3761–70. doi: 10.4161/cc.9.18.12993. [DOI] [PubMed] [Google Scholar]
- 27.Ide T, Chu K, Aaronson SA, Lee SW. GAMT joins the p53 network: branching into metabolism. Cell Cycle. 2010;9:1706–10. doi: 10.4161/cc.9.9.11473. [DOI] [PubMed] [Google Scholar]
- 28.Krüger A, Ralser M. ATM is a redox sensor linking genome stability and carbon metabolism. Sci Signal. 2011;4:pe17. doi: 10.1126/scisignal.2001959. [DOI] [PubMed] [Google Scholar]
- 29.Goldstein I, Ezra O, Rivlin N, Molchadsky A, Madar S, Goldfinger N, et al. p53, a novel regulator of lipid metabolism pathways. J Hepatol. 2012;56:656–62. doi: 10.1016/j.jhep.2011.08.022. [DOI] [PubMed] [Google Scholar]
- 30.Jiang P, Du W, Wang X, Mancuso A, Gao X, Wu M, et al. p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat Cell Biol. 2011;13:310–6. doi: 10.1038/ncb2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Feng Z, Zhang H, Levine AJ, Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci USA. 2005;102:8204–9. doi: 10.1073/pnas.0502857102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Okoshi R, Ozaki T, Yamamoto H, Ando K, Koida N, Ono S, et al. Activation of AMP-activated protein kinase induces p53-dependent apoptotic cell death in response to energetic stress. J Biol Chem. 2008;283:3979–87. doi: 10.1074/jbc.M705232200. [DOI] [PubMed] [Google Scholar]
- 33.Maddocks OD, Vousden KH, Vousden KH. Metabolic regulation by p53. J Mol Med (Berl) 2011;89:237–45. doi: 10.1007/s00109-011-0735-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–33. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Madan E, Gogna R, Bhatt M, Pati U, Kuppusamy P, Mahdi AA. Regulation of glucose metabolism by p53: emerging new roles for the tumor suppressor. Oncotarget. 2011;2:948–57. doi: 10.18632/oncotarget.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wolf A, Agnihotri S, Guha A. Targeting metabolic remodeling in glioblastoma multiforme. Oncotarget. 2010;1:552–62. doi: 10.18632/oncotarget.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schilling T, Schleithoff ES, Kairat A, Melino G, Stremmel W, Oren M, et al. TAp63α induces apoptosis by activating signaling via death receptors and mitochondria. Biochem Biophys Res Commun. 2009;387:399–404. doi: 10.1016/j.bbrc.2009.07.063. [DOI] [PubMed] [Google Scholar]
- 38.Mundt HM, Stremmel W, Melino G, Krammer PH, Schilling T, Müller M. Dominant negative (DeltaN) p63α induces drug resistance in hepatocellular carcinoma by interference with apoptosis signaling pathways. Biochem Biophys Res Commun. 2010;396:335–41. doi: 10.1016/j.bbrc.2010.04.093. [DOI] [PubMed] [Google Scholar]
- 39.Huang Y, Sen T, Nagpal J, Upadhyay S, Trink B, Ratovitski E, et al. ATM kinase is a master switch for the Δ Np63 α phosphorylation/degradation in human head and neck squamous cell carcinoma cells upon DNA damage. Cell Cycle. 2008;7:2846–55. doi: 10.4161/cc.7.18.6627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang Y, Chuang AY, Romano RA, Liegeois NJ, Sinha S, Trink B, et al. Phospho-ΔNp63α/NF-Y protein complex transcriptionally regulates DDIT3 expression in squamous cell carcinoma cells upon cisplatin exposure. Cell Cycle. 2010;9:332–42. doi: 10.4161/cc.9.2.10432. [DOI] [PubMed] [Google Scholar]
- 41.Huang Y, Ratovitski EA. Phospho-ΔNp63α/Rpn13-dependent regulation of LKB1 degradation modulates autophagy in cancer cells. Aging (Albany NY) 2010;2:959–68. doi: 10.18632/aging.100249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huang Y, Chuang A, Hao H, Talbot C, Sen T, Trink B, et al. Phospho-ΔNp63α is a key regulator of the cisplatin-induced microRNAome in cancer cells. Cell Death Differ. 2011;18:1220–30. doi: 10.1038/cdd.2010.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang Y, Chuang AY, Ratovitski EA. Phospho-ΔNp63α/miR-885-3p axis in tumor cell life and cell death upon cisplatin exposure. Cell Cycle. 