

Abstract
Levels of genetic diversity of the malaria parasites and multiclonal infections are correlated with transmission intensity. In order to monitor the effect of strengthened malaria control efforts in recent years at the China-Myanmar border area, we followed the temporal dynamics of genetic diversity of three polymorphic antigenic markers msp1, msp2, and glurp in the Plasmodium falciparum populations. Despite reduced malaria prevalence in the region, parasite populations exhibited high levels of genetic diversity. Genotyping 258 clinical samples collected in four years detected a total of 22 PCR size alleles. Multiclonal infections were detected in 45.7% of the patient samples, giving a minimum multiplicity of infection of 1.41. The majority of alleles experienced significant temporal fluctuations through the years. Haplotype diversity based on the three-locus genotypes ranged from the lowest in 2009 at 0.33 to the highest in 2010 at 0.80. Sequencing of msp1 fragments from 36 random samples of five allele size groups detected 13 different sequences, revealing an additional layer of genetic complexity. This study suggests that despite reduced prevalence of malaria infections in this region, the parasite population size and transmission intensity remained high enough to allow effective genetic recombination of the parasites and continued maintenance of genetic diversity.
Keywords: Plasmodium falciparum, antigenic diversity, border malaria, multiplicity of infection
1. Introduction
Malaria continues to be a major public health problem in the developing world. Globally, there are estimated 225 million malaria infections each year, resulting in around a million deaths (WHO, 2011). While most of malaria burden is in sub-Saharan Africa, Southeast (SE) Asia accounts for 10% of global malaria morbidity and 5% of global mortality from 2008. Within the SE Asian region, the Greater Mekong Subregion (GMS) has been one of the most serious foci of malaria, with immense geographical heterogeneity in disease endemicity, complex vector systems, and the co-existence of Plasmodium falciparum and Plasmodium vivax (Cui et al., 2012a). Furthermore, the GMS is also a hotspot of multidrug resistant P. falciparum, and recent detection of artemisinin resistance in this area has raised considerable global concern (Dondorp et al., 2009; Noedl et al., 2008). Another notable feature of malaria epidemiology in the GMS is “border malaria”, with most of the malaria cases concentrated along international borders. Cross-border human migration, which is difficult to monitor and responsible for malaria reintroduction, is an important challenge for malaria control. Therefore, knowledge of accurate malaria epidemiology at both macro and micro scales is needed for targeted malaria control, especially during the elimination phase.
Globally, malaria parasite populations exhibit great genotypic and phenotypic diversity (Kemp et al., 1990), which allows the parasites to overcome antimalarial drugs, vaccines and vector control strategies. Allelic polymorphism and sexual reproduction, which is responsible for both classical meiotic recombination and nonreciprocal recombination events, contribute to the generation of genetic diversity. Many cross-sectional studies have established a correlation between transmission intensity and genetic diversity (Hoffmann et al., 2001; Paul et al., 1998). In hyperendemic areas, patients tend to harbor more than one parasite isolate, which favors genetic recombination and generates higher genetic diversity. Conversely, in areas of low endemicity, parasite populations experience significant levels of inbreeding. In addition to transmission intensity, other factors such as selective sweeps, resulting from vaccines and drugs, can greatly influence genetic diversity. Such variability in transmission intensity and genetic diversity may have important consequences with regards to the effectiveness of control measures (such as vaccine deployment) as well as the spread of antimalarial resistance.
Historically, the China-Myanmar border areas have been considered malaria hyperendemic, exhibiting the characteristic “border malaria” phenomenon (Cui et al., 2012a). This region has also had quite different antimalarial drug policies in the past, and drug selection has resulted in parasite genotypes that are drastically different from other parts of the GMS (Meng et al., 2010; Yang et al., 2011). In recent years, with increased funding to control malaria from governments and the Global Fund to Fight against AIDS, Tuberculosis and Malaria, malaria control efforts in this region have intensified. Among the control measures are insecticide-treated bednets and artemisinin-based combination therapy (ACT) for P. falciparum malaria. As a result, malaria incidence has been significantly curtailed (Cui et al., 2012b). Whereas accurate malaria epidemiology data in the neighboring Myanmar regions are not available, malaria situation in this region is well reflected in the reduction in malaria cases in the border counties of Yunnan province in recent years (Clements et al., 2009). In addition, China is mobilizing towards malaria elimination in the near future. It is therefore critical to evaluate the impact of the control on malaria epidemiology in this region.
In this study, we assessed the genetic diversity of the P. falciparum parasites from the China-Myanmar border area near Laiza, Myanmar. Using longitudinal clinical samples, we studied the allelic diversity of genes encoding the polymorphic antigens, including merozoite surface protein (msp)-1 and -2, as well as glutamate-rich protein (glurp), and determined the temporal dynamics of their prevalence in this region.
