

Abstract
Oxidant stress caused by pathological elevation of reactive oxygen species (ROS) production in the endothelial cells lining the vascular lumen is an important component of many vascular and pulmonary disease conditions. NADPH oxidase (NOX) activated by pathological mediators including angiotensin and cytokines is a major source of endothelial ROS. In order to intercept this pathological pathway, we have encapsulated an indirect NOX inhibitor, MJ33, into immunoliposomes (Ab-MJ33/IL) targeted to endothelial marker platelet endothelial cell adhesion molecule (PECAM-1). Ab-MJ33/IL, but not control IgG-MJ33/IL specifically bound to endothelium and attenuated angiotensin-induced ROS production in vitro and in vivo. Additionally, Ab-MJ33/IL inhibited endothelial expression of the inflammatory marker vascular cell adhesion molecule (VCAM) in cells and animals challenged with the cytokine TNF. Furthermore, Ab-MJ33/IL alleviated pathological disruption of endothelial permeability barrier function in cells exposed to vascular endothelial growth factor (VEGF) and in the lungs of mice challenged with lipopolysaccharide (LPS). Of note, the latter beneficial effect has been achieved both by prophylactic and therapeutic injection of Ab-MJ33/IL in animals. Therefore, specific suppression of ROS production by NOX in endothelium, attainable by Ab-MJ33/IL targeting, may help deciphering mechanisms of vascular oxidative stress and inflammation, and potentially improve treatment of these conditions.
1. Introduction
Endothelium lining the vascular lumen is an important target for therapeutic interventions in cardiovascular, pulmonary, hematological, and other diseases [1-4]. To enhance the precision and efficacy of these interventions, therapeutics should be optimally delivered to endothelial cells. This can be achieved by conjugating drugs and their carriers (e.g., liposomes, viral and polymeric nanoparticle) with ligands of endothelial surface determinants including cell adhesion molecules such as Platelet-Endothelial Cell Adhesion Molecule, PECAM-1 (CD31) [5-8]. Recently, this strategy of vascular immunotargeting yielded several targeted interventions with potential or current translation into the clinical domain [9, 10].
The challenges of containing acute vascular oxidative stress illustrate the need and advantages of vascular immunotargeting. Elevated levels of reactive oxygen species (ROS) in endothelium are implicated in grave acute conditions including ischemia-reperfusion and acute lung injury (ALI or ARDS). In these conditions, pathological mediators including bacterial agents, cytokines and angiotensin II activate endothelial NADPH oxidase (NOX) that flux ROS superoxide anion O2.- in the vascular lumen and within intracellular compartments. In particular, O2.- flux from NOX in the lumen of endothelial endosomes results in pathologically high level of intracellular ROS, causing the NFkB-mediated inflammatory activation [11]. This pathway of vascular oxidative stress and inflammation is manifested, among other endothelial abnormalities, by disruption of vascular permeability barrier and exposure of cell adhesion molecules (e.g., VCAM-1), which further aggravate pathology via edema and mobilization of white blood cells, respectively [12, 13]. Inhibition of acute vascular oxidative stress remains an important, yet elusive, biomedical goal.
Antioxidants, including liposomal formulations of N-acetyl cysteine and long-circulating PEGylated antioxidant enzymes (AOEs) SOD and catalase, quench extracellular ROS and alleviate oxidative stress caused by activated leukocytes [14, 15]. However, these agents have relatively little access to intracellular ROS generated by endothelial NOX. As a result, they have little, if any, effect on pro-inflammatory endothelial activation mediated by endosomal ROS [11]. Vascular immunotargeting can help to overcome the challenges of localization and access. Antioxidant enzymes conjugated with antibodies to PECAM-1, but not untargeted PEG-conjugated enzymes bind to endothelium and enter endothelial endosomes, accumulate in the pulmonary, cerebral and other extended vasculatures after injection. Once there, the antioxidant enzymes quench endothelial ROS and provide protective effects in animal models of acute vascular oxidative stress that are not seen with untargeted antioxidants [11, 16, 17]. Of note, pulmonary endothelium is the privileged site for vascular immunotargeting as it represents ~30% of the total vascular surface in the body and receives >50% of total cardiac blood output. This is advantageous for the treatment of acute pulmonary disorders including ALI, which develops as an uncontrolled pro-inflammatory reaction to massive trauma, cytokines, and bacterial agents such as endotoxin (LPS). ROS produced by NOXs in the pulmonary endothelium represent especially important target for detoxification in this grave condition with high mortality [18].
Even targeted antioxidants cannot quench all produced ROS. ROS escaping detoxification react quickly with sensitive molecules in the target cell. In theory, targeted inhibition of endothelial ROS over- production may offer a helpful alternative or additive intervention. To achieve this goal, we capitalize on attractive pharmacological features of MJ33 (1-hexadecyl-3-trifluoroethylglycero-sn-2-phosphomethanol lithium, C22H43F3O6PLi, MW 498.5 Da), a transition state phospholipid analogue, and indirect suppressor of NOX activity [19]. As Figs. 1A & 1B illustrate, this small molecule (structure shown in Fig 1C) inhibits a cytosolic phospholipase 2 (PLA2) thereby blocking production of lyso-phospholipids and free fatty acids, which are needed for agonist-induced NOX activation [19]. Thus, the net effect of MJ33 in the endothelial cells is inhibition of ROS production in response to pathological mediators including cytokines, angiotensin (Ang) II and ischemia [19]. In this study we have encapsulated MJ33 into PECAM-targeted immunoliposomes (Ab-MJ33/IL) and characterized their delivery and their anti-oxidant and anti-inflammatory effects on the endothelium in vitro and in vivo.
Fig. 1. MJ33 inhibits NOX mediated ROS.
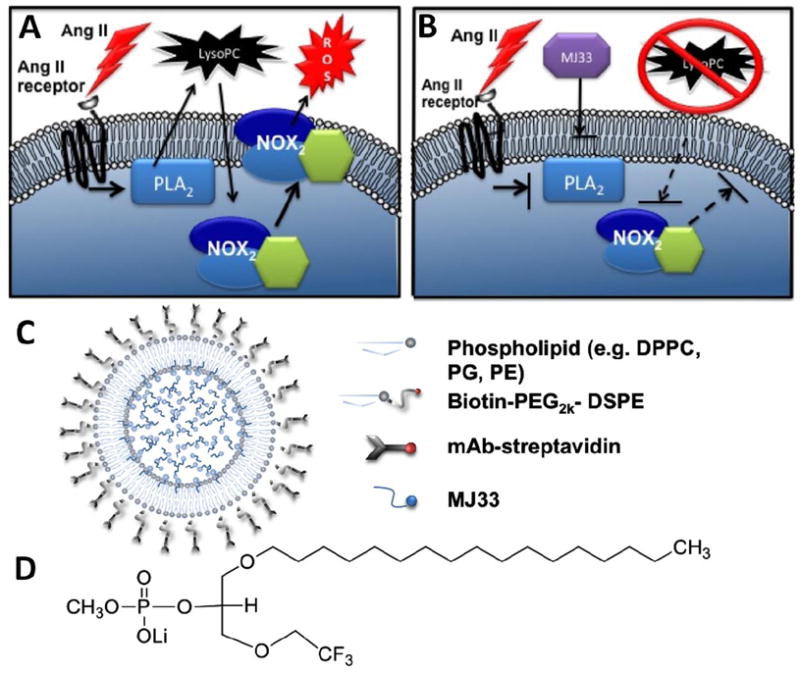
(a) Ang II actives PLA2 thereby releasing lysoPC from the plasma membrane resulting in NOX activation and release of ROS. (b) MJ33 inhibition of PLA2 activity prevents the release of lysoPC whereupon NOX2 does not translocate to the plasma membrane or release ROS. (c) PECAM targeted lipid nanoparticle structure with MJ33 cargo d) MJ33 (1-Hexadecyl-3-(trifluoroethyl)-sn-glycero-2-phosphomethanol, Li) MW 498.5 g/mol.
