

Abstract
Although the sphingolipid ceramide exhibits potent tumor suppressor effects, efforts to harness this have been hampered by poor solubility, uptake, bioavailability, and metabolic conversion. Therefore, identification of avenues to improve efficacy is necessary for development of ceramide-based therapies. In this study we used mutant p53, triple negative breast cancer (TNBC) cells, a type of breast cancer highly refractory to treatment, and cell-permeable nanoliposomal C6-ceramide in conjunction with the antiestrogen tamoxifen, which has been shown to be an effective modulator of ceramide metabolism. We show for the first time that nanoliposomal tamoxifen enhances nanoliposomal C6-ceramide cytotoxicity in cultured TNBC cells, a response that was accompanied by induction of cell cycle arrest at G1 and G2, caspase-dependent induction of DNA fragmentation, and enhanced mitochondrial and lysosomal membrane permeability, at 18 and 2 hr, respectively. Tamoxifen metabolites were also effective. Only tamoxifen promoted lysosomal membrane permeability. In addition, we demonstrate for the first time that tamoxifen inhibits acid ceramidase, as measured in intact cell assays; this effect was irreversible. Together, our findings show that tamoxifen magnifies the antiproliferative effects of C6-ceramide via combined targeting of cell cycle traverse and lysosomal and mitochondrial integrity. We adduce that C6-ceramide-induced apoptosis is amplified by tamoxifen's impact on lysosomes and perhaps accompanying inhibition of acid ceramidase, which could result in decreased levels of sphingosine 1-phosphate. This drug regimen could serve as a promising therapy for chemoresistant and triple negative types of breast cancer, and thus represents an indication for tamoxifen, irrespective of estrogen receptor status.
Keywords: ceramide, liposomes, breast cancer, antiestrogen, ceramidase
Introduction
Triple negative breast cancer (TNBC), a difficult to treat problematic subtype of breast cancer, accounts for approximately 15% of all breast cancers and disproportionally affects African American and Hispanic women. Despite improvements in the clinical outcome of breast cancer patients through use of endocrine-targeted agents and cytotoxic chemotherapies, overcoming drug resistance and the development of rational therapies for TNBC remain menacing therapeutic hurdles. The goal of the present work was to identify and evaluate new treatment combinations for TNBC, irrespective of estrogen and progesterone receptor status, HER2/neu expression, and the multidrug resistant phenotype. We have addressed this issue by formulating and evaluating novel combinatorial therapies consisting of short-chain ceramide-containing (C6-ceramide) nanoliposomes and adjuvants that are capable of regulating ceramide metabolism while affecting other independent targets as well, and propose this approach could restore breast tumor cell death programs that have been overridden by the cancer process.
Ceramide, a potent tumor suppressor lipid, can be generated in situ or administered exogenously. The sphingolipid-metabolizing machinery of cancer cells can work however to dampen ceramide's tumor-censoring effects. For example, conversion of ceramide to glucosylceramide (GC) by glucosylceramide synthase (GCS) is a main anabolic route utilized by cancer cells to neutralize ceramide downstream death signals (1-3). Ceramide cytotoxicity can also be limited by sphingomyelin synthase (SMS) and ceramide kinase (CerK) (4, 5). Ceramide hydrolysis via ceramidase is also an effective means to eliminate ceramide; however, the sphingosine generated can be phosphorylated by sphingosine kinase (SK) to yield sphingosine 1-phosphate (S1-P), a mitogenic sphingolipid with its own important place in cancer biology (6, 7). Maintaining balance between ceramide and S1-P has been viewed as paramount in maintaining ceramide's tumor suppressor properties, and to this end a number of pharmalogical and molecular approaches have been investigated to enhance ceramide's inhibition of tumor cell proliferation (8-13). Apropos here is a recent study by van Vlerken et al (14) that demonstrates, through polymer-blend nanoparticles, that combination therapy with exogenous C6-ceramide or tamoxifen, used as a GCS inhibitor, with paclitaxel, was more effective than single agent paclitaxel.
In the present study we assessed a nanoliposomal formulation of C6-ceramide, which has demonstrated enhanced activity in in vitro and in vivo models (15-18), and paired it with tamoxifen, in order to determine whether the antiproliferative effects of ceramide could be amplified. Although tamoxifen is an antiestrogen used for treatment of estrogen receptor-positive types of breast cancer, this drug has a number of estrogen receptor-independent actions, including circumvention of multidrug resistance (19), inhibition of ceramide glycosylation (20), and downregulation of survivin (21). In addition, we now show that tamoxifen inhibits acid ceramidase, an enzyme indispensible for cellular ceramide degradation. For the present study, we developed a stable formulation of tamoxifen nanoliposomes containing 30 mole percent tamoxifen and evaluated the impact of C6-ceramide-tamoxifen combinatorial regimens in TNBC cells. This combination was synergistic for reduction of cell viability, and at 24 hr the combination induced cell cycle arrest at G1 and G2, independent of retinoblastoma protein (RB) expression. One function of RB protein is to prevent excessive cell growth by inhibiting cell cycle, thus this protein can function as a tumor suppressor. Testing upstream revealed that enhanced mitochondrial and lysosomal membrane permeability were important elements of the apoptotic cascade.
Materials and Methods
Cell Lines and Reagents
Human breast cancer cell lines MDA-MB-468, MDA-MB-231, and Hs578T were obtained from the American Type Culture Collection (ATCC) (Manassas, VA). The cell lines were expanded and cryopreserved in liquid nitrogen in the investigators laboratory. The cell lines were not tested or authenticated over and above documentation provided by the ATCC, which includes antigen expression, DNA profile, and cytogenic analysis. Cells were maintained (approximately 25 passages) in RPMI-1640 Glutamax™ medium (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS) (Atlanta Biologicals, Lawrenceville, GA) and 100 units/ml each of penicillin and streptomycin plus 0.3 mg/ml L-glutamine. Cells were grown in humidified atmosphere, 95% air, 5% CO2 at 37°C and subcultured at confluence using 0.05% trypsin/0.53 mM EDTA (Invitrogen). DM102 was provided by the Department of Biomedicinal Chemistry, Institute of Advanced Chemistry of Catalonia, Barcelona, Spain.