2. Materials and methods
2.1. Collection of clinical parasite samples
P. falciparum samples were collected in 2007-2010 from patients presenting with uncomplicated malaria infection at two malaria clinics in Laiza township at the China-Myanmar border area. These malaria clinics, located ~0.8 Km apart, provide malaria diagnosis and treatment to patients in a catchment area of ~20 Km radius. In this area, malaria transmission occurs perennially but is most intense during the rainy season from April to October. Samples from these two clinics were pooled for analysis. A total of 284 P. falciparum clinical isolates from patients aged 3-66 were collected. The samples collected in different years were not from the same patients. Only patients with clinical symptoms and microscopically confirmed P. falciparum infection were enrolled in this study. Written informed consent was obtained from the participants or guardians. The human subjects protocol for this study was approved by the Institutional Review Board of Kunming Medical University. Malaria infection was diagnosed by microscopic examination of Giemsa-stained thick and thin blood films. If P. falciparum infection was confirmed, 0.2 ml of finger-prick blood was spotted on a piece of Whatman 3MM filter paper and used for molecular studies.
2.2. Allelic typing of P. falciparum msp1 and msp2, and glurp genes
For genotyping, parasite genomic DNA was extracted from the filter papers by using a QIAamp DNA microkit (Qiagen, Germany) following the manufacturer’s instructions. DNA was eluted in 80 μl of elution buffer. DNA was genotyped at msp1 (block 2), msp2, and glurp using nested PCR by previously described methods (Kaneko et al., 1997; Viriyakosol et al., 1995). The primary PCR products were used in nested reactions with allele family-specific primers for K1, MAD20, and RO33 of msp1, and for FC27 and 3D7 of msp2. Final PCR products were separated in 2% agorose gel, stained with ethidium bromide and visualized under UV light. The lengths of the PCR products were estimated based on their mobility relative to the DNA size standard (TaKaRa, Japan). DNA fragment sizes were binned into different classes of ~50 bp ranges with each bin assigned as an allele.
2.3. Estimation of parasite clone numbers per sample
The minimum number of parasite clones per sample, or the multiplicity of infection (MOI), was defined as the largest number of alleles at any one locus detected in the sample (Paganotti et al., 2004b). For this study, alleles were identified based only on type and fragment size of the amplified PCR products. If an isolate had one allele at each of the three loci, this sample was considered to have monoclonal infection. This method is conservative and may underestimate the number of clones likely to be present in the samples. Yet, this may best reflect the level of endemicity in this area. The tri-allelic haplotypes and their relative annual frequencies were assessed only from infections with no more than one locus showing multiclonal infections.
2.4. Sequence analysis of genetic polymorphism of msp1 block 2
In order to further examine the genetic diversity of the parasite population, we sequenced msp1 block 2 DNA fragments from PCR products showing similar molecular sizes in agarose gels. The PCR products were purified from the gel using a Gel extraction mini kit (Watson Biotechnologies, Inc.) and prepared for sequencing. Partial gene sequences were obtained from each end of the PCR product using the same primers for the nested PCR reactions. DNA sequences were assembled and BLAST searched in GenBank to identify identical or similar sequences.
2.5. Statistical analysis
We compared counts of samples by MOI status, genotype, year using Pearson’s χ2 test and Fisher’s exact test. The Student-Newman-Keuls test was used for a post-hoc analysis of the MOI data across the study years.
3. Results
3.1. Size polymorphisms of msp1, msp2 and glurp
Of the 284 patient samples collected, 3.2% (msp1), 4.9% (msp2), and 1.4% (glurp) failed to produce PCR results, while 258 were successfully genotyped at all three loci. The PCR fragment sizes for msp1, msp2 and glurp were 150-300 bp, 250-550 bp, and 500-1000 bp, respectively (Fig. 1). Both msp1 and msp2 showed eight size classes, whereas glurp had six size classes. For msp1 and msp2, all allele types identified earlier (K1, MAD20, and RO33 for msp1, and IC/3D7 and FC27 for msp2) were amplified, with each further subdivided into multiple size alleles. For msp1, the overall frequencies of the K1, MAD20 and RO33 allele types were 30.2%, 41.7%, 28.2%, respectively. The top three most abundant alleles were MAD20-200 bp, RO33-200 bp, and K1-200 bp, which occurred at frequencies of 36.1%, 24.3% and 19.5%, respectively (Fig. 1). The 3D7 and FC27 allele types for msp2 were about equally abundant in the samples. The most prevalent allele 3D7-525 bp occurred at 49.4%. Glurp gene had six size classes, of which the 800 bp size allele had the highest prevalence of 37.8%. It is noteworthy that there were rare size alleles for each of the three loci with frequencies below 2% (e.g., Msp1-MAD20-150, Msp2-3D7-250, Msp2-3D7-400).
Fig. 1.

Distribution of PCR fragment size alleles of msp1, msp2 and glurp. For msp1 and msp2, the alleles are also differentiated by the allele types (K1, MAD20 and RO33 for msp1, and 3D7 and FC27 for msp2). PCR allele sizes (150 – 1000) are in bp.
