

Abstract
CREBZF is a member of the mammalian ATF/CREB family of transcription factors. Here, we describe a novel functional interaction between CREBZF and the tumor suppressor p53. CREBZF was identified in a yeast two-hybrid screen using HEY1, recently characterized as an indirect p53 activator, as bait. CREBZF interacts in vitro with both HEY1 and p53, and CREBZF expression stabilizes and activates p53. Moreover, CREBZF cooperates synergistically with HEY1 to enhance p53 transcriptional activity. On the other hand, partial depletion of endogenous CREBZF diminishes p53 protein levels and inhibits HEY1-mediated activation of p53. CREBZF-positive effects on p53 signaling may reflect, at least in part, an observed induction of posttranslational modifications in p53 known to prevent its degradation. CREBZF expression protects HCT116 cells from UV radiation-induced cell death. In addition, CREBZF expression confers sensitivity to 5-fluorouracil, a p53-activating chemotherapeutic drug. Our study suggests that CREBZF may participate in the modulation of p53 tumor suppressor function.
Keywords: CREBZF, p53, ultraviolet radiation, 5-fluorouracil, DNA damage, HEY1
Introduction
The p53 tumor suppressor is a key barrier against cancer due to its role in preventing damaged cells to initiate malignant growth, hence p53 loss-of-function contributes to the tumorigenic process in human cancers. This can be caused by direct mutation of the p53 gene or by alterations in the plethora of cellular pathways, upstream or downstream of p53, which participate in p53 signaling. Accumulating evidences indicate that extensive crosstalk between the Notch and the p53 pathway contributes to the regulation of embryonic development, cell differentiation and tumor suppression (reviewed in ref. 1). To date, the complexity of both signaling pathways has made impossible to draw a simplified model that explains their reciprocal modulation, and conflicting results have been obtained on the effects of Notch signaling on p53 function, depending on the cell type or cancer stage.1 Hairy/enhancer-of-split related with YRPW motif 1 (HEY1), a well-characterized effector within the Notch signaling, has been recently identified as a new p53 co-regulator.2,3 Initially, a genome-wide functional analysis revealed that HEY1 is a positive regulator of p53 through repression of MDM2 transcription, although the detailed molecular mechanisms responsible for this regulation are still unknown.2 Subsequently, our laboratory showed that HEY1 expression impairs cell proliferation in human osteosarcoma cells (U2OS) in a p53-dependent manner and sensitizes cells to p53-activating chemotherapeutic drugs.3 HEY1-dependent activation of p53 can also promote cell cycle arrest in Ewing sarcoma family cancer cell lines.4 The human HEY1 gene maps to chromosome band 8q21. Amplification of this locus occurs in a large fraction of prostate tumors and correlates with tumor aggressiveness.5 Amplification of the q21 band of chromosome 8 also occurs in other cancer types, such as breast,6 colon,7 bone8 and pancreatic cancer.9 In addition to these genetic studies, an expression analysis found that the majority of prostate samples showed total exclusion of HEY1 protein from the nucleus,10 an abnormality that prevents HEY1-dependent p53 activation.3 Thus, alterations in HEY1 function and/or expression could contribute to the oncogenic transformation, impairing the crosstalk between Notch and p53 or other signaling pathways.
To investigate the poorly understood molecular mechanisms by which HEY1 exert its biological actions, we performed a yeast two-hybrid screen using full-length HEY1 as bait. This approach revealed a novel functional interaction between p53 and one of the HEY1-interacting proteins, CREB/ATF bZIP transcription factor (CREBZF, also known as Zhangfei and SMILE). Here, we characterize CREBZF as a novel positive regulator of p53 activity and present evidences that suggest that deregulation of CREBZF may impinge on p53 tumor suppressor functions and contribute to the origin and/or development of cancer.
Results
CREBZF interacts with HEY1
We used a yeast two-hybrid system to identify human cDNAs encoding proteins that interact with HEY1. Three identical clones found encoded amino acids 195 to 321 of CREBZF protein (HEY1-binding clone, Fig. 1A), indicating that CREBZF and HEY1 could interact in vivo. Two different CREBZF isoforms have been described that arise from the alternative usage of initiation codons within a single gene,11 hereafter named CREBZF-long (ZF-long) and CREBZF-short (ZF-short) (Fig. 1A). We confirmed by GST pull-down assays that full-length HEY1 interacts with both CREBZF isoforms (Fig. 1B). In an attempt to define HEY1 regions required for the interaction with CREBZF, we used various HEY1 deletion mutants (Fig. 1A). Our results showed that none of the isolated helical protein-protein interaction domains in HEY1 (HLH and Orange domains) were capable to interact with CREBZF (Fig. 1C). The lack of interaction does not reflect lower expression levels for the mutants, because control coomassie-stained gels shown that deletion mutants express at even higher levels than GST-HEY1 wild-type (Fig. S1).

Figure 1. In vitro interaction of CREBZF with HEY1. (A) Schematic representation of full-length CREBZF, full-length HEY1 and the deletion mutants used in this study. (B) Whole-cell extracts from COS-1 cells previously transfected with expression vectors for flag-tagged CREBZF-long (ZF-long) or CREBZF-short (ZF-short) were incubated with GST fusion proteins of HEY1 coupled with Sepharose beads. The associated proteins were detected by western blotting using anti-Flag antibody. (C) Whole-cell extracts from COS-1 cells previously transfected with expression vectors for flag-tagged ZF-long were incubated with GST fusion proteins of full-length HEY1 or deletion mutants coupled with Sepharose beads. Bound proteins were detected by western blotting using anti-Flag antibody.