C6-ceramide (N-hexanoyl-D-erythro-sphingosine) was from Avanti Polar Lipids (Alabaster, AL) and dissolved in 100% ethanol and stored as a 10 mM stock at -20°C. Tamoxifen-HCl, N-desmethyltamoxifen HCl, and (Z)-4-hydroxytamoxifen were from Sigma Chemical Co, and dissolved and stored as was C6-ceramide. Pan caspase inhibitor, Z-VADFMK, from BD Pharmigen™ (San Diego, CA), was solubilized in ethanol and stored as 40 mM stock at -20°C.
Preparation of Nanoliposomes
Pegylated nanoliposomes were prepared based upon earlier studies (22-24). Liposome formulations were made from specific lipids, at particular molar ratios (shown below), prior to nano-sizing. We used 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC), 1,2-dioleoyl-snglycero-3-phosphoethanolamine (DOPE), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (PEG(2000)-DSPE), C6-ceramide, C8-ceramide-1-succinyl[methoxy(polyethylene glycol)-750] (PEG(750)-C8), and/or tamoxifen. Briefly, lipids dissolved in chloroform, dried to a film under a stream of nitrogen, and then hydrated by addition of 0.9% NaCl. Solutions were sealed, heated at 55°C (60 min), and subjected to vortex mixing and sonicated for 5 min until light no longer diffracted through the suspension. The lipid vesicle-containing solution was quickly extruded at 55°C by passing the solution 10 times through 100 nm polycarbonate filters in an Avanti Mini-Extruder (Avanti Polar Lipids, Alabaster, AL). Nanoliposomal size for triplicate replicates of each formulation, and a neutral zeta potential were validated using a Malvern Zetasizer Nano ZS at 25°C. Nanoliposome solutions were stored at room temperature until use. All formulations were designed to deliver equal masses of total lipids. Ghost nanoliposomes were prepared from the same components excluding C6-ceramide.
| (mole percent) | DSPC | DOPE | PEG(2000)-DSPE | PEG(750)-C8 | C6 | tamoxifen | Size (nm) |
|---|---|---|---|---|---|---|---|
| Lip-Ghost | 5.66 | 2.87 | 1.47 | -- | -- | -- | 85 |
| Lip-C6 | 3.75 | 1.75 | 0.75 | 0.75 | 3.0 | -- | 86 |
| Lip-tamoxifen | 3.75 | 2.25 | 1.00 | -- | -- | 3.0 | 86 |
| Lip-C6-tamoxifen | 3.75 | 1.75 | 0.75 | 0.75 | 1.5 | 1.5 | 87 |
Cell Viability
Viability was assessed by the Cell Titer 96® AQueous One solution cell proliferation assay (Promega, Madison, WI). Cells were seeded (3,000 cells/well) in 96-well plates in medium containing 5% FBS and the following day treated with the indicated agents using 1% FBS medium (final FBS concentration, approximately 2.5%) for 72-96 hr. Viability was measured as mean (n=6) absorbance (minus ghost and/or ethanol vehicle) at 490 nm using a microplate reader FL600, Bio-Tek (Winooski, VT).
Cell Cycle and Apoptosis Analysis
Progression of cells through cell cycle and DNA fragmentation were examined using flow cytometry. DNA fragmentation was measured according to published protocol (25), with modification. MDA-MB-468 and MDA-MB-231 cells were seeded in 6-cm dishes at 200,000 cells/dish in 10% FBS medium. The following day the medium was removed and cells were treated with indicated agents in medium containing 2.5% FBS for 24 hr. Monolayers were then washed with cold PBS, and fixed in 70% ethanol. DNA was stained for 4 hr in the dark with 0.5 ml hypotonic propidium iodide buffer (0.1% sodium citrate, 50 μg/mL DNase-free RNase A, 0.1% Triton X-100). DNA content was analyzed using a FACScan, and cell-cycle analysis and subG0 (apoptosis marker) was performed using FCS Express 4 (De Novo Software).
Acid Ceramidase
AC was measured in intact cells and in cell lysates by fluorgenic assay as described (26). For intact cell assay, MDA-MB 468 cells (10,000 well) were seeded into 96-well plates in 10% FBS medium. After 24 hr, medium was removed and replaced with 5% FBS medium. Cells were preincubated at 37°C for 1 hr with either the AC inhibitor DM102, [2R, 3Z]-N-(1-hydroxyoctadec-3-en-2-yl)pivaloylamine (26) or tamoxifen (ethanol vehicles), after which, fluoregenic substrate (ethanol vehicle) was added to a final concentration of 16 μM, and the cells were incubated (125 μl final well volume) in a tissue culture incubator (37°C) for either 3 or 23 additional hr. At this point, the assay can either be continued to completion or the 96-well plates can be placed at -20°C and assayed the following day. To complete the assay, 50 μl methanol and 100 μl NaIO4 (2.5 mg/ml) in 0.1 M glycine buffer, pH 10.6, was added and the plates were placed in the dark at room temperature for 2 hr. Fluorescence was measured in the UV range (365 nm excitation/410-460 nm emission) using a GloMax® multi-detection system (Promega, Madison, WI). To measure AC in cell lysates, cultured cells were pretreated in the absence and presence of tamoxifen (5, 10 μM) for 4 hr. Cells were then washed, harvested, pelleted, briefly sonicated in 0.25 M sucrose, and centrifuged to remove debris. Cell-free assays (37 μg protein) contained sodium acetate-acetic acid buffer, pH 4.5 (25 mM), 40 μM substrate, and were incubated at 37°C for 3 hr (26). Fluorescence was then measured as above. Similar assays were conducted using cell lysates derived from tamoxifen-naïve cultures to which either DM102 or tamoxifen was then added.
Lysosomal Stability and Mitochondrial Membrane Potential (MMP)
Cells were assessed for lysosomal stability using the acridine orange (AO) relocation methods (27, 28). Acridine orange is a metachromatic fluorochrome and a weak base that exhibits red fluorescence when highly concentrated in acidic lysosomes and green fluorescence when outside the lysosomes. Lysosomal integrity was evaluated by assessing red fluorescence (FL3) by flow cytometry. Briefly, cells were stained with acridine orange (5 μg/ml) for 15 min in RPMI-1640 complete media followed by PBS washing and treatment with tamoxifen for 2 hr. After treatment, cells were trypsinized, washed with PBS, and placed on ice for flow cytometry. The shift of red fluorescence indicates lysosomal permeabilization.