3.2. Longitudinal variation
For the samples collected in 2007, 2008, 2009, and 2010, 53, 58, 123 and 24 samples were genotyped at all three loci, respectively. Most alleles were already present in earlier years and only a couple of minor new alleles were detected in successive sample years. For the msp1 allele types, K1 (P=9.403, χ2 test, df=3) and RO33 (P=100.61, χ2 test, df=6) varied significantly across the years. However, no significant annual fluctuations were detected for MAD20 (P=4.570, χ2 test, df=6). A significant increase in frequency was detected for the K1-275 bp allele in 2009. The RO33-150 bp allele occurring at 16.9% in 2007 essentially disappeared in subsequent years, whereas the minor allele RO33-200 bp gained higher frequencies, reaching 37.4% in 2009 (Fig. 2). For msp2, the IC/3D7 type alleles did not vary much across the years (P=0.740, χ2 test, df=3), and the 525 bp allele size remained the most prevalent allele throughout the study years, with frequencies close to 50% (Fig. 2). However, the frequencies of the FC27 allele sizes experienced significant annual fluctuations (P=151.5, χ2 test, df=12). Of particular interest, the predominant FC27-250 bp allele in 2008 (47.8%) was essentially undetected in subsequent years, and appeared to be gradually replaced by the 300 bp allele, which reached 47.1% in 2010. Similarly, glurp allele frequencies varied widely across the years (P=99.6, χ2 test, df=15) and allele replacement in the populations was also evident (Fig. 2). In 2010, the parasite samples had the least diversity for both msp2 and glurp, with only 2 msp2 and 3 glurp size alleles.
Fig. 2.
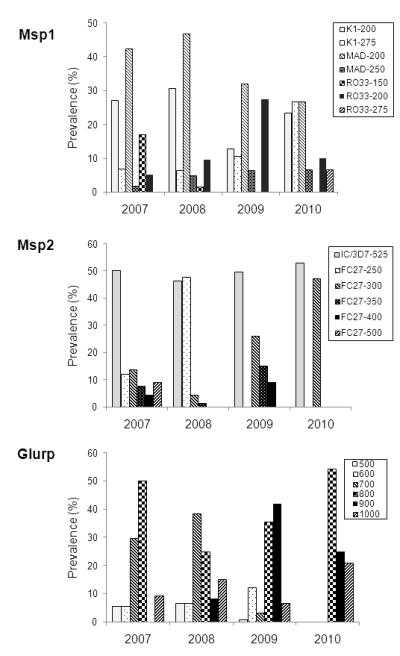
Genotype diversity of P. falciparum clinical samples from northeastern Myanmar. The prevalence of different PCR size alleles of msp1, msp2 and glurp in different years are shown. One msp1 minor allele and two msp2 minor alleles are omitted in the plots. Note the persistent prevalence of Msp1-MAD20-200, Msp2-IC/3D7-525, and Glurp-800 alleles through the years.
3.3. Multiclonal infections
Of the 258 samples successfully genotyped at the three loci, 54.3% had a single band at each of the three loci, indicating monoclonal infections by our definition. Genotyping msp1 and msp2 resulted in a significantly higher power of detecting mixed strain infections when compared to genotyping glurp (Table 1). Analysis of msp1 and msp2 alone detected 30.1% and 21.3% of the multiclonal infections, respectively, whereas analysis of glurp only detected 1.6% multiclonal infections. Combining all three markers detected a total of 45.7% multiple clonal infections, giving a minimum MOI of 1.41 (Table 1). Based on msp1, the proportions of single infections with K1, MAD20 and RO33 allelic types were 25.5 %, 32% and 10.2%, respectively. For msp1, the most common multiclonal infections had the combination of two different allele types (MAD20 and K1) instead of combination of different allele sizes within the same allele types. In the four years, the level of multiclonal infections ranged from 22.4% in 2008 to 61.8% in 2009 (Table 1). Similarly, the average MOI ranged from 1.22 to 1.62. The lowest MOI was in 2008, which was significantly lower than those in 2007 and 2009 (P=28.4, χ2 test, df=3).
Table 1.
Annual prevalence of multiclonal infections in P. falciparum clinical samples.
| Genes# | 2007 (N=53) | 2008 (N=58) | 2009 (N=123) | 2010 (N=24) | Total (N=258) |
|---|---|---|---|---|---|
| Msp1 | 6 (11.3) | 4 (6.8) | 64 (52.0) | 6 (25.0) | 80 (30.1) |
| Msp2 | 14 (26.4) | 9 (15.5) | 22 (17.9) | 10 (41.7) | 55 (21.3) |
| Glurp | 1 (1.9) | 2 (3.4) | 1 (0.8) | 0 | 4 (1.6) |
| Combined | 18 (34.0) | 13 (22.4) | 76 (61.8) | 11 (45.8) | 118 (45.7) |
|
| |||||
| MOI (mean±SD) | 1.34±0.48 | 1.22±0.42 | 1.62±0.49* | 1.46±0.51 | 1.41±0.17 |
Number (%) of mixed strain infections identified by using an individual marker (msp1, msp2 or glurp) or three markers together (combined).
Statistical analysis showed that the MOI in 2009 was significantly different from those in 2007 and 2008 (P=28.43, χ2 test, df=3).
3.4. Haplotypes defined by a combination of three polymorphic loci
Genetic diversity of the parasite populations were further estimated using a combination of three loci. Based on the three-locus genotype, a total of 112 haplotypes were differentiated; 42, 34, 58, and 20 in 2007, 2008, 2009 and 2010, respectively (Fig. 3), resulting in haplotype diversity of 0.62, 0.51, 0.33 and 0.80, respectively. The numbers of 3-locus haplotypes did appear to be proportional to the number of samples analyzed for each year. Each year there were 3-5 haplotypes occurring at much higher frequencies (5-12%) than other haplotypes (Fig. 3). Among them, three were found in more than one year: K1-200/3D7-525/700 haplotype in 2007 and 2010, the MAD20-200/3D7-525/800 haplotype in three years, and MAD20-200/3D7-525/700 in 2007 and 2008. This indicates that despite the diversity of the parasite populations, certain parasites strains were maintained in multiple years.