2011;10:3938–47. doi: 10.4161/cc.10.22.18107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang Y, Guerrero-Preston R, Ratovitski EA. Phospho-ΔNp63α-dependent regulation of autophagic signaling through transcription and micro-RNA modulation. Cell Cycle. 2012;11:1247–59. doi: 10.4161/cc.11.6.19670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roodhart JM, Daenen LG, Stigter EC, Prins HJ, Gerrits J, Houthuijzen JM, et al. Mesenchymal stem cells induce resistance to chemotherapy through the release of platinum-induced fatty acids. Cancer Cell. 2011;20:370–83. doi: 10.1016/j.ccr.2011.08.010. [DOI] [PubMed] [Google Scholar]
- 46.Düvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell. 2010;39:171–83. doi: 10.1016/j.molcel.2010.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eckel-Mahan KL, Patel VR, Mohney RP, Vignola KS, Baldi P, Sassone-Corsi P. Coordination of the transcriptome and metabolome by the circadian clock. Proc Natl Acad Sci USA. 2012;109:5541–6. doi: 10.1073/pnas.1118726109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Quijano C, Cao L, Fergusson MM, Romero H, Liu J, Gutkind S, et al. Oncogene-induced senescence results in marked metabolic and bioenergetic alterations. Cell Cycle. 2012;11:1383–92. doi: 10.4161/cc.19800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chang X, Monitto CL, Demokan S, Kim MS, Chang SS, Zhong X, et al. Identification of hypermethylated genes associated with cisplatin resistance in human cancers. Cancer Res. 2010;70:2870–9. doi: 10.1158/0008-5472.CAN-09-3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ogawa T, Liggett TE, Melnikov AA, Monitto CL, Kusuke D, Shiga K, et al. Methylation of death-associated protein kinase is associated with cetuximab and erlotinib resistance. Cell Cycle. 2012;11:1656–63. doi: 10.4161/cc.20120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang Y, Ratovitski EA. Phosphorylated TP63 induces transcription of RPN13, leading to NOS2 protein degradation. J Biol Chem. 2010;285:41422–31. doi: 10.1074/jbc.M110.158642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sen T, Sen N, Brait M, Begum S, Chatterjee A, Hoque MO, et al. DeltaNp63α confers tumor cell resistance to cisplatin through the AKT1 transcriptional regulation. Cancer Res. 2011;71:1167–76. doi: 10.1158/0008-5472.CAN-10-1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Martynova E, Pozzi S, Basile V, Dolfini D, Zambelli F, Imbriano C, et al. Gain-of-function p53 mutants have widespread genomic locations partially overlapping with p63. Oncotarget. 2012;3:132–43. doi: 10.18632/oncotarget.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stauber RH, Knauer SK, Habtemichael N, Bier C, Unruhe B, Weisheit S, et al. A combination of a ribonucleotide reductase inhibitor and histone deacetylase inhibitors downregulates EGFR and triggers BIM-dependent apoptosis in head and neck cancer. Oncotarget. 2012;3:31–43. doi: 10.18632/oncotarget.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Higgins ME, Ioannou YA. Apoptosis-induced release of mature sterol regulatory element-binding proteins activates sterol-responsive genes. J Lipid Res. 2001;42:1939–46. [PubMed] [Google Scholar]
- 56.Pediconi N, Ianari A, Costanzo A, Belloni L, Gallo R, Cimino L, et al. Differential regulation of E2F1 apoptotic target genes in response to DNA damage. Nat Cell Biol. 2003;5:552–8. doi: 10.1038/ncb998. [DOI] [PubMed] [Google Scholar]
- 57.Riley T, Sontag E, Chen P, Levine A. Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol. 2008;9:402–12. doi: 10.1038/nrm2395. [DOI] [PubMed] [Google Scholar]
- 58.Yoshihara Y, Wu D, Kubo N, Sang M, Nakagawara A, Ozaki T. Inhibitory role of E2F-1 in the regulation of tumor suppressor p53 during DNA damage response. Biochem Biophys Res Commun. 2012;421:57–63. doi: 10.1016/j.bbrc.2012.03.108. [DOI] [PubMed] [Google Scholar]
