

Abstract
PMLA nanoparticles with diameters of 150–250 nm are prepared, and their hydrolytic degradation is studied under physiological conditions. Degradation occurs by hydrolysis of the side chain methyl ester followed by cleavage of the main-chain ester group with methanol and L-malic acid as the final degradation products. No alteration of the cell viability is found after 1 h of incubation, but toxicity increases significantly after 3 d, probably due to the noxious effect of the released methanol. Anticancer drugs temozolomide and doxorubicin are encapsulated in the NPs with 20–40% efficiency, and their release is monitored using in vitro essays. Temozolomide is fully liberated within several hours, whereas doxorubicin is steadily released from the particles over a period of 1 month.
Keywords: anticancer DDS, biodegradable nanoparticles, polymalates, poly(malic acid)
Introduction
Biodegradable polymers and their copolymers are the preferred materials for the manufacture of a variety of devices that are nowadays widely applied in medicine and pharmacology.[1] One of their attractive uses is in the formulation of drug delivery systems (DDS) for parenteral administration.[2] DDS based on polymer particles, either nanoparticles (NPs) or microparticles, are clearly advantageous because (i) particle size and surface can be engineered to achieve passive or active drug targeting, (ii) drugs can be incorporated without chemical reaction, (iii) drug activity is optimally preserved, and (iv) drugs can be delivered through different routes of administration.[3] In these systems, polymers play an essential role in the therapeutic function, because in addition to act as carrier, the system can be properly designed to control the drug delivery rate and/or its selective release at a specific site of action.[4]
The application of DDS in chemotherapy has spread rapidly in the last few decades because their use allows to increasing drug concentration at a specific site and reducing systemic toxicity. Thus, DDS based on biopolymers or biodegradable synthetic polymers allow repeated treatment of patients without deposition and storage diseases. This has stimulated the modification of naturally occurring biopolymers and the development of new synthetic biodegradable polymers.[5,6] Synthetic poly(lactide-co-glycolide) (PLGA) derivatives have been the center focus of a great amount of research in the last decades due to their advantages respect to other systems.[7] Nevertheless, PLGA micro- and NPs devices still retain a number of drawbacks mainly related to their releasing pattern that largely limit their applications. New options based on biopolymers able to meet specific requirements are therefore being currently searched. In this regard, poly(β-L-malic acid) (PMLA) (Scheme 1) and its derivatives constitute a family of promising candidates.
Scheme 1.
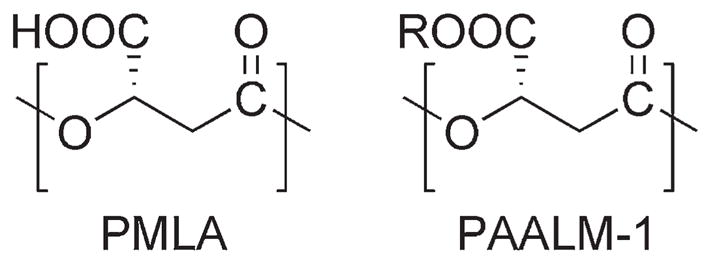
Repeating unit of PMLA and its methyl ester (R = CH3).
PMLA is a water-soluble, biodegradable, bioabsorbable, and non-immunogenic polyester[8] that can be produced either by chemical synthesis[6,9] or by fermentation of certain microorganisms.[8] The properties and functionality of PMLA are adjustable by chemical derivatization.[10] In fact, the carboxylic side group of PMLA can be modified to modulate the overall hydrophobicity of the polymer or to introduce bioactive ligands.[11] Recently, we have reported the methylation of PMLA by diazomethane[10] to create hydrophobic PMLA methyl esters suitable for the formulation of drug delivery microparticles.[12]
In cancer therapy, the treatment of tumor cells must proceed with minimal side effects to normal cells and non-toxic drug formulations should be applied.[3] The use of polymeric DDS in such therapy is increasing in popularity since they are less immunogenic than viral vectors.[11] Furthermore, DDS based on NPs offer clear advantages compared to microparticles for their potential ability to cross cell membranes. Once the polymer is selected, generation of NPs with the desired size and encapsulation efficiency are the foremost requirements to fulfill DDS design.