Fig. 3.

The prevalence (%) of different 3-locus genotypes in the clinical samples. The graph shows the diversity and prevalence (%) of different 3-locus genotypes in the four years. Only genotypes with >5% prevalence are shown in the legends and marked in the columns. Note that the three genotypes with >5% prevalence and present in more than one year are highlighted in bold.
3.5. Increased genetic diversity from sequencing analysis
To determine the genetic diversity within the same allele bins, we selected msp2 block 2 PCR products from 36 monoclonal infections for sequencing, which included five different alleles. Sequencing analysis revealed an additional layer of genetic complexity, even though many of the sequences appeared to have similar fragment lengths and were binned together. While sequenced fragments were chosen from the 150 bp and 200 bp bins, the sequence data showed that the actual fragment lengths were 159 and 168 bp for the 150 bp bin, and 186 bp and 195 bp for the 200 bp bin (Table 2). From the 36 PCR products, 13 different genotypes were identified. For K1-150, MAD20-200, and RO33-150 alleles, the prevalent genotypes were PF-K1-2, PF-M7 and PF-R1, representing 6/8, 9/16, and 6/9 of allele fragments sequenced, respectively (Table 2).
Table 2.
Pfmsp1 haplotype diversity revealed by sequencing
| Type | Estimated PCR fragment size (bp) |
# isolates sequenced |
Genotype (#) | Actual fragment size (bp) |
Reported identical sequences* |
Origin | Reference |
|---|---|---|---|---|---|---|---|
| K1 | PF-K1-1 (2) | 159 | AB276017 | Thailand | (Tanabe et al., 2007) | ||
|
|
|||||||
| 150 | 8 | PF-K1-2 (6) | 168 | M77730 | Thailand | (Jongwutiwes et al., 1992) | |
|
| |||||||
| 200 | 2 | PF-K1-3 (2) | 195 | GQ861443.1 | Myanmar | (Kang et al., 2010) | |
|
| |||||||
| MAD20 | PF-M1 (2) | 159 | AF509634 | Brazil | (Ferreira et al., 2003) | ||
|
|
|||||||
| 150 | 4 | PF-M2 (2) | 168 | EU032232.1 | Senegal | (Noranate et al., 2009) | |
|
| |||||||
| 200 | 16 | PF-M3 (2) | 186 | JF460927.1 | India | U | |
|
| |||||||
| PF-M4 (2) | 186 | EU445563.1 | Myanmar | U | |||
|
| |||||||
| PF-M5 (1) | 186 | EU445556.1 | Myanmar | U | |||
|
| |||||||
| PF-M6 (1) | 186 | U | |||||
|
| |||||||
| PF-M7 (9) | 195 | EU445557 | Myanmar | U | |||
|
| |||||||
| PF-M8 (1) | 195 | AF061141 | Tanzania | (Jiang et al., 2000) | |||
|
| |||||||
| RO33 | PF-R1 (6) | 159 | M32112 | Brazil | U | ||
|
|
|||||||
| 150 | 9 | PF-R2 (3) | 159 | AB276005.1 | Ghana | (Tanabe et al., 2007) | |
GenBank accession numbers; U – unpublished.
Most sequence variations in block 2 of MSP1 were created by rearrangement of a limited number of building blocks (Fig. 4A). In the K1 type alleles, the tripeptide repeat region begins with SAQ and terminates with SGT, and differs in the number of tripeptide repeats. In the MAD20-type alleles, the repeat region mostly starts with one of two different tripeptide sequences SGG or SKG, and in many cases ends with a hexapeptide sequence SVASGG. The diversity of the MAD20 allele type was also primarily due to differences in repeats of SGG, SVT and SVA. A BLAST search of the GenBank showed that 12 of the 13 sequences were identical to those reported earlier. Specifically, the K1 sequences matched those identified from the GMS (Jongwutiwes et al., 1992; Kang et al., 2010; Tanabe et al., 2007), whereas the MAD20 sequences were identified earlier in Myanmar, South Asia, South America, and Africa (Ferreira et al., 2003; Jiang et al., 2000; Noranate et al., 2009). In comparison, the two RO33 sequences were identified earlier in South America and Africa. PF-M6 was a new sequence found in this study, most closely related to M77722 and AAF18429 in the GenBank, which was identified from Thailand and Indonesia, respectively (Fig. 4B). The PF-M6 sequence had an extra SVA repeat when compared with the Thai isolate, and different from the Indonesian isolate by a single G/S substitution.
Fig. 4.

Alignment of Msp1 Block 2 sequences. (A) Alignment of protein sequences of the respective types, K1, MAD20 and RO33 summarized in Table 2. (B) Alignment of PF-M6 sequence with two most closely related sequences in samples from Thailand (M77722) and Indonesia (AAF18429). The single amino acid substitutions are boxed, and the central repeat elements are shaded.
