

Abstract
We have examined CD40-dependent signals in endothelial cells (EC) mediating the expression of vascular endothelial growth factor (VEGF) and VEGF-induced angiogenesis. We treated confluent cultures of EC with soluble CD40L (sCD40L), and by Western blot found a marked increase in the phosphorylation of Akt, 4EBP-1, and S6K1, compared with untreated cells. EC were transfected with a full-length VEGF promoter-luciferase construct and cultured in the absence or presence of rapamycin and sCD40L. We found that rapamycin, which blocks mTORC1 and mTORC2 signaling, inhibited sCD40L-mediated transactivation of VEGF. In addition, by Western blot, we found that the transfection of EC with small interfering RNA (siRNA) to rictor (to inhibit mTORC2), and not raptor (to inhibit mTORC1), inhibited sCD40L-dependent protein expression of VEGF. In addition, we found that basal levels of phosphorylated Akt as well as VEGF were increased in EC transfected with the raptor siRNA. Also, rapamycin failed to inhibit VEGF promoter activation, as well as VEGF protein expression in EC transfected with a constitutively active construct of Akt, further demonstrating that mTORC1 is not necessary for CD40- and Akt-induced expression of VEGF. Finally, we injected human CD40L-transfected fibroblasts or mock transfectants into human skin on SCID mice. We found that the injection of CD40L transfectants, but not mock cells, resulted in VEGF expression and mediated a marked angiogenesis reaction, and this response was reduced in mice treated with rapamycin. Together, these observations indicate that mTORC2 and Akt facilitate CD40-inducible expression of VEGF in EC, which is of clinical importance in tumor growth and the progression of chronic inflammatory diseases.
CD40 is a 50-kDa member of the TNF receptor superfamily of molecules that has a wide distribution of expression on different cell types of hematopoietic and nonhematopoietic lineages, including mature T cells, B cells, monocyte/ macrophages, vascular endothelial cells (EC)4 and epithelial cells (1, 2). Its ligand, CD40L (also called CD154, gp39, TRAP-1, TBAM), is a 39-kDa membrane protein expressed by many cell types within the immune system, but is most notable on activated T cells and platelets (2–4). Interactions between CD40L and CD40 are well established to promote T and B cell activation and the maturation of myeloid-derived APCs and to facilitate both cellular and humoral immune responses (2, 5–7). Indeed, studies using CD40- and CD40L-deficient mice have confirmed a major role for CD40 in immunity as well as in several immune inflammatory diseases (8–10). Furthermore, humans with genetic mutations of CD40L, rendering the protein incapable of binding to CD40, have been found to have reduced circulating levels of immunoglobulins and have defects in T cell function and cell-mediated immunity (11). Collectively, these observations have defined CD40L-CD40 interactions to be crucial for adaptive immune responses.
Nevertheless, beyond its function on immune cells, CD40-induced responses have also been found to mediate angiogenesis (12–15). We previously reported that the ligation of CD40 on EC results in the induced expression of vascular endothelial growth factor (VEGF) in vitro and in vivo (12, 13, 16, 17), and we found that blockade of VEGF inhibits CD40-induced EC proliferation in vitro (12), and angiogenesis in vivo (12, 13). We concluded that CD40L-CD40 interactions and the associated expression of VEGF represent a mechanistic link between immunity and the development of angiogenesis. Moreover, others have suggested that the effect of CD40-induced signaling on EC proliferation, migration and tube formation may also be independent of VEGF, and relate to direct effect(s) on intracellular signaling mediated by the kinase Akt (18, 19).
Recently, Chiodoni et al. (14, 15) demonstrated that CD40-inducible responses in EC, mediated by platelet CD40L, are also of functional importance in early tumor growth. Using a transgenic mouse mammary tumor model, it was observed that tumor growth was significantly delayed in CD40-deficient mice. Moreover, using bone marrow transplantation of wild-type immune cells into CD40-deficient animals (and CD40 deficient marrow into wild-type mice), it was demonstrated that the reduced tumor growth in CD40-deficient animals was independent of an effect on immunity, but rather was dependent on CD40 expression by local vascular endothelial cells. In light of these recent observations, it was suggested that interruption of CD40L-CD40 interactions, CD40-inducible expression of VEGF and/or angiogenesis will have significant therapeutic implications for the prevention of the progression of breast cancer (14, 15).
Therapeutically, the blockade of CD40-inducible responses have the potential to inhibit pathological angiogenesis (13, 15). Indeed, agents that inhibit platelet CD40L expression inhibit local CD40-inducible angiogenesis (14). CD40-inducible signals in EC leading to the expression of VEGF involve the activation of Ras, PI3K, and Akt (16, 18). mTOR, the mammalian target of rapamycin, is a serine/threonine kinase that exists in mammalian cells as two distinct protein complexes, mTORC1 and mTORC2. mTORC1 is composed of mTOR, mLST8, and raptor and regulates a number of major cellular processes including nutrient metabolism, ribosome biogenesis, and mRNA translation (20). The activation of Akt and Akt-induced signals result in the phosphorylation of mTOR and mTORC1-induced responses. In contrast, mTORC2 which is composed of mTOR, mLST8, rictor, sin1, protor1, protor2 and PRR5 regulates Akt activity as well as actin polymerization (21–24). Thus, both of these mTOR complexes and their associated signals are intricately associated with the activation of Akt, and Akt-induced responses. The association between raptor or rictor and mTOR has been shown to be critical for the function of mTORC1 and mTORC2, respectively (20, 21). These observations indicate that the assembly of each of these complexes is the rate limiting step for select mTOR signaling responses.
In this study, we describe a role for mTOR in CD40L-CD40-induced activation of Akt, CD40-inducible expression of VEGF as well as the associated angiogenesis reaction. Our findings provide a rationale for the use of mTOR inhibitors therapeutically (alone or in combination with platelet inhibitors (14, 15)) in diseases in which the stimulation of CD40 in EC and the associated angiogenesis reaction are known to be of functional consequence.
Materials and Methods
Reagents
Soluble CD40L was purchased from Ancell. Abs against S6K1, phosphorylated S6K1, 4E-BP1, phosphorylated 4E-BP1, Akt, phosphorylated Akt, mTOR, and raptor were purchased from Cell Signaling Technologies, anti-VEGF was obtained from Santa Cruz Biotechnology, and the anti-rictor Ab was purchased from Bethyl Laboratories as well as Cell Signaling Technologies. LY294002 was purchased from Calbiochem and rapamycin was gifted to the laboratory by Wyeth-Ayest Research.
