

Abstract
Cutaneous T-cell lymphoma (CTCL) is a heterogeneous group of neoplastic disorders characterized by clonally derived and skin-homing malignant T-cells that express high level of chemokine receptor CCR4, which is associated with their skin-homing capacity. CCR4 is also highly expressed on T-regulatory cells (Tregs) that can migrate to several different types of chemotactic ligand CCL17 and CCL22 secreting tumors to facilitate tumor cell evasion from immune surveillance. Thus, its high level expression on CTCL cells and Tregs makes CCR4 a potential ideal target for antibody-based immunotherapy for CTCL and other types of solid tumors. Here we performed humanization and affinity optimization of a murine anti-CCR4 monoclonal antibody (mAb), mAb1567, that recognizes both the N-terminal and extracellular domains of CCR4 with high affinity and inhibits chemotaxis of CCR4+ CTCL cells. In a mouse CTCL tumor model, mAb1567 exhibited a potent anti-tumor effect and in vitro mechanistic studies showed that both complement-dependent cytotoxicity (CDC) and neutrophil-mediated antibody-dependent cellular cytotoxicity (ADCC) likely mediated this effect. MAb1567 also exerts human NK cell-mediated ADCC activity in vitro. Moreover, mAb1567 also effectively inhibits chemotaxis of CD4+CD25high Tregs via CCL22 and abrogates Treg suppression activity in vitro. An affinity optimized variant of humanized mAb1567, mAb2-3, was selected for further preclinical development based on its higher binding affinity and more potent ADCC and CDC activities. Taken together, this high affinity humanized mAb2-3 with potent anti-tumor effect and a broad range of mechanisms of action may provide a novel immunotherapy for CTCL and other solid tumors.
Keywords: CCR4, monoclonal antibody, lymphoma, humanization, immunotherapy
Introduction
Cutaneous T-cell lymphoma (CTCL) is the second most common extranodal non-Hodgkin lymphoma in adults, characterized by primary accumulation of clonally derived malignant CD4+ T-cells in the skin. There are 13 clinically and histologically distinct types of CTCL, and the majority falls into three classes: mycosis fungoides (MF), the most common type of CTCL, accounts for almost 50% of all primary cutaneous lymphomas; primary cutaneous CD30+ lymphoproliferative disorders—more specifically primary cutaneous anaplastic large cell lymphoma (PC-ALCL)—the second most common CTCL, account for circa 30%; and Sézary syndrome (SS) – the most aggressive type of CTCL makes up circa 5% (1, 2). Other forms of CTCL include Adult T-cell leukemia/lymphoma (ATLL) (2–5%) and peripheral T-cell lymphoma (PTCL) (2%). Although CTCL usually has indolent clinical behavior, in advanced stages it can progress into an aggressive phenotype with poor prognosis and survival (1, 3, 4) with severe immunodeficiency characteristically developing during disease progression (5). Current hypotheses maintain that the malignant T-cells drive this evolving immunodeficiency through the constitutive secretion of immunosuppressive cytokines, dysregulated expression of immunoregulatory proteins by the malignant T-cells, and loss of TCR repertoire complexity due to competitive replacement of normal T-cells by the clonally expanded malignant T-cells. In addition, a subset of the malignant T-cells may act as regulatory T-cells (Tregs) to suppress antitumor responses in some CTCL patients (6–8). Current therapies for CTCLs do not prevent new lesions from emerging (9) and durable long-term remissions are rare (10). Therefore, disease specific and more effective therapeutics that can decrease toxicity profiles and induce durable responses will greatly benefit CTCL patients.
Normal skin-homing CD4+ T-cells express cutaneous lymphocyte-associated antigen (CLA) and CC chemokine receptors (CCRs) CCR4, CCR6, CCR7 and CCR10 (11–14). Among these, only CCR4 is universally expressed at high levels on the malignant skin-homing T-cells, and its surface expression is closely associated with the enhanced skin-homing characteristics of CTCL cells and unfavorable disease outcome (15, 16). Regulatory T-cells also selectively express high levels of CCR4 compared to other T-cell subsets. The two specific ligands for CCR4, chemokines CCL17 and CCL22, produced by tumor cells and cells of the tumor microenvironment, attract CCR4+ Tregs to the tumor, where they suppress host immune responses against tumor and create a favorable environment for cancer cells to grow (17, 18). Thus, the high-level expression of CCR4 on CTCL cells and it’s preferential expression on Tregs (19) make CCR4 not only a potential ideal therapeutic target for CTCLs, but also for other type of cancers for which CCR4+ Tregs are involved in their immune-evasion.
In this study, we characterized and humanized a mouse anti-CCR4 monoclonal antibody, mAb1567 that recognizes both the N-terminal (NT) and the extracellular domains of CCR4. The antibody exhibited potent anti-tumor effects in a CTCL mouse model and its mechanism(s) of action, including complement-dependent cytotoxicity (CDC), neutrophil- and NK-mediated antibody-dependent cellular cytotoxicity (ADCC), were elucidated by a number of in vitro studies. In addition, mAb1567 could not only inhibit Tregs migration toward CCR4 ligand, CCL22, but also abrogate suppression by Tregs in the T-cell proliferation assay. Finally, after the affinity maturation of humanized mAb1567, the resulting mAb2-3 was further improved in affinity and showed stronger CDC and ADCC activities against CCR4+ tumor cells.
Materials and Methods
Cells
Mac-1 cell line was isolated from a patient with PC-ALCL, one of CTCLs (20), obtained from Dr. Thomas S. Kupper and cultured in 10% FBS RPMI1640. Luciferase-expressed Mac-1 cells were stably transduced with a luciferase reporter retrovirus and authenticated by detecting luminescence. 293F cell line was purchased from Invitrogen™. 293T (CRL-11268) and Cf2Th (CRL-1430) cell lines were purchased from American Type Culture Collection and incubated in 10% FBS DMEM. No additional authentication of these cell lines was conducted by the authors.
Antibodies and flow cytometry analysis
MAb1567 was purchased from R&D systems and the other 1567 variant antibodies were produced as described previously (21). Briefly, scFv-Fcs were constructed by cloning the single-chain variable region (scFv) into pcDNA3.1-Hinge vector in frame with human IgG1 Fc region. IgG1 was generated by cloning heavy chain variable region (VH) and light chain variable region (VL) into TCAE5.3 vector (22). Antibodies were produced in 293T or 293F cells and purified by proteinA-Sepharose (Amersham) affinity chromatography. For staining, Mac-1 was stained with anti-CCR4 antibodies, detected by FITC-conjugated goat-anti-human IgG or anti-mouse IgG antibodies (Sigma), and analyzed with FACSCalibur and CellQuest software.