2. Materials and methods
2.1. Reagents
Lipids including DPPC (1,2-dipalmitoyl-sn-glycero-3-phosphocholine), PC (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine, PG (1,2-di-O-tetradecyl-sn-glycero-3-phospho-(1’-rac-glycerol)) , and cholesterol, MJ33 (1-Hexadecyl-3-(trifluoroethyl)-sn-glycero-2-phosphomethanol, Li), and lipopolysaccharide (LPS, from E. coli B4), ammonium acetate, methanol and chloroform were purchased from Sigma Aldrich (St Louis, MO). Biotinylated PEG 2000 DSPE (1,2-distearoyl -sn-glycero-3-phosphoethanolamine-N-[biotinyl (polyethylene glycol)-2000] and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(carboxyfluorescein)(CF-PE), and 1-heptadecanoyl-2-hydroxy-sn-glycero-3-phosphocholine (17:0 Lyso PC), used as internal standard for LC-MS analysis was purchased from Avanti Polar Lipids (Alabaster, AL). Gases were supplied by BOC Gases (Lebanon, NJ, USA). Kinetex C8 column was from Phenomenex (Torrace, CA, USA). 3H-DPPC (phosphatidylcholin, L-α-dipalmitoyl) American Radiolabeled Chemicals, Inc (St Louis, MO). Succinimidyl 4-[N-maleimidomethyl] cyclohexane-1-carboxylate, N-succinimidyl-S-acetylthioacetate, and N-succinimidyl-biotin (SMCC, SATA, NHS-biotin, respectively) were from Pierce Biotechnology (Rockford, IL). Fluorescent dyes dihydrodichlorofluorescein (H2DCF) diacetate and Amplex Red were purchased from Invitrogen (Carlsbad, CA). Angiotensin II (Ang II) was from Bachem Bioscience (Torrance, CA). Mouse anti-PECAM MEC13.3 was purchased from BD Bioscience (San Jose, CA); monoclonal antibody (mAb 62) against human anti-PECAM was provided by Dr Marian Nakada (Centoor, Malvern, PA). Vascular endothelial growth factor, VEGF came from R&D Systems (Minneapolis, MN). Whole molecule rat IgG, HPLC-grade water, acetonitrile and other reagents were obtained from Fisher Scientific (Pittsburg, PA).
2.2. Cell Culture and Treatment
Human umbilical vein endothelial cells (HUVECs) were purchased at first passage from Lonza Walkersville (Walkersville, MD), and were grown in Falcon tissue culture flasks (BD Biosciences, San Jose, CA) coated with 1% gelatin (Sigma Aldrich) in EGM-BulletKit media (Lonza Walkersville) containing 10% v/v fetal bovine serum (FBS). Passages between 4-6 were used throughout the studies. Confluent HUVECs (105cells/cm2) were used for the binding and the TNF activation studies, 3×105 cells/cm2 were used for the permeability assay. Mouse pulmonary microvascular endothelial cells (MPVECs) were isolated from wild type mice as described [19] and were grown in Dulbecco’s Modified Eagle’s Medium (DMEM) with 10% vol/vol FBS and were used in the MJ33 activity assay. Pancreatic islet mouse endothelial cells (MS1; ATCC, Manassas, VA) were grown in Dulbecco’s Modified Eagle’s Medium (DMEM) with 5% vol/vol FBS and used in the Ang II ROS inhibition study.
2.3. Biotinylated liposome preparation
Liposomes were prepared as previously described [20] but were modified by the addition of the biotinylated PEG2000-DSPE (1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[biotinyl (polyethylene glycol)-2000]) to facilitate Ab conjugation. Briefly, 100 μl of 73.4 mg/ml DPPC, 36.7 μl of 100 mg/ml PC, 148 μl of 10 gm/ml PG, 100 μl of 11.6 mg/ml cholesterol, and 64.3 μl of 10 mg/ml b-DSPE-PEG2000 (all in HPLC grade chloroform) were combined in a glass tube and dried to form a thin film under a steady stream of N2 (5 L/min). For experiments using tracing of radioactivity 0.1 mol % 20 μCi of 3H-DPPC in chloroform was included or 125I-labeled IgG streptavidin (SA), and for those using fluorescent imaging 0.1 mol % 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(carboxyfluorescein)(CF-PE) was included. The thin films were hydrated with 1.0 ml of either MJ33 (concentrations 0.05-0.2 mM in PBS) or PBS by agitation, followed by three freeze-thaw cycles using liquid nitrogen and a 50°C water bath (Branson 1510, Branson Ultrasonics Corporation, Woodbury CT). The liposomes were extruded 10 times through 200 μm polycarbonate filters (Avanti Polar Lipids, Mini-Extruder; Alabaster, Alabama), and with a 1:100 dilution in DI, were measured for hydrodynamic diameter and particle distribution by DLS (90Plus Particle Sizer, Brookhaven Instruments; Holtsville, NY). Efficiency of phospholipid particle formulation was measured by radiotracing 3H-DPPC using a liquid scintillation analyzer (Packard Tri-carb 2900TR, Perkin Elmer, Waltham MA).
2.4. Antibody-streptavidin conjugate preparation
The antibody conjugates were prepared as previously described [21]. Briefly, SMCC was used to introduce stable malemide groups onto SA using a 40-fold excess at room temperature for 1 hour. Simultaneously, sulfhydryls were introduced onto the antibody through primary amine directed chemistry using SATA. A 6-fold excess of SATA was added to the antibodies at room temperature for 30 minutes to achieve 1 sulfhydryl group per IgG molecule. Acetylated sulfhydryls were deprotected using hydroxylamine (50 mM final concentration) and antibodies were conjugated with activated SA at 2:1 molar ratio of IgG:SA. Unreacted components were removed at each step using desalting columns (Thermo Scientific Zeba spin columns; Rockford, IL.)
2.5. Binding of anti-PECAM/SA or IgG/SA conjugates to biotinylated liposomes
Biotinylated liposomes and Ab conjugates were combined and rotated gently at room temperature for 1 h to bind (Grant-Bio PTR-30, Boekel Scientific, Feasterville, PA). Unbound protein and free lipids were separated from Ab conjugated liposomes by ultra centrifuging at 35k rcf for 60 min (Thermo Scientific Sorvall WX Ultraseries Centrifuge Ultra 80, Waltham MA.) Efficiency of binding was measured by radiotracing a 10% addition of 125I-rIgG-SA to the anti-PECAM-SA. Antibody radiolabeling was performed using iodination beads per the manufacturer’s protocol (Pierce Iodination Beads, Thermo Scientific.) Extent of radiolabeling of antibody was measured using a standard triachloroacetic acid (TCA) assay. A 2 l aliquot of labeled rat IgG, 1 ml 3% BSA and 0.2 ml TCA were vortexed and incubated at room temperature for 15 min. Precipitated protein was separated from free iodine supernatant by centrifugation (15 min, 4°C, 2300 g). The size of the biotinylated and Ab bound liposomes was measured by dynamic light scattering (DLS, 90Plus Particle Sizer, Brookhaven Instruments, Holtsville, NY, USA) as previously described [16].
2.6. MJ33 PLA2 activity
Inhibition of PLA2 activity by MJ33 liposomes was measured by tracing release of radiolabeled free fatty acid from 3H-DPPC relative to palmitic acid as a known standard. PLA2 activity in mouse lung homogenate was with radiolabeled liposomes +/- anti-PECAM mAbs as described [20, 22]. Mouse lung homogenate was incubated in Ca2+-free buffer (40mM sodium acetate and 5mM EDTA) at pH 4.0 for 1 h. Lipids were extracted and radiolabeled free fatty acid product was separated by thin layer chromatography using hexane/ether/acetic acid, and analyzed by scintillation counting [20, 22]. To evaluate inhibitors, MJ33 was added to the liposomes at 1 mol%. PLA2 activity was expressed as nmol/hr/mg protein.
2.7. Binding of anti-PECAM/SA or IgG/SA conjugates to endothelial cells
For studies monitoring EC binding by radiotracing of 125I-rIgG-SA included with anti-PECAM Abs on particle surfaces increasing quantities of Ab conjugated radiolabeled liposomes were incubated with confluent HUVECs for 1 hour at 37°C. After incubation with targeted or non-specific rIgG control liposomes, confluent HUVECs were rinsed and lysed (1% Triton X100 in 1N NaOH). Radioactivity in cell lysate was measured and compared to the amount of added activity (Wizard 1470 gamma counter, Wallac, Oy, Turku, Finland). Particle number bound per cell calculations derived from a liposome concentration of 2 × 1013 #/ml [23] and an EC density of 105 cells/well on a 24 well plate.
2.8. Biodistribution of endothelial targeted liposomes
Animal experiments were performed according to the protocol approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania. For tracing, 10% 125I-radiolabeded rIgG-SA were included with targeting mouse anti-PECAM-SA Abs (Mec13.3) bound to 0.2 mM MJ33 containing liposomes. Control liposomes contained only rIgG-SA with an equivalent dosing of tracing isotope.