Cell culture
Human umbilical endothelial cells were isolated from umbilical cords as previously described (12) and cultured in M199 (Cambrex), supplemented with 20% FBS (Life Technologies-BRL Products), endothelial growth supplement, 1% penicillin/streptomycin, L-glutamine, and heparin. Cultured EC were used for our studies at passages 2–4.
Plasmids
A VEGF promoter luciferase construct containing the 2.6 kb full-length VEGF promoter sequence was gifted to the laboratory by Debabrata Mukhopadhyay, (Mayo Clinic, Minneapolis, MN) and was used as we previously described (16, 17). The dominant negative mutant of PI3K, wild-type Akt construct, dominant negative mutant Akt (T308A, S473A) were gifted to the laboratory and used as described (16, 25). The constitutive dominant active mutant of Akt (T308D, S473D) was generated and used as we described (24). In pilot studies, we found that the transfection of either 100 ng or 1 μg of plasmid DNA resulted in high levels of expression of each construct within transfected EC. All current studies were performed using 100 ng of DNA. In addition, in each experiment, EC were transfected with the empty expression vector (pcDNA3) as a control. In some controls, expression of the desired protein (PI3K or Akt) in transfected cells was examined by Western blot.
VEGF protein assays
In pilot studies, we found that the quantification of secreted VEGF in our serum-starved EC cell cultures using ELISA gave inconsistent results, even in circumstances where secreted VEGF was functional for endothelial proliferation (12). In contrast, in previous studies, we have found that the quantification of VEGF protein expression in our cultured EC is optimal using Western blot (16). Thus, in these studies, we have evaluated the regulation of VEGF protein expression using Western blot. EC were washed in PBS and lysed with ice-cold RIPA buffer (Boston Bioproducts) as described (16, 24). Lysates were centrifuged at 4°C for 30 min and samples containing 50 μg of protein were separated on a polyacrylamide gel with Tris-glycine-SDS running buffer (Bio-Rad), and were subsequently transferred onto a polyvinylidene difluoride membrane (NEN) for 1 h. Membranes were blocked with 5% milk in TBS-Tween 20 and incubated with the primary Ab overnight. Membranes were washed and incubated with a secondary peroxidase linked Ab and the reactive bands were detected by chemiluminescence (Pierce).
Transfection assays
Transfection assays were performed using the Effectene transfection reagent according to the manufacturer’s instructions (Qiagen) and as described (25). Promoter luciferase activity was measured using a standard assay kit (Promega), and the difference between experimental samples were calculated as the fold change in luciferase counts, compared with untreated cells. As a positive control for transfection efficiency, in occasional cultures, we transfected cells with the β-galactosidase gene under control of cytomegalovirus immediate early promoter and assessed β-galactosidase activity as we described (16).
siRNA
mTOR small interfering RNA (siRNA) and control siRNA were purchased from Qiagen. Rictor and raptor were purchased from Dharmacon. Transfection of EC with siRNA (40 nM) was performed using lipofectamine 2000 according to the manufacturer’s instructions (Invitrogen). In brief, subconfluent EC, grown in six-well plates, were cultured in serum free medium with siRNA and after 6 h of transfection, the cells were washed one time and cultured in regular medium. Expression of target genes were assessed by Western blot to confirm the efficiency of siRNA transfection and knockdown vs control siRNA transfected cells.
In vivo assessment of angiogenesis
Full-thickness human neonatal foreskin grafts were transplanted onto CB17 SCID beige mice (Taconic Farms) as previously described (13, 26). Following engraftment for 4 – 6 wk, the human skin was injected intracutaneously with 35 μl of growth factor reduced Matrigel (BD Biosciences) mixed with either 2.5 × 106 murine fibroblasts stably transfected with human CD40L (CD40L cells) or mock transfectants (gifted by Cees Van Kooten, University of Leiden, Leiden, The Netherlands) each suspended in 35 μl saline (total volume of 70 μl). Before injection, the transfectants were irradiated at 7500 rads. Rapamycin (0.3 mg/kg/day or 1.5 mg/kg/day) was administered to the SCID mice by i.p. injection and the human skin grafts were harvested 7 days later. Immunohistochemistry was performed for the evaluation of vWF as described (13, 26).
Immunohistochemistry
Four micron cryostat cut frozen sections were fixed in acetone for 5 min and incubated with a rabbit anti-human vWF Ab (DakoCytomation) in PBS with 3% human serum for 1 h at room temperature. Subsequently, the specimens were incubated with a species-specific peroxidase-conjugated secondary Ab (Jackson Immuno Research) and were developed with 0.25 mg/ml 3-amino-ethylcarbazole in 2% N,N-dimethylformamide and 0.1 mol/L sodium acetate buffer (pH 5.2) with 0.03% hydrogen peroxide as described (13, 26). Finally, sections were counterstained in hematoxylin and mounted in glycerol gelatin.
Quantification of angiogenesis
Vessels identified by vWF staining were counted using a standard grid-counting method at ×400 magnification by light microscopy, as previously described (13, 26). For each group, the vessels within four to six adjacent nonoverlapping fields of each skin specimen were counted by two independent observers in a blinded manner, and the mean vessel counts for each specimen was calculated.
Statistics
Data were compared using the Student’s t test or the Mann-Whitney U test as indicated. p values <0.05 were considered statistically significant.
Results
Ligation of CD40 by CD40L activates mTORC1 in endothelial cells
We first evaluated the phosphorylation of mTOR (Ser2448) as well as 4E-BP1 (Ser 65) and S6K1 (Thr 389), (two well-established downstream targets of mTORC1 (27)) in EC following stimulation by soluble CD40L (sCD40L). By Western blot, we found that the phosphorylation of mTOR increased following treatment of EC with sCD40L, peaking in expression after 15 min and persisting for greater than 1 h. In contrast, the phosphorylation of 4E-BP1 and S6K1 increased after 15 min, peaked after 30 min and decreased 60 min following treatment (Fig. 1A). To demonstrate that the phosphorylation of 4E-BP1 and S6K1 was mediated by mTORC1, we pretreated EC with the mTOR inhibitor rapamycin before stimulation with sCD40L, and found that it prevented sCD40L-mediated expression of p4E-BP1 and pS6K1 as assessed by Western blot (Fig. 1B).