CREBZF positively regulates p53 transcriptional activity
HEY1 is a positive regulator of p53-dependent transcription2,3 (Fig. 2A). However, the detailed mechanisms mediating this effect have not been characterized. We have now found that full-length HEY1 fused to GST is able to interact with p53 in vitro (Fig. 2B). In view of this result, we tested in a similar pull-down assay whether CREBZF, a HEY1-interacting protein, was also able to interact directly with p53. Interestingly, we found that both CREBZF isoforms indeed interact with p53 (Fig. 2C). A series of CREBZF deletion mutants fused to GST revealed that the middle bZIP region in CREBZF, also present in the HEY1-interacting clone found in the yeast two-hybrid, was a key determinant for the interaction (Fig. 2C). Coomassie-stained gels showed the stability of the GST fusion proteins used in the pull-down assay (Fig. S2). These results prompted us to investigate a possible role for CREBZF as a modulator of p53-dependent transcription. For this, we performed reporter assays in p53-competent HCT116 human colon carcinoma cells using two luciferase reporter plasmids containing natural p53-responsive promoters (PIG3-LUC and NOXA-LUC). The expression of both CREBZF isoforms greatly enhanced transcriptional activity from both p53-dependent reporters in a dose-dependent manner (Fig. 2D). Interestingly, ZF-short was a more potent p53 activator in these assays (Fig. 2D and F). Identical results were obtained when performing similar transfection experiments in the p53-competent U2OS human osteosarcoma cells (data not shown). To analyze a putative cooperation between HEY1 and CREBZF in p53 activation, we co-transfected suboptimal amounts of ZF-short and HEY1 expression plasmids, either independently or in combination, together with the PIG3-LUC reporter. In these conditions, we observed a synergistic activation of PIG3-LUC reporter activity (Fig. 2E), suggesting that both proteins may cooperate to activate p53.

Figure 2. CREBZF positively regulates p53-dependent transactivation. (A) HEY1 greatly enhances PIG3-luciferase reporter activity. U2OS or HCT116 cells were transfected with 100 ng of empty pGL3 plasmid or PIG3-Luc in the presence or absence of 200 ng of expression vector for HEY1. (B) In vitro interaction of HEY1 with p53. Whole-cell extracts from COS-1 cells previously transfected with an expression vector for p53 were incubated with GST fusion proteins of HEY1 coupled with Sepharose beads. The associated proteins were detected by western blotting using anti-p53 antibody. (C) In vitro interaction of CREBZF with p53. Whole-cell extracts from COS-1 cells previously transfected with expression vectors for p53 were incubated with GST fusion proteins of CREBZF-long, CREBZF-short or deletion mutants coupled with Sepharose beads. The associated proteins were detected by western blotting using anti-p53 antibody. (D) HCT116 cells were transfected with 100 ng of PIG3-Luc reporter or 100 ng of NOXA-Luc reporter in the presence of increasing amounts (indicated in ng) of expression vector for CREBZF-long or Short (ZF-long or ZF-short). (E) HCT116 cells were transfected with 100 ng of PIG3-Luc in the presence or absence of 25 ng of expression vector for ZF-short, HEY1 or both together. (F) HCT116 cells were transfected with 100 ng of PIG3-Luc, p21-Luc or PUMA-LUC in the presence or absence of 100 ng of expression vector for ZF-long or ZF-short. (G) Deletion of the p53 binding site in PIG3 impairs CREBZF positive effect. U2OS cells were transfected with 100 ng of PIG3-Luc or PIG3 delF-Luc and 200 ng of expression vector for ZF-long or ZF-short. (H) CREBZF positive effect on PIG3 promoter depends on the p53 status. HCT116 p53−/− cells were transfected with 100 ng PIG3-Luc and expression vectors for ZF-long or ZF-short (200 ng) and p53 (5 ng) as indicated. In every case, after transfection, cells were washed and incubated 24 h. Subsequently, cell lysates were assayed using a dual luciferase reporter system. Normalized values are expressed relative to the activity of the reporter in the absence of HEY1 (A) or relative to the activity of the reporter in the absence of CREBZF (D–F) or relative to the activity of reporter in the absence CREBZF and p53 (G). The results shown represent the averages of results of three independent experiments assayed in duplicate + s.d.
Independent transfection experiments with three other natural p53-responsive luciferase reporters (APAF, p21 and PUMA) demonstrated that both CREBZF isoforms behave as activators of p53-dependent activity (Fig. 2F), although the potency of the effect was promoter-dependent, ranging from 2-fold activation (PUMA-LUC) to 25-fold (PIG3). To analyze the requirement of p53 binding to the PIG3 reporter for CREBZF-positive effects, we used a PIG3 reporter plasmid lacking the p53-binding site, PIG3-delF.12 The absence of the p53-binding site greatly diminished the reporter induction upon CREBZF co-transfection in U2OS cells (Fig. 2G). We then performed similar transfection experiments using HCT116 p53−/− cells, which are genetically identical to HCT116 p53+/+ except for the p53 status. CREBZF failed to activate PIG3-LUC reporter in the absence of endogenous p53 protein (Fig. 2H). Expression of p53 in HCT116 p53−/− cells induced p53-reporter activity and co-transfection of CREBZF further stimulate the p53-responsive promoter (Fig. 2H). Taken together, these results indicate that CREBZF’s effects require the binding of p53 to its cognate response element in the PIG3 reporter.
CREBZF expression stabilizes and activates p53
To investigate whether CREBZF also regulates the expression of endogenous p53-target genes, we transfected ZF-short into p53-competent human HCT116 cells. We observed that ZF-short expression resulted in increased endogenous p53 protein levels (Fig. 3A). Accordingly, CREBZF also increased the expression of p21, a well-characterized p53-target gene (Fig. 3A). To better characterize the functional interaction between p53 and CREBZF, we generated tetracycline-inducible pools of HCT116 and U2OS cells stably expressing either flag-tagged ZF-short or ZF-long (HCT116-ZF-long, HCT116-ZF-short, U2OS-ZF-short and U2OS-ZF-long). Exogenous Flag-tagged CREBZF isoforms were predominantly nuclear as previously described13 and co-localized with p53 (Fig. S3). In agreement with previous results, induction of CREBZF expression increased both p53 and p21 protein levels in all four stable cell lines (Fig. 3B, E and F and data not shown). The increase in p53 protein levels can be detected as early as 4 h following ZF-short expression; however it does not correlate with an increase in p53 mRNA levels (Fig. 3D), suggesting that p53 induction is mediated by post-transcriptional mechanisms. In keeping with that, immunoblotting experiments showed that induction of CREBZF expression, both in HCT116 and U2OS cells, strongly increases the levels of p53 Lys-382 acetylation, an event that prevents MDM2-mediated p53 degradation (Fig. 3C, E and F).14 Also, ZF-short expression in HCT116 cells induces phosphorylation of all p53 N-terminal residues tested (Ser-20, Ser-15 AND Ser-46) (Fig. 3C), protein modifications also known to stabilize and activate p53.14 Conversely, p21 protein increase does reflect a clear rise in its mRNA (Fig. S4). Similarly, PIG3 mRNA levels were also clearly induced upon CREBZF expression both in HCT116 (Fig. 3D) and U2OS cells (Fig S4), thus implying that the upregulation of both p21 and PIG3 genes is a direct consequence of increased p53-dependent transcriptional activity. Although induction of the cell cycle inhibitor p21 results frequently in cell cycle arrest, CREBZF did not affect U2OS cell proliferation, and it only moderately attenuated HCT116 proliferation rate (Fig. S5). Hence, our results suggest that CREBZF-mediated stabilization and activation of p53 does not result in the most typical p53 cellular responses: cell cycle arrest or apoptosis.