Chemotaxis
Mac-1 cells (1×106/well) were placed in Transwell-migration wells (Corning) with or without mAb1567 for 3hrs at 37°C. Migrated cells harvested from the bottom chamber containing 50ng/ml human CCL17 or CCL22 (R&D Systems) were enumerated by FACS. Human CD4+ T-cells were isolated by CD4+ T-cell isolation kit (Miltenyi Biotech) and placed in Transwell-migration assays with c1567IgG. Migrated cells (CD4+CD25high) were enumerated as above in response to 100ng/ml CCL22. Percentages of migrated cells were calculated by dividing the number of transmigrated Mac-1 or CD4+CD25high cells by the number of input cells.
Antibody-dependent cell cytotoxicity assay
For LDH release assay, SCID/Beige mouse neutrophils, human peripheral blood mononucleated cells (PBMCs), or human NK cells and neutrophils were used as effector cells and Mac-1, Cf2Th-CCR4, or Cf2Th were used as target cells. Target cells (1×104/well) were plated into 96-well plates and antibodies were added. After one-hour, effector cells were added at an appropriate effector/target (E/T) ratio and incubated (PBMCs, NK and neutrophils for 4, 16 and 6 hours, respectively). The supernatants were recovered by centrifugation at 300×g and measured using nonradioactive cytotoxicity assay kits (Promega) at 490 nm. For 51Cr release assay, 1×106 Mac-1 were labeled with 100 μCi (3.7 MBq) of Na51Cr (Amersham International), washed, and used as targets. 51Cr-labeled target cells (5000/well) were seeded into 96-well plates and the release of 51Cr into supernatants was determined. The cytotoxicity was calculated by the following formula: ; E, released LDH from E/T culture with antibody; SE, spontaneous released LDH from effectors; ST, spontaneous released LDH from targets; M, the maximum released LDH from lysed targets.
Complement dependent cytotoxicity assay
5×104 Mac-1/well resuspended with medium containing rabbit serum (Cedarlane Laboratories) or mouse serum (IMS-COMPL, Innovative Research) were plated in 96-well plates with anti-CCR4 antibodies. After four-hour incubation, the supernatants were recovered and detected by LDH release assay, calculating as: .
Regulatory T-cell suppression assay
CD4+CD25high and CD4+CD25− T-cells were sorted by Beckman CoulterMoFlo sorter using mouse-anti-human CD4-PE-Cy5 (RPA-T4) and anti-human CD25-PE (M-A251) antibodies (BD Pharmigen). CD4+CD25− Teffs (2500) were cultured with or without CD4+CD25high Tregs (1250) in 96-well plates with 25,000 irradiated (3000rad) CD3-depleted PBMCs. Cells were stimulated with 0.05μg/ml plate-bound anti-CD3 (UCHT1) and 1μg/ml soluble anti-CD28 (CD28.2) antibodies (BD Pharmigen). Anti-CCR4 antibodies were included in the appropriate cultures. The cultures were pulsed on day 5 after culture initiation with 1μCi 3H-labelled thymidine/well (Perkin Elmer). Proliferation of cultures was measured in terms of incorporation of 3H-thymidine by reading counts in a scintillation counter (Perkin Elmer).
CCR4+ CTCL tumor-bearing mouse model
2×106 luciferase-Mac-1 or 1×107 Mac-1 cells were injected subcutaneously into the dorsolateral flank in 6-week SCID/Beige mice (Charles River). After 24hrs of injection, mice were randomly assigned into different groups and treated with 3mg/kg of mAb1567 and mouse IgG2b (twice a week for three weeks) or 5mg/kg of control-scFv-Fc, c1567-scFv-Fc and h1567-scFv-Fc (twice a week for four weeks) by intraperitoneal (i.p.) injection. Body weight and tumor size were measured using digital caliper and Xenogen imaging. Tumor volumes were calculated as length × (width)2 × 0.52. Animal care was carried out in accordance with the guidelines of Animal Care and Use Committee of Dana-Farber Cancer Institute.
Statistical analyses
Data was analyzed using two-sided unpaired Student’s t-test. “*”, “**”, and “***” indicate p < 0.05, 0.01 and 0.001, respectively. All values and bars are represented as mean ± standard deviation.
Results
Characterization of a mouse anti-CCR4 mAb, mAb1567, in vitro and in vivo
CCR4 has four regions exposed at the cell surface: the NT (~30–50 aa) and three extracellular domains loops (ECLs, each of ~10–30 aa), which are important for ligand binding, intracellular signaling and other biological functions. In this study, two commercially available murine anti-CCR4 mAbs, mAb1567 (R&D systems) and 1G1 (BD Pharmingen), both generated by immunizing mouse with full-length human CCR4 (hCCR4) expressing cells (13, 23), were initially selected for evaluation. Both mAb1567 and 1G1 showed specific binding activity in FACS analysis to hCCR4 expressing Cf2Th-CCR4 cells but not to the parental Cf2Th cells. By comparison, mAb1567 had relatively higher affinity than 1G1 under the same antibody concentrations tested (data not shown). Therefore, we selected only mAb1567 for further characterization.
Binding of mAb1567 was further tested using the CCR4+ Mac-1 cells by FACS analysis and the half maximal effective binding concentration (EC50) is about 0.45 nM (Fig. 1A). Chemotaxis inhibition assay showed that mAb1567 effectively inhibited chemotaxis of Mac-1 cells in a dose dependent manner toward both CCR4 ligands, CCL17 and CCL22 (Fig. 1B). We next examined the epitope recognized by mAb1567, in particular whether it recognizes solely the NT or a nonlinear conformational dependent epitope comprising of both NT and ECL, by using hCCR4 and hCCR8 NT swapping chimeras that either contained CCR8-NT/CCR4-ECLs (Chi#1) or CCR4-NT/CCR8-ECLs (Chi#2) (24). As shown in Supplementary Fig. S1A, all constructs encoding wild type or chimeras CCR4 and CCR8 expressed to similar levels on the cell surface as validated by antibody staining against the HA tag. MAb1567 specifically recognized cell surface full-length hCCR4 but not hCCR8. It bound to Chi#1 and Chi#2 to similar level as wild type CCR4 indicating that the epitope of mAb1567 is not solely a linear epitope on NT of CCR4, rather both ECLs and NT contribute to the binding of mAb1567 with CCR4. However, the CCR4-Nt alone is also sufficient for some degree of mAb1567 binding to CCR4-Nt-Fc as determined in Elisa studies to plate-bound mAb1567 (Supplementary Fig. S1B-D). Moreover, mAb1567 showed high specificity for CCR4 rather than CCR5, the most similar CCR molecule to CCR4 (Supplementary Fig. S1E).
Figure 1. Overexpression of functional CCR4 on cutaneous T-cell lymphoma and mouse anti-CCR4 mAb1567 inhibits tumor formation.