FIGURE 1.

Ligation of CD40 on EC activates mTOR and mediates mTORC1 signaling. A, Confluent cultures of EC were serum starved over-night and treated with 3 μg/ml sCD40L for the indicated times. Cells lysates were prepared and Western blotting was performed for total and phosphorylated mTOR, 4E-BP1, and S6K1. B, Confluent cultures of EC were serum starved overnight and treated with 10 ng/ml rapamycin 1 h before stimulation with 3 μg/ml sCD40L for 30 min. Western blotting analysis was performed using Abs to total and phophorylated 4E-BP1 and S6K1. C, upper panel, EC were transfected with a dominant negative mutant of PI3K (DN PI3K, 100 ng) or the empty vector (pcDNA3) as a control. After 48 h, the cells were serum starved overnight and treated with sCD40L (3 μg/ml) for 30 min. A Western blot illustrating total and phosphorylated S6K1 is shown. Lower panel, Protein expression of PI3K and Actin in EC following transfection with control pcDNA3 or the DN PI3K, evaluated by Western blot. D, EC were transfected with a dominant negative mutant of Akt (DN Akt, 100 ng) or pcDNA3 as a control and were treated with sCD40L as in C. A Western blot was performed to evaluate the expression of total and phosphorylated S6K1. Lower panel, The expression of Akt and Actin were evaluated by Western blot in empty vector (pcDNA3) and DN Akt-transfected EC. E, Serum starved EC were cultured with the PI3K inhibitor LY294002 (20 μM) for 1 h and stimulated with sCD40L (3 μg/ml) for 30 min. A Western blot of total and phosphorylated S6K1 was performed. The illustrated blots are representative of at least three experiments, all with similar results.
Because it is known that treatment of EC with sCD40L results in the activation of PI3K and Akt (16, 18), we next wished to evaluate whether these kinases are intermediaries in CD40-inducible mTORC1 activation. We transiently transfected EC with dominant negative (DN) mutants of either PI3K or Akt, and subsequently treated the cells with sCD40L and evaluated S6K1 phosphorylation by Western blot. As illustrated in Fig. 1, C and D, we found that each DN mutant significantly reduced sCD40L-inducible phosphorylation of S6K1 as compared with the response in EC transfected with the empty expression vector (pcDNA3). In addition, we treated EC with LY294002, a pharmacological PI3K inhibitor, before treatment with sCD40L, and again we found a reduction in S6K1 phosphorylation (Fig. 1E). Therefore, CD40-induced signaling in EC results in the activation of PI-3K and Akt, and mediates signaling via mTORC1.
mTORC2 and CD40-mediated signaling in EC
We have recently reported that the treatment of EC with the mTOR inhibitor rapamycin reduces Akt phosphorylation as well as its kinase activity via inhibition of mTORC2 (24). Furthermore, it is known that rapamycin can inhibit mTORC1-mediated responses in EC (19, 24). Next, we used siRNAs to knockdown rictor or raptor and disrupt mTORC2 or mTORC1 assembly respectively, before treatment with sCD40L. Following transfection of EC with each siRNA and stimulation with sCD40L, the phosphorylation of Akt was evaluated by Western blot. As expected (16), we observed that treatment with sCD40L resulted in an induction in Akt phosphorylation in control siRNA transfected EC (Fig. 2A). Moreover, we found that knockdown of mTOR and rictor, but not raptor, markedly blocked basal levels of pAkt expression as well as sCD40L-induced phosphorylation of Akt (Fig. 2A). The efficiency of knockdown following transfection with our siRNAs (including raptor) was confirmed in control blots (Fig. 2B). Therefore, we interpret these observations to indicate that mTORC2, and not mTORC1, mediates the activation of Akt in response to CD40 signaling. Furthermore, as illustrated in Fig. 2C, we found that pretreatment of EC with rapamycin for 12 h (to disrupt assembly of mTORC2; Ref. 24) inhibited the phosphorylation of Akt, including both basal and sCD40L-inducible levels of expression. Thus, the ligation of CD40 on EC results in rictor-dependent (and thus, mTORC2-associated) phosphorylation of Akt, thereby eliciting Akt-induced responses.
FIGURE 2.

mTORC2 is critical for CD40-inducible activation of Akt. A, EC were transfected with control, mTOR, rictor or raptor siRNA. Thirty-six hours following transfection, the EC were serum starved for 12 h and subsequently stimulated with sCD40L (3 μg/ml) for 30 min. EC were lysed and subjected to Western blot analysis using anti-phospho Akt (pAktS473) or anti-Akt. Western blots of siRNA transfected cells confirmed efficient knockdown of mTOR, rictor and raptor in our EC (data not shown). B, Control blot in which EC were transfected with control, mTOR, rictor, or raptor siRNA, and after 48 h, Western blot analysis was performed using anti-mTOR, anti-rictor, or anti-raptor to confirm efficiency of knockdown. C, EC were serum starved for 12 h and treated with rapamycin (10 ng/ml) for an additional 12 h before stimulation with sCD40L (3 μg/ml) for 30 min. Illustrated is a representative Western blot using anti-phospho-Akt (pAktS473) and anti-Akt. One of three experiments with similar findings.
CD40-induced expression of VEGF involves mTORC2-dependent signals
As discussed above, the ligation of CD40 on EC results in mTORC1- and mTORC2-dependent signaling. Further, these signals result in VEGF expression and VEGF-dependent angiogenesis (12). To next investigate the function of mTORC1 and mTORC2 in CD40-inducible expression of VEGF, EC were transfected with our full-length VEGF promoter-luciferase construct (12, 17) and cultured in the absence or presence of rapamycin and sCD40L. As expected (12), treatment with sCD40L augmented VEGF promoter activity, and we found that this activity was totally blocked by rapamycin (Fig. 3A). Similarly, by Western blot, we observed that rapamycin blocked sCD40L-mediated induction of VEGF protein expression (Fig. 3B). To further evaluate which mTOR complex regulates the expression of VEGF, we next transfected EC with mTOR, rictor, or raptor siRNAs and cultured them in the absence or presence of sCD40L. Similar to that observed with rapamycin, we found that sCD40L failed to induce VEGF expression in EC transfected with the mTOR siRNA (Fig. 3C). In addition, by Western blot, we found that knockdown of rictor significantly inhibited sCD40L-mediated expression VEGF. However, sCD40-induced expression of VEGF was similar to controls in EC transfected with the raptor siRNA (Fig. 3C and data not shown). We also found that the endogenous expression of VEGF was reduced in EC transfected with the rictor siRNA. In contrast, we repeatedly observed that basal levels of VEGF were increased in EC transfected with the raptor siRNA (Fig. 3C). These data indicate that mTORC2, and not mTORC1 is functional in both basal as well as CD40-inducible expression of VEGF. Furthermore, these observations suggest that mTORC1 may regulate a negative signaling loop for endogenous VEGF expression.