MMP (ΔΨm) was measured using JC-1 (Cell Technology, Mountain View, CA). Cells were plated as in the apoptosis experiments and treated the following day with agents for 18 hr. JC-1 is a ratiometric dye that exists as a monomer in the cytosol (green) and also accumulates as aggregates in the mitochondria which stain red. Quantitative analysis of ΔΨm was detected by JC-1 at FL-1 (green) and FL-2 (red) using flow cytometry.
Statistical Analysis
The results are expressed as means ± SE and were analyzed by ANOVA. Differences among the treatment groups were assessed by Tukey's Post Hoc Test. Differences were considered significant at P < 0.05.
Results
Unless otherwise stated, all experiments were carried out using nanoliposomal formulations. The first experiments were conducted to assess the effects of combinatorial C6-ceramide and tamoxifen on cell viability. As shown in Fig. 1, these combinations were more effective than single agents in reducing viability in all cell lines. In some instances, the effects were supradditive. For example, in Hs578T cells (Fig. 1A), whereas tamoxifen did not effect viability, and C6-ceramide reduced viability to approximately 60% of control, concomitant exposure elicited amplified antiproliferative responses (viability < 20%). Similarly, in MDA-MB-231 cells (Fig. 1B), C6-ceramide, tamoxifen, and the combination reduced viability to 70, 90, and 40% of control, respectively, with like results in MDA-MB-468 cells (Fig. 1C). Interestingly, N-desmethyltamoxifen, the primary metabolite of tamoxifen, and 4-hydroxytamoxifen, a metabolite with more potent antiestrogenic activity that the parent drug, were also affective when co-administered with C6-ceramide (Fig. 1D), albeit N-desmethyltamoxifen was the most effective. We also compared the activity of free ceramide (ethanol vehicle) with nanoliposomal C6-ceramide and found that at a dose of 2.0 μM, MDA-MD-468 cell viability was 72 ± 6.27 and 2 ± 10.28 % of control after 96 hr exposure to the respective ceramide forms. These findings are similar with observations using free and nanoformulated C6-ceramide in a murine mammary adenocarcinoma cell line model (29).
Figure 1.

Effect of tamoxifen on C6-ceramide cytotoxicity in TNBC. A-D. Cells were seeded in 96-well plates (3,000 cells/well) in RPMI-1640 medium containing 10% FBS and cultured at 37°C. The next day, growth medium was replaced with 2.5 % FBS medium, agents were added and cells were incubated for 96 hr. D. MDA-MB-468 cells treated with N-desmethyltamoxifen (dme-tam) and 4-hydroxytamoxifen (4-OH tam). Viability was evaluated by MTS assay. [μM] concentrations. Data are the mean (n=6) ± SE, relative to vehicle controls and/or the ghost. Experiments were repeated and yielded similar results.
Because of the strong antiproliferative responses demonstrated with C6-ceramidetamoxifen combinations, we next sought to determine whether treatment impacted cell cycle. In MDA-MB-468 cells, which do not express RB protein, the drug duo promoted cell cycle arrest at G1 and G2 (Fig. 2A). Similar results were obtained in MDA-MB-231 cells (Fig. 2B), which express RB-protein. With single agents, the data demonstrate that C6-ceramide principally influenced G1 as did tamoxifen, only although slightly; the combination also increased cells in G2 (Fig. 2B), indicating that the combination induces cell cycle arrest independent of RB expression.
Figure 2.

Effect of tamoxifen and C6-ceramide on cell cycle progression in TNBC. Cells were treated with C6-ceramide, tamoxifen, and the mix for 24 hr, collected and fixed in ethanol, stained with propidium iodide, and analyzed by flow cytometry as detailed in Methods. The distribution of cells in the different phases of the cell cycle was analyzed using FCS express software. Data are mean ± SE from three independent experiments. [μM] concentrations.
We next investigated the influence of single agents and combinatorial regimens on the type of cell death. It is well known that ceramide produces many of its tumor suppressor effects through the induction of apoptosis. Using DNA fragmentation as a gauge, the data in Fig. 3A show that C6-ceramide, as opposed to tamoxifen, was the major player in eliciting apoptosis in MDA-MB-468 cells; however, concurrent administration produced a response that was supra-additive. For example, whereas 5.0 μM tamoxifen had no significant effect, and 2.5 μM C6-ceramide increased DNA fragmentation to 13% (7% over control), the combination yielded 29% fragmentation (23% over control). A similar trend was evidenced in MDA-MB-231 cells (Fig. 3B). Apoptosis was caspase-dependent as shown by inhibition of DNA fragmentation in MDAMB-468 cells exposed to C6-ceramide-tamoxifen in the presence of a pan caspase inhibitor (Fig. 3A). N-desmethyltamoxifen also effectively enhanced DNA fragmentation when co-administered with C6-ceramide (Fig. 3C).
Figure 3.

Influence of C6-ceramide, tamoxifen, and caspase inhibition on apoptosis in TNBC. A. MDA-MB-468 cells. B. MDA-MB-231 cells. C. MDA-MB-468 cells with N-desmethyltamoxifen. Cells were treated with C6-ceramide, tamoxifen, N-desmethyltamoxifen, or the combination for 24 hr. Cells were then trypsinized, fixed in ethanol, stained with propidium iodide, and analyzed for DNA fragmentation by flow cytometry. SubG0 (apoptosis marker) was analyzed using FCS express software. Data are means ± SE from three independent experiments. Concentrations [μM] noted in brackets. CaspI [50 μM], caspase inhibitor.