200 μl aliquots of 1.4 mg total lipid at 1500 cpm/μl were injected intravenously in ketamine/xylazine anesthetized (100/10 mg/kg) C57BL/6 male mice (The Jackson Laboratory, Bar Harbor, ME). After the allotted time the organs were harvested, rinsed with saline, blotted dry and weighed. Radioactivity in organs and 100 μl samples of blood were measured with a Walac 1470 Wizard gamma counter (Perkin Elmer Life and Analytical Sciences-Wallac Oy, Turku, Finland). The results of the 125I measurements were used to calculate the tissue biodistribution injected dose per gram.
2.9. MJ33 pulmonary uptake measured by mass spectroscopy
The amount of MJ33 delivered by targeted ILs to mouse lungs was quantifed by liquid chromatography- mass spectroscopy (LC-MS) from lung homogenates. 200 μl aliquots of MJ33 ILs with 1.4 mg total lipid and pre-purification mass of 20 μmol MJ33 were injected intravenously in anesthetized C57BL/6 mice. After 1 h the mice were euthanized and the lungs were perfused with buffered saline to flush out the blood. Lungs were immediately frozen in liquid nitrogen. Frozen lungs were thawed and homogenized in Tris-HCl buffer with 1% Triton X100, pH 8.3, at 4°C, at a ratio of 1:10 (wt/vol) at medium speed for 1 min/sample (TissueMiser, Fisher Scientific), centrifuged for 10 min at 1000g, whereupon the supernatant was collected.
2.10. Lipids extraction
Lung homogenate (0.5 mL) was transferred to a 10 mL glass test tube containing 1 mL of PBS buffer (1 M, pH 6.8) spiked with 2 ng of 17:0 Lyso PC (20 μL × 1 ng/ μL sol in methanol). 17:0 Lyso PC was chosen as an internal standard (ISTD) since the molecules share a similar molecular weight and the Lyso PC standard has an odd numbered carbon chain (C17) that would not be naturally occurring in the tissues. The retention time of the MJ33 was always determined relatively to the retention time of the ISTD. A control of the tissue extracted without the added 17:0 Lyso PC showed no peak corresponding to 17:0 Lyso PC in the corresponding MS transition (data not shown). For control, 0.5 mL of water was added instead of the lung homogenate, and the sample underwent same procedures as the lung samples. As expected, no MJ33 was detected in the control sample (data not shown).
After adding 2 mL of methanol, the samples were vortexed for 30 sec. Then 4 mL of chloroform was added, and the samples shaken for 30 min. After centrifugation at 2500 RPM for 5 min, the organic layer was transferred to a clean glass tube, and then evaporated to dryness under nitrogen. The dry samples were kept at -80 °C overnight under nitrogen.
2.11. LC-MS conditions
Reverse-phase chromatography for LC-MS experiments were performed using a Waters Alliance 2690 HPLC system (Waters Corp., Milford, MA, USA). Gradient elution was performed in the linear mode. A Kinetex C8 (100 × 2.6 mm i.d., 2.6 μm) was employed with a flow rate of 0.2 mL/min. Solvent A was water with 10 mM ammonium acetate and solvent B was 95% acetonitrile with 10 mM ammonium acetate. The linear gradient was as follows: 30% B at 0 min, 30% B at 2 min, 100 % B at 8 min, 100 % B at 16 min, 30 % B at 16 min, and 30 % B at 25 min. The separation was performed at 30°C. After extraction the samples were re-suspended in 100 μL of methanol and were maintained at 4 °C in the autosampler tray; injections of 20 μL were made.
Mass spectrometry was conducted on a Thermo Finnigan TSQ Quantum Ultra AM mass spectrometer (Thermo Fisher, San Jose, CA) equipped with an electrospray ionization (ESI) source operated in the positive ion mode. Unit resolution was maintained for both parent and product ions for multiple reaction monitoring (MRM) analyses. Operating conditions were as follows: spray voltage was 3500 V, vaporizer temperature was 300 °C, and heated capillary temperature was 280 °C. Nitrogen was used for the sheath gas and auxiliary gas set at 25 and 10 (in arbitrary units), respectively. Collision induced dissociation (CID) was performed using argon as the collision gas at 1.5 mTorr in the second (rf-only) quadrupole. An additional dc offset voltage was applied to the region of the second multipole ion guide (Q0) at 5 V. The following SRM transitions were monitored: MJ33, m/z 493 → 113 (collision energy 18 eV) as qualifier, m/z 493 → 139 (collision energy 15 eV) as quantifier, 17:0 Lyso PC, m/z 510 → 104 (collision energy 18 eV) as qualifier and m/z 510 → 184 (collision energy 15 eV) as quantifier. All data analysis was performed using Xcalibur software, version 2.0 SR2 (Thermo Electron Corporation) from raw mass spectral data.
2.12. Targeted inhibition of Ang II mediated ROS generation by MJ33 in endothelial cells
MS1 cells were grown to confluence on 8 well chamber slides (Ibidi, Martinsried, Germany) and then incubated with +/- Ab-M33/IL for 1 hour in serum free RPMI. Control cells were incubated with equivalent concentrations of PBS liposomes. A 15 min incubation with 10 μM DCFDA (fluorescent dye) and rinse was followed by an addition 100 μM Ang II. ROS production in MS1 cells was measured by fluorescence microscopy in dye-loaded cells. Fluorescent imaging was carried out with limited and equivalent exposure to light and the resulting images were analyzed for fluorescence intensity as a correlation to ROS generation using ImageJ software (Rasband, W.S., ImageJ, U. S. National Institutes of Health, Bethesda, Maryland, USA, 1997-2011.).
2.13. Targeted inhibition of Ang II mediated ROS generation by MJ33 in isolated perfused lungs
In order to test effect of Ab-MJ33/IL in a more physiologically relevant model, we used an isolated perfused mouse lung technique described previously [24]. Intact mice were injected IV with Ab-MJ33/IL vs IgG-MJ33/IL and 30 min later animals were sacrificed. Lungs were isolated and perfused for 60 min with the buffer containing an ROS fluorescent probe Amplex Red and Ang II. 30 min post IV administration of +/- Ab-MJ33/IL mice were anesthetized with 50 mg/kg intraperitoneal sodium pentobarbital, and the lungs were cleared of blood and then removed from the thorax and placed in a perfusion chamber. Lungs were continuously ventilated through a tracheal cannula with 5% CO2 in air (BOC, Murray Hill, NJ) and perfused with recirculating Krebs-Ringer bicarbonate solution supplemented with 10 mM glucose and 3% bovine serum albumin. H2O2 generation was measured by the addition of Amplex Red (50 μM) plus HRP (50 μg/ml) to the perfusate; this fluorophore does not permeate the cell membrane and thus detects extracellular H2O2. Ang II (50 μM) was added to the lung perfusate as a NOX2 agonist. Aliquots of the perfusate were removed during 15-min intervals, and fluorescence intensity was measured (λem/λex = 545/610) using a spectrofluorimeter (Photon Technology International, Inc., Birmingham, NJ) and expressed as arbitrary fluorescence units.
2.14. Western blot analysis
For Western blot cell culture analysis of VCAM in HUVECs, 24 well culture dishes (~105 cells/well) were washed twice with PBS and lysed in 100 ul of sample buffer for sodium dodecyl sulfate polyacrylamide gel electrophoresis. For lung homogenate VCAM detection, lungs were homogenized in a protease inhibitor in PBS for 6 mins with a 5 mm steel ball (TissueLyser II; Qiagen, Valencia, CA), lysed with a solution of 10% Triton X100 and 10% SDS for 1 hr rotating at 4°C, sonicated for 20 sec, and then centrifuged for 10 min at 16k rcf at 4°C. The resulting lung homogenate was removed as supernatant. Lysed cells or lung homogenate were added to a 4-15% gradient gel. Gels were transferred to a PVDF membrane (Millipore, Billerica, MA) that was subsequently blocked with 3% nonfat dry milk in TBS-T (100 mM Tris, pH 7.5; 150 mMNaCl; and 0.1% Tween 20) for 1 h, followed by incubations with primary and secondary antibodies in blocking solution. The blot was detected using ECL Plus reagents (GE Healthcare, New York, NY). Quantification of blots was done using standard densiometry methods (Biorad Fluor-SM, Biorad Laboratories Hercules, CA)
2.15. Endothelial permeability to fluorescein iosothiocyanate-dextran assay
HUVECs were seeded onto gelatin-coated, 24 well transwell 3.0 μm pore-sized culture inserts (Corning Life Sciences, Lowell MA) at ~105 cells/well and were cultured for 72 h to form restrictive endothelial monolayers. Cells were starved overnight and exposed to MJ33 anti-PECAM liposomes or IgG controls with and without VEGF stimulation to induce permeability. After treatment, fluorescein iosothiocyanate (FITC)-dextran (30 kDa, Sigma Aldrich, St. Louis, MO) dissolved in starvation media was added to the apical compartment at a final concentration of 500 μg/ml and allowed to equilibrate for 2 hr. Samples were taken from both apical and basolateral chambers to measure fluorescence. Results were expressed as a percentage of FITC-dextran influx across the HUVEC monolayers.