Figure 3. CREBZF expression stabilizes and activates p53. (A) HCT116 cells were transfected with expression vector for ZF-short and the levels of the indicated proteins were determined 24 h later by immunobloting. (B) Immunoblot expression of ZF-short, p53, p21 and β-actin in stable HCT116 cell lines expressing inducible CREBZF-short (HCT116-ZF-short) treated with 1 µg/ml tetracycline (Tet) for 4, 8, 12 or 24 h. (C) Induction of p53 phosphorylation and acetylation by ZF-short expression. HCT116-ZF-short cells were treated with 1 µg/ml tetracycline (Tet) for 4, 8, 12 or 24 h and proteins were analyzed by immunobloting with the indicated p53 phospho-specific antibodies, anti-Ac-Lys382-p53 or anti-β-actin. (D) Quantitative RT-PCR analysis for the expression of p53 and PIG3 transcripts in HCT116-ZF-short cells treated with 1 µg/ml tetracycline (Tet) for the indicated times. (E) Immunoblot expression of ZF-long, p53, Ac-Lys382-p53, p21 and β-actin in stable U2OS cell lines expressing inducible CREBZF-long (U2OS-ZF-long) treated with 1 µg/ml tetracycline (Tet) for 4, 8, 12 or 24 h. (F) Immunoblot expression of ZF-short, p53, Ac-Lys382-p53, p21 and β-actin in stable U2OS cell lines expressing inducible CREBZF-short (U2OS-short) treated with 1 µg/ml tetracycline (Tet) for 4, 8, 12 or 24 h.
HEY1 expression reduces MDM2 protein levels,2,3 providing a molecular mechanism to explain, at least in part, HEY1-induced p53 stabilization. However, induction of ZF-short expression in U2OS or HCT116 cells did not downregulate MDM2 expression at mRNA or protein level (Fig. S6 and data not shown), suggesting that CREBZF-dependent stabilization of p53 relies on different molecular mechanisms than those triggered by HEY1.
CREBZF partial depletion diminishes p53 protein levels
Quantitative RT-PCR and immunoblotting experiments revealed that both U2OS and HCT116 cell lines express endogenous CREBZF (Figs. 4 and 6). We made use of CREBZF siRNAs to downregulate CREBZF at both mRNA and protein levels (Fig. 4A and B). We observed that, in accordance with previous results, even a relatively modest decrease of CREBZF mRNA and endogenous ZF-long protein levels (the only isoform detected in U2OS cells) causes a noticeable reduction of endogenous p53 protein levels (Fig. 4B). We had previously shown that HEY1 expression induces p53-dependent cell-cycle arrest in tetracycline-inducible U2OS cells stably expressing HEY1.3 To analyze whether HEY1’s effects are mediated by CREBZF, U2OS-HEY1 cells were transfected with a CREBZF-specific siRNA or non-specific control siRNA, and cell proliferation after induction of HEY1 was determined. CREBZF knockdown only partly rescued the proliferation arrest caused by induction of HEY1 in U2OS cells (Fig. 4C) indicating that the activation of p53 signaling by HEY1 is, at least in part, mediated by CREBZF.
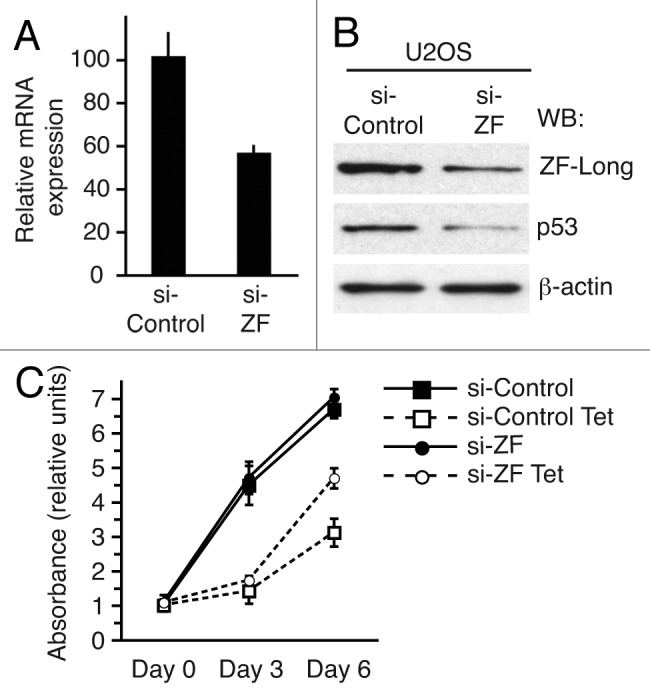
Figure 4. Partial CREBZF depletion diminishes cellular p53 protein levels. (A) Quantitative RT-PCR analysis for the expression of CREBZF transcript in U2OS cells 48 h after transfection with either human CREBZF on-target plus smart pool siRNA (si-ZF) or with non-targeting pool negative control siRNA (si-Control). (B) Immunoblot expression of ZF-long, p53, and β-actin in U2OS cells 48 h after transfection with either human CREBZF on-target plus smart pool siRNA (si-ZF) or with non-targeting pool negative control siRNA (si-Control). (C) U2OS-HEY1 cells transfected with CREBZF-specific on-target plus smart pool siRNA (si-ZF) or non-targeting pool negative control siRNA (si-Control) were untreated or treated with 1 µg/ml tetracycline (Tet) to induce HEY1 expression. Cell proliferation was monitored at different time points by using MTS assay. Results from a representative experiment assayed in quadruplicate are shown.