FIGURE 3.

mTORC2 regulates CD40-inducible expression of VEGF in EC. A, EC were transfected with a full-length 2.6-kb VEGF promoter-luciferase construct. Twenty-four hours following transfection, the cells were stimulated with sCD40L (3 μg/ml) in the absence or presence of rapamycin (10 ng/ml) for an additional 24 h. The cells were lysed and promoter activity was calculated as the fold change in luciferase counts from each group of cells, compared with untreated cells. Illustrated are the mean results of three independent experiments (± 1 SD). p values were calculated using the Student’s t test. Panel B, EC were cultured for 12 h in the absence or presence of rapamycin (10 ng/ml) before stimulation with sCD40L (3 μg/ml) for an additional 24 h. The cells were lysed and the expression of VEGF protein was analyzed by Western blot. The illustrated blot is representative of three experiments with similar results. The expression of β-tubulin served as an internal control. C, EC were transfected with control, mTOR, rictor, or raptor siRNA, and after 24 h, were cultured in the absence or presence of sCD40L (3 μg/ml) for an additional 24 h. Subsequently, the cells were lysed and the expression of VEGF was examined by Western blot (labeled WB). As a control for siRNA transfection and knockdown, each lysate was also simultaneously evaluated for the expression of either mTOR, rictor or raptor by Western blot, as indicated. The expression of actin served as an internal control. Each blot is representative of four with similar results.
To further determine the function(s) of mTORC2 and Akt in CD40L-CD40-inducible expression of VEGF, we first cotransfected EC with our full-length VEGF promoter-luciferase construct and a DN mutant of Akt, or an empty vector as a control. We found that the DN mutant of Akt significantly inhibited the ability of sCD40L to increase VEGF transcriptional activity (Fig. 4A). Furthermore, by Western blot, we found that sCD40L-inducible protein expression of VEGF was reduced in EC transfected with the DN mutant of Akt, as compared with empty vector transfected cells (Fig. 4B). We next transiently transfected EC with an Akt expression vector in which the S473 and T308 sites of Akt were mutated to encode a constitutively active (and mTORC2-independent) form of the kinase (called 2DAkt) (24). As illustrated in Fig. 4C, we found that the transfection of EC with the 2DAkt construct resulted in the transactivation of VEGF, and further, we found that this response was not inhibited by rapamycin. By Western blot, we additionally observed that the transfection of EC with 2DAkt increased VEGF protein expression, and that rapamycin failed to inhibit the response (Fig. 4D). Thus, while mTORC1 may be sufficient to mediate VEGF expression, for instance under conditions of hypoxia (28, 29), it is not necessary for endogenous or CD40-inducible expression.
FIGURE 4.

CD40-inducible expression of VEGF is mediated by mTORC2-Akt interactions. A, EC were cotransfected with the full-length 2.6-kb VEGF promoter-luciferase construct and either a dominant negative mutant of Akt (DN Akt) or an empty expression vector (pcDNA3) as a control. Twenty-four hours later, transfected EC were treated with sCD40L (3 μg/ml) as indicated. Promoter activity was assessed as the relative fold change in luciferase counts from each group of cells as compared with untreated empty vector transfected cells. Illustrated are the mean results of three independent experiments (± 1 SD). B, EC were transfected with the DN mutant of Akt or control pcDNA3, rested for 24 h and treated with sCD40L (3 μg/ml) for an additional 24 h. The cells were subsequently lysed and subjected to Western blot for the analysis of VEGF expression, or β-tubulin as a control. Representative of three experiments with similar findings. C, EC were cotransfected with the VEGF promoter-luciferase construct and a dominant active mutant of Akt (2D Akt) or the empty expression vector (pcDNA3) as a control (labeled ‘c’). Twenty-four hours later, transfected EC were cultured in the absence or presence of rapamycin (10 ng/ml) for an additional 24 h. VEGF promoter activity was assessed as the relative fold change in luciferase counts from each group of cells as compared with untreated empty vector transfected cells. Illustrated are the mean results of three experiments (± 1 SD). D, Thirty six hours following transfection of EC with the dominant active 2DAkt construct or control pcDNA3 (labeled ‘c’), the cells were treated with rapamycin (10 ng/ml) for an additional 24 h. Cells were lysed and analyzed by Western blot for the expression of VEGF, and β-tubulin as an internal control. One of three independent experiments is illustrated. A and C, p values were calculated using the Student’s t test.
Function of mTOR in CD40-inducible angiogenesis in vivo
To evaluate the physiological relevance of our in vitro observations, we next assessed whether pharmacological inhibition of mTOR in vivo prevents CD40L-mediated angiogenesis. For these studies, we used an established model of human angiogenesis in human skin (13, 26). Neonatal foreskin was transplanted onto SCID beige mice, using standard procedures and was allowed to heal for 4 – 6 wk. We have previously found that this period of healing is sufficient to enable stabilization of the healing angiogenesis reaction in the skin graft, and that ~50% of EC in the healed skin remain of human origin (30). Untransfected murine fibroblasts (mock cells) or murine fibroblasts stably expressing human CD40L (CD40L cells) were injected intracutaneously into the human skin on the SCID mice, and the mice were divided into three groups: untreated or treated with rapamycin 0.3 mg/kg/day or 1.5 mg/kg/day (Fig. 5, A and B). The skins were harvested after 7 days and were evaluated for the development of angiogenesis by immunostaining with anti-vWF and standard quantitative grid counting analysis. We found that the mean vessel density in the skins injected with mock cells was identical with that observed in untreated skins (data not shown and Ref. 13). In addition, as expected, mean vessel density was significantly increased in the skins injected with CD40L (1.7 fold increase vs mock cells, p < 0.05, Fig. 5B), whereas the vessel density observed in skins harvested from SCID mice treated with rapamycin (1.5 mg/kg/ day) was similar to that found in untreated mice and negative controls ( p < 0.05 vs untreated mice, Fig. 5B***********). Lower doses of rapamycin (0.3 mg/kg/day) did not visibly inhibit the angiogenesis reaction. Finally, we evaluated the expression of VEGF by Western blot in skins following injection of mock or CD40L-expressing cells. As illustrated in Fig. 5C, we found that VEGF expression was marginally increased in skins injected with CD40L cells, as compared with those injected with mock cells. Moreover, we found that both baseline as well as induced expression of VEGF was decreased in the skins harvested from mice that were treated with rapamycin (1.5 mg/kg/d). We conclude that the inhibition of mTOR with rapamycin is sufficient to block both CD40L-inducible expression of VEGF and CD40L-mediated angiogenesis in vivo.