2.16. Analysis of pulmonary edematous injury by protein level bronchoalveolar lavage
To induce acute pulmonary injury, lipopolysaccharide (LPS) was administered intratracheally, at 1 mg/kg in 50 μl of sterile PBS followed by 100 μl of air, either 15 min after or 60 before intrajugular injections of MJ33 liposomes bearing either anti-PECAM or IgG antibodies. After 24 hours, bronchoalveolar lavage (BAL) was performed by exposing and cannulating the trachea with a 20 gauge angiocatheter (BD Biosciences, Sandy, UT) and then lavaging 3x with a 0.5 ml PBS containing a protease inhibitor cocktail (Sigma Aldrich, St. Louis, MO) at 10 ml/ml [25]. The lavage fluid was centrifuged at 2000 rpm for 4 min, and the supernatant was collected and frozen at -80°C. Protein concentration was measured using a standard BCA assay (Pierce Chemicals; Rockford, IL).
2.17. Statistical analysis
Experimental data were analyszed using a two variable heteroscedastic student t-test. Differences were determined significant at p<0.05.
3. Results
3.1. Synthesis and characterization of PECAM-targeted immunoliposomes encapsulating MJ33
MJ33-containing liposomes were synthesized in which 4.1 mol% of DSPE phospholipid contained biotinylated PEG chains. Efficiency of liposome formation from the phospholipids, measured tracing 3H-DPPC, was 90±4%. Dynamic light scattering (DLS) measurements showed that liposomes have a mean diameter of ~140-150 nm with a polydispersity index of 0.08-0.15. Mass spectrometric analysis showed that concentration of MJ33 (structure shown in Fig. 1D) in IL approached 0.28 mM (50% loading efficiency) (Fig. S2 and Table S1).
Subsequent attachment of antibody/streptavidin (Ab/SA) conjugate via the PEG spacer created immunoliposomes (schematic structure is shown in Fig. 1C). 125I-labeled IgG/SA specifically bound to biotin-PEG-liposomes vs biotin-free PEG-liposomes, to a maximal surface density ~2800 IgG molecules per square micron of the liposome surface, or ~200 IgG molecules per liposome (Fig. 2A). Antibody-coated IL were slightly bigger than their uncoated counterpart, ~155-170 nm (Fig. 2B). The increase in hydrodynamic diameter (~20 nm) corresponds to the addition of IgG layer to the surface. The formulation retained stable size during two months storage in the dark at 4°C, although Ab/IL showed a trend to modest enlargement to ~190 nm (Fig. 2B). MJ33 encapsulated into non-targeted liposomes and Ab-coated ILs exerted equal potency for PLA2 inhibition (Fig. 2C).
Fig. 2. MJ33 immunoliposome characterization.
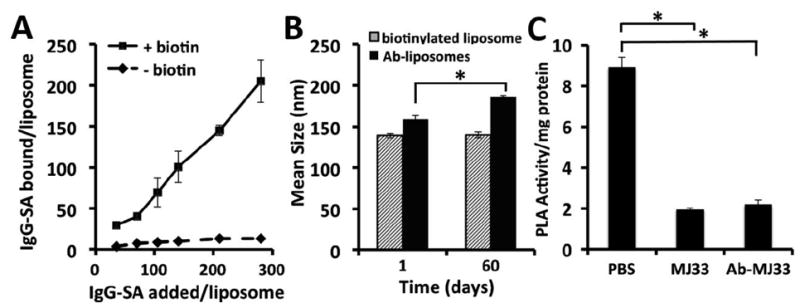
(a) DLS mean hydrodynamic diameter of liposomes measured at 1:40 dilution in ultrapurified DI water. Data shows stability of particle size of the IgG-SA coated liposomes size initially and at two months stored at 4°C. (b) Varying concentrations of radiolabeled IgG-SA were incubated with biotinylated liposomes for 1 hr at RT with unbound IgG-SA separated from ILs by ultracentrifugation. Binding was calculated by comparison of cpm in purified samples relative to cpm added. (c) MJ33 PLA2 activity retained in antibody conjugated liposome formulation. Activity represents cleavage of FFA. Error is standard deviation, n>=3, for all data. Significance p<0.001.
3.2. PECAM-directed endothelial targeting of anti-PECAM-MJ33/IL
Binding to endothelial cells was first tested in cell culture. In the experiment shown in Fig. 3A & B fluorescently labeled Ab/IL or IgG/IL were incubated with mouse pulmonary vascular endothelial cells (MPVECs) for a hour (1-10 ×103 IL/cell added) and after washing, cells were inspected with a fluorescent microscope. This qualitative assay revealed binding of the Ab/IL with negligible binding of the IgG/IL. The liposome targeting was characterized quantitatively through radiotracing. A 10% fraction of 125I-IgG-SA was mixed to the remaining 90% unlabeled IgG-SA or Ab-SA prior to conjugation to liposomes. This approach allows quantitative measurement of cellular uptake and level in organs of the immunoliposomes, while tracing the non-targeted component of the drug delivery system that helps to avoid false-positive results (e.g., due to endothelial binding of detached Ab-SA) [3, 21]. Radioisotope tracing affirmed highly specific endothelial targeting: Ab/IL, but not IgG/IL bound to endothelial cells in a dose-dependent manner, to the level of ~200 liposomes bound per cell (Fig. 3C).
Fig. 3. Anti-PECAM conjugated liposomes bind specifically to endothelial cell targets in vitro.
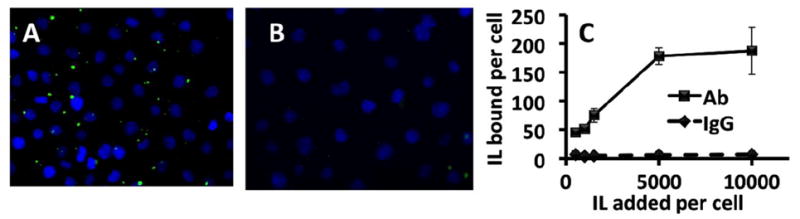
(a) Binding of Ab-MJ33/IL fluorescently labeled with the inclusion of CF-DSPE compared to (b) IgG-ILs. (c) Binding of anti-PECAM (Ab62) and IgG liposomes to cultured HUVECs quantified by radiotracing 125I labeled IgG dosed with unlabeled mAbs at 10% by mass. Expressed data was calculated by comparing radioactivity measures per well of lysed cells post incubation normalized to cell number relative to the activity of known activity and concentration of the liposome aliquot added. n=4, Error bars represent standard deviation.
Next, we studied distribution of Ab/IL injected intravenously in intact mice. As noted previously, the pulmonary vasculature is the preferential site for accumulation of compounds with affinity to the endothelium such as anti-PECAM conjugates, and, therefore, the level of pulmonary uptake of Ab/IL vs IgG/IL characterizes endothelial targeting in vivo [11, 26]. One hour after injection, the pulmonary uptake of Ab/IL was ~200% ID/g (this parameter can exceed 100% since mass of mouse lungs is ~0.1-0.15 g), whereas uptake of non-targeted control IgG/IL did not exceed 10%ID/g (Fig. 4A).
Fig. 4. Biodistribution of anti-PECAM MJ33 liposomes vs IgG control liposomes.