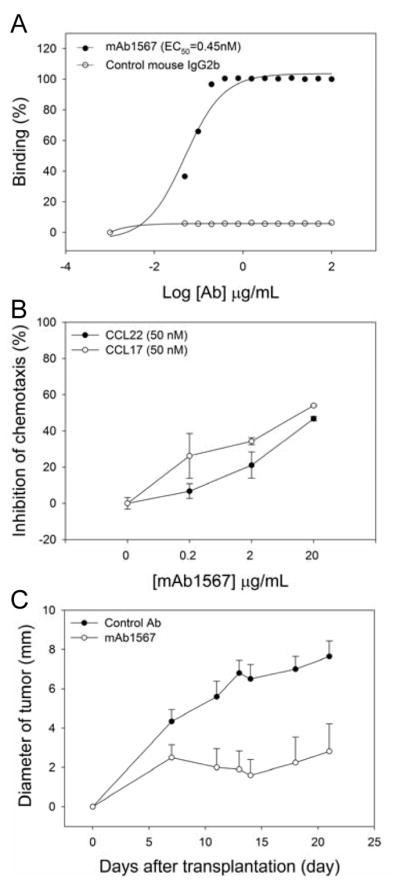
A, Dose-dependent binding curve of mAb1567 to CCR4+ Mac-1 cells by FACS analysis. The EC50 was generated using SigmaPlot software. B, MAb1567 effectively inhibited chemotaxis of Mac-1 cells to CCR4 ligands, CCRL22 and CCL17. C, The antitumor effect of mAb1567 in SCID/Beige mice bearing Mac-1 xenografts.
We then tested the antitumor effect of mAb1567 in vivo in a CTCL model using immunodeficient SCID/Beige mice that lack T- and B-cells and have defective NK function. SCID/Beige mice implanted with Mac-1 cells can efficiently form subcutaneous tumors (25). As shown in Fig. 1C, the tumor size in the mAb1567 treated group was 3 to 4-fold smaller than seen in the control group. None of the mice showed mAb1567 treatment related toxicity.
MAb1567 mediates against Mac-1 cells both CDC in the presence of mouse and rabbit complement and neutrophil-ADCC
To further understand the mechanism underlying the anti-tumor effect of mAb1567 seen in the SCID/Beige mice, we tested if mAb1567 can mediate CDC and/or neutrophil-mediated ADCC effects against CCR4+ tumor cells in vitro. MAb1567 induced a significant lysis of Mac-1 cells in a dose-dependent manner in the presence of mouse complement as compared to the mouse IgG2b isotype control antibody (Fig. 2A). Rabbit complement was also tested and mAb1567 mediated a much more potent CDC activity, reached 80% of target cell lysis (Fig. 2B). Next, neutrophils isolated from the SCID/Beige mice were tested in an in vitro ADCC assay. As shown in Fig. 2C, mAb1567 specifically mediated ~20% lysis via mouse neutrophils as compared to control at E/T (neutrophils/Mac-1) ratio of 80:1. These results show that mAb1567 can directly mediate not only CDC but also mouse neutrophil-induced ADCC activities.
Figure 2. MAb1567 mediates against Mac-1 cells both CDC and neutrophil-ADCC.

A, MAb1567 mediated CDC activity via mouse complement. Figure shown is one experiment that is representative of at least three independent experiments. B, MAb1567 mediated CDC activity with rabbit complement. C, Neutrophils from SCID-Beige mice mediated-mAb1567-dependent ADCC.
Cloning, expression and activity of chimeric mAb1567
To humanize mAb1567 for further pre-clinical studies, the cDNAs encoding the VH and VL genes from the hybridoma cell line were individually recovered by RT-PCR using primers specific for mouse antibody variable genes. The VH and VL of mAb1567 belong to mouse VH1 (IGHV1S56*01) and VK8 (IGKV8-27*01) families and were rearranged with the JH1 (IGHJ1*01) and JK2 (IGKJ2*01) segments, respectively. The cloned, rearranged VH and VL genes were then assembled as a single chain antibody variable region fragment (scFv) using a (G4S)3 linker. Binding of the recombinant mAb1567 to CCR4 was verified in both scFv-Fc IgG1 minibody (c1567-scFv-Fc) (Fig. 3A) and full-length chimeric IgG1 form (c1567-IgG) (data not shown).
Figure 3. Humanization of mAb 1567 and function analysis.

A, Comparative binding analysis of c1567 and h1567 scFv-Fcs. B and C, ADCC activity mediated by c1567 scFv-Fc. Either PBMCs (B) or NK cells (C) from healthy donors were used as effector cells. Target cell lysis was measured either by Cr51 (B) or LDH release (C). Data were calculated from triplicate wells of one experiment and are representative of three independent experiments. D, Amino acid sequence alignment of the rearranged mouse and humanized variable heavy (VH) and variable light kappa (VK) domains. Residues in magenta indicate framework residues that were changed for humanization.
As NK cell-mediated ADCC is one of the most important mechanisms of action for immunotherapy with human IgG1 Abs, we further tested if recombinant mAb1567 can mediate ADCC via NK cells. Chimeric 1567 in both scFv-Fc or IgG1 forms were highly effective in killing Mac-1 cells in an in vitro ADCC assay using human PBMCs (Fig. 3B) or purified NK (CD56+CD16+) cells (Fig. 3C) from healthy donors as effector cells at different E/T ratios.
Humanization of mAb1567 and related biological studies
Next, the structure-guided complementarity-determining region (CDR) grafting approach was employed to humanize mAb1567. Homology three-dimensional modeling of the VH and VL chains of mAb1567 using Web Antibody Modeling program (WAM) (26) was generated to known antibody structures in the PDB database. For selecting the human acceptor framework template for CDR-grafting, the VH and VL amino acid sequences of mAb1567 were separately compared to human Ab sequences in the IGBLAST database to identify the most similar human Ab and Ig germline VH and VL sequences (Fig 3D). The human VH (McAb Ctm01, PDB:1ae6H) and VL (Genebank #ABG38372) share 82% and 84% amino acid sequence homology to the VH and VL of mAb1567, respectively; the best matched human Ig germline V sequences are IGHV1-3*01 (67% homology to mAb1567-VH) and IGKV4-1*01 (83% homology to mAb1567-VL). The framework residues of mAb1567 were manually changed to the selected human framework residues to generate the humanized mAb1567 (h1567). GROMOS force field energy minimization parameter was then applied to homology model h1567 using DeepView program (27). Examination of this energy minimized homology model of h1567 was performed to ensure no residues that had distorted geometry or steric clashes with other residues.