4. Discussion
To better understand the population structure of the malaria parasites at the China-Myanmar border area, we performed longitudinal analysis of genetic diversity of the P. falciparum populations. Genotyping field isolates from this area by allele-specific PCR for msp1 (block 2), msp2 (block 3) and glurp revealed extensive genetic diversity and a high degree of MOI. The extensive polymorphisms in these vaccine candidates are a major impediment for subunit vaccines against the malaria parasites. Both msp1 and msp2 had eight PCR size alleles, as compared to six alleles for glurp. For msp1, MAD20 was more common than K1 and RO33, similar to other parts of the GMS such as Yunnan Province, China (Zhu et al., 1999), Laos (Khaminsou et al., 2011), and Thailand (Snounou et al., 1999). However, the result is drastically different from a recent report from central Myanmar, where the RO33 allele type was not detected (Kang et al., 2010). For msp2, 3D7 and FC27 were about equally prevalent, which is similar to parasite populations from the GMS such as Thailand (Snounou et al., 1999) and Laos (Khaminsou et al., 2011), and from nations within South Asia such as India and Pakistan (Najia et al., 2010; Rout et al., 2009), but different from parasite populations from other parts of SE Asia such as Malaysia (Atroosh et al., 2011). The similarity of parasite genetic makeup in the GMS and neighboring regions may be due to shared or similar demographic histories of the parasite populations and/or population mixing from neighboring areas. Since the GMS and neighboring areas are quite heterogeneous in terms of malaria endemicity, and since these reports spanned across more than a decade, the distribution of the allelic types does not seem associated with levels of endemicity, as reported earlier from Africa (Paganotti et al., 2004a). Though malaria control efforts have decreased malaria transmission in this area over time (Clements et al., 2009), the extensive genetic diversity detected in recent years might be the result of previously high levels of malaria endemicity.
Overall, genotyping the msp1 and msp2 loci offers a convenient assessment of parasite population diversity. In addition, this study identified six glurp alleles, and in some endemic areas adding this marker is necessary to achieve adequate levels of parasite differentiation (Gatton and Cheng, 2008). However, this marker seemed to offer little power in detecting multiclonal infections in this study area. Whereas allele-specific PCR alone allows identification of a dozen of allele types, the power of detection could be enhanced by high-resolution genotyping (Schoepflin et al., 2009), because sequences within the same fragment size bins can differ by ~10 bp. Furthermore, when 3-locus genotypes are considered, at least 112 different parasite isolates could be differentiated from the 258 patient samples successfully genotyped. At this sensitivity level, there were multiple clusters of parasites showing identical 3-locus genotypes, suggesting of high relatedness of these parasite isolates. This result is consistent with the parasite populations at the Thai-Myanmar border area, where significant relatedness between parasite isolates was found by genotyping multiple microsatellite loci (Anderson et al., 2010). This may indicate that clusters of common parasite clones are circulating in this endemic area, which might also be the result of selection by antimalarial drugs.
The level of genetic diversity and multiplicity of infections generally reflect the level of transmission intensity, albeit their relationship is not linear (Hoffmann et al., 2001; Paul et al., 1998). In this study, the proportion of patients exhibiting multiclonal infections ranged from 22.4 to 61.8%, which appeared unproportionally high compared with the current hypoendemic status of this region. The high levels of genetic diversity persistent through the years suggest that the parasite population size remained large enough to allow effective mixing of genotypes. In addition, extensive human migration in the border area should increase genetic diversity by introducing additional parasite genotypes (Cui et al., 2012b). Although the lower number of alleles detected in 2010 might reflect stronger selection on the parasite population as a result of enhanced control, it was most likely related to the smaller sample size analyzed in 2010 (since the MOI remained consistently high). Whereas the large variation in the MOI among the years might truly reflect the dynamics of mixed infections in different years, other factors such as parasite collection sites and time also might be partially responsible. In the future more accurate quantification of MOI may be achieved through next generation sequencing of polymorphic genes from patient samples, which offers an unparalleled opportunity for accurate assessment of genetic diversity within individual patients (Gandhi et al., 2012).
Plasmodium parasite merozoite surface antigens are under strong selection from the host immune system. Infections generate a form of strain-specific immune response against the respective antigens, which biases against reinfection by parasites carrying identical and closely-related forms of the antigens. As a result, temporal variation in the prevalence of specific genotypes of the antigens is expected within parasite populations. For example, at both the parasite population and individual host levels, significant levels of temporal replacement of the msp2 alleles have been reported in several endemic sites (Eisen et al., 1998; Eisen et al., 2002; Putaporntip et al., 2008). This study showed that some genotypes (such as msp1/RO33, msp2/FC27, and glurp) experienced significant fluctuations across study years, whereas other alleles were more or less stable. Similarly, most 3-locus haplotypes were highly dynamic through the years, while some haplotypes remained the most abundant across several years. The study area undergoes seasonal fluctuations in malaria incidence, with most cases detected in the rainy season from April through October. How the parasite diversity is retained through the “bottleneck” dry season is unknown, but it seems to also occur in other endemic areas with seasonal transmission dynamics (Babiker et al., 1998; Zwetyenga et al., 1999).