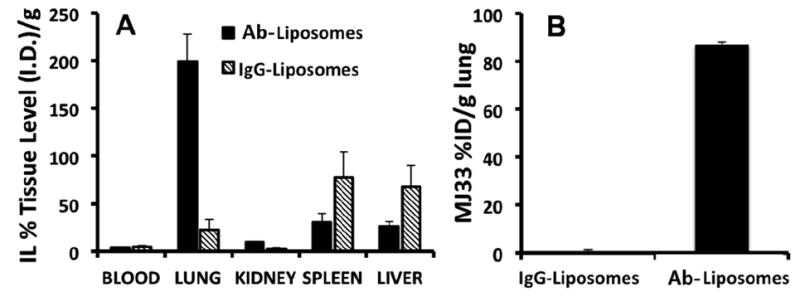
(a) Tissue localization of anti-PECAM targeted liposomes versus IgG controls 60 mins post IV administration. Individual organs were weighed and gamma counted. Biodistribution data shown are calculated as cpm in the organ divided by the injected dose and normalized by mass of organ (%ID/g) (b) Tissue level of MJ33 found in lung homogenate measured by mass spectroscopy.
To detect delivery of the cargo, the level of MJ33 in the lungs of mice injected IV with either IgG-MJ33/IL or Ab-MJ33/IL loaded with MJ33 was determined by liquid chromatography-mass-spectroscopy (LC-MS). The analyses showed that Ab-MJ33/IL delivered 1.63 μg of MJ33 per gram of lung tissue, approximately 200 times more than IgG-MJ33/IL (Figs. 4B, S1, Table S1).
3.3. Inhibition of endothelial ROS production by anti-PECAM-MJ33/IL
The effect of targeting MJ33 on ROS production induced in endotheliacells by a pro-inflammatory vasoactive peptide Ang II was tested in two complementary model systems. In the first, cultured mouse endothelial cells have been sequentially pre-loaded with a fluorescent ROS probe DCFDA, incubated for 30 min with PECAM-targeted or non-targeted liposomes loaded with MJ33, washed to eliminate unbound materials, and then stimulated by Ang II, as previously described [19]. Ang II caused ROS production in endothelium and this reaction was inhibited by Ab-MJ33/IL more effectively than by non-targeted counterpart (Fig. 5A & B).
Fig. 5. Inhibition of ROS production with NOX2 agonist Ang II in vitro and in vivo.
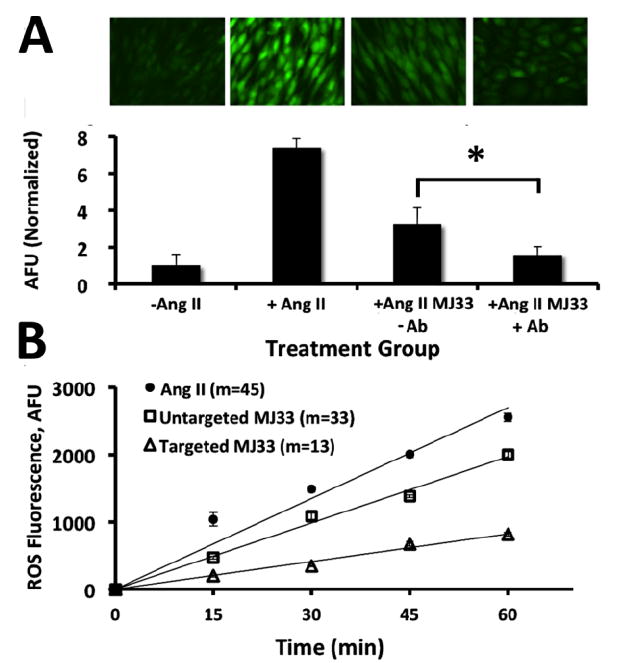
(a) Fluorescence from DCFDA dye representing ROS generation in Ang II treated MS1 cells. Representative fluorescent images in each treatment group taken 10 mins after adminstration of Ang II to ensure equivalance of potential ROS generation. Images left to right: (1) Untreated; (2) Ang II treatment, no MJ33; (3) Ang II + untargeted MJ33 liposomes;(4) Ang II + PECAM targeted MJ33 liposomes (b) Corresponding quantifications of fluorescence. Normalized relative to the untreated control. Quantification based on no less than 6 images. Error bars represent standard deviation. Significance between targeted and untargeted *p < 0.001. (c) Inhibition of ROS production in Ang II (50 mM) treated isolated perfused lung model. Reduction of ROS 2.5 fold by targeted MJ33 liposomes over untargeted from lung perfusate. Fluorescence generated from reaction of Amplex Red with ROS. Values shown represent slopes of the linear regression of the time vs FL units relative to untreated controls. Error bars represent standard deviation. n=3.
In order to test the protective efficacy of Ab-MJ33/IL in a more physiologically relevant model, intact mice were injected IV with Ab-MJ33/IL versus uncoated MJ33 liposomes, and 30 min later animals were sacrificed. Lungs were isolated and perfused for 60 min with the buffer containing an ROS fluorescent probe Amplex Red and Ang II, as we described [19]. Measurement of fluorescent signal in the perfusion buffer of mouse lungs revealed Ang II-induced production of ROS in the pulmonary vasculature. Ang II-induced ROS production was suppressed to a markedly greater extent (nearly 3 fold) in lungs obtained from mice injected with Ab-MJ33/IL vs non-targeted MJ33 liposomes (Fig. 5C). We observed therefore, that targeted delivery of MJ33 to endothelial cells both in vitro and in vivo Ab inhibits ROS production induced by Ang II.
3.4. Inhibition of VCAM expression in the activated endothelium by anti-PECAM-MJ33/IL in vitro and in vivo
To test whether inhibition of endothelial ROS production by Ab-MJ33/IL results in anti-inflammatory effects, we exposed cultured endothelial cells to a potent cytokine, tissue necrosis factor (TNF), that induces synthesis of inducible cell adhesion molecules including VCAM-1 [27]. VCAM-1 detection by Western-blotting showed that TNF caused a profound increase in VCAM-1 that had markedly greater inhibition by Ab-MJ33/IL than by IgG-MJ33/IL (Fig. 6A). Furthermore, the extent of inhibition of VCAM-1 expression was proportional to the dose of targeted liposomes (Fig. S3A).
Fig. 6. Inhibition of pro-inflammatory endothelial activation by targeted MJ33 liposomes in vitro and in vivo.
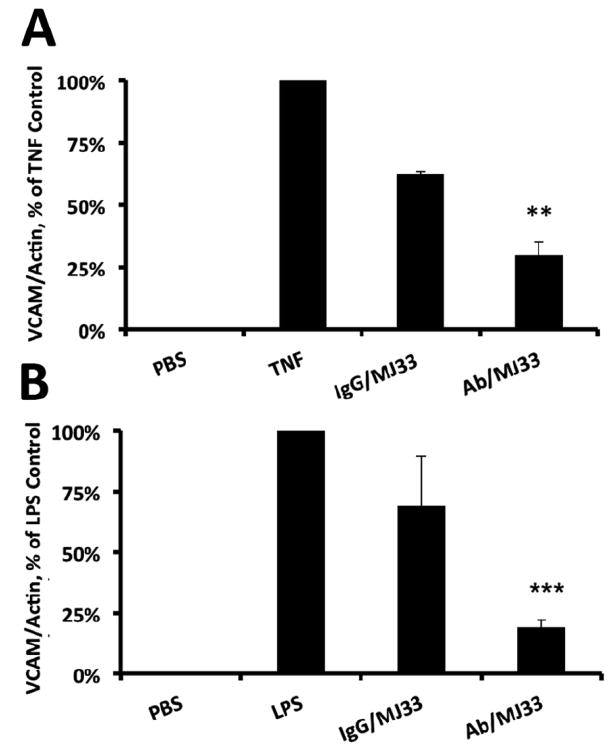
(a) Quantification of VCAM expression normalized to actin from cells treated +/- anti-PECAM (Ab62) ILs challenged by 10 ng/ml TNF for 4 hr. Resulting cell lysates were measured for VCAM by Western blot. Error bars represent standard deviation. **p < 0.005. (b) Quantification of VCAM expression normalized to actin in mice lung homogenates +/- anti-PECAM (Mec13) ILs challenged by intratracheally administered LPS (I mg/kg) 24 hr prior to lung harvest. Resulting lung homogenates were measured for VCAM by Western blot. Error bars represent standard deviation, n=3 for targeted and untargeted ILs. **p<0.001
Cell culture studies permitting prolonged exposures of static cells to liposomes provide less stringent tests for the targeting than studies in intact animals. To affirm the anti-inflammatory effect of MJ33 targeting to the pulmonary endothelium, we used an in vivo model in which an acute inflammatory reaction in the murine lungs was caused by intratracheal administration of bacterial endotoxin LPS, leading to a dramatic elevation of VCAM-1 level in lung tissue (Fig. 6B, supplemental Fig. S3B). In this model, pro-inflammatory VCAM-1 expression has been markedly and statistically significantly suppressed in the animals injected IV by Ab-MJ33/IL one hour prior to LPS challenge, whereas injection of MJ33 loaded in IgG/IL produced a marginal effect at this dose (Fig. 6B).