FIGURE 5.
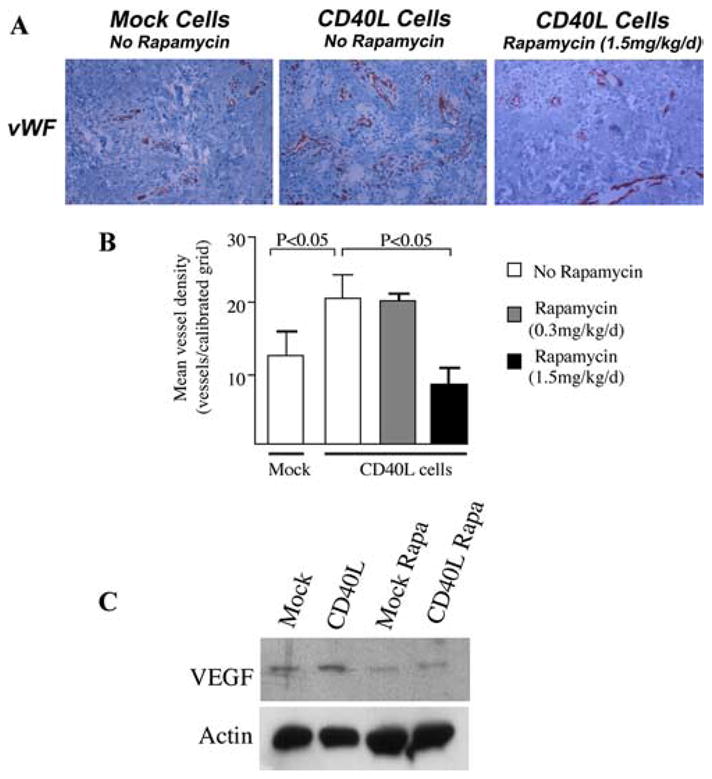
Function of mTOR in CD40L-mediated angiogenesis in vivo. SCID mice bearing human skin transplants received intracutaneous injections of mock transfectant cells (Mock Cells) or CD40L-expressing murine fibroblasts (CD40L Cells), and were left untreated or were treated with rapamycin (0.3 mg/kg/day or 1.5 mg/kg/day) by i.p injection. The human skin was harvested after 7 days and evaluated for angiogenesis by immunohistochemical staining for vWF (A and B), and for VEGF expression by Western blot (C). A, Representative photomicrographs of vWF staining of endothelial cells in Mock- or CD40L-injected skins (magnification ×400) from untreated or rapamycin treated mice. B, Mean vessel density (± 1 SD) in skins following injection of Mock cells (n = 5), CD40L cells in untreated animals (n = 11), or in animals treated with rapamycin (0.3 mg/kg/d, n = 10) or rapamycin (1.5 mg/kg/d, n = 8). p values were calculated using the Mann-Whitney U test. C, Representative Western blot of VEGF expression in Mock- and CD40L-injected skins harvested from mice untreated or treated with rapamycin (Rapa, 1.5 mg/kg/d). Representative of three with similar results.
Discussion
In this report, we define mTOR and mTOR-dependent regulation of Akt in EC as a critical signal for basal as well as CD40L-CD40-inducible expression of VEGF in vitro. Further, we demonstrate the possibility of a feedback signaling loop whereby mTORC1 negatively regulates both Akt activity and VEGF overexpression. A model of our observations is illustrated as a cartoon in Fig. 6. We also find that rapamycin (which targets mTORC2 and Akt in EC; Ref. 24) inhibits sCD40L-inducible expression of VEGF as well as the associated angiogenesis reaction in vivo. Collectively, these observations define mTOR as a critical signaling molecule for CD40L-CD40 mediated angiogenesis, which is of clinical importance in tumor growth and in the progression of chronic inflammatory diseases.
FIGURE 6.

Model illustrating CD40-induced signals that mediate the expression of VEGF in endothelial cells. Our previous report (16) demonstrated that the ligation of CD40 mediates VEGF expression through the Ras and PI3K signaling pathway. In these studies, we found a critical role for mTOR and Rictor (mTORC2) in CD40-induced phosphorylation of Akt (arrow 1), a function for Akt in VEGF expression (arrow 2) and a function for mTORC2 in CD40-induced expression of VEGF (arrow 3). In addition, following activation with a dominant active construct of Akt, we found that a pharmacological mTOR inhibitor did not block VEGF expression. Rather, inhibition of mTORC1 via knockdown of raptor resulted in an increased phosphorylation of Akt (arrow 4) and an increase in endogenous expression of VEGF (arrow 5). Our data provide for a model in which mTORC1 activity provides negative feedback for endogenous VEGF expression, but CD40-inducible expression is dependent on mTORC2-Akt signaling.