In this work, we wish to report the preparation of NPs from poly(α-methyl-β-L-malate) (PAALM-1), their in vitro degradation under physiological conditions, and their capacity for encapsulation and delivery of drugs to treat brain cancer. A major factor limiting intracranial therapeutic levels of systemically administered active agents is the restriction of permeability imposed by the blood/brain barrier (BBB).[13] Temozolomide (TMZ) is widely recognized as one of the most effective antineoplastic agents for glial tumor, in great part due to its ability to cross the BBB. Some detrimental side effects have been observed however upon its prolonged systemic administration because high dosages have to be given in order to achieve the required therapeutic effect.[14] A platform based on PMLA for the design of a water soluble nanoconjugate for brain tumor treatment has been described.[15] This nanoconjugate is known to cross the brain/tumor barrier (BTB) by transcytosis using an attached antibody that binds to transferrin receptor. A similar nanobioconjugate based on PMLA for the delivery of TMZ has been recently published.[16]
Experimental Section
Materials
The PMLA used in this work was produced by cultivation of Physarum polycephalum and purified as described elsewhere.[17] Final PMLA was spectroscopically pure by NMR and had a weight-average molecular weight of 34 kDa with a polydispersity index (PDI) of 1.08 as determined by gel permeation chromatography (GPC). TMZ (3,4-dihydro-3-methyl-4-oxoimidazo[5,1-d]-as-tetra-zine-8-carboxamide) and doxorubicin [(7S,9S)-7-[(2R,4S,5S,6S)-4-amino-5-hydroxy-6-methyloxan-2-yl]oxy-6,9,11-trihydroxy-9-(2-hydroxyacetyl)-4-methoxy-8,10-dihydro-7H-tetracene-5,12-dione] (DOX) were obtained from AKSci (Union City, CA, USA). All organic solvents were either analytical or HPLC grade and used without further purification. PAALM-1 was prepared by methylation of PMLA with diazomethane in dry acetone as described previously.[10]
NP Preparation and Drug Encapsulation
The solvent evaporation method was used to produce NPs of PAALM-1. Briefly, 20 mg of PAALM-1 were dissolved in 1 mL of dichloromethane (DCM) and the solution was then emulsified with 10 mL of water containing 0.2, 0.5, 1, or 2% of an emulsifier or without emulsifier. Poly(vinyl alcohol) (PVA) of two different molecular weights (2 and 14 kDa) and PMLA with a molecular weight of 34 kDa were used as emulsifiers. Emulsions were generated by sonication with a tip-probe sonicator (Bandelin, Berlin, Germany, Sonoplus, 200 W) operating at 50% of amplitude during 45 s followed by rotary evaporation under reduced pressure to remove the organic solvent. NPs were recovered by centrifugation, washed three times with distilled water to eliminate the excess emulsifier and freeze-dried for storage. Particle morphology was monitored by scanning electron microscopy (SEM) while ζ-potential and average hydrodynamic diameters were determined by light scattering. The same procedure was used for drug encapsulation with both TMZ and DOX; the drug (at a concentration 10 wt% with respect to PAALM-1) was added into DCM previous to polymer dissolution, and emulsification was then performed without using emulsifier to avoid subsequent washes. In the case of DOX, addition of triethylamine (TEA) prior to emulsification was necessary to render the drug solubilization in DCM.
Hydrolytic Degradation
Hydrolytic degradation rate was monitored by measuring the molecular weight as a function of incubation time. Degradation essays were carried out in vials with 1 mg of PAALM-1, either as powder or NPs, immersed in 1 mL of phosphate buffer at pH = 7.4 and 37 °C. Vials were collected at scheduled times and freeze-dried for further GPC analysis. The process was followed in parallel by NMR to investigate the degradation mechanism. For this, 10 mg of polymer were placed in NMR tubes containing 1 mL of deuterated water and incubated at 37 and 60 °C. The supernatant was analyzed by 1H NMR at scheduled times to identify the soluble products that generated upon hydrolysis.