3.5. Ab-MJ33/IL targeting attenuates pathological endothelial permeability
In the next series, we tested the capacity of PECAM targeted Ab-MJ33/IL to alleviate abnormal vascular permeability, such as induced by VEGF in inflammation. For this purpose, we measured trans-endothelial flux of FITC-dextran across a confluent HUVEC monolayer. VEGF caused marked elevation of permeability across the endothelial barrier. This pathological change has been attenuated to near basal level by targeted Ab-MJ33/IL, but not by IgG-MJ33/IL (Fig. 7A).
Fig. 7. Attenuation of permeability by targeted delivery of MJ33 in vitro and in vivo.
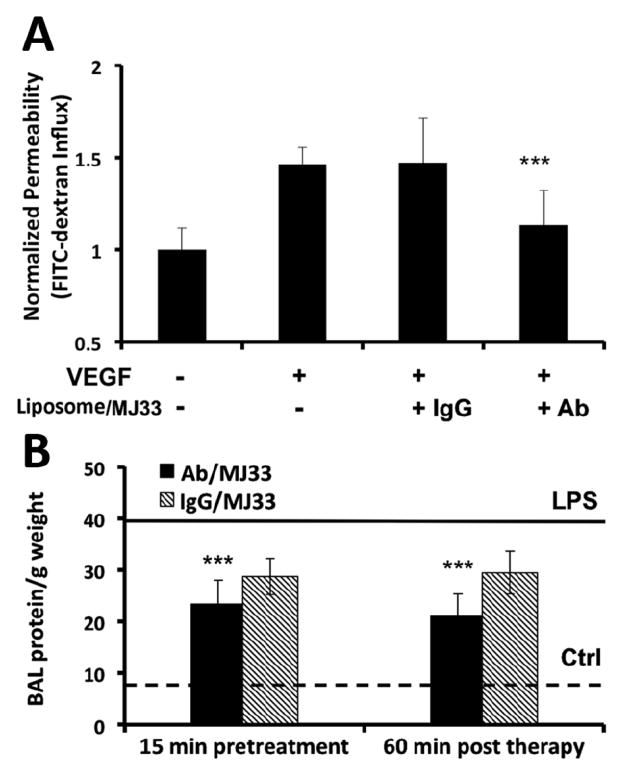
(a) HUVECs incubated with anti-PECAM (Ab62) Ils IgG ILs +/- VEGF challenge were evaluated for permeability by measuring leakage of FITC-dextran across the endothelial monolayer in transwell culture dishes. Fluorescence in top and bottom wells ere measured. Values shown are normalized percentages of the total FITC-dextran crossing the cells relative to that of the untreated controls. Error bars represent st dev. (n=4) ** p<0.001 vs VEGF controls. B. BAL protein with MJ33 treatment 15 mins before and 60 mins post LPS treatment (1 mg/kg intratracheally). The bottom dotted line represents BAL protein/ g weight in unchallenged animals that received sham PBS injections. The top solid line represents BAL protein/g weight LPS challenged animals untreated by ILs. Error bars represent st dev. n>= 4 for all groups. **p<0.001 vs LPS treated controls.
Finally, we tested the effect of both a prophylactic and a therapeutic injection of Ab-MJ33/IL in a model of acute lung injury induced by bacterial LPS installed via trachea in anesthetized mice. Analysis of the BALF showed that one day after LPS challenge the protein level in the BALF is elevated ~4-fold, which reflected disruption of the vascular barrier and alveolar edema (Fig. 7B). Untargeted MJ33 liposome, injected either prior or after the LPS challenge, showed the trend to attenuation of the pulmonary edema, yet the protective effect did not reach significance. However, both prophylactic and therapeutic injection of the same dose of MJ33 encapsulated in Ab-MJ33/IL afforded markedly more potent and statistically significant protective effect, attenuating alveolar edema by approximately 50% (Fig. 7B).
4. DISCUSSION
Vascular immunotargeting of drug carriers utilizing antibodies and their fragments binding to endothelial surface determinants including cell adhesion molecules is a hot area of translational research [28-32]. Thus, endothelial targeting of antioxidant and anti-inflammatory agents is being explored as a means for more specific and effective treatment of vascular ischemia, oxidative stress and inflammation [33-37]. In the present study, we have combined two well-characterized elements of endothelial immunotargeting (PECAM antibodies and PEG-liposomes), in order to devise targeted delivery of a novel pharmacological cargo, MJ33, which offers a new antioxidant mechanism, namely, indirect inhibition of cellular ROS production.
Enzymes of the PLA family are known to hydrolyze phospholipids in cellular membranes, causing release of an array of degradation products [38]. One of the products of phospholipid breakdown, lyso-phospholipids, is known to mediate agonist-induced assembly and activation of constitutively silent NOX2 in leukocytes and endothelium, inducing ROS production in these cells [39]. These ROS constitute an important part of the response to infection, serving as bactericidal agents and components of inflammatory signaling cascades. However, ROS overzealously produced by NOX2 in non-septic pro-inflammatory conditions (e.g., massive trauma, bleeding or cytokine release) contribute to acute vascular oxidative stress and may promote the development of disease states such as acute lung injury (ALI) and the adult respiratory distress syndrome (ARDS) [40].
MJ33 competitively inhibits PLA2 activity [41] and, via this mechanism, suppresses ROS production by NOX, as Fig. 1 illustrates [19]. MJ33 has been shown to suppress production of ROS induced by diverse agonists in endothelial and white blood cells in cell cultures and in vivo [42, 43]. In theory, MJ33 may more fully suppress oxidative and signaling effects of the PLA2-NOX system than ROS quenching by alternative antioxidant interventions, including delivery or transfection of AOEs. Mechanistically, MJ33-based therapeutics are specific for ROS generated by NOX2, whereas enzymatic and non-enzymatic antioxidants decompose ROS produced by xanthine oxidase, mitochondria, and other enzymatic systems. Specific suppression of PLA2-NOX system may help to dissect the role of this pathway in the pathophysiology of acute vascular oxidative injury. From a translational perspective, the combination of AOE-based and MJ33-based therapeutics -- which inhibit ROS via distinct mechanisms -- may have a synergistic effect in preventing ALI and ARDS.
Initial studies of pharmacological properties of MJ33 for the alleviation of pulmonary oxidative stress employed its delivery via the airways using classical PEG-free phospholipid liposomes to achieve delivery to alveolar cells [19]. Resident alveolar macrophages and migrant leukocytes are the predominant source of ROS in this compartment and represent the target cells in this application. Indeed, this approach of PLA2 inhibition by liposomal MJ33 provided a significant protection against acute oxidative stress in the lungs of animals exposed to hyperoxia [44]. This compound does not appear to cause acute toxic effects; experiments in mice have shown that animals tolerate up to 2 μmol of MJ33, a dose 1,000 times greater than what is needed to provide antioxidant effects [45].
In this study, we devised vascular immunotargeting of MJ33 encapsulated in PEG-liposomes to endothelial cells, as an alternative to alveolar delivery. Endothelial cells play a key role in pulmonary oxidative stress and inflammation, typical of the ALI/ARDS, ischemia/reperfusion and some other pulmonary and cardiovascular maladies. Using antibodies to PECAM-1 conjugated to the end groups of PEG-chains on the liposomes, we achieved targeted delivery of MJ33 to endothelium in vitro and in vivo after intravascular administration (Figs. S3 and 4A). This strategy markedly enhanced potency of MJ33 in terms of inhibition of endothelial ROS production induced by Ang II (Fig. 5) and alleviation of endothelial pro-inflammatory changes manifested by VCAM1 expression (Fig. 6) and barrier disruption (Fig. 7). The targeting and functional features of Ab-MJ33/IL have been tested both in vitro and in vivo, demonstrating attenuation of LPS-induced pulmonary edema in both prophylactic and therapeutic modes of intervention (Fig 7B).
Of note, in some cases in vivo testing revealed more profound augmentation of antioxidant effect of MJ33 by targeting vs. corresponding testing in cell culture (Fig. 6B). Most likely, this reflects more stringent limitations on non-targeted delivery to endothelium in vivo, mediated, among other factors, by elimination of drug from circulation and detachment of untargeted liposomes from endothelium by hemodynamic forces. Taken together, results of this study unequivocally indicate that endothelial targeting significantly improves the protective effects of MJ33 in animal models of acute pulmonary oxidative stress and inflammation.