Mitochondria are the orchestrators of the intrinsic pathway of apoptosis, and ceramide can elicit this process via mitochondrial involved avenues (30, 31). One way that ceramide impacts mitochondria is by increasing outer membrane permeabilization through induction of mitochondrial transmembrane depolarization (ΔΨm); this promotes leakage of apoptotic proteins. In order to determine if the C6-ceramide-tamoxifen regimen targeted the mitochondrial pathway, we measured ΔΨm in response to treatment. The data in Fig. 4A demonstrate a robust increase in ΔΨm in MDA-MB-468 cells in response to combination C6-ceramide-tamoxifen and C6-ceramide-N-desmethyltamoxifen (Fig. 4B). It is noteworthy that ΔΨm was evident as early as 18 hr and was promoted using concentrations of C6-ceramide and tamoxifen similar to those that affected decreased viability at 96 hr (see Fig. 1C). Evaluating the effects of single agents on ΔΨm revealed that C6-ceramide was the predominant player in eliciting altered mitochondrial membrane integrity (Fig. 4A). Although ΔΨm constitutes a major checkpoint in apoptotic cell death, lysosomal membrane permeabilization (LMP) has also been shown, in certain circumstances, to trigger cell death. LMP causes release of proteases into the cytosol with deleterious consequences. The experiment in Fig. 4C shows that tamoxifen affected enhanced LMP in MDA-MB-468 cells after only a 2 hr exposure (quantification data on right); the same was not produced upon exposure to C6-ceramide. Thus, LMP appears to be an early event contributing to cell death that is chiefly regulated by tamoxifen.
Figure 4.

Effect of C6-ceramide and tamoxifen on mitochondrial and lysosomal integrity. A. Effect of C6-ceramide on mitochondrial depolarization. B. Effect of C6-ceramide and N-desmethyltamoxifen on mitochondrial depolarization. MDA-MB-468 cells were treated with C6-ceramide, tamoxifen, N-desmethyltamoxifen, or the combination for 18 hr. Cells were then stained with JC-1 for 15 min in phenol red-free RPMI, trypsinized, washed with PBS, and placed on ice until quantitation by FACS. Quantitative analysis of Δψm was detected by JC-1 at FL-1 (green) and FL-2 (red). Data are mean from 3-4 independent experiments. C. Effect of C6-ceramide and tamoxifen on lysosomal permeabilization. MDA-MB-468 cultures were stained with Acridine orange for 15 minutes followed by PBS washing, and treated with tamoxifen for 2 hr. After treatment, cells were trypsinized, washed with PBS, and placed on ice until FACS analysis. A shift in red fluorescence (FL-3) indicates lysosomal peremeabilization. Data was analyzed using FCS express software. Data are means ± SE from at least three independent experiments.
The structure of tamoxifen is similar to other amphiphilic agents that have been shown to inhibit acid ceramidase (AC) (32), the lysosomal enzyme indispensable for cellular ceramide degradation. We were therefore interested in determining whether tamoxifen influenced AC activity. Here we demonstrate for the first time that tamoxifen inhibits AC activity as assessed in intact cells. Inhibition was dose-dependent (Table 1, 4 hr) and N-desmethyltamoxifen and 4-hydroxytamoxifen were as effective as tamoxifen (Table 1, 24 hr). Free tamoxifen (ethanol vehicle) and tamoxifen nanoliposomes were equally effective. As negative control we assessed triphenylbutene, which is the tamoxifen nucleus devoid of the dimethylaminoethanol function; it did not inhibit AC activity in MDA-BM-468 cells when tested at concentrations of 5 – 20 μM (10 μM shown). Two other tricyclic cationic amphiphilic agents with a nitrogen-containing side group similar to tamoxifen, chloropromazine and desipramine, inhibited AC activity in intact MDA-MB-468 cells by 66 and 42%, respectively (at 10 μM) (data not shown). The action of the cysteine protease, cathepsin B, has been shown to exert a role in inhibition of AC (32). To determine whether AC inhibition by tamoxifen could be linked to degradation of AC protein by lysosomal cathepsin, we employed CA074ME, a cathepsin B/L inhibitor; however, when introduced over a concentration range of 5 – 20 μM (1 hr pre-incubation), CA074ME failed to reverse the inhibitory effects of tamoxifen on AC. These data indicate that inhibition of AC by tamoxifen is not related to cathepsin degradation of the enzyme. Lastly, we determined the effect of tamoxifen on AC activity using MDA-MB-468 cell lysates. For this, two types of experiments were conducted: i) pre-exposure of intact cells to tamoxifen followed by cell disruption and assay, ii) cell disruption and addition of tamoxifen or DM102 to the cell-free assay. As shown in Fig. 5A, preincubation of intact cells with tamoxifen resulted in inhibition of AC activity as assayed in the resultant lysates. On the other hand, tamoxifen did not inhibit AC activity when added directly to the cell lysates (Fig. 5B), demonstrating that the mechanism underlying enzyme inhibition requires intact cells. DM102, an AC substrate mimic, inhibited cell-free AC activity.
Table 1.
The effect of tamoxifen, tamoxifen metabolites, and other agents on AC activity in intact MDA-MB-468 cells
| 4 hr incubation |
24 hr incubation* |
||||
|---|---|---|---|---|---|
| Conditions | mean FU | Activity | Conditions | mean FU | Activity |
| control | 7367 | 100% | control | 16958 | 100% |
| tam 5 μM | 5367 | 73% | tam (EtOH) | 3891 | 23% |
| 10 | 3167 | 43% | tam (nano) | 4458 | 26% |
| 20 | 2067 | 28% | desMe-tam | 3925 | 23% |
| 4-OH-tam | 3891 | 23% | |||
| Triphenylbutene | 17733 | 105% | |||
| NH4G [20 μM] | 15262 | 90% | |||
| **pre-tam [20 μM] | 6892 | 41% | |||
Tamoxifen and metabolites were tested at 10 μM, except where noted.
Cells were exposed (pre-tam) to tamoxifen for 3 hr, rinsed and reincubated in tamoxifen-free medium (10% FBS) for 24 hr before AC activity was determined. Each data point is the mean of triplicate cultures. Repeated experiments yielded similar results. Error was within ± 6% of the mean. EtOH, ethanol vehicle; tam (nano), nanoliposomal tamoxifen; dme-tam, N –desmethyltamoxifen; 4-OH-tam, 4-hydroxytamoxifen; FU, fluorescence units.
Figure 5.