Cell Lines and Culture Media
Cell lines used in cytotoxicity studies were primary human glioma U-87 MG and T98G, and human invasive breast carcinoma MDA-MB-231 and MDA-MB-468, obtained from American Type Culture Collection (ATCC, Manassas, VA, USA). U-87 MG and T98G cells were cultured in minimal essential medium (MEM) supplemented with the following ingredients (final concentrations): 10% fetal bovine serum, 1% MEM non essential amino acids (NEAA), 10−3 M sodium pyruvate, and 2 × 10−3 M L-glutamine. For MDA-MB-231 and MDA-MB-468, Leibovitz’s L-15 medium with 10% final concentration fetal bovine serum was used. Cells were seeded at 10 000 cells per well (0.1 mL) in 96-well flat-bottomed plates and incubated overnight at 37 °C in humid atmosphere with 5% CO2 (breast cancer cell lines MDA-MB-231 and MDA-MB-468 were incubated without CO2).
Cytotoxicity studies were performed on cells incubated for 1, 12, 24, and 72 h with freshly prepared NPs suspensions, previously filtered on a 0.20 μm sterile filter, at increasing concentrations of polymer ranging from 1 to 1 000 μg · mL−1. After the scheduled incubation time, the medium was removed and the cellular viability was measured using the CellTiter 96 Aqueous One Solution Cell Proliferation Assay kit (Promega Co, Madison, WI, USA). Yellow 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium inner salt (MTS) is bioreduced by cells into formazan that is soluble in the tissue culture medium.[18] The absorbance reading at 490 nm from the 96-well plates was directly proportional to the number of living cells. The viability of the untreated cells was referenced as 100%.
In vitro Drug Release Studies
In vitro TMZ and DOX release was assessed by dialysis method. Briefly, 5 mg of freeze-dried TMZ-loaded NPs were resuspended in 1 mL of PBS at pH = 6.8 and 7.4 and transferred into a dialysis tube with 8 kDa molecular cut-off (Spectrum Laboratories, Rancho Domínguez, CA, USA). For dialysis, the tube was immersed into 15 mL of PBS and incubated at 37 °C. For measurement, aliquots of 0.5 mL of the releasing media were taken at scheduled times and the volume replaced by fresh medium every time. Drug concentration was determined by HPLC at 330 and 480 nm for TMZ and DOX, respectively. Since TMZ is hydrolytically labile its degradation product 5-aminoimidazole-4-carboxamide (AIC) absorbing at 254 nm was also measured.
Measurements
SEM images were taken with a field-emission JEOL JSM-7001F instrument (JEOL, Japan) from uncoated samples. Particle size measurements, based on intensity, and ζ-potential were performed with a ZetaSizer NS (Malvern Instruments, UK) after particles have been suspended in deionized water. NMR spectra were recorded on a Bruker AMX-300 instrument, from samples immersed in D2O. 1H NMR spectra where recorded at 25 °C operating at 300.1 MHz, 128 scans were acquired with 32-K data points and relaxations of 2 s.
GPC was done using a Waters 515 HPLC pump with a Waters 410 differential refractometer detector and a Waters Styragel HR 5E column (7.8 × 300 mm2, Waters, Massachusetts, USA). Samples were chromatographed using 0.05 M sodium trifluoroacetate in hexafluoroisopropyl alcohol at 0.5 mL · min−1 flow rate. Chromatograms were calibrated against poly(methyl methacrylate) standards (Varian, California, USA). High performance liquid chromatography (HPLC) was performed with a Waters 600 system with a Waters 996 photodiode array detector provided with a GL Sciences (California, USA) Inertsil ODS-3V column (5 μm, 4.6 × 250 mm2). Mobil phase used for TMZ was methanol in 0.5% aqueous acetic acid (10:90) pumped at a rate of 1 mL · min−1, and for DOX, a mixture of 0.02 M sodium hydrogen phosphate and acetonitrile (60:40).
Results and Discussion
Synthesis and Characterization of PAALM-1
PMLA was methylated to 100% degree with diazomethane in dry acetone. This reaction allowed us to obtaining a complete esterification of the carboxylic side groups of PMLA without significant reduction in molecular weight. PAALM-1 precipitated from the reaction medium and was purified by repeated dissolution-precipitation in chloroform/ether. The final PAALM-1 was NMR spectroscopically pure and its weight-average molecular weight and PDI determined by GPC were 33 kDa and 1.4, respectively.