It is interesting to note that non-targeted MJ33/IL also produced detectable protective effects. At the doses and regimens used in the present study, these effects were inferior to those afforded by targeted formulation, but favorable toxicity profile allows wide range of MJ33 doses, providing significant effects in models of oxidative stress. The effective dose of M33 has been shown to be 1000x less than that of other PLA2 inhibitors [45]. Most likely, the source of action of IgG-MJ33/IL is via inhibition of NOX in leukocytes or resident macrophages of the reticulo-endothelial system (RES). High levels of hepatic and splenic uptake (Fig. 4A) indicate that these cells take up the large fraction of circulating nanoparticles - likely via phagocytosis as well as Fc-receptor mediated mechanisms. From the translational standpoint, replacing whole targeting antibody by Fc-lacking fragments will further enhance the specificity of delivery and effect.
In addition to having translational potential as an antioxidant and anti-inflammatory intervention, we believe that targeted MJ33 ILs represent an important tool for deciphering the role of the endothelial PLA2/NOX system in human disease. Since the precise contribution of endothelial and leukocyte NOX to vascular oxidative stress is not clear, it is difficult to speculate at the present time whether MJ33 delivery to endothelial cells, as described in this paper, will have a decisive advantage over untargeted liposomes. Most likely, every approach in antioxidant protection has some utility, dictated by the role of specific ROS-mediated mechanisms in given pathologies. Thus, suppression of leukocyte ROS production and detoxification of extracellular ROS may provide additional or alternative protective effects to those afforded by endothelium-targeted antioxidants. Systematic comparative studies of targeted vs. untargeted MJ33 delivery in various models of human vascular oxidative stress seem warranted to better understand mechanism of action and evaluate translational potential of these novel therapeutics.
5. Conclusion
In this study we have devised targeting of the experimental antioxidant agent, MJ33, to endothelial cells and demonstrated that this approach markedly augments protective effects of MJ33 against vascular oxidative stress mediated by reactive oxygen species produced in response to diverse pathological mediators including Ang II, TNF, VEGF and LPS in vitro and in animals. Therefore, targeting of the agent(s) suppressing ROS production by NOX in endothelium represents a specific tool for deciphering mechanisms of vascular oxidative stress and inflammation, as well as prospective translational approach for management of disease conditions involving this pathological pathway.
Supplementary Material
Acknowledgments
EDH, CFG, JH, and VRM acknowledge NIH grants R01 HL087036 and R01 HL073940. CM and IB acknowledge NIH grant P30ES013508. ABF and CD acknowledge RO1-HL105509.
Footnotes
None of the authors have any conflicts of interest to declare
The work in this manuscript was done at the University of Pennsylvania, Philadelphia PA, 19104
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Simone E, Ding BS, Muzykantov V. Targeted delivery of therapeutics to endothelium. Cell Tissue Res. 2009;335:283–300. doi: 10.1007/s00441-008-0676-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Muro S, Garnacho C, Champion JA, Leferovich J, Gajewski C, Schuchman EH, Mitragotri S, Muzykantov VR. Control of endothelial targeting and intracellular delivery of therapeutic enzymes by modulating the size and shape of ICAM-1-targeted carriers. Mol Ther. 2008;16:1450–1458. doi: 10.1038/mt.2008.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shuvaev VV, Ilies MA, Simone E, Zaitsev S, Kim Y, Cai S, Mahmud A, Dziubla T, Muro S, Discher DE, Muzykantov VR. Endothelial Targeting of Antibody-Decorated Polymeric Filomicelles. ACS Nano. 2011;5:6991–6999. doi: 10.1021/nn2015453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reynolds PN, Zinn KR, Gavrilyuk VD, Balyasnikova IV, Rogers BE, Buchsbaum DJ, Wang MH, Miletich DJ, Grizzle WE, Douglas JT, Danilov SM, Curiel DT. A Targetable, Injectable Adenoviral Vector for Selective Gene Delivery to Pulmonary Endothelium in Vivo. Mol Ther. 2000;2:562–578. doi: 10.1006/mthe.2000.0205. [DOI] [PubMed] [Google Scholar]
- 5.Atochina EN, Balyasnikova IV, Danilov SM, Granger DN, Fisher AB, Muzykantov VR. Immunotargeting of catalase to ACE or ICAM-1 protects perfused rat lungs against oxidative stress. Am J Physiol. 1998;275:L806–817. doi: 10.1152/ajplung.1998.275.4.L806. [DOI] [PubMed] [Google Scholar]
- 6.Massey KA, Schnitzer JE. Targeting and Imaging Signature Caveolar Molecules in Lungs. Proceedings of the American Thoracic Society. 2009;6:419–430. doi: 10.1513/pats.200903-011AW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pasqualini R, McDonald DM, Arap W. Vascular targeting and antigen presentation. Nat Immunol. 2001;2:567–568. doi: 10.1038/89704. [DOI] [PubMed] [Google Scholar]
- 8.Scherpereel A, Wiewrodt R, Christofidou-Solomidou M, Gervais R, Murciano JC, Albelda SM, Muzykantov VR. Cell-selective intracellular delivery of a foreign enzyme to endothelium in vivo using vascular immunotargeting. FASEB J. 2001;15:416–426. doi: 10.1096/fj.00-0022com. [DOI] [PubMed] [Google Scholar]
- 9.Giordano RJ, Edwards JK, Tuder RM, Arap W, Pasqualini R. Combinatorial Ligand-directed Lung Targeting. Proceedings of the American Thoracic Society. 2009;6:411–415. doi: 10.1513/pats.200903-014AW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ding BS, Gottstein C, Grunow A, Kuo A, Ganguly K, Albelda SM, Cines DB, Muzykantov VR. Endothelial targeting of a recombinant construct fusing a PECAM-1 single-chain variable antibody fragment (scFv) with prourokinase facilitates prophylactic thrombolysis in the pulmonary vasculature. Blood. 2005;106:4191–4198. doi: 10.1182/blood-2005-05-2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shuvaev VV, Han J, Yu KJ, Huang S, Hawkins BJ, Madesh M, Nakada M, Muzykantov VR. PECAM-targeted delivery of SOD inhibits endothelial inflammatory response. FASEB J. 2011;25:348–357. doi: 10.1096/fj.10-169789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vandenbroucke E, Mehta D, Minshall R, Malik AB. Regulation of Endothelial Junctional Permeability. Annals of the New York Academy of Sciences. 2008;1123:134–145. doi: 10.1196/annals.1420.016. [DOI] [PubMed] [Google Scholar]
- 13.Fu P, Birukov KG. Oxidized phospholipids in control of inflammation and endothelial barrier. Translational Research. 2009;153:166–176. doi: 10.1016/j.trsl.2008.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fan J, Shek PN, Suntres ZE, Li YH, Oreopoulos GD, Rotstein OD. Liposomal antioxidants provide prolonged protection against acute respiratory distress syndrome. Surgery. 2000;128:332–338. doi: 10.1067/msy.2000.108060. [DOI] [PubMed] [Google Scholar]