It is well established that Akt-induced responses in EC are potent to facilitate angiogenesis (19, 31). Although some reports have partially elucidated a role for mTOR and mTORC1 in the inducible expression of VEGF (28), our studies have demonstrated that the CD40-induced response involves mTORC2. Because the ligation of CD40 on EC mediates the activation of Akt, and since Akt induces the proliferation of EC in part via mTORC1 and S6K, one possibility is that some of the proangiogenic effects of CD40 may also involve mTORC1-inducible signals. mTORC1 has been found to regulate HIF-1α, which faciliates the transactivation of VEGF expression (28). However, our previous studies determined that CD40-inducible transactivation of VEGF is independent of HIF-1α (17). Using VEGF promoter deletion constructs, we found that CD40-inducible activation of VEGF was similar using either our full-length VEGF promoter construct or the HIF-1 promoter deletion construct. Further, using promoter deletion constructs, we found that CD40-inducible expression of VEGF involves other transactivators, and/or involves novel signaling molecules such as the transcriptional repressor protein MeCP2 (17). Our findings in this report are supportive of this mechanism, and demonstrate that factor(s) leading to the transactivation of VEGF are regulated by mTORC2 and Akt, and not mTORC1. Knockdown of rictor using siRNA (to inhibit mTORC2 assembly), but not knockdown of raptor (to inhibit mTORC1), prevented sCD40L-mediated VEGF expression. Therefore, although mTORC1 may be sufficient to mediate VEGF expression (for instance via HIF-1α and/or other transcription factors; Refs. 28, 29), we suggest that mTORC2 is both necessary and sufficient for CD40-inducible expression. Further, our observations indicate that this mTORC2-mediated response is also dependent on the activation of Akt, and that Akt-inducible signaling is of major importance for both baseline as well as inducible expression of VEGF.
Interestingly, we found that inhibition of mTORC1-mediated responses increased basal levels of pAkt, as well as VEGF protein expression. Also, although not as notable, there was also some minimal effect on CD40-inducible levels of VEGF expression (Figs. 2 and 3, and data not shown). Although this observation is again consistent with a critical role for mTORC2 and Akt in the inducible expression of VEGF, it also suggests that mTORC1 regulates negative feedback signaling loops (illustrated in Fig. 6). For instance, blockade of mTORC1 may result in the inhibition of S6K-mediated down-regulation of IRS-1 activity, which positively regulates PI3K and Akt-inducible signaling, and subsequently results in an increase in pAkt levels and VEGF expression. Alternatively, it is possible that blockade of mTORC1 may feed back to inhibit other regulators of Akt activation (such as PTEN), so that the phosphorylation of Akt is increased. Nevertheless, these findings are consistent with our observations that mTORC2 and Akt are the key regulatory signaling molecules for CD40-induced expression of VEGF.
Activated T cells and platelets express CD40L, and CD40 is expressed on many cell types within most tissue beds (1–4). In our previous studies using our human skin transplant/SCID mouse model, we observed that CD40 is expressed on resident EC, macrophages, and keratinocytes, as well as other cell types within human skin grafts (13). Therefore, it is possible that the local injection of CD40L-expressing cells into the human skin results in interactions between CD40L and CD40 on non-EC. Nevertheless, we found that the associated angiogenesis reaction is dependent on CD40L-inducible expression of VEGF, since treatment of the SCID mice with a blocking anti-human VEGF Ab significantly inhibited the effect of CD40L on the response (13). In the current study, we find that treatment of mice with rapamycin inhibits both CD40L-inducible expression of VEGF in vivo and the associated angiogenesis response. Therefore, we suggest that the local infiltration of tissues with CD40L-expressing cells can mediate VEGF expression and angiogenesis via mTOR-dependent signaling pathways in both EC and non-EC.
The observations identified in this study may have significant therapeutic implications. For instance, CD40L blockade (using the platelet inhibitor clodipogel) has been shown to inhibit angiogenesis and to slow the growth of breast tumors by inhibiting CD40-dependent responses in EC (14, 15). Our findings are suggestive that mTOR inhibitors alone or in combination with platelet antagonists may be additive in this effect, and thus may warrant consideration in the development of future therapeutics. In addition, several chronic immune inflammatory diseases (such as arthritis, inflammatory bowel disease, asthma, and chronic allograft rejection) are known to be associated with increases in local CD40 and VEGF expression (32–38), and blockade of VEGF has been found to attenuate disease activity (39 – 42). Thus, the identification of mTOR as a key intermediary in CD40-inducible and proangiogenic signaling in EC (13) could also lead to clinically relevant therapeutic interventions for immune-mediated angiogenic diseases in the future.
Acknowledgments
We thank Joren Madsen for ongoing constructive comments about these studies. In addition, we acknowledge Debabrata Mukhopadhyay for the gift of VEGF constructs and for helpful comments and insight, and we thank Aninda Basu for helpful discussions about the techniques used in this report. Finally, we thank the maternity staff of South Shore Hospital (Weymouth, MA) for help in the supply of umbilical cords used for the generation of endothelial cells.
Footnotes
This work was supported by National Institutes of Health Grant AI046756 (to D.M.B.). O.D. was supported by a Transplantation Fellowship from Swiss House for Advanced Research and Education-Novartis and the SICPA foundation.
Abbreviations used in this paper: EC, endothelial cell; mTOR, mammalian target of rapamycin; mTORC, mammalian target of rapamycin complex; VEGF, vascular endothelial growth factor; CD40L, CD40 ligand; sCD40L soluble CD40L; DN, dominant negative.
Disclosures
D.M.B. has received investigator-originated research grants from Roche and Wyeth Pharmaceuticals (ROTRF, ROFAR, and Wyeth I.O.P. grants). A.G.C. was supported by a Novartis Fellowship grant from the American Society of Transplant Surgeons. O.D. was supported by a Fellowship grant from Novartis.