NP Formation and Characterization
Spherical NPs were obtained by the emulsion-evaporation method using three emulsifiers at four different concentrations. We were also able to obtain NPs without using emulsifier by fast evaporation of the organic phase under reduced pressure; under such conditions particle could be formed before coalescence of the disperse phase occurred. The overall appearance of the obtained nanospheres is shown in the SEM pictures of Figure 1. Light scattering measurements indicated that they have diameters with average values ranging between 100 and 350 nm, the smallest ones being those prepared in the absence of emulsifier, and a satisfactory polydispersity.
Figure 1.

SEM micrographs of PAALM-1 NPs prepared using different emulsifiers (a) PVA-2 kDa, (b) PVA-14 kDa, (c) PMLA, and (d) without emulsifier.
As it is seen in Figure 2, all samples show a unimodal distribution of sizes when analyzed by light scattering. The effect of emulsifier concentration on the size distribution profiles were not significant, which can be attributed to the governing effect of the solvent evaporation process. On the contrary, the influence of the applied sonication time was clearly noticeable. As demonstrated in Figure 2, size distribution profiles were displaced to the left and became narrower with increasing sonication time indicating that particles became smaller and less polydisperse in size. Variation in particle size as a function of sonication time is plotted in Figure 3 showing that the particle diameter decreases with time approaching asymptotically to a value close to 220 nm when an emulsifier was used for preparation and around 175 nm in the absence of emulsifier. According to the previous work carried out on NPs preparation using this method,[19] such effect appears because the size of dispersed drops decreases with sonication time approaching to a minimum critical size, which is determined by the relative phase viscosities, interfacial tension between the phases and the magnitude of the emulsion generating force. In our case, where a rapid organic phase evaporation occurs and mean particle size is not affected by increasing emulsifier concentrations, it seems that it is emulsion droplet size (related to sonication time) rather than emulsion stability (related to emulsifier concentration) which determines the final particle size. This is fully consistent with the fact that NPs could be obtained in the absence of emulsifier and also with the different effect that emulsifier concentration and sonication times exerted on particle size. Particles sonicated for 45 s were chosen for the subsequent assays since their size differences are minimum. The characteristics of these NPs are compared in Table 1. The ζ-potentials of the particles have average values between −20 and −34 mV with the highest negative value observed for NPs that were prepared without using emulsifier. As the ζ-potential is related to surface charges, it directly affects particle’s suspension stability. Usually a ζ-potential higher than 25 mV (positive or negative) is taken as the minimum to maintain the system in a stable disperse state. Our results indicate that NPs prepared using PVA as emulsifier do not reach such value whereas those prepared without emulsifier will be able to form well stable dispersions.
Figure 2.

Particle size distribution as a function of sonication time for particles prepared with and without emulsifier, as indicated.
Figure 3.

Decrease of mean particle diameter with sonication time. Error bars stand for standard deviations.
Table 1.
Mean diameter, polydispersity index, and ζ-potential of NPs used for degradation.
| Nanoparticle | Emulsifier | Diameter [nm] | Polydispersity Indexa) | ζ-Potential [mV] |
|---|---|---|---|---|
| NP-PVA2 | PVA 2 kDa | 222 | 0.033 | −23.3 |
| NP-PVA14 | PVA 14 kDa | 231 | 0.193 | −20.6 |
| NP-PMLA | PMLA | 238 | 0.063 | −25.7 |
| NP | – | 207 | 0.236 | −33.9 |
Defined as the ratio of weight average to the number average particle radius.