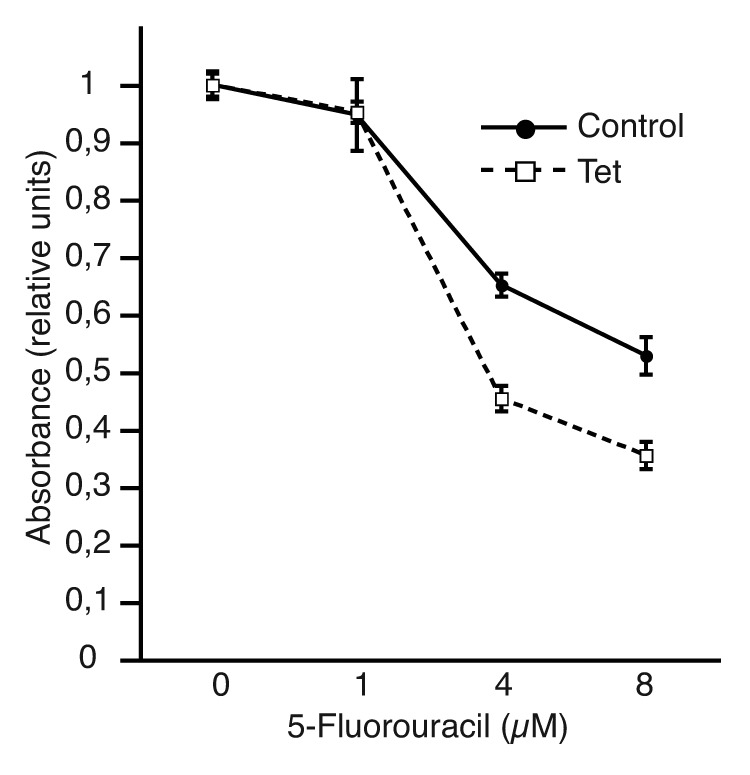
Figure 6. The expression of ZF-short leads to increased sensitivity to the chemotherapy agent 5-fluorouracil. U2OS-ZF-short cells were untreated (control) or treated with 1 µg/ml tetracycline (Tet) to induce ZF-short expression. After 24 h, the cells were cultured with varying concentrations of 5-fluorouracil for another 72 h in the presence or absence of tetracycline. Subsequently, cell viability was assayed using an MTS-based assay. Data were plotted relative to the drug-free controls. The results shown represent the averages of results of two independent experiments assayed in quadruplicate ± s.d.
CREBZF expression promotes cell survival upon UV light radiation
Activation of p53 signaling is a key cellular event upon UVC damage, and Notch signaling has been reported to downregulate p53-dependent apoptosis induced upon UVC irradiation.15 The results shown above demonstrate that CREBZF interacts with the Notch effector HEY1, and that, like HEY1, CREBZF expression activates p53 signaling. To explore whether CREBZF expression influences the rate of UV-induced cell death, we induced ZF-short expression in HCT116 cells for 24 h prior to UVC irradiation (30 mJ/cm2). Following UVC irradiation, HCT116-ZF-short cells were incubated for 4 additional days, and cell survival was determined by cell counting. Interestingly, we found that CREBZF expression significantly protected the cells from UVC-induced apoptosis, increasing the survival rate from 10% to 30% (p < 0.05) (Fig. 5).
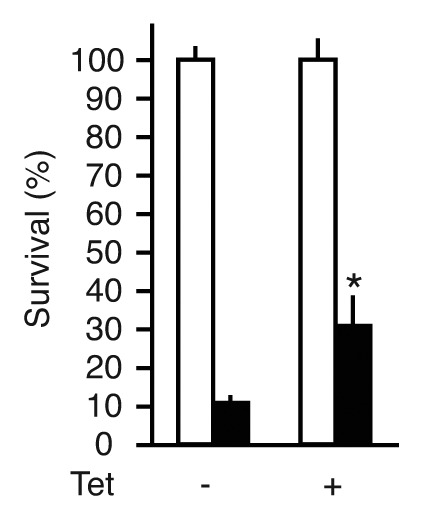
Figure 5. CREBZF expression promotes cell survival upon UV light radiation. HCT116-ZF-short cells were untreated or treated with 1 µg/ml tetracycline (Tet) to induce ZF-short expression. Twenty-four hours after tetracycline treatment cells were seeded at low density and irradiated with 30 mJ/cm2 UV light-C (UVC) as described in Materials and Methods. The cells were cultured for another 4 d in the presence or absence of tetracycline and cell survival was determined counting the number of control cells (white bars) or UVC-irradiated cells (black bars). Normalized values are expressed relative to non-irradiated cells. The results shown represent the averages of results of three independent experiments assayed in quadruplicate + s.d. *p < 0.05 relative to control (Student’s t-test).
CREBZF expression sensitizes U2OS cells to the chemotherapeutic drug 5-fluorouracil
p53 signaling has a key role in the sensitivity to several chemotherapeutic drugs, including 5-fluorouracil.16,17 Besides, a genome-wide screening has revealed that the cytogenetic region comprising the human CREBZF gene (11q14.1) may encode genes that modulate drug resistance to 5-fluorouracil-based drugs.18 To explore whether CREBZF, via regulation of p53 activity, could influence the response of cells to 5-fluorouracil, we performed cytotoxicity assays. Interestingly, we observed that induction of ZF-short expression in both U2OS-ZF-short cells (Fig. 6) and HCT116-ZF-short cells (Fig. S7) resulted in an increase in sensitivity to 5-fluorouracil, suggesting that altered CREBZF expression might be one of the causes influencing drug resistance in human tumors.