- 59.Huang Y, Jeong JS, Okamura J, Sook-Kim M, Zhu H, Guerrero-Preston R, et al. Global tumor protein p53/p63 interactome: making a case for cisplatin chemoresistance. Cell Cycle. 2012;11:2367–79. doi: 10.4161/cc.20863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.de Pablo MA, Susin SA, Jacotot E, Larochette N, Costantini P, Ravagnan L, et al. Palmitate induces apoptosis via a direct effect on mitochondria. Apoptosis. 1999;4:81–7. doi: 10.1023/A:1009694124241. [DOI] [PubMed] [Google Scholar]
- 61.Yi CH, Pan H, Seebacher J, Jang IH, Hyberts SG, Heffron GJ, et al. Metabolic regulation of protein N-alpha-acetylation by Bcl-xL promotes cell survival. Cell. 2011;146:607–20. doi: 10.1016/j.cell.2011.06.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pisano C, Vesci L, Milazzo FM, Guglielmi MB, Foderà R, Barbarino M, et al. Metabolic approach to the enhancement of antitumor effect of chemotherapy: a key role of acetyl-L-carnitine. Clin Cancer Res. 2010;16:3944–53. doi: 10.1158/1078-0432.CCR-10-0964. [DOI] [PubMed] [Google Scholar]
- 63.Mazzarelli P, Pucci S, Bonanno E, Sesti F, Calvani M, Spagnoli LG. Carnitine palmitoyltransferase I in human carcinomas: a novel role in histone deacetylation? Cancer Biol Ther. 2007;6:1606–13. doi: 10.4161/cbt.6.10.4742. [DOI] [PubMed] [Google Scholar]
- 64.Maxwell SA, Davis GE. Differential gene expression in p53-mediated apoptosis-resistant vs. apoptosis-sensitive tumor cell lines. Proc Natl Acad Sci USA. 2000;97:13009–14. doi: 10.1073/pnas.230445997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kuo MT, Savaraj N, Feun LG. Targeted cellular metabolism for cancer chemotherapy with recombinant arginine-degrading enzymes. Oncotarget. 2010;1:246–51. doi: 10.18632/oncotarget.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Patterson SD, Spahr CS, Daugas E, Susin SA, Irinopoulou T, Koehler C, et al. Mass spectrometric identification of proteins released from mitochondria undergoing permeability transition. Cell Death Differ. 2000;7:137–44. doi: 10.1038/sj.cdd.4400640. [DOI] [PubMed] [Google Scholar]
- 67.Cho YE, Singh TS, Lee HC, Moon PG, Lee JE, Lee MH, et al. In-depth identification of pathways related to cisplatin-induced hepatotoxicity through an integrative method based on an informatics-assisted label-free protein quantitation and microarray gene expression approach. Mol Cell Proteomics. 2012;11:M111–, 010884. doi: 10.1074/mcp.M111.010884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Polimeni M, Voena C, Kopecka J, Riganti C, Pescarmona G, Bosia A, et al. Modulation of doxorubicin resistance by the glucose-6-phosphate dehydrogenase activity. Biochem J. 2011;439:141–9. doi: 10.1042/BJ20102016. [DOI] [PubMed] [Google Scholar]
- 69.Zhang Z, Liew CW, Handy DE, Zhang Y, Leopold JA, Hu J, et al. High glucose inhibits glucose-6-phosphate dehydrogenase, leading to increased oxidative stress and β-cell apoptosis. FASEB J. 2010;24:1497–505. doi: 10.1096/fj.09-136572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fico A, Paglialunga F, Cigliano L, Abrescia P, Verde P, Martini G, et al. Glucose-6-phosphate dehydrogenase plays a crucial role in protection from redox-stress-induced apoptosis. Cell Death Differ. 2004;11:823–31. doi: 10.1038/sj.cdd.4401420. [DOI] [PubMed] [Google Scholar]
- 71.Zecchin KG, Rossato FA, Raposo HF, Melo DR, Alberici LC, Oliveira HC, et al. Inhibition of fatty acid synthase in melanoma cells activates the intrinsic pathway of apoptosis. Lab Invest. 2011;91:232–40. doi: 10.1038/labinvest.2010.157. [DOI] [PubMed] [Google Scholar]
- 72.Bandyopadhyay S, Zhan R, Wang Y, Pai SK, Hirota S, Hosobe S, et al. Mechanism of apoptosis induced by the inhibition of fatty acid synthase in breast cancer cells. Cancer Res. 2006;66:5934–40. doi: 10.1158/0008-5472.CAN-05-3197. [DOI] [PubMed] [Google Scholar]