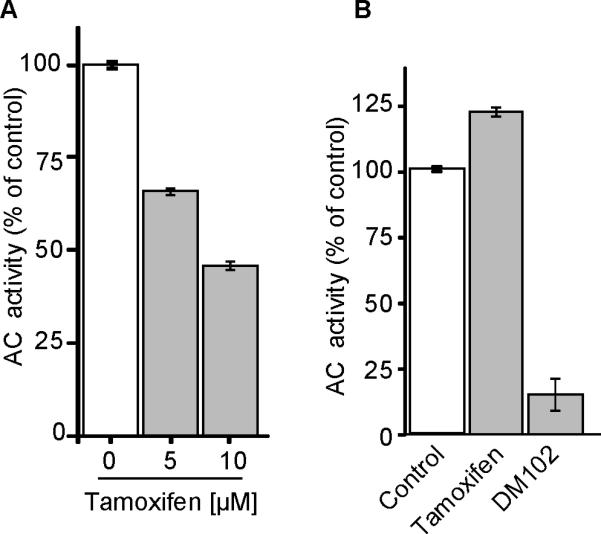
Effect of tamoxifen and DM102 on AC activity in MDA-MB-468 cell lysates. A. Cell-free assay. Cells were preincubated with tamoxifen for 4 hr, washed, lysed, and assayed for AC activity as described in Methods. B. Cell-free assays with tamoxifen [20 μM] and DM102 [20 μM] added after lysis. Data are means ± SE, n = 3 from two independent experiments.
Discussion
This promise of ceramide-based therapeutics has largely been boosted by the use of short-chain ceramides, which are more hydrophilic, and by the employ of nanoliposomal formulations, which demonstrate enhanced bioavailability (15, 17, 33), few side effects (17), and efficacy via anti-proliferative and pro-apoptotic properties (15, 18, 29). In some instances, nanoliposomal ceramides demonstrate enhanced activity when co-administered with other anticancer agents such as sorafenib or gemcitabine, or the GCS inhibitor D-threo-1-phenyl-2-decanoylamino-3-morpholino-1-propanol (PDMP) (16, 22). The discovery that tamoxifen inhibits ceramide metabolism at the step of glycosylation (20) suggests that this antiestrogen could have estrogen receptor-independent utility in the extant of ceramide therapies. A recent study from our group, using ethanol vehicle, showed the value of combination C6-ceramide-tamoxifen for limiting proliferation in several human cancer cell lines; however, neither the mechanism nor the targets were defined (34). In the present study we have focused on breast cancer, and specifically, because we have employed tamoxifen, on TNBC, which is devoid of estrogen receptor and thus not amiable to antiestrogen therapy.
The present work shows that a C6-ceramide-tamoxifen regimen produced cell cycle arrest in G1 and G2, phases in which growth and preparation for mitosis occur. This regimen was effective in RB-expressing and in RB-non-expressing breast cancer cells. This is important because RB, which maintains cell proliferative balance, is functionally inactivated, mutated, or lost in most human neoplasms. From data accrued, it appears that both agents contribute (see Fig. 2). Ceramide and tamoxifen have been shown to induce cell cycle arrest (35-37). We have also established that cell death is by caspase-dependent apoptosis, but whereas C6-ceramide was the major single agent player, the addition of tamoxifen provided enhanced effects (see Fig. 3A). Interestingly, in mammary tumor studies, Mandlekar et al (38) showed that N-desmethyltamoxifen as well as tamoxifen and 4-hydroxytamoxifen, induced caspase-dependent apoptosis, and in a comparative study in multidrug resistant human leukemia cells, both tamoxifen and N-desmethyltamoxifen functioned equally in restoring daunorubicin sensitivity. Tamoxifen is also a P-glycoprotein antagonist (39), and in this capacity functions to retard ceramide glycosylation by virtue of its competence to block sphingolipid trafficking at the Golgi (40, 41).
With regard to subcellular targets, we documented a clear impact on mitochondria, as gauged by an increase in ΔΨm in response to C6-ceramide-tamoxifen. An interesting aspect here, as seen in the viability studies, was that N-desmethyltamoxifen was as effective as tamoxifen when co-administered with C6-ceramide. This has clinical relevance as N-desmethyltamoxifen is the major in vivo tamoxifen metabolite in humans. Short-chain ceramides induce cytochrome c release from mitochondria (42), augment curcumin-induced cell death in melanoma via mitochondrial apoptosis (43), and induce cell death through induction of hari-kari gene (HRK)-mediated mitochondrial dysfunction (44). Our studies with nanoliposomal ceramide also demonstrate that mitochondrial targeting perpetuates cell death.
Lysosomal destabilization was the earliest upstream event that we detected. Using LMP as a measure, lysosomal destabilization was seen at 2 hr and was initiated by tamoxifen, dose dependently. C6-ceramide at 2 hr had no effect. LMP occurs in response to a myriad of stimuli, and this process is a factor in the regulation of apoptosis (45) through release of lysosomal constituents such as destructive cathepsins. For this reason, lysosomes are promising therapeutic targets in cancer. Because tamoxifen induced LMP, we were interested in learning whether tamoxifen affected AC activity, as AC is localized in the lysosomal lumen, and AC is another important target in cancer (46-49). Herein we show that tamoxifen as well as tamoxifen metabolites inhibited AC activity in intact MDA-MB-468 cells at concentrations that induced LMP. Tamoxifen did not inhibit AC when added directly to the cell lysate, whereas cell preincubation with tamoxifen was effective in the inhibition of AC when tested in the resultant lysate. Tamoxifen is a cationic amphilic drug, as are chlorpromazine and desipramine, which we demonstrated also inhibited AC activity. Along these lines, Elojeimy et al (32) demonstrated that desipramine inhibited AC in prostate cancer cells, based on downregulation of AC protein, and that CA074ME, a cathepsin B/C inhibitor, blocked the effect on AC. In our system, CA074ME failed to block tamoxifen-regulated inhibition of AC, suggesting that cathepsin hydrolysis of AC protein is not a factor in the mechanism of enzyme inhibition. Further, exposure of cells to NH4Cl, which neutralizes lysosomal pH, did not affect cellular AC activity. Although we have not defined the mechanism by which tamoxifen inhibits AC, in light of the above discussion we propose that lysosomal destabilization is a factor involved in inhibition of AC by tamoxifen, and that this response requires intact cell machinery.