Hydrolytic Degradation
The hydrolytic degradation rate of PAALM-1, both in powder form and as NPs, was comparatively estimated by following the evolution of the molecular weight with incubation time (Figure 4). PAALM-1 NPs showed a degradation rate lower than the powder. After 18 weeks of incubation, when the powder appeared completely degraded, the polymer in the NPs still retained between 25 and 75% of its original molecular weight depending on the procedure applied for emulsification. A problem associated to the use of emulsifier is its binding on the particles surface. Since emulsifier stays at the oil/water interface during solvent evaporation, it presumably remains attached to the surface of the particle altering thereby the surface composition and consequently the degradation rate. The occurrence of irreversible binding of PVA on particle surface of PLGA at the water/DCM interphase has been reported by several authors,[20,21] and the slow degradation rate observed for such particles was related to the relatively low digestibility of the PVA coating.[22] As it is shown in Figure 4, NPs prepared either without emulsifier or with PMLA emulsifier, degraded much faster than those prepared using PVA, which strongly supports that also in our case, PVA attached to NPs surface acts as a hydrolysis protecting coat. Nevertheless, what it is really worthy to note is that PAALM-1 NPs degrade considerably faster than those made of poly(α-benzyl β-malate), which has been reported to undergo only 40% of molecular weight reduction after 20 weeks of incubation.[2]
Figure 4.

Hydrolytic degradation of PAALM-1 powder and NPs in PBS pH = 7.4 at 37 °C. NPs were prepared using three different emulsifiers or without emulsifier.
Hydrolytic degradation mechanism of PAALM-1 was studied in deuterated water at 60 °C by NMR analysis of the incubating medium using powder samples (Figure 5a). At the physiological temperature of 37 °C, hydrolysis rate was so low that signals arising from degraded products were almost undetectable in the first months of incubation. Conversely, a singlet signal corresponding to methanol, which is released in the hydrolysis of the ester side group, started to be observed after only 1 week of incubation at 60 °C. This signal increased continuously with time until complete hydrolysis of the methoxycarbonyl group. Conversely, the signal corresponding to the methyl group attached to the polymer chain appeared after 3 weeks indicating the presence of soluble chain fragments in the supernatant. This solubilized material may be oligomers or partially side chain hydrolyzed PAALM-1 long fragments. As the polymer began to be water soluble, the hydrolysis rate increased so the spectra recorded after 5 weeks of incubation showed signals corresponding to a mixture of degradation compounds including methanol, malic acid, and more or less methylated oligomers.
Figure 5.

1H NMR spectra of the supernatant. Degradation in deuterated water at 60 °C of PAALM-1: (a) powder, (b) NP-PVA14, recorded at the indicated incubation times. *Residual solvent.
A similar NMR analysis carried out with PAALM-1 NPs revealed that the degradation mechanism was essentially the same as observed with powder but with noticeable differences in the timing of signal appearance. 1H NMR spectra recorded at increasing incubation times from the supernatant of the degradation of PAALM-1 NPs prepared by adding PVA 14 kDa, are compared in Figure 5b. As it happened with the powder, the earliest evidence on degradation was the presence of methanol, which also appeared in this case within the first week of incubation. The intensity of this signal increased continuously until the 15 week when hydrolysis of the methoxycarbonyl group was almost over. Signals arising from solubilized chain fragments were detected at the fifth week but it was not until after 7 weeks of incubation when malic acid arrived to be detected. After 20 weeks, no residual sample was perceivable and the relative intensity of NMR signals was stabilized indicating full degradation of the PAALM-1. The NMR results obtained in the degradation essays carried out on NPs made with PMLA as emulsifier or without emulsifier (not shown), were very similar to the case of the powder. On the basis of the collected NMR and GPC data, and in agreement with previous results reported by us on partially methylated PMLA,[12] the basic mechanism that can be outlined for the hydrolytic degradation of PMLA is depicted in Scheme 2.
Scheme 2.

Hydrolytic degradation mechanism of PAALM-1 at 37 °C.
Cytotoxicity
For its potential use as a biomaterial it was mandatory to evaluate the toxicity of NPs made of fully methylated PMLA. Thus, an in vitro study of PAALM-1 NPs cytotoxicity on human glioma cell lines U-87 MG and T98G, and invasive human breast carcinoma cell lines MDA-MB-231 and MDA-MB-468, was performed. Cellular viability was measured as a function of polymer concentration using the MTT test for contact times of 1, 12, 24, and 72 h between polymer and cells. As it is expected, results obtained for all cell lines indicated that the percentage of viability decreased when contact time and concentration of polymer increased (Figure 6). The effect of the NPs on cell viability depended also on cell line type so that glioma U-87MG and breast cancer MDA-MB-231 cells were more affected than glioma T98G cells and breast cancer MDA-MB-468 cells.