References
- 1.Schonbeck U, Libby P. The CD40/CD154 receptor/ligand dyad. Cell Mol Life Sci. 2001;58:4–43. doi: 10.1007/PL00000776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grewal IS, Flavell RA. The CD40 ligand: at the center of the immune universe? Immunol Res. 1997;16:59–70. doi: 10.1007/BF02786323. [DOI] [PubMed] [Google Scholar]
- 3.van Essen D, Kikutani H, Gray D. CD40 ligand-transduced co-stimulation of T cells in the development of helper function. Nature. 1995;378:620–623. doi: 10.1038/378620a0. [DOI] [PubMed] [Google Scholar]
- 4.Henn V, Slupsky JR, Grafe M, Anagnostopoulos I, Forster R, Muller-Berghaus G, Kroczek RA. CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature. 1998;391:591–594. doi: 10.1038/35393. [DOI] [PubMed] [Google Scholar]
- 5.Grewal IS, Foellmer HG, Grewal KD, Xu J, Hardardottir F, Baron JL, Janeway CA, Jr, Flavell RA. Requirement for CD40 ligand in costimulation induction, T cell activation, and experimental allergic encephalomyelitis. Science. 1996;273:1864–1867. doi: 10.1126/science.273.5283.1864. [DOI] [PubMed] [Google Scholar]
- 6.Stout RD, Suttles J, Xu J, Grewal IS, Flavell RA. Impaired T cell-mediated macrophage activation in CD40 ligand-deficient mice. J Immunol. 1996;156:8–11. [PubMed] [Google Scholar]
- 7.Buhlmann JE, Foy TM, Aruffo A, Crassi KM, Ledbetter JA, Green WR, Xu JC, Shultz LD, Roopesian D, Flavell RA, et al. In the absence of a CD40 signal, B cells are tolerogenic. Immunity. 1995;2:645–653. doi: 10.1016/1074-7613(95)90009-8. [DOI] [PubMed] [Google Scholar]
- 8.Xu J, Foy TM, Laman JD, Elliott EA, Dunn JJ, Waldschmidt TJ, Elsemore J, Noelle RJ, Flavell RA. Mice deficient for the CD40 ligand. Immunity. 1994;1:423–431. doi: 10.1016/1074-7613(94)90073-6. [DOI] [PubMed] [Google Scholar]
- 9.Grewal IS, Xu J, Flavell RA. Impairment of antigen-specific T-cell priming in mice lacking CD40 ligand. Nature. 1995;378:617–620. doi: 10.1038/378617a0. [DOI] [PubMed] [Google Scholar]
- 10.Borrow P, Tishon A, Lee S, Xu J, Grewal IS, Oldstone MB, Flavell RA. CD40L-deficient mice show deficits in antiviral immunity and have an impaired memory CD8+ CTL response. J Exp Med. 1996;183:2129–2142. doi: 10.1084/jem.183.5.2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aruffo A, Farrington M, Hollenbaugh D, Li X, Milatovich A, Nonoyama S, Bajorath J, Grosmaire LS, Stenkamp R, Neubauer M, et al. The CD40 ligand, gp39, is defective in activated T cells from patients with X-linked hyper-IgM syndrome. Cell. 1993;72:291–300. doi: 10.1016/0092-8674(93)90668-g. [DOI] [PubMed] [Google Scholar]
- 12.Melter M, Reinders ME, Sho M, Pal S, Geehan C, Denton MD, Mukhopadhyay D, Briscoe DM. Ligation of CD40 induces the expression of vascular endothelial growth factor by endothelial cells and monocytes and promotes angiogenesis in vivo. Blood. 2000;96:3801–3808. [PubMed] [Google Scholar]
- 13.Reinders ME, Sho M, Robertson SW, Geehan CS, Briscoe DM. Proangiogenic function of CD40 ligand-CD40 interactions. J Immunol. 2003;171:1534–1541. doi: 10.4049/jimmunol.171.3.1534. [DOI] [PubMed] [Google Scholar]
- 14.Chiodoni C, Iezzi M, Guiducci C, Sangaletti S, Alessandrini I, Ratti C, Tiboni F, Musiani P, Granger DN, Colombo MP. Triggering CD40 on endothelial cells contributes to tumor growth. J Exp Med. 2006;203:2441–2450. doi: 10.1084/jem.20060844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bergmann S, Pandolfi PP. Giving blood: a new role for CD40 in tumorigenesis. J Exp Med. 2006;203:2409–2412. doi: 10.1084/jem.20061754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Flaxenburg JA, Melter M, Lapchak PH, Briscoe DM, Pal S. The CD40-induced signaling pathway in endothelial cells resulting in the overexpression of vascular endothelial growth factor involves Ras and phosphatidylinositol 3-kinase. J Immunol. 2004;172:7503–7509. doi: 10.4049/jimmunol.172.12.7503. [DOI] [PubMed] [Google Scholar]
- 17.Lapchak PH, Melter M, Pal S, Flaxenburg JA, Geehan C, Frank MH, Mukhopadhyay D, Briscoe DM. CD40-induced transcriptional activation of vascular endothelial growth factor involves a 68-bp region of the promoter containing a CpG island. Am J Physiol. 2004;287:F512–F520. doi: 10.1152/ajprenal.00070.2004. [DOI] [PubMed] [Google Scholar]
- 18.Deregibus MC, Buttiglieri S, Russo S, Bussolati B, Camussi G. CD40-dependent activation of phosphatidylinositol 3-kinase/Akt pathway mediates endothelial cell survival and in vitro angiogenesis. J Biol Chem. 2003;278:18008–18014. doi: 10.1074/jbc.M300711200. [DOI] [PubMed] [Google Scholar]
- 19.Phung TL, Ziv K, Dabydeen D, Eyiah-Mensah G, Riveros M, Perruzzi C, Sun J, Monahan-Earley RA, Shiojima I, Nagy JA, et al. Pathological angiogenesis is induced by sustained Akt signaling and inhibited by rapamycin. Cancer Cell. 2006;10:159–170. doi: 10.1016/j.ccr.2006.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110:163–175. doi: 10.1016/s0092-8674(02)00808-5. [DOI] [PubMed] [Google Scholar]
- 21.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 22.Pearce LR, Huang X, Boudeau J, Pawlowski R, Wullschleger S, Deak M, Ibrahim AF, Gourlay R, Magnuson MA, Alessi DR. Identification of Protor as a novel Rictor-binding component of mTOR complex-2. Biochem J. 2007;405:513–522. doi: 10.1042/BJ20070540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Woo SY, Kim DH, Jun CB, Kim YM, Haar EV, Lee SI, Hegg JW, Bandhakavi S, Griffin TJ. PRR5, a novel component of mTOR complex 2, regulates platelet-derived growth factor receptor β expression and signaling. J Biol Chem. 2007;282:25604–25612. doi: 10.1074/jbc.M704343200. [DOI] [PubMed] [Google Scholar]