To our knowledge this is the first report showing that tamoxifen inhibits AC activity, an action we suggest is related to LMP. In MCF-7 cells, Hwang et al (50) showed that tamoxifen-induced cell death was accompanied by increased oxidative stress and LMP. We have previously demonstrated that tamoxifen blocks C6-ceramide glycosylation (34) which would, in our case, extend the intracellular residence time of C6-ceramide and thus amplify cellular responses. That tamoxifen also inhibits AC, leads to a number of interesting avenues for pursuit. For the present, we offer the following scenario (Fig. 6). Increasing the levels or residence time of intracellular C6-ceramide by virtue of tamoxifen's inhibitory effect on ceramide metabolism may not be a chief factor in cellular response. We propose that tamoxifen and C6-ceramide each have common as well as distinct targets, and this perpetuates the perfect storm. We suggest that initial targeting of lysosomes by tamoxifen initiates cellular demise. This action can directly engage apoptosis and/or elicit ΔΨm, which could be driven by lysosomal cathepsins. The overall C6-ceramide mitochondrial response could be magnified by tamoxifen's indirect impact on ΔΨm through LMP. Thus, apoptosis can be driven by a trio of events, which as shown, enlist lysosomal, mitochondrial, and anti-mitogenic responses.
Figure 6.
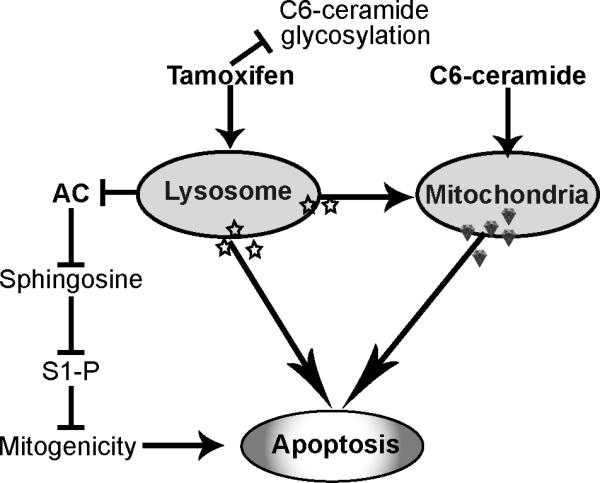
Scheme for C6-ceramide-tamoxifen elicitation of apoptosis. Tamoxifen can block C6-ceramide glycosylation and hydrolysis, the latter mediated via enhanced LMP. LMP can also impact mitochondria as can C6-ceramide, directly. Blocking AC dampens S1-P mitogenic responses and potentiates lysosomal and mitochondrial driven apoptosis. AC, acid ceramidase; S1-P, sphingosine 1-phosphate.
Acknowledgments
This research was supported by the Association for Breast and Prostate Cancer Studies (Los Angeles), the Fashion Footwear Association of New York Charitable Foundation (New York, NY), National Institute of General Medical Sciences (grant no GM77391).
Footnotes
Disclosure of Potential Conflicts of Interest
The Penn State Research Foundation has licensed ceramide nanoliposomes and other nanoliposomal nanotechnology to Keystone Nano Inc (State College, PA). MK is the Chief Medical Officer of Keystone Nano Inc.
References
- 1.Liu YY, Han TY, Giuliano AE, Ichikawa S, Hirabayashi Y, Cabot MC. Glycosylation of ceramide potentiates cellular resistance to tumor necrosis factor-alpha-induced apoptosis. Exp Cell Res. 1999;252:464–70. doi: 10.1006/excr.1999.4649. [DOI] [PubMed] [Google Scholar]
- 2.Bleicher RJ, Cabot MC. Glucosylceramide synthase and apoptosis. Biochim Biophys Acta. 2002;1585:172–8. doi: 10.1016/s1388-1981(02)00338-4. [DOI] [PubMed] [Google Scholar]
- 3.Liu YY, Han TY, Giuliano AE, Cabot MC. Expression of glucosylceramide synthase, converting ceramide to glucosylceramide, confers adriamycin resistance in human breast cancer cells. J Biol Chem. 1999;274:1140–6. doi: 10.1074/jbc.274.2.1140. [DOI] [PubMed] [Google Scholar]
- 4.Meng A, Luberto C, Meier P, Bai A, Yang X, Hannun YA, et al. Sphingomyelin synthase as a potential target for D609-induced apoptosis in U937 human monocytic leukemia cells. Exp Cell Res. 2004;292:385–92. doi: 10.1016/j.yexcr.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 5.Gomez-Munoz A, Gangoiti P, Granado MH, Arana L, Ouro A. Ceramide-1-phosphate in cell survival and inflammatory signaling. Adv Exp Med Biol. 2010;688:118–30. doi: 10.1007/978-1-4419-6741-1_8. [DOI] [PubMed] [Google Scholar]
- 6.Pitson SM. Regulation of sphingosine kinase and sphingolipid signaling. Trends Biochem Sci. 2011;36:97–107. doi: 10.1016/j.tibs.2010.08.001. [DOI] [PubMed] [Google Scholar]
- 7.Pyne NJ, Pyne S. Sphingosine 1-phosphate and cancer. Nat Rev Cancer. 2010;10:489–503. doi: 10.1038/nrc2875. [DOI] [PubMed] [Google Scholar]
- 8.Oskouian B, Saba JD. Cancer treatment strategies targeting sphingolipid metabolism. Adv Exp Med Biol. 2010;688:185–205. doi: 10.1007/978-1-4419-6741-1_13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Graf C, Klumpp M, Habig M, Rovina P, Billich A, Baumruker T, et al. Targeting ceramide metabolism with a potent and specific ceramide kinase inhibitor. Mol Pharmacol. 2008;74:925–32. doi: 10.1124/mol.108.048652. [DOI] [PubMed] [Google Scholar]
- 10.Senchenkov A, Litvak DA, Cabot MC. Targeting ceramide metabolism--a strategy for overcoming drug resistance. J Natl Cancer Inst. 2001;93:347–57. doi: 10.1093/jnci/93.5.347. [DOI] [PubMed] [Google Scholar]