The h1567 sequence shown in Fig. 3D has 21 and 11 amino acid differences in the framework regions compared to the mouse VH and VL, respectively. The humanized VH and VL gene were de novo synthesized and codon-optimized for mammalian cell expression. The binding affinity of h1567 and c1567 scFv-Fcs to CCR4 was then compared by FACS with Mac-1 cells. The h1567 had ~2-fold decrease in binding as compared to c1567 but both are in the nanomolar range, with EC50 of 2.2 nM and 1 nM, respectively (Fig. 3A). The humanized h1567 scFv-Fc maintained potent NK-mediated ADCC killing of Mac-1 cells compared to c1567 (Fig 3C).
To test the in vivo anti-tumor effect of h1567, the luciferase-expressing Mac-1 cells were subcutaneously implanted into the dorsolateral flank of SCID/Beige mice, and mice were i.p. treated with 5 mg/kg of control-scFv-Fc, c1567-scFv-Fc, h1567-scFv-Fc or equivalent volumes of saline. Tumor growth in mice was monitored for luciferase intensity by IVIS imaging. All mice were sacrificed on day 28 and tumors were excised for photographing and measuring tumor weight. As shown in Fig. 4, tumors were significantly reduced in size at day 21 in c1567 and h1567 treated groups but not in control-scFv-Fc or PBS treated groups as measured by IVIS imaging (Fig. 4A-top and C), size of the excised tumors (Fig. 4A-bottom), tumor volume (Fig. 4B) and tumor weight (Fig. 4D).
Figure 4. Humanized 1567 in tumor treatment.

A, Mice bearing Mac-1 tumors were imaged using an IVIS imaging system. Luciferase signal (top) and tumor size (bottom) in mice treated with anti-CCR4 antibodies. Color scale, luminescent signal intensity: blue, least intense signal; red, most intense signal. Bar scale, 1 cm. B, Tumor sizes, image intensity (C), and tumor weight (D) were measured.
ADCC and CDC activities of higher-affinity h1567 variants
Although the h1567 exhibited similar biological activity as its murine counterpart in both in vitro and in vivo, the relative apparent binding affinity of h1567 is 2-fold lower than c1567 (Fig. 3A). In order to further affinity mature the h1567, we performed VL-chain shuffling and alanine scanning to identify key residues in CDRs, followed by selection and screening of phage display library constructed by random mutagenesis of key residues in the CDRs (see Supplementary method and Supplementary Fig. S2 for details).
The two affinity improved h1567 variants, mAbs 1-44 and 2-3 that showed higher binding affinity to Mac-1 cells than parental h1567 with EC50 of 1.47 and 1.39 nM, respectively (Supplementary Fig. S2F), were further evaluated for their capacity to mediate ADCC activity using human NK cells. The result showed that improvement in ADCC activity of the h1567 variants is correlated with their binding affinity, 2-3-scFv-Fc exhibited the best human NK-mediated ADCC activity for both Mac-1 cells (Fig. 5A) and Cf2Th-CCR4 cells but not to negative control Cf2Th (Supplementary Fig. S3). Moreover, since parental mAb1567 could induce mouse neutrophil-mediated ADCC, h1567 and 2-3 were tested for human neutrophil-mediated ADCC assay and mAb2-3 showed enhanced cytotoxic activity (Fig. 5B) compared with h1567. Furthermore, slightly improved CDC activity against Mac-1 cells was also observed for both 1-44 and 2-3 variants, but more for the 2-3-scFv-Fc (Supplementary Fig. S4).
Figure 5. Humanized 1567 variants with improved binding affinity, ADCC and CDC activity.

A, H1567 and its variants 1-44 and 2-3 were tested the ADCC activity. Data shown in the box and whiskers graph represent three independent experiments and each was performed with NK cells from a different healthy donor; the box extends from lowest percentile to the highest percentile, with a line at the median. The whiskers above and below the box indicate the 95th and 5th percentiles. B, ADCC activities on Mac-1 with human neutrophils. C, ADCC activities on Mac-1 with human NK cells. D, CDC of wild-type Fc antibodies (scFv-Fcs) and mutant Fc antibodies (scFv-mFcs) against Mac-1.
Fc engineering was also performed on mutant Abs 1-44 and 2-3 to further enhance ADCC activity by mutating three residues (S239D, A330L and I332E) in CH2 domain, which have been shown to increase human IgG1 antibody’s ADCC effect (28). As shown in Fig. 5C, ADCC mediated by 1-44- and 2-3-scFv-mFcs was significantly enhanced as compared to their wild type Fc counterparts or the wild type h1567. However as the A330L mutation in the Fc domain can ablate CDC function (29), we also tested and confirmed that CDC activity for the scFv-mFcs forms of 1-44 and 2-3 scFv-mFcs was completely abolished (Fig. 5D). Taken together, the affinity optimized variants of humanized 1567, in particular the 2-3 variant, demonstrated improved ADCC and CDC effector functions, and NK cell-mediated ADCC activity can be further enhanced through Fc engineering.
1567 inhibits Tregs chemotaxis and partially abrogates Tregs’ suppressive activity in vitro
Finally, as the majority (94%) of freshly isolated CD4+CD25high Tregs from peripheral blood express high level of surface CCR4 (30) and they have been reported to migrate to tumors secreting CCL22 (31), we investigated whether Ab1567 could have an anti-tumor role by modulating the chemotactic recruitment and suppressive activity of human CD4+ Tregs. First, we confirmed that CD4+CD25high Tregs migrated toward CCL22 much more effectively than CD4+CD25− T-cells (Fig. 6A). Next, using peripheral blood CD4+ T-cells in combination with examining the Treg phenotype of the migrated cells, we confirmed that c1567 completely inhibited the migration of CD4+CD25high Tregs in a transwell chemotaxis assay at concentrations greater than 2 μg/mL (Fig. 6B).
Figure 6. Anti-CCR4 antibody abrogates suppression by T-regulatory cells.
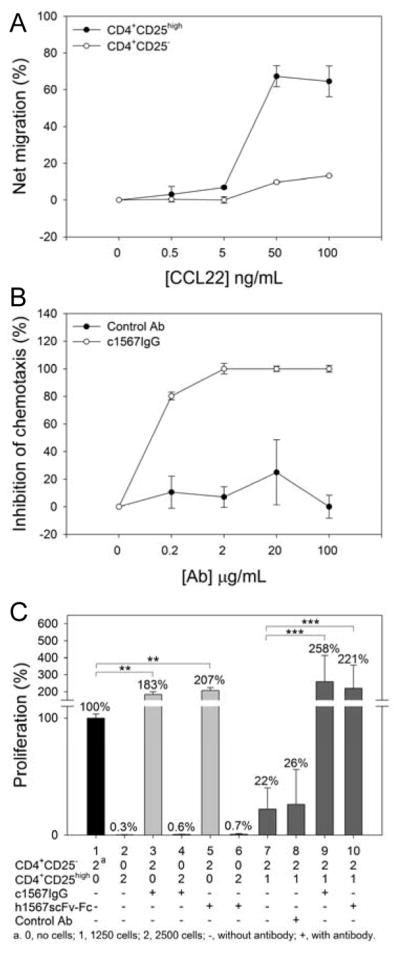
A, CD4+CD25high T-cells showed demonstrable chemotactic responses toward CCL22. B, Chimeric mAb1567 effectively inhibited chemotaxis of Tregs to CCL22. C, The effect of anti-CCR4 antibodies on proliferation of Teffs and the abrogation of the suppressive function of Tregs. The percent proliferation was normalized to CD4+CD25− Teffs without antibody treatment.