- 24.Dormond O, Madsen JC, Briscoe DM. The effects of mTOR-Akt interactions on anti-apoptotic signaling in vascular endothelial cells. J Biol Chem. 2007;282:23679–23686. doi: 10.1074/jbc.M700563200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boulday G, Haskova Z, Reinders ME, Pal S, Briscoe DM. Vascular endothelial growth factor-induced signaling pathways in endothelial cells that mediate overexpression of the chemokine IFN-γ-inducible protein of 10 kDa in vitro and in vivo. J Immunol. 2006;176:3098–3107. doi: 10.4049/jimmunol.176.5.3098. [DOI] [PubMed] [Google Scholar]
- 26.Moulton KS, Melder RJ, Dharnidharka VR, Hardin-Young J, Jain RK, Briscoe DM. Angiogenesis in the huPBL-SCID model of human transplant rejection. Transplantation. 1999;67:1626–1631. doi: 10.1097/00007890-199906270-00020. [DOI] [PubMed] [Google Scholar]
- 27.Fingar DC, Salama S, Tsou C, Harlow E, Blenis J. Mammalian cell size is controlled by mTOR and its downstream targets S6K1 and 4EBP1/ eIF4E. Genes Dev. 2002;16:1472–1487. doi: 10.1101/gad.995802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hudson CC, Liu M, Chiang GG, Otterness DM, Loomis DC, Kaper F, Giaccia AJ, Abraham RT. Regulation of hypoxia-inducible factor 1α expression and function by the mammalian target of rapamycin. Mol Cell Biol. 2002;22:7004–7014. doi: 10.1128/MCB.22.20.7004-7014.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Land SC, Tee AR. Hypoxia-inducible factor 1α is regulated by the mammalian target of rapamycin (mTOR) via an mTOR signaling motif. J Biol Chem. 2007;282:20534–20543. doi: 10.1074/jbc.M611782200. [DOI] [PubMed] [Google Scholar]
- 30.Briscoe DM, V, Dharnidharka R, Isaacs C, Downing G, Prosky S, Shaw P, Parenteau NL, Hardin-Young J. The allogeneic response to cultured human skin equivalent in the hu-PBL-SCID mouse model of skin rejection. Transplantation. 1999;67:1590–1599. doi: 10.1097/00007890-199906270-00014. [DOI] [PubMed] [Google Scholar]
- 31.Phung TL, Eyiah-Mensah G, O’Donnell RK, Bieniek R, Shechter S, Walsh K, Kuperwasser C, Benjamin LE. Endothelial Akt signaling is rate-limiting for rapamycin inhibition of mouse mammary tumor progression. Cancer Res. 2007;67:5070–5075. doi: 10.1158/0008-5472.CAN-06-3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Durie FH, Fava RA, Foy TM, Aruffo A, Ledbetter JA, Noelle RJ. Prevention of collagen-induced arthritis with an antibody to gp39, the ligand for CD40. Science. 1993;261:1328–1330. doi: 10.1126/science.7689748. [DOI] [PubMed] [Google Scholar]
- 33.Danese S, Scaldaferri F, Vetrano S, Stefanelli T, Graziani C, Repici A, Ricci R, Straface G, Sgambato A, Malesci A, et al. Critical role of the CD40 CD40-ligand pathway in regulating mucosal inflammation-driven angiogenesis in inflammatory bowel disease. Gut. 2007;56:1248–1256. doi: 10.1136/gut.2006.111989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reul RM, Fang JC, Denton MD, Geehan C, Long C, Mitchell RN, Ganz P, Briscoe DM. CD40 and CD40 ligand (CD154) are coexpressed on microvessels in vivo in human cardiac allograft rejection. Transplantation. 1997;64:1765–1774. doi: 10.1097/00007890-199712270-00025. [DOI] [PubMed] [Google Scholar]
- 35.Reinders ME, Fang JC, Wong W, Ganz P, Briscoe DM. Expression patterns of vascular endothelial growth factor in human cardiac allografts: association with rejection. Transplantation. 2003;76:224–230. doi: 10.1097/01.TP.0000071363.55007.D0. [DOI] [PubMed] [Google Scholar]
- 36.Lemstrom KB, Krebs R, Nykanen AI, Tikkanen JM, Sihvola RK, Aaltola EM, Hayry PJ, Wood J, Alitalo K, Yla-Herttuala S, Koskinen PK. Vascular endothelial growth factor enhances cardiac allograft arteriosclerosis. Circulation. 2002;105:2524–2530. doi: 10.1161/01.cir.0000016821.76177.d2. [DOI] [PubMed] [Google Scholar]
- 37.Kanazawa S, Tsunoda T, Onuma E, Majima T, Kagiyama M, Kikuchi K. VEGF, basic-FGF, and TGF-β in Crohn’s disease and ulcerative colitis: a novel mechanism of chronic intestinal inflammation. Am J Gastroenterol. 2001 Mar;96:822–828. doi: 10.1111/j.1572-0241.2001.03527.x. [DOI] [PubMed] [Google Scholar]
- 38.Hoshino M, Takahashi M, Aoike N. Expression of vascular endothelial growth factor, basic fibroblast growth factor, and angiogenin immunore-activity in asthmatic airways and its relationship to angiogenesis. J Allergy Clin Immunol. 2001;107:295–301. doi: 10.1067/mai.2001.111928. [DOI] [PubMed] [Google Scholar]
- 39.Ferrara N. The role of VEGF in the regulation of physiological and pathological angiogenesis. EXS. 2005;94:209–231. doi: 10.1007/3-7643-7311-3_15. [DOI] [PubMed] [Google Scholar]
- 40.Sone H, Kawakami Y, Sakauchi M, Nakamura Y, Takahashi A, Shimano H, Okuda Y, Segawa T, Suzuki H, Yamada N. Neutralization of vascular endothelial growth factor prevents collagen-induced arthritis and ameliorates established disease in mice. Biochem Biophys Res Commun. 2001;281:562–568. doi: 10.1006/bbrc.2001.4395. [DOI] [PubMed] [Google Scholar]
- 41.Miotla J, Maciewicz R, Kendrew J, Feldmann M, Paleolog E. Treatment with soluble VEGF receptor reduces disease severity in murine collagen-induced arthritis. Lab Invest. 2000;80:1195–1205. doi: 10.1038/labinvest.3780127. [DOI] [PubMed] [Google Scholar]
- 42.Oliver SJ, Cheng TP, Banquerigo ML, Brahn E. Suppression of collagen-induced arthritis by an angiogenesis inhibitor, AGM-1470, in combination with cyclosporin: reduction of vascular endothelial growth factor (VEGF) Cell Immunol. 1995;166:196–206. doi: 10.1006/cimm.1995.9978. [DOI] [PubMed] [Google Scholar]