As demonstrated in numerous cases, sequencing the polymorphic regions of the antigens provides the best estimate of genetic complexity (Eisen et al., 1998; Weisman et al., 2001). Sequencing analysis of selected msp1 block 2 fragments from 36 patients from the study population further differentiated five size alleles into 13 sequence genotypes. It is noteworthy that most sequences identified in this study are identical not only to those from neighboring regions in SE Asia, but also to those from other continents. The high level of sequence diversity of block 2 is expected, since it is a principal target of human immunity, and antibodies against this domain are strongly associated with protection from P. falciparum malaria (Conway et al., 2000). It is likely that the diversity of block 2 is maintained by balancing selection from host immunity whereby multiple different alleles are kept in the population. Therefore, it would be interesting to determine the level and dynamics of strain-specific antibodies to block 2 and its relationship with the dynamics of msp1 genotypes in the parasite populations.
Taken together, in spite of intensified malaria control and the resulting reduction in malaria incidence in the China-Myanmar border area, the P. falciparum parasite population remained highly diverse at the three antigenic loci analyzed in this study: msp1, msp2 and glurp. Likewise, the high MOI suggests effective genetic recombination of the parasites, and the continued maintenance of genetic diversity. Throughout the years of this survey, parasite genotype frequencies underwent significant fluctuation, which may be due to selection pressure from host immunity and/or drugs, as well as contraction of parasite population size resulting from malaria control. Continued surveys of the parasite population genetics in this region are necessary for monitoring the effect of the malaria control and elimination campaign, which are now being unfolded in this region. It is also noteworthy that the samples were from symptomatic patients attending local malaria clinics, while asymptomatic parasite carriers were missed. Future studies from active case detection might be needed to include both populations in order to obtain more accurate assessment of the genetic complexity of the parasites.
Highlights.
P. falciparum at China-Myanmar border is highly diverse at three antigenic markers.
The annual prevalence of most genotypes varies considerably between the years.
Additional genetic complexity is revealed by sequencing of msp1 block 2.
High multiplicity of infection favors effective maintenance of genetic diversity.
Acknowledgements
This study was partially supported by National Institutes of Health (NIH) international grant (1R01AI075429) and the National Natural Science Foundation of China (no. 30960050, 81161120421) and U19AI089672 from National Institute of Allergy and Infectious Diseases, NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anderson TJ, Williams JT, Nair S, Sudimack D, Barends M, Jaidee A, Price RN, Nosten F. Inferred relatedness and heritability in malaria parasites. Proceedings of the Royal Society B. 2010;277:2531–2540. doi: 10.1098/rspb.2010.0196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atroosh WM, Al-Mekhlafi HM, Mahdy MAK, Saif-Ali R, Al-Mekhlafi AM, Surin J, Sana’a Y. Genetic diversity of Plasmodium falciparum isolates from Pahang, Malaysia based on MSP-1 and MSP-2 genes. Parasites & Vectors. 2011;4:233. doi: 10.1186/1756-3305-4-233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babiker HA, Abdel-Muhsin AM, Ranford-Cartwright LC, Satti G, Walliker D. Characteristics of Plasmodium falciparum parasites that survive the lengthy dry season in eastern Sudan where malaria transmission is markedly seasonal. The American Journal of Tropical Medicine and Hygiene. 1998;59:582–590. doi: 10.4269/ajtmh.1998.59.582. [DOI] [PubMed] [Google Scholar]
- Clements AC, Barnett AG, Cheng ZW, Snow RW, Zhou HN. Space-time variation of malaria incidence in Yunnan province, China. Malaria journal. 2009;8:180. doi: 10.1186/1475-2875-8-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway DJ, Cavanagh DR, Tanabe K, Roper C, Mikes ZS, Sakihama N, Bojang KA, Oduola AM, Kremsner PG, Arnot DE, Greenwood BM, McBride JS. A principal target of human immunity to malaria identified by molecular population genetic and immunological analyses. Nature Medicine. 2000;6:689–692. doi: 10.1038/76272. [DOI] [PubMed] [Google Scholar]