- 11.Shida D, Takabe K, Kapitonov D, Milstien S, Spiegel S. Targeting SphK1 as a new strategy against cancer. Curr Drug Targets. 2008;9:662–73. doi: 10.2174/138945008785132402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beckham TH, Elojeimy S, Cheng JC, Turner LS, Hoffman SR, Norris JS, et al. Targeting sphingolipid metabolism in head and neck cancer: rational therapeutic potentials. Expert Opin Ther Targets. 2010;14:529–39. doi: 10.1517/14728221003752768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Canals D, Perry DM, Jenkins RW, Hannun YA. Drug targeting of sphingolipid metabolism: sphingomyelinases and ceramidases. Br J Pharmacol. 2011;163:694–712. doi: 10.1111/j.1476-5381.2011.01279.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Vlerken LE, Duan Z, Little SR, Seiden MV, Amiji MM. Augmentation of therapeutic efficacy in drug-resistant tumor models using ceramide coadministration in temporal-controlled polymer-blend nanoparticle delivery systems. AAPS J. 2010;12:171–80. doi: 10.1208/s12248-010-9174-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stover T, Kester M. Liposomal delivery enhances short-chain ceramide-induced apoptosis of breast cancer cells. J Pharmacol Exp Ther. 2003;307:468–75. doi: 10.1124/jpet.103.054056. [DOI] [PubMed] [Google Scholar]
- 16.Tran MA, Smith CD, Kester M, Robertson GP. Combining nanoliposomal ceramide with sorafenib synergistically inhibits melanoma and breast cancer cell survival to decrease tumor development. Clin Cancer Res. 2008;14:3571–81. doi: 10.1158/1078-0432.CCR-07-4881. [DOI] [PubMed] [Google Scholar]
- 17.Zolnik BS, Stern ST, Kaiser JM, Heakal Y, Clogston JD, Kester M, et al. Rapid distribution of liposomal short-chain ceramide in vitro and in vivo. Drug Metab Dispos. 2008;36:1709–15. doi: 10.1124/dmd.107.019679. [DOI] [PubMed] [Google Scholar]
- 18.Shabbits JA, Mayer LD. Intracellular delivery of ceramide lipids via liposomes enhances apoptosis in vitro. Biochim Biophys Acta. 2003;1612:98–106. doi: 10.1016/s0005-2736(03)00108-1. [DOI] [PubMed] [Google Scholar]
- 19.Hotta T, Tanimura H, Yamaue H, Iwahashi M, Tani M, Tsunoda T, et al. Tamoxifen circumvents the multidrug resistance in fresh human gastrointestinal cancer cells. J Surg Res. 1996;66:31–5. doi: 10.1006/jsre.1996.0368. [DOI] [PubMed] [Google Scholar]
- 20.Lavie Y, Cao H, Volner A, Lucci A, Han TY, Geffen V, et al. Agents that reverse multidrug resistance, tamoxifen, verapamil, and cyclosporin A, block glycosphingolipid metabolism by inhibiting ceramide glycosylation in human cancer cells. J Biol Chem. 1997;272:1682–7. doi: 10.1074/jbc.272.3.1682. [DOI] [PubMed] [Google Scholar]
- 21.Guo R, Huang Z, Shu Y, Jin S, Ge H. Tamoxifen inhibits proliferation and induces apoptosis in human hepatocellular carcinoma cell line HepG2 via down-regulation of survivin expression. Biomed Pharmacother. 2009;63:375–9. doi: 10.1016/j.biopha.2008.09.010. [DOI] [PubMed] [Google Scholar]
- 22.Jiang Y, Divittore NA, Kaiser JM, Shanmugavelandy SS, Fritz JL, Heakal Y, et al. Combinatorial therapies improve the therapeutic efficacy of nanoliposomal ceramide for pancreatic cancer. Cancer Biol Ther. 2011;12 doi: 10.4161/cbt.12.7.15971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tagaram HR, Divittore NA, Barth BM, Kaiser JM, Avella D, Kimchi ET, et al. Nanoliposomal ceramide prevents in vivo growth of hepatocellular carcinoma. Gut. 2011;60:695–701. doi: 10.1136/gut.2010.216671. [DOI] [PubMed] [Google Scholar]
- 24.Liu X, Ryland L, Yang J, Liao A, Aliaga C, Watts R, et al. Targeting of survivin by nanoliposomal ceramide induces complete remission in a rat model of NK-LGL leukemia. Blood. 2010;116:4192–201. doi: 10.1182/blood-2010-02-271080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Riccardi C, Nicoletti I. Analysis of apoptosis by propidium iodide staining and flow cytometry. Nat Protoc. 2006;1:1458–61. doi: 10.1038/nprot.2006.238. [DOI] [PubMed] [Google Scholar]
- 26.Bedia C, Casas J, Garcia V, Levade T, Fabrias G. Synthesis of a novel ceramide analogue and its use in a high-throughput fluorogenic assay for ceramidases. Chembiochem. 2007;8:642–8. doi: 10.1002/cbic.200600533. [DOI] [PubMed] [Google Scholar]
- 27.Kirkegaard T, Roth AG, Petersen NH, Mahalka AK, Olsen OD, Moilanen I, et al. Hsp70 stabilizes lysosomes and reverts Niemann-Pick disease-associated lysosomal pathology. Nature. 2010;463:549–53. doi: 10.1038/nature08710. [DOI] [PubMed] [Google Scholar]
- 28.Boya P, Kroemer G. Lysosomal membrane permeabilization in cell death. Oncogene. 2008;27:6434–51. doi: 10.1038/onc.2008.310. [DOI] [PubMed] [Google Scholar]
- 29.Stover TC, Sharma A, Robertson GP, Kester M. Systemic delivery of liposomal short-chain ceramide limits solid tumor growth in murine models of breast adenocarcinoma. Clin Cancer Res. 2005;11:3465–74. doi: 10.1158/1078-0432.CCR-04-1770. [DOI] [PubMed] [Google Scholar]
- 30.Colombini M. Ceramide channels and their role in mitochondria-mediated apoptosis. Biochim Biophys Acta. 2010;1797:1239–44. doi: 10.1016/j.bbabio.2010.01.021. [DOI] [PubMed] [Google Scholar]