Discussion
The p53 protein is a key tumor suppressor, as reflected by the fact that inactivation of the p53 pathway occurs in most human cancers. Thus, the discovery and characterization of novel p53 regulators can provide invaluable information about oncogenesis and uncover novel potential cancer biomarkers. We report here that CREBZF, a member of the mammalian ATF/CREB family of transcription factors is a positive regulator of p53 activity. The ATF/CREB family of proteins comprises a large and heterogeneous group of transcription factors that share the ability to bind to the consensus ATF/CRE site.19 CREBZF was identified as a protein that interacts with host cell factor C1 (HCFC1), a transcriptional factor that is required for transcription activation of herpes simplex virus genes.13 Unlike most ATF/CREB proteins, CREBZF does not bind ATF/CRE consensus sites by itself,13 although it can bind to CRE reporters, forming heterodimers with other members of the ATF family.20 Subsequently, CREBZF was shown to function as a corepressor of nuclear receptors, modulating their interaction with corepressor and coactivator complexes.11,21 Little is known about CREBZF’s biological role, although it has been recently suggested that it could play a role in cellular responses to various forms of stress, such as the unfolded protein response22,23 and amino acid deprivation.24 In line with these reports, our results indicate that CREBZF could also participate in DNA damage stress responses by regulating the p53 pathway. We have found that CREBZF expression stabilizes p53 and activate its transcriptional activity. Contrasting with HEY1-induced increase in p53 stability, which depends on inhibition of MDM2 expression, CREBZF expression does not reduce cellular MDM2 levels, and p53 stabilization may be mediated, at least in part, by induction of posttranslational modifications in p53 that prevent its degradation.
HEY1 and CREBZF interact with each other and with p53, and expression of both proteins activates p53-dependent transcription; however, both stimuli had very different biological outcomes in U2OS cell lines: HEY1 expression induces p53-dependent cell cycle arrest and aberrant neuron-like differentiation,3 but CREBZF expression neither affects U2OS proliferation rate nor induces visible morphological changes. Notwithstanding this clear dissimilarity, our results suggest a role for CREBZF in the regulation of HEY1 activity, because CREBZF synergizes with HEY1 in the activation of p53 transcriptional activity. In addition, reduction of endogenous CREBZF levels partly rescues HEY1-induced cell cycle arrest. Of note, HEY1 expression results in cell cycle arrest despite causing a strong downregulation of the cell cycle inhibitor p21,3 whereas the robust induction of p21 expression caused by CREBZF does not inhibit cell proliferation. These observations most likely reflect the great complexity of p53 signaling and the variety of cellular responses induced by its activation depending on the type of the stimuli and cellular context.
A possible role for CREBZF in modulating p53 activation in response to cellular stress is reinforced by our data showing that CREBZF expression protects HCT116 cells against apoptosis induced by UV light. Interestingly, in keeping with our results, a phosphoproteome profiling of human skin fibroblasts revealed that X-ray irradiation induced phosphorylation of several serine and threonine residues in CREBZF.25 The characterization of the possible effects of those modifications on the stability of CREBZF and its functional interaction with p53 could help to unravel novel mechanisms that fine-tune p53 stress responses.
The functional interaction between HEY1 and CREBZF suggests that CREBZF might participate in the modulation of Notch signaling. However, to address this regulatory functional interaction, it will be necessary to elucidate the molecular mechanisms responsible for CREBZF effects on p53 activity and to study other possible effects of CREBZF expression on Notch signaling independent of p53 function. Another member of ATF/CREB family, ATF3, was also recently shown to be an activator of p53 activity. ATF3 interacts with the C-terminal region of p53 and prevents MDM2-mediated ubiquitination. Thus, ATF3 expression protects against MDM2-directed p53 degradation in vivo when co-transfected in p53-null human lung adenocarcinoma H1299 cells.26 However, in similar experiments, CREBZF did not protect p53 from MDM2-mediated degradation (Fig. S8). Therefore, the stabilization of p53 observed in p53-competent cell lines upon induction of CREBZF expression may reflect activation of p53 phosphorylation-acetylation cascades that prevent its interaction with MDM2 instead of physically blocking MDM2-p53 interaction.
We had previously shown that HEY1-dependent p53 induction sensitized U2OS cells to the chemotherapeutic agents cisplatin and doxorubicin. However, expression of CREBZF in U2OS or HCT116 cells does not affect their sensitivity to those drugs (data not shown), once more indicating a different outcome for both p53-activating proteins. Nonetheless, CREBZF expression was able to enhance chemosensitivity to the anticancer drug 5-fluorouracil, which activates p53 by interfering with the MDM2-p53 feedback circuit, resulting in cell cycle arrest and/or apoptosis.27 In humans, CREBZF maps to chromosome band 11q14.1, and, based on a genetic screening of 27 human cancer xenografts, it has been proposed that this locus could encode genes that influence sensitivity to 5-fluorouracil.18 Our results suggest that CREBZF could be one of the candidate genes responsible for those effects. Loss of 11q14.1 chromosomic region has been observed in several types of cancer, including malignant melanoma,28 neuroblastoma,29 head and neck squamous cell carcinomas,30 chronic lymphocytic leukemia31 and lung carcinoids.32 Moreover, pooled genome linkage also suggested that chromosome 11q14.1 contains genes, which, when mutated, predispose men to develop a more aggressive prostate cancer.33 These genetic evidences, together with CREBZF role in the regulation of p53, suggest that CREBZF could have a tumor-suppressive activity and, therefore, alterations in the expression or function of this protein might contribute to tumor progression. To confirm this possibility, it will be required to further characterize the molecular mechanisms by which CREBZF regulates p53 signaling, to address how different types of cellular stress could modulate CREBZF expression and, ultimately, to study whether loss or mutation of CREBZF gene contributes to malignant cell transformation.