- Cui L, Yan G, Sattabongkot J, Cao Y, Chen B, Chen X, Fan Q, Fang Q, Jongwutiwes S, Parker D, Sirichaisinthop J, Kyaw MP, Su XZ, Yang H, Yang Z, Wang B, Xu J, Zheng B, Zhong D, Zhou G. Malaria in the Greater Mekong Subregion: Heterogeneity and complexity. Acta Tropica. 2012a;121:227–239. doi: 10.1016/j.actatropica.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui L, Yan G, Sattabongkot J, Chen B, Cao Y, Fan Q, Parker D, Sirichaisinthop J, Su XZ, Yang H, Yang Z, Wang B, Zhou G. Challenges and prospects for malaria elimination in the Greater Mekong Subregion. Acta Tropica. 2012b;121:240–245. doi: 10.1016/j.actatropica.2011.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dondorp AM, Nosten F, Yi P, Das D, Phyo AP, Tarning J, Lwin KM, Ariey F, Hanpithakpong W, Lee SJ, Ringwald P, Silamut K, Imwong M, Chotivanich K, Lim P, Herdman T, An SS, Yeung S, Singhasivanon P, Day NP, Lindegardh N, Socheat D, White NJ. Artemisinin resistance in Plasmodium falciparum malaria. The New England Journal of Medicine. 2009;361:455–467. doi: 10.1056/NEJMoa0808859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen D, Billman-Jacobe H, Marshall VF, Fryauff D, Coppel RL. Temporal variation of the merozoite surface protein-2 gene of Plasmodium falciparum. Infection and Immunity. 1998;66:239–246. doi: 10.1128/iai.66.1.239-246.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen DP, Saul A, Fryauff DJ, Reeder JC, Coppel RL. Alterations in Plasmodium falciparum genotypes during sequential infections suggest the presence of strain specific immunity. The American Journal of Tropical Medicine and Hygiene. 2002;67:8–16. doi: 10.4269/ajtmh.2002.67.8. [DOI] [PubMed] [Google Scholar]
- Ferreira MU, Ribeiro WL, Tonon AP, Kawamoto F, Rich SM. Sequence diversity and evolution of the malaria vaccine candidate merozoite surface protein-1 (MSP-1) of Plasmodium falciparum. Gene. 2003;304:65–75. doi: 10.1016/s0378-1119(02)01180-0. [DOI] [PubMed] [Google Scholar]
- Gandhi K, Thera MA, Coulibaly D, Traore K, Guindo AB, Doumbo OK, Takala-Harrison S, Plowe CV. Next generation sequencing to detect variation in the Plasmodium falciparum circumsporozoite protein. The American Journal of Tropical Medicine and Hygiene. 2012;86:775–781. doi: 10.4269/ajtmh.2012.11-0478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatton ML, Cheng Q. Can estimates of antimalarial efficacy from field studies be improved? Trends in Parasitology. 2008;24:68–73. doi: 10.1016/j.pt.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann EH, da Silveira LA, Tonhosolo R, Pereira FJ, Ribeiro WL, Tonon AP, Kawamoto F, Ferreira MU. Geographical patterns of allelic diversity in the Plasmodium falciparum malaria-vaccine candidate, merozoite surface protein-2. Annals of Tropical Medicine and Parasitology. 2001;95:117–132. doi: 10.1080/00034980120045833. [DOI] [PubMed] [Google Scholar]
- Jiang G, Daubenberger C, Huber W, Matile H, Tanner M, Pluschke G. Sequence diversity of the merozoite surface protein 1 of Plasmodium falciparum in clinical isolates from the Kilombero District, Tanzania. Acta tropica. 2000;74:51–61. doi: 10.1016/s0001-706x(99)00045-5. [DOI] [PubMed] [Google Scholar]
- Jongwutiwes S, Tanabe K, Nakazawa S, Yanagi T, Kanbara H. Sequence variation in the tripeptide repeats and T cell epitopes in P190 (MSA-1) of Plasmodium falciparum from field isolates. Molecular and Biochemical Parasitology. 1992;51:81–89. doi: 10.1016/0166-6851(92)90203-v. [DOI] [PubMed] [Google Scholar]
- Kaneko O, Kimura M, Kawamoto F, Ferreira MU, Tanabe K. Plasmodium falciparum: allelic variation in the merozoite surface protein 1 gene in wild isolates from southern Vietnam. Experimental Parasitology. 1997;86:45–57. doi: 10.1006/expr.1997.4147. [DOI] [PubMed] [Google Scholar]
- Kang JM, Moon SU, Kim JY, Cho SH, Lin K, Sohn WM, Kim TS, Na BK. Genetic polymorphism of merozoite surface protein-1 and merozoite surface protein-2 in Plasmodium falciparum field isolates from Myanmar. Malaria Journal. 2010;9:131. doi: 10.1186/1475-2875-9-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp DJ, Cowman AF, Walliker D. Genetic diversity in Plasmodium falciparum. Advances in Parasitology. 1990;29:75–149. doi: 10.1016/s0065-308x(08)60105-0. [DOI] [PubMed] [Google Scholar]
- Khaminsou N, Kritpetcharat O, Daduang J, Charerntanyarak L, Kritpetcharat P. Genetic analysis of the merozoite surface protein-1 block 2 allelic types in Plasmodium falciparum clinical isolates from Lao PDR. Malaria Journal. 2011;10:371. doi: 10.1186/1475-2875-10-371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng H, Zhang R, Yang H, Fan Q, Su X, Miao J, Cui L, Yang Z. In vitro sensitivity of Plasmodium falciparum clinical isolates from the China-Myanmar border area to quinine and association with polymorphism in the Na+/H+ exchanger. Antimicrobial Agents and Chemotherapy. 2010;54:4306–4313. doi: 10.1128/AAC.00321-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najia G, Andreas M, Sana J, Sándor B, Rabia H, Mohammad B. Genetic diversity among Plasmodium falciparum field isolates in Pakistan measured with PCR genotyping of the merozoite surface protein 1 and 2. Malaria Journal. 2010;9 doi: 10.1186/1475-2875-9-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noedl H, Se Y, Schaecher K, Smith BL, Socheat D, Fukuda MM. Evidence of artemisinin-resistant malaria in western Cambodia. The New England Journal of Medicine. 2008;359:2619–2620. doi: 10.1056/NEJMc0805011. [DOI] [PubMed] [Google Scholar]