In addition, as we are unaware of any published data on the role of CCR4 in Treg function, we also examined whether 1567 engagement of CCR4 could result in modulation of Treg suppression activity in an in vitro Treg suppression assay. As shown in Fig. 6C, the proliferation of CD4+ T effector cells (Teffs alone, lane 1) was inhibited by highly purified CD4+CD25high Tregs (1:2 ratio) by 78 % (lane 7), which is our typical Treg suppression effect on Teffs (32). Surprisingly, in the presence of c1567IgG or h1567scFv-Fc, the proliferation of Teff was stimulated to 183% and 207%, respectively, (lane 3 and 5) but there was no stimulatory effect on Treg (lane 4 and 6). In the Treg/Teff co-culture (1:2 ratio), the proliferation of Teff was inhibited directly (lane 7) and there was no reversal of this inhibition by control mAb (lane 8). Moreover, T-cell proliferation was restored with a net positive response to 258% and 221% (lane 9 and 10) in the presence of c1567IgG or h156scFv-Fc, respectively.
The increase in CD4+CD25− T-cell proliferation in the presence of anti-CCR4 antibodies was further observed by flow cytometry. We detected CD4+CD25− T-cell proliferation on the basis of CFSE fluorescence intensity of labeled CD4+CD25− T-cells in the Treg/Teff coculture. The analysis revealed that over the 7 day study, CD4+CD25− T-cell proliferation responded to stimulation of anti-CCR4 antibodies in a time-dependent manner and was ~50% higher than with control antibody treated cells which showed no proliferation (Supplementary Fig. S5). Importantly, Treg mediated suppression of Teff proliferation occurred even in the present of anti-CD3/CD28 costimulation but could be reversed in the presence of anti-CCR4 antibody resulting in an increased proliferative capacity of CD4+CD25− T-cells.
Discussion
In this study, we humanized a mouse anti-CCR4 antibody, mAb1567, which recognizes both the NT and the ECLs of CCR4 with high affinity and inhibits migration of CCR4+ tumor cells towards its two ligands, CCL22 and CCL17. The antibody exhibited potent anti-tumor effect in a CTCL mouse model with the mechanisms of CDC and neutrophil-mediated ADCC likely involved (Figs. 1 and 2). The chimeric or humanized mAb1567 also showed potent CDC and human NK cell-mediated ADCC activity in vitro and therapeutic effects in vivo (Figs. 3 and 4). In addition, this antibody effectively inhibited the chemotaxis of CD4+CD25high Tregs to CCL22. Interestingly, it also stimulated CD4+CD25− cell proliferation and inhibited Tregs’ immune-suppressive activity (Fig. 6 and Supplementary Fig. S5). The affinity of h1567 was further improved by employing a targeted mutagenesis strategy in combination with phage-display library selection and the affinity optimized h1567 variant mAb2-3 showed better CDC and ADCC activities against CCR4+ tumor cells in vitro (Fig. 5). These studies support that the affinity selected mAb2-3 may provide a novel immunotherapy option not only to directly kill the CCR4+ tumor cells, but also may have a role in other cancers by suppressing Treg trafficking and overcoming the suppressive effect of CCR4+ Tregs to enhance host anti-tumor immune responses.
A number of studies have been reported on a humanized anti-CCR4 antibody, KM-0761 (33, 34). These studies have not only demonstrated KM-0761’s effect in CTCL tumor animal models and it’s mechanism of action, but also convincing support that CCR4 is an important antibody-based immunotherapy target for relapsed CCR4+ ATL or PTCL (35, 36). The KM-0761 antibody was originally raised in mouse by immunizing with the CCR4’s NT(12-29) peptide (34, 37), it works in killing tumor cells by ADCC mainly through NK and/or macrophages (33, 38, 39), and augmentation of FcγR engagement and ADCC killing is achieved by defucosylation (34). In contrast, mAb1567 was generated with full-length CCR4, it recognizes an epitope comprising of both NT and ECLs of CCR4, and ADCC enhanced killing is achieved through both affinity maturation and Fc engineering (29). In addition, mAb1567 demonstrated both CDC and ADCC activity. The disparity in CDC activities between these two IgG1 antibodies is likely due to different recognition of the CCR4 epitope(s). CDC is often dependent on the distance between the plasma membrane and the constant region of the sensitizing antibody that mediates effector functions (40). It is possible the CDC activity mediated by mAb1567 is related to the position and orientation of the recognized epitope, when bound the mAb1567 can position its Fc region more proximal to the membrane to recruit complement efficiently. Another possibility is that binding to the different epitope may promote more efficient cross-linking of CCR4, and thus increasing the binding avidity (41).
Anti-tumor effect mediated by cancer cell-directed antibodies can generally be attributed to ADCC, CDC or direct anti-proliferation. We found that mAb1567 did not show any inhibition of cell proliferation (data not shown). Effector cells that can mediate ADCC are NK cells, neutrophils and monocytes/macrophages. The SCID/Biege mice used in our in vivo studies not only lack T- and B-lymphocytes but are also NK cell defective. Thus, we surmised that the anti-tumor activity of mAb1567 seen in SCID/Beige mice might be due to effector cells other than NK cell-mediated ADCC and/or to CDC. In vitro CDC experiments indeed showed that mAb1567 had CDC activity as compared to control Ab though the level of lysis target Mac-1 cells was low, up to ~20%. Most mouse strains (including the mouse stains tested here) have exceptionally low complement activity, much lower than that of humans or other animals, including rabbits, guinea pigs and hamsters (42). CDC assay with rabbit complement indeed showed dramatically improved lysis of target cells by mAb1567 (Fig. 2B) up to ~80%. Furthermore, mAb1567 could mediate ADCC activity (~25%) via neutrophils isolated from SCID/Beige mice. Due to limited number of neutrophils available, only a single dose of mAb1567 and isotype control mAb at 50 μg/mL was tested (Fig. 2C). These results suggest that the antitumor activity of mAb1567 in the mouse model is likely due to a combination of both CDC and through neutrophil-mediated ADCC although it remains to be tested if mouse monocytes/macrophages may also play a role.