Figure 6.

Cell viability, of human glioma cell lines U-87 MG and T98G and invasive breast carcinoma cell lines MDA-MB-231 and MDA-MB-468 after different contact times between polymer and cells as a function of polymer concentration.
Toxicity caused by physical damage due to membrane disruption may be neglected since NPs effect on cell viability is not manifested in the time scale of 1 h. In a way similar to that observed for partial hydrolyzed PAALM-1 derivatives,[23] it is highly probable that the low toxicity observed was due to the effect of methanol that is released during polymer degradation. Both methanol and L-malic acid are generated in the hydrolysis of PAALM-1 but whereas L-malic acid is converted into water and carbon dioxide in the tricarboxylic acid cycle, methanol is known to adversely affect the living cells. Similar results have been reported for NPs made of other PMLA derivatives, where cytotoxicity was also related to the degradation products generated in the cell culture media.[5] Nevertheless, the toxicity observed for exposure times over 12 h may be considered negligible, because in DDS applications residence times for NPs in the human body will be only a few hours before they are cleared from blood through the renal system.
Drug Encapsulation and In vitro Release
TMZ and DOX encapsulation was made in PAALM-1 without using emulsifiers in order to avoid exhaustive washing and minimize drug losses. Although both drugs were encapsulated by the same method, DOX was encapsulated with a higher efficiency than TMZ (Table 2), due to the higher solubility of TMZ in water or the poor solvent compatibility between the drug and the polymer. Both drugs display a remarkable affinity for polar solvents which causes a diffusion of these compounds from the organic phase to the aqueous phase during emulsion’s generation. In the case of DOX this phenomena could be reduced by the addition of TEA to modify DOX solubility and increase its encapsulation efficiency.
Table 2.
Poly(α-methyl-β, L-malate) (PAALM-1) NPs encapsulation of TMZ and DOX.
| Substance | Encapsulationa) [wt%] | Encapsulation efficiencyb) [wt%] |
|---|---|---|
| TMZ | 2.18 ± 0.17 | 21.75 ± 1.7 |
| DOX | 4.20 ± 0.62 | 42.06 ± 6.2 |
Percentage of drug contained in the NPs upon encapsulation;
Percentage of the drug that is encapsulated.
The releasing profiles of TMZ from PAALM-1 NPs obtained at different pH are shown in Figure 7. The analysis of TMZ release under physiological conditions is complex because above pH = 7.0 it undergoes fast hydrolysis yielding AIC together with the methyldiazonium ion, which is the chemotherapeutically active molecule.[24,25] For a correct evaluation of the in vitro TMZ release, it will be therefore necessary to quantify the delivery of both compounds. A releasing assay carried out at pH = 6.8 revealed that a significant decomposition took place even at this pH. Nevertheless, there is meaningful differences in the AIC release profiles generated at pH = 7.4 and 6.8, which clearly indicate that the formation of AIC follows the release and decomposition of TMZ from the particles. The release of TMZ seems to be independent of pH, but its half-life time appeared to be longer at pH = 6.8, as it was expected. Thus, firstly TMZ was delivered from PAALM-1 NPs and then it decomposed forming AIC with the consequent release of the methyldiazonium ion.
Figure 7.

Profiles of TMZ in vitro release from PAALM-1 NPs and formation of AIC from released TMZ at pH = 7.4 and 6.4.
Under physiological conditions the release of DOX followed a much lower rate than TMZ. Whereas only a few hours were required for a complete release of TMZ the complete liberation of DOX needed more than 1 month (Figure 8). Since the releasing profile of DOX follows more or less closely the degradation profile of PAALM-1, it can be reasonably concluded that the delivery of this drug is governed by the hydrolysis of the polymer. On the contrary, the fast release of TMZ must happen by diffusion during the first stages of the NPs degradation. The different chemical nature of TMZ and DOX account for the remarkable differences they display in their releasing from PAALM-1 NPs.
Figure 8.

DOX in vitro release from PAALM-1 NPs at pH = 7.4.