Materials and Methods
Yeast two-hybrid screen
The screen and data analysis were performed by Hybrigenics S.A. The coding sequence for full-length human HEY1 was cloned into the pB29 plasmid as an N-terminal fusion to LexA and used as a bait to screen at saturation a high-complexity random-primed human placenta cDNA library as previously described.34 The selectivity of the HIS3 reporter gene was modulated with 0.5 mM 3-aminotriazole. We obtained 62 cDNA clones, three of which encoded amino acids 195 to 321 of CREBZF (genebank accession number NM_001039618.2).
Plasmids
The following plasmids have been described: pSG5-HEY1, GST-HEY1 (full-length and deletion mutants ΔY, amino acids 1–285; ΔY+O, amino acids 1–115; ΔY+O+H, amino acids 1–49; ΔHLH, amino acids 116–299).10 PIG3-LUC, PIG3-delF, NOXA-LUC, p21-LUC, APAF-LUC, PUMA-LUC and pCDNA-p53.3
The complete open reading frame of the full-length CREBZF-long and CREBZF-short were amplified by PCR from cDNA obtained from human MCF-7 cells and subcloned into pSG5-Flag10 or pGEX-6P-1 (Amersham Pharmacia Biotech). The CREBZF deletion mutants GST-ZF 269–354, GST-ZF 1–268 and GST-ZF 1–207 were generated subcloning into pGEX-6P-1 the corresponding cDNA regions amplified by PCR.
Cell culture and transient transfections
COS-1, U2OS and HCT116 cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum. Twenty-four hours before transfection, cells were plated in 24-well plates (50,000 U2OS cells per well or 100,000 HCT116 cells per well). Cells were transfected using Lipofectamine LTX (Life Technologies). Transfected plasmids are detailed in the figure legends. pRL-TK (Promega) was used as internal control for transfection efficiency. The amounts of plasmid DNA used were: pGL3, PIG3-LUC, NOXA-LUC, APAF-LUC, p21-LUC, PUMA-LUC, delF-LUC (100 ng/well), pRL-TK (10 ng/well), pSG5-HEY1 (100 ng/well), pSG5-CREBZF-short and -long (100 ng/well unless otherwise indicated), pCDNA-p53 (5 ng/well). Cell extracts were assayed for luciferase activity as described previously.10 ON-TARGETplus SMARTpool-Human CREBZF siRNAs (Dharmacon, L-013938) were trasfected into U2OS cells using Lipofectamine LTX (100 nM final siRNA concentration).
GST pull-down assays
Expression vectors were transfected into COS-1 cells using Lipofectamine LTX, Glutathione S-transferase (GST) fusion proteins were induced, purified, bound to Sepharose beads (GE Healthcare) and incubated with COS-1 whole-cell extracts as described previously35 in NETN buffer (20 mM TRIS-HCl (pH 8.0), 1 mM EDTA, 0.5 Nonidet P-40, 100 mM NaCl). After extensive washing, the samples were separated on SDS-10% polyacrylamide gels. Gels were blotted onto nitrocellulose and probed with antibodies.
Antibodies and immunoblotting
The antibodies used were anti-FLAG M2 (Sigma-Aldrich), anti-p53 (DO-1, Santa Cruz Biotechnology), anti-β-actin (AC-15, Sigma-Aldrich), anti-p21 (C-19, Santa Cruz Biotechnology), anti-CREBZF (Abcam ab28700), anti-Cleaved Caspase 3 Asp 175, (Cell Signaling 9661), anti-Acetyl-p53 Lys379 (Cell signaling 2570), anti-Phospho-p53 Ser15 (Cell Signaling 9286), anti-Phospho-p53 Ser20 (Cell Signaling 9287), anti-Phospho-p53 Ser 46 (Cell Signaling 2521). Immunoblotting was performed as previously described.10
Generation of U2OS and HCT116 cells expressing tetracycline-inducible CREBZF-long or CREBZF-short
Lentiviral vectors encoding flag-tagged ZF-short or ZF-long were used to generate lentivirus expressing ZF-long or ZF-short by the ViraPower™ T-REx™ system following manufacturer’s instructions (Invitrogen). Cells were co-transduced with Tet-repressor-lentivirus and either ZF-long- or ZF-short-lentivirus. Selection of stably co-transduced cells was achieved with Zeocin (650 µg/ml for HCT116 and 1000 µg/ml for U2OS) and Blasticidin (6 µg/ml for HCT116 and 4 µg/ml for U2OS). To induce ZF-long or ZF-short expression, 1 µg/ml of tetracycline was added to the media.
Quantitative real-time PCRs
Total RNA was extracted using TRI-reagent (Sigma-Aldrich) and treated with RNase-free DNase (Invitrogen). Gene expression was analyzed by real-time RT-PCR using Brilliant SYBR-Green QPCR Master-Mix (Stratagene). The human-specific primers used were: CREBZF-forward; GCTCCGTTGTAGGGGTTACA, CREBZF-reverse; AAAAACAGCAGAAGCCCTGA, p21-forward; CCTGTCACTGTCTTGTACCCT, p21-reverse; GCGTTTGGAGTGGTAGAAATCT, PIG3-forward; AATGCAGAGACAAGGCCAGTA, PIG3-reverse; TCCCCGATCTTCCAGTGTCC.
The constitutively expressed L19 ribosomal gene was used as control to normalize mRNA expression (L19-forward; 5′-GCGGAAGGGTACAGCCAAT-3′, L19-reverse; GCAGCCGGCGCAAA).
Cytotoxicity assays
U2OS cells expressing tetracycline-inducible ZF-short were plated into 96-well plates (5,000 cells/well). ZF-short expression was induced by adding 1 µg/ml of tetracycline and 24 h later were incubated with varying concentrations of 5-fluorouracil (Sigma, F6627) for 72 h. Cell viability was determined in quadruplicate using the CellTiter One-Solution-Assay (Promega) and reading absorbance at 490 nm. Proliferation assays with U2OS-HEY1 cells were performed as previously described.3
UV light irradiation assay
To analyze the effect of ZF-short expression on UV-C sensitivity HCT116-ZF-short cells untreated or treated with 1 µg/ml of tetracycline for 24 h to induce ZF-short expression were plated into 60 mm plates (120.000 cells per plate) and 24 h later both groups (± tet) were sham- or UV-C-irradiated as described above. After the treatment fresh medium was added, with or without tetracycline, and cell survival was determined, in quadruplicate, counting the number of cells 4 d later.