Many therapies for cancer are accompanied by adverse side effects and dose-dependent toxicities. The development of more effective cancer immunotherapy with better discrimination between tumor cells and normal cells is the most important goal of current anticancer research. CCR4 is universally expressed at high levels among most CTCL cells; however, a subset of normal T-cells also can express CCR4. It was reported that the CCR4 is selectively expressed on Th2 cells, but not Th1 cells, which are the precursor of memory T-cells (43). Of particular importance to the in vivo expression of CCR4 on Th2 cells would be a concern of bystander cytotoxicity in response to anti-CCR4 antibody treatment. However, potential bystander effects can be managed as shown in the recent clinical trial reports on the anti-CCR4 mAb, KW-0761, for treatment of ATLL and PTCL where good clinical activity was seen without serious side effects (35, 36). This may be due, at least in part, to the CCR4-expressing CTCL patient cells showing a significantly 4-fold higher expression level than T cells from healthy donors (44). This high CCR4 expression in CTCL patients may result in most anti-CCR4 antibody binding to tumor T-cells but not normal T-cells. We are also aware of reports that CCR4 is expressed on platelets (45, 46) but again in the KW-0761 clinical trial (35) grade 3 or 4 thrombocytopenia was only seen in 5/28 patients (18%) and not associated with bleeding.
CCR4 is also highly expressed on the majority (94%) of CD4+CD25highFoxP3+ Tregs (30) that are considered the most potent inhibitors of anti-tumor immunity and the greatest barrier to successful immunotherapy (31, 47). CCR4 also plays an important role in Treg recruitment to the site of action. In some solid tumors including breast cancer (17, 48), ovarian cancer (18) and oral squamous cell carcinoma (49), increased numbers of recruited Tregs that are chemoattracted through the CCR4-CCL22 axis likely play a significant role in the suppression of host anti-tumor immunity. This trafficking pattern makes CCR4 an even more attractive therapeutic target for a broader range of tumors. MAb1567 was tested for inhibition of Treg migration and showed complete inhibition of chemotaxis of CD4+CD25high Tregs (Fig. 6A). Interestingly, it appeared that mAb1567 can also abrogate the immune suppressive function of CD4+CD25high Tregs, while stimulating the proliferation of CD4+CD25− Teffs (Fig. 6C and Supplementary Fig. S5). It has been proposed that Tregs exert their function by multiple suppressive mechanisms including cell-cell contact, including competitive consumption of IL-2, production of the immunosuppressive cytokines IL-10 and TGF-β, cytolysis, metabolic disruption and modulation of the function of antigen-presenting cells (5, 47). However, CCR4 is not known to play a role in CD4+CD25− cell proliferation and although a previous study described that CCR4−/− Tregs are defective in suppressive activity in a model of colitis, this was due to their lack of chemotactic recruitment to the mesenteric lymph nodes since their in vitro suppressive function was intact (50). It will be important to perform additional studies to further confirm these findings and elucidate the mechanism(s) by which anti-CCR4 mAb1567 could act through CCR4 on these cells to exercise different functions - proliferative effect on CD4+CD25− Teffs and partial to complete abrogation of Tregs’ suppression.
In summary, we have humanized and affinity maturated murine anti-CCR4 antibody 1567 and demonstrated its in vitro and in vivo antitumor activity against CTCL cells. The mechanisms of tumor cell killing by mAb1567 appear to be multiple including CDC as well as neutrophil-and NK cell-mediated ADCC. The affinity matured mAb2-3 promoted more potent CTCL lysis by augmentation of both NK cell-mediated ADCC and CDC activities. Remarkably, the activity of mAb1567 also extended to the normal T-cell compartment with demonstrable proliferative effect on CD4+CD25− Teffs and abrogation of Treg suppression. These findings support further evaluation of our anti-CCR4 antibody as an immunotherapy for CTCL and other cancers where augmentation of Teff functions against the tumor cells are likely to have a therapeutic benefit.
Supplementary Material
Acknowledgments
We thank Maryam Ali, Hong Tao and Erin M. McConocha for technical support, and National Foundation of Cancer Research (CFCR) for contribution of equipment used in this study.
Grant Support
This work was funded by Skin Cancer Score project 2P50CA093683 to T.S. Kupper, J. Campbell and W.A. Marasco and NIH AI058804 to Q. Zhu.
Footnotes
Disclosure of Conflicts of Interest: All authors declare no conflicts of interest.
References
- 1.Willemze R, Jaffe ES, Burg G, Cerroni L, Berti E, Swerdlow SH, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768–85. doi: 10.1182/blood-2004-09-3502. [DOI] [PubMed] [Google Scholar]
- 2.Clark RA, Watanabe R, Teague JE, Schlapbach C, Tawa MC, Adams N, et al. Skin Effector Memory T Cells Do Not Recirculate and Provide Immune Protection in Alemtuzumab-Treated CTCL Patients. Sci Transl Med. 2012;4:117ra7. doi: 10.1126/scitranslmed.3003008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim YH, Liu HL, Mraz-Gernhard S, Varghese A, Hoppe RT. Long-term outcome of 525 patients with mycosis fungoides and Sezary syndrome: clinical prognostic factors and risk for disease progression. Arch Dermatol. 2003;139:857–66. doi: 10.1001/archderm.139.7.857. [DOI] [PubMed] [Google Scholar]
- 4.Kadin ME, Carpenter C. Systemic and primary cutaneous anaplastic large cell lymphomas. Semin Hematol. 2003;40:244–56. doi: 10.1016/s0037-1963(03)00138-0. [DOI] [PubMed] [Google Scholar]
- 5.Krejsgaard T, Odum N, Geisler C, Wasik MA, Woetmann A. Regulatory T cells and immunodeficiency in mycosis fungoides and Sezary syndrome. Leukemia: official journal of the Leukemia Society of America, Leukemia Research Fund, UK. 2011;26:424–32. doi: 10.1038/leu.2011.237. [DOI] [PubMed] [Google Scholar]