- 31.Morales A, Lee H, Goni FM, Kolesnick R, Fernandez-Checa JC. Sphingolipids and cell death. Apoptosis. 2007;12:923–39. doi: 10.1007/s10495-007-0721-0. [DOI] [PubMed] [Google Scholar]
- 32.Elojeimy S, Holman DH, Liu X, El-Zawahry A, Villani M, Cheng JC, et al. New insights on the use of desipramine as an inhibitor for acid ceramidase. FEBS Lett. 2006;580:4751–6. doi: 10.1016/j.febslet.2006.07.071. [DOI] [PubMed] [Google Scholar]
- 33.Struckhoff AP, Bittman R, Burow ME, Clejan S, Elliott S, Hammond T, et al. Novel ceramide analogs as potential chemotherapeutic agents in breast cancer. J Pharmacol Exp Ther. 2004;309:523–32. doi: 10.1124/jpet.103.062760. [DOI] [PubMed] [Google Scholar]
- 34.Chapman JV, Gouaze-Andersson V, Messner MC, Flowers M, Karimi R, Kester M, et al. Metabolism of short-chain ceramide by human cancer cells--implications for therapeutic approaches. Biochem Pharmacol. 2010;80:308–15. doi: 10.1016/j.bcp.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee JY, Bielawska AE, Obeid LM. Regulation of cyclin-dependent kinase 2 activity by ceramide. Exp Cell Res. 2000;261:303–11. doi: 10.1006/excr.2000.5028. [DOI] [PubMed] [Google Scholar]
- 36.Phillips DC, Hunt JT, Moneypenny CG, Maclean KH, McKenzie PP, Harris LC, et al. Ceramide-induced G2 arrest in rhabdomyosarcoma (RMS) cells requires p21Cip1/Waf1 induction and is prevented by MDM2 overexpression. Cell Death Differ. 2007;14:1780–91. doi: 10.1038/sj.cdd.4402198. [DOI] [PubMed] [Google Scholar]
- 37.Ichikawa A, Ando J, Suda K. G1 arrest and expression of cyclin-dependent kinase inhibitors in tamoxifen-treated MCF-7 human breast cancer cells. Hum Cell. 2008;21:28–37. doi: 10.1111/j.1749-0774.2008.00048.x. [DOI] [PubMed] [Google Scholar]
- 38.Mandlekar S, Hebbar V, Christov K, Kong AN. Pharmacodynamics of tamoxifen and its 4-hydroxy and N-desmethyl metabolites: activation of caspases and induction of apoptosis in rat mammary tumors and in human breast cancer cell lines. Cancer Res. 2000;60:6601–6. [PubMed] [Google Scholar]
- 39.Callaghan R, Higgins CF. Interaction of tamoxifen with the multidrug resistance P-glycoprotein. Br J Cancer. 1995;71:294–9. doi: 10.1038/bjc.1995.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Borst P, Zelcer N, van Helvoort A. ABC transporters in lipid transport. Biochim Biophys Acta. 2000;1486:128–44. doi: 10.1016/s1388-1981(00)00053-6. [DOI] [PubMed] [Google Scholar]
- 41.Chapman JV, Gouaze-Andersson V, Cabot MC. Expression of P-glycoprotein in HeLa cells confers resistance to ceramide cytotoxicity. Int J Oncol. 2010;37:1591–7. doi: 10.3892/ijo_00000813. [DOI] [PubMed] [Google Scholar]
- 42.Richter C, Ghafourifar P. Ceramide induces cytochrome c release from isolated mitochondria. Biochem Soc Symp. 1999;66:27–31. doi: 10.1042/bss0660027. [DOI] [PubMed] [Google Scholar]
- 43.Yu T, Li J, Sun H. C6 ceramide potentiates curcumin-induced cell death and apoptosis in melanoma cell lines in vitro. Cancer Chemother Pharmacol. 2010;66:999–1003. doi: 10.1007/s00280-010-1374-1. [DOI] [PubMed] [Google Scholar]
- 44.Rizvi F, Heimann T, Herrnreiter A, O'Brien WJ. Mitochondrial dysfunction links ceramide activated HRK expression and cell death. PLoS One. 2011;6:e18137. doi: 10.1371/journal.pone.0018137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Johansson AC, Appelqvist H, Nilsson C, Kagedal K, Roberg K, Ollinger K. Regulation of apoptosis-associated lysosomal membrane permeabilization. Apoptosis. 2010;15:527–40. doi: 10.1007/s10495-009-0452-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu X, Cheng JC, Turner LS, Elojeimy S, Beckham TH, Bielawska A, et al. Acid ceramidase upregulation in prostate cancer: role in tumor development and implications for therapy. Expert Opin Ther Targets. 2009;13:1449–58. doi: 10.1517/14728220903357512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu X, Elojeimy S, Turner LS, Mahdy AE, Zeidan YH, Bielawska A, et al. Acid ceramidase inhibition: a novel target for cancer therapy. Front Biosci. 2008;13:2293–8. doi: 10.2741/2843. [DOI] [PubMed] [Google Scholar]
- 48.Flowers M, Fabrias G, Delgado A, Casas J, Abad JL, Cabot MC. C6-Ceramide and targeted inhibition of acid ceramidase induce synergistic decreases in breast cancer cell growth. Breast Cancer Res Treat. 2011 doi: 10.1007/s10549-011-1768-8. [DOI] [PubMed] [Google Scholar]
- 49.Gouaze-Andersson V, Flowers M, Karimi R, Fabrias G, Delgado A, Casas J, et al. Inhibition of acid ceramidase by a 2-substituted aminoethanol amide synergistically sensitizes prostate cancer cells to N-(4-hydroxyphenyl) retinamide. Prostate. 2011;71:1064–73. doi: 10.1002/pros.21321. [DOI] [PubMed] [Google Scholar]
- 50.Hwang JJ, Kim HN, Kim J, Cho DH, Kim MJ, Kim YS, et al. Zinc(II) ion mediates tamoxifen-induced autophagy and cell death in MCF-7 breast cancer cell line. Biometals. 2010;23:997–1013. doi: 10.1007/s10534-010-9346-9. [DOI] [PubMed] [Google Scholar]