- Noranate N, Prugnolle F, Jouin H, Tall A, Marrama L, Sokhna C, Ekala MT, Guillotte M, Bischoff E, Bouchier C, Patarapotikul J, Ohashi J, Trape JF, Rogier C, Mercereau-Puijalon O. Population diversity and antibody selective pressure to Plasmodium falciparum MSP1 block2 locus in an African malaria-endemic setting. BMC Microbiology. 2009;9:219. doi: 10.1186/1471-2180-9-219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paganotti GM, Babiker HA, Modiano D, Sirima BS, Verra F, Konaté A, Ouedraogo AL, Diarra A, Mackinnon MJ, Coluzzi M. Genetic complexity of Plasmodium falciparum in two ethnic groups of Burkina Faso with marked differences in susceptibility to malaria. The American Journal of Tropical Medicine and Hygiene. 2004a;71:173–178. [PubMed] [Google Scholar]
- Paganotti GM, Babiker HA, Modiano D, Sirima BS, Verra F, Konate A, Ouedraogo AL, Diarra A, Mackinnon MJ, Coluzzi M, Walliker D. Genetic complexity of Plasmodium falciparum in two ethnic groups of Burkina Faso with marked differences in susceptibility to malaria. The American Journal of Tropical Medicine and Hygiene. 2004b;71:173–178. [PubMed] [Google Scholar]
- Paul RE, Hackford I, Brockman A, Muller-Graf C, Price R, Luxemburger C, White NJ, Nosten F, Day KP. Transmission intensity and Plasmodium falciparum diversity on the northwestern border of Thailand. The American Journal of Tropical Medicine and Hygiene. 1998;58:195–203. doi: 10.4269/ajtmh.1998.58.195. [DOI] [PubMed] [Google Scholar]
- Putaporntip C, Jongwutiwes S, Hughes AL. Differential selective pressures on the merozoite surface protein 2 locus of Plasmodium falciparum in a low endemic area. Gene. 2008;427:51–57. doi: 10.1016/j.gene.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rout R, Mohapatra BN, Kar SK, Ranjit M. Genetic complexity and transmissibility of Plasmodium falciparum parasites causing severe malaria in central-east coast India. Tropical Biomedicine. 2009;26:165–172. [PubMed] [Google Scholar]
- Schoepflin S, Valsangiacomo F, Lin E, Kiniboro B, Mueller I, Felger I. Comparison of Plasmodium falciparum allelic frequency distribution in different endemic settings by high-resolution genotyping. Malaria journal. 2009;8:250. doi: 10.1186/1475-2875-8-250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snounou G, Zhu X, Siripoon N, Jarra W, Thaithong S, Brown KN, Viriyakosol S. Biased distribution of msp1 and msp2 allelic variants in Plasmodium falciparum populations in Thailand. Transactions of the Royal Society of Tropical Medicine and Hygiene. 1999;93:369–374. doi: 10.1016/s0035-9203(99)90120-7. [DOI] [PubMed] [Google Scholar]
- Tanabe K, Sakihama N, Walliker D, Babiker H, Abdel-Muhsin AMA, Bakote’e B, Ohmae H, Arisue N, Horii T, Rooth I. Allelic dimorphism-associated restriction of recombination in Plasmodium falciparum msp1. Gene. 2007;397:153–160. doi: 10.1016/j.gene.2007.04.033. [DOI] [PubMed] [Google Scholar]
- Viriyakosol S, Siripoon N, Petcharapirat C, Petcharapirat P, Jarra W, Thaithong S, Brown KN, Snounou G. Genotyping of Plasmodium falciparum isolates by the polymerase chain reaction and potential uses in epidemiological studies. Bulletin of the World Health Organization. 1995;73:85–95. [PMC free article] [PubMed] [Google Scholar]
- Weisman S, Wang L, Billman-Jacobe H, Nhan DH, Richie TL, Coppel RL. Antibody responses to infections with strains of Plasmodium falciparum expressing diverse forms of merozoite surface protein 2. Infection and Immunity. 2001;69:959–967. doi: 10.1128/IAI.69.2.959-967.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO Wolrd Malaria Report 2010. 2011. p. 248.
- Yang Z, Li C, Miao M, Zhang Z, Sun X, Meng H, Li J, Fan Q, Cui L. Multidrug-resistant genotypes of Plasmodium falciparum, Myanmar. Emerging Infectious Diseases. 2011;17:498–501. doi: 10.3201/eid1703.100870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Zhou L, Liu Q, Gao X. Genotype and sequence analysis of merozoite surface protein 1 of Plasmodium falciparum isolates in Yunnan province. Chinese Journal of Parasitology & Parasitic Diseases. 1999;17:155–158. [PubMed] [Google Scholar]
- Zwetyenga J, Rogier C, Spiegel A, Fontenille D, Trape JF, Mercereau-Puijalon O. A cohort study of Plasmodium falciparum diversity during the dry season in Ndiop, a Senegalese village with seasonal, mesoendemic malaria. Transactions of the Royal Society of Tropical Medicine and Hygiene. 1999;93:375–380. doi: 10.1016/s0035-9203(99)90122-0. [DOI] [PubMed] [Google Scholar]