- 6.Zhang Q, Nowak I, Vonderheid EC, Rook AH, Kadin ME, Nowell PC, et al. Activation of Jak/STAT proteins involved in signal transduction pathway mediated by receptor for interleukin 2 in malignant T lymphocytes derived from cutaneous anaplastic large T-cell lymphoma and Sezary syndrome. Proc Natl Acad Sci U S A. 1996;93:9148–53. doi: 10.1073/pnas.93.17.9148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kasprzycka M, Zhang Q, Witkiewicz A, Marzec M, Potoczek M, Liu X, et al. Gamma c-signaling cytokines induce a regulatory T cell phenotype in malignant CD4+ T lymphocytes. J Immunol. 2008;181:2506–12. doi: 10.4049/jimmunol.181.4.2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yawalkar N, Ferenczi K, Jones DA, Yamanaka K, Suh KY, Sadat S, et al. Profound loss of T-cell receptor repertoire complexity in cutaneous T-cell lymphoma. Blood. 2003;102:4059–66. doi: 10.1182/blood-2003-04-1044. [DOI] [PubMed] [Google Scholar]
- 9.Liu HL, Hoppe RT, Kohler S, Harvell JD, Reddy S, Kim YH. CD30+ cutaneous lymphoproliferative disorders: the Stanford experience in lymphomatoid papulosis and primary cutaneous anaplastic large cell lymphoma. J Am Acad Dermatol. 2003;49:1049–58. doi: 10.1016/s0190-9622(03)02484-8. [DOI] [PubMed] [Google Scholar]
- 10.Querfeld C, Rosen ST, Guitart J, Kuzel TM. The spectrum of cutaneous T-cell lymphomas: new insights into biology and therapy. Curr Opin Hematol. 2005;12:273–8. doi: 10.1097/01.moh.0000166498.64515.03. [DOI] [PubMed] [Google Scholar]
- 11.Kupper TS, Fuhlbrigge RC. Immune surveillance in the skin: mechanisms and clinical consequences. Nat Rev Immunol. 2004;4:211–22. doi: 10.1038/nri1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kunkel EJ, Boisvert J, Murphy K, Vierra MA, Genovese MC, Wardlaw AJ, et al. Expression of the chemokine receptors CCR4, CCR5, and CXCR3 by human tissue-infiltrating lymphocytes. Am J Pathol. 2002;160:347–55. doi: 10.1016/S0002-9440(10)64378-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Campbell JJ, Haraldsen G, Pan J, Rottman J, Qin S, Ponath P, et al. The chemokine receptor CCR4 in vascular recognition by cutaneous but not intestinal memory T cells. Nature. 1999;400:776–80. doi: 10.1038/23495. [DOI] [PubMed] [Google Scholar]
- 14.Campbell JJ, Clark RA, Watanabe R, Kupper TS. Sezary syndrome and mycosis fungoides arise from distinct T-cell subsets: a biologic rationale for their distinct clinical behaviors. Blood. 2010;116:767–71. doi: 10.1182/blood-2009-11-251926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu XS, Lonsdorf AS, Hwang ST. Cutaneous T-cell lymphoma: roles for chemokines and chemokine receptors. J Invest Dermatol. 2009;129:1115–9. doi: 10.1038/jid.2009.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tokura Y, Sugita K, Yagi H, Shimauchi T, Kabashima K, Takigawa M. Primary cutaneous anaplastic large cell lymphoma with fatal leukemic outcome in association with CLA and CCR4-negative conversion. J Am Acad Dermatol. 2007;57:S92–6. doi: 10.1016/j.jaad.2006.08.053. [DOI] [PubMed] [Google Scholar]
- 17.Olkhanud PB, Baatar D, Bodogai M, Hakim F, Gress R, Anderson RL, et al. Breast cancer lung metastasis requires expression of chemokine receptor CCR4 and regulatory T cells. Cancer Res. 2009;69:5996–6004. doi: 10.1158/0008-5472.CAN-08-4619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–9. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 19.Ishida T, Iida S, Akatsuka Y, Ishii T, Miyazaki M, Komatsu H, et al. The CC chemokine receptor 4 as a novel specific molecular target for immunotherapy in adult T-Cell leukemia/lymphoma. Clin Cancer Res. 2004;10:7529–39. doi: 10.1158/1078-0432.CCR-04-0983. [DOI] [PubMed] [Google Scholar]
- 20.Kadin ME, Cavaille-Coll MW, Gertz R, Massague J, Cheifetz S, George D. Loss of receptors for transforming growth factor beta in human T-cell malignancies. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:6002–6. doi: 10.1073/pnas.91.13.6002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sui J, Li W, Murakami A, Tamin A, Matthews LJ, Wong SK, et al. Potent neutralization of severe acute respiratory syndrome (SARS) coronavirus by a human mAb to S1 protein that blocks receptor association. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:2536–41. doi: 10.1073/pnas.0307140101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reff ME, Carner K, Chambers KS, Chinn PC, Leonard JE, Raab R, et al. Depletion of B cells in vivo by a chimeric mouse human monoclonal antibody to CD20. Blood. 1994;83:435–45. [PubMed] [Google Scholar]
- 23.[cited; Available from: www.rndsystems.com/pdf/fab1567a.pdf
- 24.Jopling LA, Sabroe I, Andrew DP, Mitchell TJ, Li Y, Hodge MR, et al. The identification, characterization, and distribution of guinea pig CCR4 and epitope mapping of a blocking antibody. The Journal of biological chemistry. 2002;277:6864–73. doi: 10.1074/jbc.M109974200. [DOI] [PubMed] [Google Scholar]
- 25.Pfeifer W, Levi E, Petrogiannis-Haliotis T, Lehmann L, Wang Z, Kadin ME. A murine xenograft model for human CD30+ anaplastic large cell lymphoma. Successful growth inhibition with an anti-CD30 antibody (HeFi-1) Am J Pathol. 1999;155:1353–9. doi: 10.1016/S0002-9440(10)65237-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Whitelegg NR, Rees AR. WAM: an improved algorithm for modelling antibodies on the WEB. Protein Eng. 2000;13:819–24. doi: 10.1093/protein/13.12.819. [DOI] [PubMed] [Google Scholar]
- 27.Daura X, Oliva B, Querol E, Aviles FX, Tapia O. On the sensitivity of MD trajectories to changes in water-protein interaction parameters: the potato carboxypeptidase inhibitor in water as a test case for the GROMOS force field. Proteins. 1996;25:89–103. doi: 10.1002/(SICI)1097-0134(199605)25:1<89::AID-PROT7>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 28.Carter PJ. Potent antibody therapeutics by design. Nat Rev Immunol. 2006;6:343–57. doi: 10.1038/nri1837. [DOI] [PubMed] [Google Scholar]