Supplementary Material
Acknowledgments
We thank Dr. Vikram Misra for gifts of plasmids and Linn Markert for technical help. This research was supported by the Ministerio de Ciencia e Innovación (SAF2010–21013).
Glossary
Abbreviations:
- APAF
apoptotic peptidase activating factor 1
- ATF/CREB
activating transcription factors/cAMP response element binding
- ATF3
activating transcription factor 3
- bZIP
basic leucine zipper domain
- CREBZF
CREB/ATF bZIP transcription factor
- GST
glutathione S-transferase
- HCT116-ZF-long
HCT116 cells expressing inducible CREBZF-long isoform
- HCT116-ZF-short
HCT116 cells expressing inducible CREBZF-short isoform
- HEY1
hairy/enhancer-of-split related with YRPW motif 1
- HLH
helix-loop-helix domain
- MDM2
p53 E3 ubiquitin protein ligase homolog (mouse)
- p21
cyclin-dependent kinase inhibitor 1A (CDKN1A, Cip1)
- PCR
polymerase chain reaction
- PIG3
p53-induced gene 3
- PUMA
p53 upregulated modulator of apoptosis
- RT-PCR
real-time PCR
- U2OS-ZF-long
U2OS cells expressing inducible CREBZF-long isoform
- U2OS-ZF-short
U2OS cells expressing inducible CREBZF-short isoform
- UV
ultraviolet radiation
- ZF-short
CREBZF-short isoform
- ZF-long
CREBZF-long isoform
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/cc/article/22133
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/22133
References
- 1.Dotto GP. Crosstalk of Notch with p53 and p63 in cancer growth control. Nat Rev Cancer. 2009;9:587–95. doi: 10.1038/nrc2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huang Q, Raya A, DeJesus P, Chao SH, Quon KC, Caldwell JS, et al. Identification of p53 regulators by genome-wide functional analysis. Proc Natl Acad Sci USA. 2004;101:3456–61. doi: 10.1073/pnas.0308562100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Villaronga MA, Lavery DN, Bevan CL, Llanos S, Belandia B. HEY1 Leu94Met gene polymorphism dramatically modifies its biological functions. Oncogene. 2010;29:411–20. doi: 10.1038/onc.2009.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ban J, Bennani-Baiti IM, Kauer M, Schaefer KL, Poremba C, Jug G, et al. EWS-FLI1 suppresses NOTCH-activated p53 in Ewing’s sarcoma. Cancer Res. 2008;68:7100–9. doi: 10.1158/0008-5472.CAN-07-6145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.DeMarzo AM, Nelson WG, Isaacs WB, Epstein JI. Pathological and molecular aspects of prostate cancer. Lancet. 2003;361:955–64. doi: 10.1016/S0140-6736(03)12779-1. [DOI] [PubMed] [Google Scholar]
- 6.Rodriguez V, Chen Y, Elkahloun A, Dutra A, Pak E, Chandrasekharappa S. Chromosome 8 BAC array comparative genomic hybridization and expression analysis identify amplification and overexpression of TRMT12 in breast cancer. Genes Chromosomes Cancer. 2007;46:694–707. doi: 10.1002/gcc.20454. [DOI] [PubMed] [Google Scholar]
- 7.Augenlicht LH, Wadler S, Corner G, Richards C, Ryan L, Multani AS, et al. Low-level c-myc amplification in human colonic carcinoma cell lines and tumors: a frequent, p53-independent mutation associated with improved outcome in a randomized multi-institutional trial. Cancer Res. 1997;57:1769–75. [PubMed] [Google Scholar]
- 8.Stock C, Kager L, Fink FM, Gadner H, Ambros PF. Chromosomal regions involved in the pathogenesis of osteosarcomas. Genes Chromosomes Cancer. 2000;28:329–36. doi: 10.1002/1098-2264(200007)28:3<329::AID-GCC11>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 9.Loukopoulos P, Shibata T, Katoh H, Kokubu A, Sakamoto M, Yamazaki K, et al. Genome-wide array-based comparative genomic hybridization analysis of pancreatic adenocarcinoma: identification of genetic indicators that predict patient outcome. Cancer Sci. 2007;98:392–400. doi: 10.1111/j.1349-7006.2007.00395.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Belandia B, Powell SM, García-Pedrero JM, Walker MM, Bevan CL, Parker MG. Hey1, a mediator of notch signaling, is an androgen receptor corepressor. Mol Cell Biol. 2005;25:1425–36. doi: 10.1128/MCB.25.4.1425-1436.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xie YB, Lee OH, Nedumaran B, Seong HA, Lee KM, Ha H, et al. SMILE, a new orphan nuclear receptor SHP-interacting protein, regulates SHP-repressed estrogen receptor transactivation. Biochem J. 2008;416:463–73. doi: 10.1042/BJ20080782. [DOI] [PubMed] [Google Scholar]
- 12.Contente A, Dittmer A, Koch MC, Roth J, Dobbelstein M. A polymorphic microsatellite that mediates induction of PIG3 by p53. Nat Genet. 2002;30:315–20. doi: 10.1038/ng836. [DOI] [PubMed] [Google Scholar]
- 13.Lu R, Misra V. Zhangfei: a second cellular protein interacts with herpes simplex virus accessory factor HCF in a manner similar to Luman and VP16. Nucleic Acids Res. 2000;28:2446–54. doi: 10.1093/nar/28.12.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kruse JP, Gu W. Modes of p53 regulation. Cell. 2009;137:609–22. doi: 10.1016/j.cell.2009.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim SB, Chae GW, Lee J, Park J, Tak H, Chung JH, et al. Activated Notch1 interacts with p53 to inhibit its phosphorylation and transactivation. Cell Death Differ. 2007;14:982–91. doi: 10.1038/sj.cdd.4402083. [DOI] [PubMed] [Google Scholar]
- 16.Longley DB, Harkin DP, Johnston PG. 5-fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer. 2003;3:330–8. doi: 10.1038/nrc1074. [DOI] [PubMed] [Google Scholar]