Conclusion
Fully methylated polymalic acid obtained by methylation of fungal PMLA is a biodegradable polyester that can be used to produce nanospheres with an average diameter of around 200 nm and ζ-potential of −20 to −35 mV depending of the kind of emulsifier used in the preparation. PAALM-1 NPs prepared using polyvinylalcohol as emulsifier degraded slower than without emulsifier indicating that PVA acts as a hydrolysis protecting coat. In aqueous buffer particles hydrolyze releasing methanol followed by main chain ester bond cleavage. DOX and TMZ can be encapsulated in the NPs and released upon incubation under physiological conditions. TMZ was released within a few hours with subsequent hydrolytic pH-dependent activation resulting in AIC, while DOX was released in a time scale of days. These differences agree with the different chemical nature of both drugs. The particles described (in the absence of loaded TMZ or DOX) did not show a sign of toxicity after a few hours of administration but cell viability is significantly altered after days of contact. Although PAALM-1 NPs are suitable for either short or long time DDSs depending on the chemical nature of the drug, long residence times are expected to result in undesired side effects.
Acknowledgments
This work was funded by MICINN (Spain) with project MAT2009-14053-CO2-01 to S. M.-G. CONACyT (México) and Programa AlBan to A. L.-L. NIH (R01 CA123495, R01 CA 136841 and U01 CA151815) to J. Y. L., a Winnick Family Foundation clinical grant to J. Y. L.
Contributor Information
Alberto Lanz-Landázuri, Departament d’Enginyeria Química, Universitat Politécnica de Catalunya, ETSEIB, Diagonal 647, 08028 Barcelona, Spain.
Montserrat García-Alvarez, Departament d’Enginyeria Química, Universitat Politécnica de Catalunya, ETSEIB, Diagonal 647, 08028 Barcelona, Spain.
José Portilla-Arias, Department of Neurosurgery, Cedars-Sinai Medical Center, 8631 W. Third Street, Suite 800E, Los Angeles, CA 90048, USA.
Antxon Martínez de Ilarduya, Departament d’Enginyeria Química, Universitat Politécnica de Catalunya, ETSEIB, Diagonal 647, 08028 Barcelona, Spain.
Rameshwar Patil, Department of Neurosurgery, Cedars-Sinai Medical Center, 8631 W. Third Street, Suite 800E, Los Angeles, CA 90048, USA.
Eggehard Holler, Department of Neurosurgery, Cedars-Sinai Medical Center, 8631 W. Third Street, Suite 800E, Los Angeles, CA 90048, USA.
Julia Y. Ljubimova, Department of Neurosurgery, Cedars-Sinai Medical Center, 8631 W. Third Street, Suite 800E, Los Angeles, CA 90048, USA
Sebastián Muñoz-Guerra, Email: sebastian.munoz@upc.edu, Departament d’Enginyeria Química, Universitat Politécnica de Catalunya, ETSEIB, Diagonal 647, 08028 Barcelona, Spain.
References
- 1.Zambaux MF, Bonneaux F, Gref R, Maincent P, Dellacherie E, Alonso MJ, Labrude P, Vigneron C. J Controlled Release. 1998;50:31. doi: 10.1016/s0168-3659(97)00106-5. [DOI] [PubMed] [Google Scholar]
- 2.Stolnik S, Garnett MC, Davies MC, Illum L, Davis SS. J Mater Sci: Mater Med. 1996;7:161. [Google Scholar]
- 3.Manocha B, Margaritis A. Crit Rev Biotechnol. 2008;28:83. doi: 10.1080/07388550802107483. [DOI] [PubMed] [Google Scholar]
- 4.Soppimath KS, Aminabhavi TM, Kulkarni AR, Rudzinski WE. J Controlled Release. 2001;70:1. doi: 10.1016/s0168-3659(00)00339-4. [DOI] [PubMed] [Google Scholar]