- 29.Lazar GA, Dang W, Karki S, Vafa O, Peng JS, Hyun L, et al. Engineered antibody Fc variants with enhanced effector function. Proc Natl Acad Sci U S A. 2006;103:4005–10. doi: 10.1073/pnas.0508123103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baatar D, Olkhanud P, Sumitomo K, Taub D, Gress R, Biragyn A. Human peripheral blood T regulatory cells (Tregs), functionally primed CCR4+ Tregs and unprimed CCR4- Tregs, regulate effector T cells using FasL. J Immunol. 2007;178:4891–900. doi: 10.4049/jimmunol.178.8.4891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Byrne WL, Mills KH, Lederer JA, O’Sullivan GC. Targeting regulatory T cells in cancer. Cancer research. 2011;71:6915–20. doi: 10.1158/0008-5472.CAN-11-1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hirahara K, Liu L, Clark RA, Yamanaka K, Fuhlbrigge RC, Kupper TS. The majority of human peripheral blood CD4+CD25highFoxp3+ regulatory T cells bear functional skin-homing receptors. Journal of immunology. 2006;177:4488–94. doi: 10.4049/jimmunol.177.7.4488. [DOI] [PubMed] [Google Scholar]
- 33.Ishida T, Ishii T, Inagaki A, Yano H, Kusumoto S, Ri M, et al. The CCR4 as a novel-specific molecular target for immunotherapy in Hodgkin lymphoma. Leukemia. 2006;20:2162–8. doi: 10.1038/sj.leu.2404415. [DOI] [PubMed] [Google Scholar]
- 34.Niwa R, Shoji-Hosaka E, Sakurada M, Shinkawa T, Uchida K, Nakamura K, et al. Defucosylated chimeric anti-CC chemokine receptor 4 IgG1 with enhanced antibody-dependent cellular cytotoxicity shows potent therapeutic activity to T-cell leukemia and lymphoma. Cancer Res. 2004;64:2127–33. doi: 10.1158/0008-5472.can-03-2068. [DOI] [PubMed] [Google Scholar]
- 35.Ishida T, Joh T, Uike N, Yamamoto K, Utsunomiya A, Yoshida S, et al. Defucosylated anti-CCR4 monoclonal antibody (KW-0761) for relapsed adult T-cell leukemia-lymphoma: a multicenter phase II study. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2012;30:837–42. doi: 10.1200/JCO.2011.37.3472. [DOI] [PubMed] [Google Scholar]
- 36.Yamamoto K, Utsunomiya A, Tobinai K, Tsukasaki K, Uike N, Uozumi K, et al. Phase I study of KW-0761, a defucosylated humanized anti-CCR4 antibody, in relapsed patients with adult T-cell leukemia-lymphoma and peripheral T-cell lymphoma. J Clin Oncol. 2010;28:1591–8. doi: 10.1200/JCO.2009.25.3575. [DOI] [PubMed] [Google Scholar]
- 37.Ishida T, Utsunomiya A, Iida S, Inagaki H, Takatsuka Y, Kusumoto S, et al. Clinical significance of CCR4 expression in adult T-cell leukemia/lymphoma: its close association with skin involvement and unfavorable outcome. Clin Cancer Res. 2003;9:3625–34. [PubMed] [Google Scholar]
- 38.Yano H, Ishida T, Inagaki A, Ishii T, Ding J, Kusumoto S, et al. Defucosylated anti CC chemokine receptor 4 monoclonal antibody combined with immunomodulatory cytokines: a novel immunotherapy for aggressive/refractory Mycosis fungoides and Sezary syndrome. Clin Cancer Res. 2007;13:6494–500. doi: 10.1158/1078-0432.CCR-07-1324. [DOI] [PubMed] [Google Scholar]
- 39.Yano H, Ishida T, Imada K, Sakai T, Ishii T, Inagaki A, et al. Augmentation of antitumour activity of defucosylated chimeric anti-CCR4 monoclonal antibody in SCID mouse model of adult T-cell leukaemia/lymphoma using G-CSF. British journal of haematology. 2008;140:586–9. doi: 10.1111/j.1365-2141.2007.06947.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bindon CI, Hale G, Bruggemann M, Waldmann H. Human monoclonal IgG isotypes differ in complement activating function at the level of C4 as well as C1q. J Exp Med. 1988;168:127–42. doi: 10.1084/jem.168.1.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Teeling JL, Mackus WJ, Wiegman LJ, van den Brakel JH, Beers SA, French RR, et al. The biological activity of human CD20 monoclonal antibodies is linked to unique epitopes on CD20. J Immunol. 2006;177:362–71. doi: 10.4049/jimmunol.177.1.362. [DOI] [PubMed] [Google Scholar]
- 42.Ong GL, Mattes MJ. Mouse strains with typical mammalian levels of complement activity. J Immunol Methods. 1989;125:147–58. doi: 10.1016/0022-1759(89)90088-4. [DOI] [PubMed] [Google Scholar]
- 43.Lloyd CM, Delaney T, Nguyen T, Tian J, Martinez AC, Coyle AJ, et al. CC chemokine receptor (CCR)3/eotaxin is followed by CCR4/monocyte-derived chemokine in mediating pulmonary T helper lymphocyte type 2 recruitment after serial antigen challenge in vivo. The Journal of experimental medicine. 2000;191:265–74. doi: 10.1084/jem.191.2.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ferenczi K, Fuhlbrigge RC, Pinkus J, Pinkus GS, Kupper TS. Increased CCR4 expression in cutaneous T cell lymphoma. The Journal of investigative dermatology. 2002;119:1405–10. doi: 10.1046/j.1523-1747.2002.19610.x. [DOI] [PubMed] [Google Scholar]
- 45.Gear AR, Camerini D. Platelet chemokines and chemokine receptors: linking hemostasis, inflammation, and host defense. Microcirculation. 2003;10:335–50. doi: 10.1038/sj.mn.7800198. [DOI] [PubMed] [Google Scholar]
- 46.Abi-Younes S, Si-Tahar M, Luster AD. The CC chemokines MDC and TARC induce platelet activation via CCR4. Thromb Res. 2001;101:279–89. doi: 10.1016/s0049-3848(00)00402-3. [DOI] [PubMed] [Google Scholar]
- 47.Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol. 2006;6:295–307. doi: 10.1038/nri1806. [DOI] [PubMed] [Google Scholar]
- 48.Hong H, Gu Y, Zhang H, Simon AK, Chen X, Wu C, et al. Depletion of CD4+CD25+ regulatory T cells enhances natural killer T cell-mediated anti-tumour immunity in a murine mammary breast cancer model. Clin Exp Immunol. 2010;159:93–9. doi: 10.1111/j.1365-2249.2009.04018.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Watanabe Y, Katou F, Ohtani H, Nakayama T, Yoshie O, Hashimoto K. Tumor-infiltrating lymphocytes, particularly the balance between CD8(+) T cells and CCR4(+) regulatory T cells, affect the survival of patients with oral squamous cell carcinoma. Oral surgery, oral medicine, oral pathology, oral radiology, and endodontics. 2010;109:744–52. doi: 10.1016/j.tripleo.2009.12.015. [DOI] [PubMed] [Google Scholar]
- 50.Yuan Q, Bromley SK, Means TK, Jones KJ, Hayashi F, Bhan AK, et al. CCR4-dependent regulatory T cell function in inflammatory bowel disease. The Journal of experimental medicine. 2007;204:1327–34. doi: 10.1084/jem.20062076. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.