- 17.Lu C, El-Deiry WS. Targeting p53 for enhanced radio- and chemo-sensitivity. Apoptosis. 2009;14:597–606. doi: 10.1007/s10495-009-0330-1. [DOI] [PubMed] [Google Scholar]
- 18.Ooyama A, Okayama Y, Takechi T, Sugimoto Y, Oka T, Fukushima M. Genome-wide screening of loci associated with drug resistance to 5-fluorouracil-based drugs. Cancer Sci. 2007;98:577–83. doi: 10.1111/j.1349-7006.2007.00424.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hai T, Hartman MG. The molecular biology and nomenclature of the activating transcription factor/cAMP responsive element binding family of transcription factors: activating transcription factor proteins and homeostasis. Gene. 2001;273:1–11. doi: 10.1016/S0378-1119(01)00551-0. [DOI] [PubMed] [Google Scholar]
- 20.Hogan MR, Cockram GP, Lu R. Cooperative interaction of Zhangfei and ATF4 in transactivation of the cyclic AMP response element. FEBS Lett. 2006;580:58–62. doi: 10.1016/j.febslet.2005.11.046. [DOI] [PubMed] [Google Scholar]
- 21.Xie YB, Nedumaran B, Choi HS. Molecular characterization of SMILE as a novel corepressor of nuclear receptors. Nucleic Acids Res. 2009;37:4100–15. doi: 10.1093/nar/gkp333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bergeron T, Zhang R, Elliot K, Rapin N, Macdonald V, Linn K, et al. The effect of Zhangfei on the unfolded protein response and growth of cells derived from canine and human osteosarcomas. Vet Comp Oncol. 2012 doi: 10.1111/j.1476-5829.2011.00310.x. [DOI] [PubMed] [Google Scholar]
- 23.Misra J, Chanda D, Kim DK, Li T, Koo SH, Back SH, et al. Curcumin differentially regulates endoplasmic reticulum stress through transcriptional corepressor SMILE (small heterodimer partner-interacting leucine zipper protein)-mediated inhibition of CREBH (cAMP responsive element-binding protein H) J Biol Chem. 2011;286:41972–84. doi: 10.1074/jbc.M111.274514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Y, Jin Y, Williams TA, Burtenshaw SM, Martyn AC, Lu R. Amino acid deprivation induces CREBZF/Zhangfei expression via an AARE-like element in the promoter. Biochem Biophys Res Commun. 2010;391:1352–7. doi: 10.1016/j.bbrc.2009.12.059. [DOI] [PubMed] [Google Scholar]
- 25.Yang F, Stenoien DL, Strittmatter EF, Wang J, Ding L, Lipton MS, et al. Phosphoproteome profiling of human skin fibroblast cells in response to low- and high-dose irradiation. J Proteome Res. 2006;5:1252–60. doi: 10.1021/pr060028v. [DOI] [PubMed] [Google Scholar]
- 26.Yan C, Lu D, Hai T, Boyd DD. Activating transcription factor 3, a stress sensor, activates p53 by blocking its ubiquitination. EMBO J. 2005;24:2425–35. doi: 10.1038/sj.emboj.7600712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sun XX, Dai MS, Lu H. 5-fluorouracil activation of p53 involves an MDM2-ribosomal protein interaction. J Biol Chem. 2007;282:8052–9. doi: 10.1074/jbc.M610621200. [DOI] [PubMed] [Google Scholar]
- 28.Jönsson G, Dahl C, Staaf J, Sandberg T, Bendahl PO, Ringnér M, et al. Genomic profiling of malignant melanoma using tiling-resolution arrayCGH. Oncogene. 2007;26:4738–48. doi: 10.1038/sj.onc.1210252. [DOI] [PubMed] [Google Scholar]
- 29.Maris JM, Guo C, White PS, Hogarty MD, Thompson PM, Stram DO, et al. Allelic deletion at chromosome bands 11q14-23 is common in neuroblastoma. Med Pediatr Oncol. 2001;36:24–7. doi: 10.1002/1096-911X(20010101)36:1<24::AID-MPO1007>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 30.Smeets SJ, Braakhuis BJ, Abbas S, Snijders PJ, Ylstra B, van de Wiel MA, et al. Genome-wide DNA copy number alterations in head and neck squamous cell carcinomas with or without oncogene-expressing human papillomavirus. Oncogene. 2006;25:2558–64. doi: 10.1038/sj.onc.1209275. [DOI] [PubMed] [Google Scholar]
- 31.Karhu R, Knuutila S, Kallioniemi OP, Siltonen S, Aine R, Vilpo L, et al. Tampere CLL Group Frequent loss of the 11q14-24 region in chronic lymphocytic leukemia: a study by comparative genomic hybridization. Genes Chromosomes Cancer. 1997;19:286–90. doi: 10.1002/(SICI)1098-2264(199708)19:4<286::AID-GCC12>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 32.Swarts DR, Claessen SM, Jonkers YM, van Suylen RJ, Dingemans AM, de Herder WW, et al. Deletions of 11q22.3-q25 are associated with atypical lung carcinoids and poor clinical outcome. Am J Pathol. 2011;179:1129–37. doi: 10.1016/j.ajpath.2011.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schaid DJ, McDonnell SK, Zarfas KE, Cunningham JM, Hebbring S, Thibodeau SN, et al. Pooled genome linkage scan of aggressive prostate cancer: results from the International Consortium for Prostate Cancer Genetics. Hum Genet. 2006;120:471–85. doi: 10.1007/s00439-006-0219-9. [DOI] [PubMed] [Google Scholar]
- 34.Rain JC, Selig L, De Reuse H, Battaglia V, Reverdy C, Simon S, et al. The protein-protein interaction map of Helicobacter pylori. Nature. 2001;409:211–5. doi: 10.1038/35051615. [DOI] [PubMed] [Google Scholar]
- 35.Belandia B, Orford RL, Hurst HC, Parker MG. Targeting of SWI/SNF chromatin remodelling complexes to estrogen-responsive genes. EMBO J. 2002;21:4094–103. doi: 10.1093/emboj/cdf412. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.