- 15.Mitsopoulos P, Suntres ZE. Protective Effects of Liposomal N-Acetylcysteine against Paraquat-Induced Cytotoxicity and Gene Expression. Journal of Toxicology. 2011;2011 doi: 10.1155/2011/808967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chorny M, Hood E, Levy RJ, Muzykantov VR. Endothelial delivery of antioxidant enzymes loaded into non-polymeric magnetic nanoparticles. J Control Release. 2010;146:144–151. doi: 10.1016/j.jconrel.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Han J, Shuvaev VV, Muzykantov VR. Catalase and SOD conjugated with PECAM antibody distinctly alleviate abnormal endothelial permeability caused by exogenous ROS and vascular endothelial growth factor. J Pharmacol Exp Ther. 2011 doi: 10.1124/jpet.111.180620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frey RS, Ushio-Fukai M, Malik AB. NADPH oxidase-dependent signaling in endothelial cells: role in physiology and pathophysiology. Antioxid Redox Signal. 2009;11:791–810. doi: 10.1089/ars.2008.2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chatterjee S, Feinstein SI, Dodia C, Sorokina E, Lien YC, Nguyen S, Debolt K, Speicher D, Fisher AB. Peroxiredoxin 6 phosphorylation and subsequent phospholipase A2 activity are required for agonist-mediated activation of NADPH oxidase in mouse pulmonary microvascular endothelium and alveolar macrophages. J Biol Chem. 2011;286:11696–11706. doi: 10.1074/jbc.M110.206623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fisher AB, Dodia C, Feinstein SI, Ho YS. Altered lung phospholipid metabolism in mice with targeted deletion of lysosomal-type phospholipase A2. J Lipid Res. 2005;46:1248–1256. doi: 10.1194/jlr.M400499-JLR200. [DOI] [PubMed] [Google Scholar]
- 21.Dziubla TD, Shuvaev VV, Hong NK, Hawkins BJ, Madesh M, Takano H, Simone E, Nakada MT, Fisher A, Albelda SM, Muzykantov VR. Endothelial targeting of semi-permeable polymer nanocarriers for enzyme therapies. Biomaterials. 2008;29:215–227. doi: 10.1016/j.biomaterials.2007.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fisher AB, Dodia C. Role of phospholipase A2 enzymes in degradation of dipalmitoylphosphatidylcholine by granular pneumocytes. J Lipid Res. 1996;37:1057–1064. [PubMed] [Google Scholar]
- 23.Hansen CB, Kao GY, Moase EH, Zalipsky S, Allen TM. Attachment of antibodies to sterically stabilized liposomes: evaluation, comparison and optimization of coupling procedures. Biochim Biophys Acta. 1995;1239:133–144. doi: 10.1016/0005-2736(95)00138-s. [DOI] [PubMed] [Google Scholar]
- 24.Zhang Q, Matsuzaki I, Chatterjee S, Fisher AB. Activation of endothelial NADPH oxidase during normoxic lung ischemia is KATP channel dependent. Am J Physiol Lung Cell Mol Physiol. 2005;289:L954–961. doi: 10.1152/ajplung.00210.2005. [DOI] [PubMed] [Google Scholar]
- 25.Kinniry P, Amrani Y, Vachani A, Solomides CC, Arguiri E, Workman A, Carter J, Christofidou-Solomidou M. Dietary Flaxseed Supplementation Ameliorates Inflammation and Oxidative Tissue Damage in Experimental Models of Acute Lung Injury in Mice. The Journal of Nutrition. 2006;136:1545–1551. doi: 10.1093/jn/136.6.1545. [DOI] [PubMed] [Google Scholar]
- 26.Muzykantov VR, Christofidou-Solomidou M, Balyasnikova I, Harshaw DW, Schultz L, Fisher AB, Albelda SM. Streptavidin facilitates internalization and pulmonary targeting of an anti-endothelial cell antibody (platelet-endothelial cell adhesion molecule 1): a strategy for vascular immunotargeting of drugs. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:2379–2384. doi: 10.1073/pnas.96.5.2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thomas SR, Witting PK, Drummond GR. Redox Control of Endothelial Function and Dysfunction: Molecular Mechanisms and Therapeutic Opportunities Antioxidants & Redox Signaling. 2008;10:1713–1766. doi: 10.1089/ars.2008.2027. [DOI] [PubMed] [Google Scholar]
- 28.Chrastina A, Valadon P, Massey KA, Schnitzer JE. Lung vascular targeting using antibody to aminopeptidase P: CT-SPECT imaging, biodistribution and pharmacokinetic analysis. Journal of vascular research. 2010;47:531–543. doi: 10.1159/000313880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chittasupho C, Xie SX, Baoum A, Yakovleva T, Siahaan TJ, Berkland CJ. ICAM-1 targeting of doxorubicin-loaded PLGA nanoparticles to lung epithelial cells. European journal of pharmaceutical sciences : official journal of the European Federation for Pharmaceutical Sciences. 2009;37:141–150. doi: 10.1016/j.ejps.2009.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fakhari A, Baoum A, Siahaan TJ, Le KB, Berkland C. Controlling ligand surface density optimizes nanoparticle binding to ICAM-1. Journal of pharmaceutical sciences. 2011;100:1045–1056. doi: 10.1002/jps.22342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Karjalainen K, Jaalouk DE, Bueso-Ramos CE, Zurita AJ, Kuniyasu A, Eckhardt BL, Marini FC, Lichtiger B, O’Brien S, Kantarjian HM, Cortes JE, Koivunen E, Arap W, Pasqualini R. Targeting neuropilin-1 in human leukemia and lymphoma. Blood. 2011;117:920–927. doi: 10.1182/blood-2010-05-282921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Staquicini FI, Moeller BJ, Arap W, Pasqualini R. Combinatorial vascular targeting in translational medicine. Proteomics. Clinical applications. 2010;4:626–632. doi: 10.1002/prca.200900213. [DOI] [PubMed] [Google Scholar]
- 33.Klyachko NL, Manickam DS, Brynskikh AM, Uglanova SV, Li S, Higginbotham SM, Bronich TK, Batrakova EV, Kabanov AV. Cross-linked antioxidant nanozymes for improved delivery to CNS. Nanomedicine : nanotechnology, biology, and medicine. 2012;8:119–129. doi: 10.1016/j.nano.2011.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hegeman MA, Cobelens PM, Kamps J, Hennus MP, Jansen NJG, Schultz MJ, van Vught AJ, Molema G, Heijnen CJ. Liposome-encapsulated dexamethasone attenuates ventilator-induced lung inflammation. British Journal of Pharmacology. 2011;163:1048–1058. doi: 10.1111/j.1476-5381.2011.01314.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nowak K, Weih S, Metzger R, Albrecht RF, 2nd, Post S, Hohenberger P, Gebhard MM, Danilov SM. Immunotargeting of catalase to lung endothelium via anti-angiotensin-converting enzyme antibodies attenuates ischemia-reperfusion injury of the lung in vivo. Am J Physiol Lung Cell Mol Physiol. 2007;293:L162–169. doi: 10.1152/ajplung.00001.2007. [DOI] [PubMed] [Google Scholar]
- 36.Muzykantov VR. Delivery of antioxidant enzyme proteins to the lung. Antioxid Redox Signal. 2001;3:39–62. doi: 10.1089/152308601750100489. [DOI] [PubMed] [Google Scholar]
- 37.Muzykantov VR. Targeting of superoxide dismutase and catalase to vascular endothelium. J Control Release. 2001;71:1–21. doi: 10.1016/s0168-3659(01)00215-2. [DOI] [PubMed] [Google Scholar]
- 38.Kumar Jain M, Gelb MH, Rogers J, Berg OG. [21] Kinetic basis for interfacial catalysis by phospholipase A2. In: Daniel LP, editor. Methods in enzymology. Academic Press; 1995. pp. 567–614. [DOI] [PubMed] [Google Scholar]
- 39.Henderson LM, Chappell JB, Jones OTG. The superoxide-generating NADPH oxidase of human-neutrophiles is electrogenic and associated with an H+ channel. Biochem J. 1987;246:325–329. doi: 10.1042/bj2460325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Babior BM. The NADPH oxidase of endothelial cells. IUBMB life. 2000;50:267–269. doi: 10.1080/713803730. [DOI] [PubMed] [Google Scholar]
- 41.al-Mehdi AB, Dodia C, Jain MK, Fisher AB. A phospholipase A2 inhibitor decreases generation of thiobarbituric acid reactive substance during lung ischemia-reperfusion. Biochim Biophys Acta. 1993;1167:56–62. doi: 10.1016/0005-2760(93)90217-w. [DOI] [PubMed] [Google Scholar]
- 42.Fisher AB, Dodia C, Chander A, Jain M. A competitive inhibitor of phospholipase A2 decreases surfactant phosphatidylcholine degradation by the rat lung. Biochem J. 1992;288(Pt 2):407–411. doi: 10.1042/bj2880407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fisher AB, Dodia C. Lysosomal-type PLA2 and turnover of alveolar DPPC. Am J Physiol Lung Cell Mol Physiol. 2001;280:L748–754. doi: 10.1152/ajplung.2001.280.4.L748. [DOI] [PubMed] [Google Scholar]
- 44.Manevich Y, Fisher AB. Peroxiredoxin 6, a 1-Cys peroxiredoxin, functions in antioxidant defense and lung phospholipid metabolism. Free Radic Biol Med. 2005;38:1422–1432. doi: 10.1016/j.freeradbiomed.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 45.Lee I, Zagorski J, Dodia C, Feinstein S, Fisher AB. Safety evaluation of MJ33 as a potential in vivo inhibitor of NADPH oxidase (NOX2) activation. The FASEB Journal. 2012;26:658–656. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.