- 5.Martinez-Barbosa ME, Cammas S, Appel M, Ponchel G. Biomacromolecules. 2004;5:137. doi: 10.1021/bm0300608. [DOI] [PubMed] [Google Scholar]
- 6.Coulembier O, Degée P, Hedrick JL, Dubois P. Prog Polym Sci. 2006;31:723. [Google Scholar]
- 7.Mundargi RC, Ramesh Babu V, Rangaswamy V, Patel P, Aminabhavi TM. J Controlled Release. 2008;125:193. doi: 10.1016/j.jconrel.2007.09.013. [DOI] [PubMed] [Google Scholar]
- 8.Lee BS, Vert M, Holler E. Water-soluble Aliphatic Polyesters: Poly(malic acid)s, Volume 3a: Polyesters I. In: Doi Y, Steinbuechel A, editors. Biopolymers. Wiley-VCH; Weinheim: 2002. pp. 75–103. [Google Scholar]
- 9.Vert M. Polym Degrad Stab. 1998;59:169. [Google Scholar]
- 10.Fernández CE, Mancera M, Holler E, Galbis JA, Muñoz-Guerra S. Polymer. 2006;47:6501. [Google Scholar]
- 11.Lee BS, Fujita M, Khazenzon NM, Wawrowsky KA, Wachsmann-Hogiu S, Farkas DL, Black KL, Ljubimova JY, Holler E. Bioconjugate Chem. 2006;17:317. doi: 10.1021/bc0502457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Portilla-Arias J, García-Álvarez M, Martínez de Ilarduya A, Holler E, Galbis JA, Muñoz-Guerra S. Macromol Biosci. 2008;8:540. doi: 10.1002/mabi.200700248. [DOI] [PubMed] [Google Scholar]
- 13.Huynh GH, Deen DF, Szoka FC., Jr J Controlled Release. 2006;110:236. doi: 10.1016/j.jconrel.2005.09.053. [DOI] [PubMed] [Google Scholar]
- 14.Zhang H, Gao S. Int J Pharm. 2007;329:122. doi: 10.1016/j.ijpharm.2006.08.027. [DOI] [PubMed] [Google Scholar]
- 15.Ding H, Inoue S, Ljubimov AV, Patil R, Portilla-Arias J, Hu J, Konda B, Wawrowsky KA, Fujita M, Karabalin N, Sasaki T, Black KL, Holler E, Ljubimova JY. PNAS. 2010;107:18143. doi: 10.1073/pnas.1003919107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Patil R, Portilla-Arias J, Ding H, Inoue S, Konda B, Hu J, Wawrowsky KA, Shin PK, Black KL, Holler E, Ljubimova J. Pharm Res. 2010;27:2317. doi: 10.1007/s11095-010-0091-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Holler E. In: Handbook of Engineering Polymeric Materials. Cheremisinoff NP, editor. Marcel Dekker; New York: 1997. pp. 93–103. [Google Scholar]
- 18.Mosmann T. J Immunol Methods. 1983;65:55. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 19.Freitas S, Merkle HP, Gander B. J Controlled Release. 2005;102:313. doi: 10.1016/j.jconrel.2004.10.015. [DOI] [PubMed] [Google Scholar]
- 20.Boury F, Ivanova T, Panaiotov I, Proust JE, Bois A, Richou J. J Colloid Interface Sci. 1995;169:380. [Google Scholar]
- 21.Lee SC, Oh JT, Jang MH, Chung SI. J Controlled Release. 1999;59:123. doi: 10.1016/s0168-3659(98)00185-0. [DOI] [PubMed] [Google Scholar]
- 22.Landry FB, Bazile DV, Spenlehauer G, Veillard M, Kreuter J. Biomaterials. 1996;17:715. doi: 10.1016/0142-9612(96)86742-1. [DOI] [PubMed] [Google Scholar]
- 23.Portilla-Arias J, Patil R, Hu J, Ding H, Black KL, García-Alvarez M, Muñoz-Guerra S, Ljubimova JY, Holler E. J Nanomater. 2010:825363. doi: 10.1155/2010/825363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arrowsmith J, Jennings SA, Clark AS, Stevens MFG. J Med Chem. 2002;45:5458. doi: 10.1021/jm020936d. [DOI] [PubMed] [Google Scholar]
- 25.Baker SD, Wirth M, Statkevich P, Reidenberg P, Alton K, Sartorius SE, Dugan M, Cutler D, Batra V, Grochow LB, Donehower RC, Rowinsky EK. Clin Cancer Res. 1999;5:309. [PubMed] [Google Scholar]