- 73.Liu H, Liu Y, Zhang JT. A new mechanism of drug resistance in breast cancer cells: fatty acid synthase overexpression-mediated palmitate overproduction. Mol Cancer Ther. 2008;7:263–70. doi: 10.1158/1535-7163.MCT-07-0445. [DOI] [PubMed] [Google Scholar]
- 74.D’Erchia AM, Tullo A, Lefkimmiatis K, Saccone C, Sbisà E. The fatty acid synthase gene is a conserved p53 family target from worm to human. Cell Cycle. 2006;5:750–8. doi: 10.4161/cc.5.7.2622. [DOI] [PubMed] [Google Scholar]
- 75.Sabbisetti V, Di Napoli A, Seeley A, Amato AM, O’Regan E, Ghebremichael M, et al. p63 promotes cell survival through fatty acid synthase. PLoS ONE. 2009;4:e5877. doi: 10.1371/journal.pone.0005877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jeon TI, Osborne TF. SREBPs: metabolic integrators in physiology and metabolism. Trends Endocrinol Metab. 2012;23:65–72. doi: 10.1016/j.tem.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Seo YK, Jeon TI, Chong HK, Biesinger J, Xie X, Osborne TF. Genome-wide localization of SREBP-2 in hepatic chromatin predicts a role in autophagy. Cell Metab. 2011;13:367–75. doi: 10.1016/j.cmet.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Logette E, Le Jossic-Corcos C, Masson D, Solier S, Sequeira-Legrand A, Dugail I, et al. Caspase-2, a novel lipid sensor under the control of sterol regulatory element binding protein 2. Mol Cell Biol. 2005;25:9621–31. doi: 10.1128/MCB.25.21.9621-9631.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gibot L, Follet J, Metges JP, Auvray P, Simon B, Corcos L, et al. Human caspase 7 is positively controlled by SREBP-1 and SREBP-2. Biochem J. 2009;420:473–83. doi: 10.1042/BJ20082057. [DOI] [PubMed] [Google Scholar]
- 80.Lhoták S, Sood S, Brimble E, Carlisle RE, Colgan SM, Mazzetti A, et al. ER stress contributes to renal proximal tubule injury by increasing SREBP-2-mediated lipid accumulation and apoptotic cell death. Am J Physiol Renal Physiol. 2012;303:F266–78. doi: 10.1152/ajprenal.00482.2011. [DOI] [PubMed] [Google Scholar]
- 81.Cavill R, Kamburov A, Ellis JK, Athersuch TJ, Blagrove MS, Herwig R, et al. Consensus-phenotype integration of transcriptomic and metabolomic data implies a role for metabolism in the chemosensitivity of tumour cells. PLoS Comput Biol. 2011;7:e1001113. doi: 10.1371/journal.pcbi.1001113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mauro C, Leow SC, Anso E, Rocha S, Thotakura AK, Tornatore L, et al. NF-κB controls energy homeostasis and metabolic adaptation by upregulating mitochondrial respiration. Nat Cell Biol. 2011;13:1272–9. doi: 10.1038/ncb2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zawacka-Pankau J, Grinkevich VV, Hünten S, Nikulenkov F, Gluch A, Li H, et al. Inhibition of glycolytic enzymes mediated by pharmacologically activated p53: targeting Warburg effect to fight cancer. J Biol Chem. 2011;286:41600–15. doi: 10.1074/jbc.M111.240812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.McDonald PC, Winum JY, Supuran CT, Dedhar S. Recent developments in targeting carbonic anhydrase IX for cancer therapeutics. Oncotarget. 2012;3:84–97. doi: 10.18632/oncotarget.422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Evans AM, DeHaven CD, Barrett T, Mitchell M, Milgram E. Integrated, nontargeted ultrahigh performance liquid chromatography/electrospray ionization tandem mass spectrometry platform for the identification and relative quantification of the small-molecule complement of biological systems. Anal Chem. 2009;81:6656–67. doi: 10.1021/ac901536h. [DOI] [PubMed] [Google Scholar]
- 86.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci USA. 2003;100:9440–5. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.