

Summary
The malignant transformation of mesenchymal cells within the bone leads to the development of osteosarcoma (OS), but the genetic underpinnings of these events are not understood. From a clinical perspective, primary tumour management can be achieved successfully in most patients. However, the development of metastasis to the lungs represents the most common cause of death in OS patients. A clearer understanding of metastasis biology is required to improve cancer mortality and improve outcomes. Modelling the genetics, biology and therapy of OS can be accomplished through research involving a number of species. Most notable is the naturally occurring form of OS that develops in dogs. Through a cross-species and comparative approach important questions can be asked within specific and suitable models to advance our understanding of this disease and its common metastatic outcome. A comparative perspective on the problem of OS metastasis that utilizes a cross-species approach may offer unique opportunities to assist in this prioritization and generate new hypotheses related to this important clinical problem.
Keywords: dog, metastasis, osteosarcoma, protein translation
Osteosarcoma
Osteosarcoma (OS) is an uncommon tumour of bone, arising from the malignant transformation of mesenchymal cells, which differentiate towards the formation of osteoid and bone (Malawar et al., 2005; Kansara and Thomas, 2007). While OS can arise in any bone, the most common sites of primary tumours are the distal femur, proximal tibia and proximal humerus. Beyond primary tumour growth in the appendicular and less commonly the axial skeleton, a defining feature of OS biology is its high propensity for pulmonary metastasis (90% of metastases are to the lung; Krishnan et al., 2005). These defining features of OS are shared between human and canine OS patients. This cross-species review describes the disease with an emphasis on metastasis biology and our interest in the potential linkage between efficient protein translation and metastasis.
Human Osteosarcoma in Paediatric Patients
OS may occur at any age; however, it has a peak incidence in the second decade of life with a second smaller peak of incidence in the elderly population (>50 years of age) (Hayden and Hoang, 2006; Kansara and Thomas, 2007). Although rare, it is the most common paediatric bone malignancy in the USA (Gurney et al., 1999). Approximately 800–1,000 cases of OS are diagnosed in the USA each year and about half of these patients are teenagers (Wang, 2005; Kansara and Thomas, 2007). OS occurs most frequently during the adolescent growth spurt in areas of rapid bone growth, suggesting a relationship between tumour formation and rapid growth and/or growth factors expressed at this time in life (American Cancer Society, http://www.cancer.org/docroot/home/index.asp). OS may occur more frequently in males than females (60% versus 40%) and slightly more often in African Americans than Caucasians (Gurney et al., 1999).
Fifty years ago, when surgery was the only available treatment, a diagnosis of OS was often fatal. Patients had only a 15–20% chance of cure (Dahlin and Coventry, 1967; Hayden and Hoang, 2006). Fortunately, advances in chemotherapy, including the introduction of single agent and combination chemotherapy, and advanced orthopaedic surgical techniques in the 1980s and 1990s increased survival rates dramatically. Today, patients with localized disease at presentation have a 65–70% chance of 5-year relapse-free survival (Bielack et al., 2002; Hayden and Hoang, 2006; Kansara and Thomas, 2007; Meyers, 2009). Unfortunately, the remaining patients will relapse (with pulmonary metastasis) within the first 5 years, attesting to the fact that these patients have undetectable metastatic disease at diagnosis. Indeed, up to 20–25% of newly diagnosed patients have detectable metastases at diagnosis (Hattinger et al., 2010). Patients presenting with metastases have a poor prognosis, with long-term survival rates of 10–30% (Meyers, 2009). Other prognostic indicators for early relapse include the size and location of the primary tumour, the response to preoperative (neoadjuvant) chemotherapy and surgical success (Bielack et al., 2002). Current chemotherapy treatment plans are based on neoadjuvant chemotherapy followed by surgical removal of the tumour and post-operative (adjuvant) chemotherapy. The use of neoadjuvant chemotherapy has the advantage of down-staging the size of the primary tumour and as such providing a more successful opportunity for primary tumour resection with limb salvage. Pathological analysis of the definitive resection provides an opportunity to evaluate histologically chemotherapy-induced tumour necrosis in the resected specimen.
The Huvos grading system (Huvos et al., 1977) is a non-invasive quantitative estimate of the percentage of tumour cell necrosis (death) observed in the tumour after surgery. Patients showing tumour necrosis in at least 90% of the resected tumour specimen are classified as good responders (Huvos score 3 or 4) and have more favourable prognoses than patients with less necrosis. Patients with low necrosis scores (Huvos score of 1 or 2) are often selected to receive alternative and more aggressive chemotherapy protocols in the adjuvant setting. Patients who achieve a good histological response to preoperative chemotherapy have considerably better survival than those who have a poor response. Five-year survival for those with good response is approximately 75–80%, compared with 45–55% for those with poor response (Whelan et al., 2000; Bielack et al., 2002). Exemplary of this approach, the European and American Osteosarcoma Study (EURAMOS) group, has launched, and now nearly completed, the EURAMOS I study. In this study histological response (Huvos score) following preoperative chemotherapy (http://www.ctu.mrc.ac.uk/euramos) is used to assign patients to distinct treatment groups. The current treatment backbone used as first-line therapy in patients includes cisplatin, doxorubicin and methotrexate. The EURAMOS objectives are: (1) to examine whether the addition of ifosfamide and etoposide (IE) to postoperative chemotherapy improves event-free survival for patients with a poor histological response to preoperative chemotherapy and (2) to examine whether the addition of interferon (IFN)-α as maintenance therapy after post-operative chemotherapy improves event-free survival for patients with a good histological response to pre-operative chemotherapy.
Despite sophistication of trial designs and multimodality treatments for OS, improvements in long-term survival in the last 20 years have been modest at best. The primary cause of death for patients continues to be the development of metastasis. In order for patient outcomes to improve, we must improve our understanding of the biology of metastasis. Given the complexity of metastasis, there remains a need to develop or to identify reliable animal models of OS. An ideal model would include development of spontaneous primary bone tumour and pulmonary metastases within an immunocompetent host. This type of model would provide the best opportunity to identify key regulatory pathways involved in the development and metastatic progression of OS, as well as the ability to investigate activities of novel antimetastatic therapeutics.
Comparative Studies in Animals
Cross-species comparative opportunities to understand OS biology and therapy are strong. Current animal models of OS include rats, mice and dogs. Each model provides unique strengths. Rat OS models play an important role in evaluating new surgical and molecular methods of treatment for appendicular OS (Blouin et al., 2005). Mouse models of OS are used commonly to study tumour biology and develop new approaches for treating OS. Transplantable mouse OS models include syngeneic (genetically identical or closely related, so as to allow successful tissue transplant), xenograft (derived or obtained from an organism of a different species, as a tissue graft; human explants), heterotopic (pertaining to the injection of cancer cells/tissue to a place it is not normally found; e.g. non-osseous tissues) and orthotopic (pertaining to the transplantation of cancer cells/tissue into its normal anatomical position; e.g. bone microenvironment). Mouse syngeneic models of OS are especially valuable for studying primary tumour and metastatic progression. These models utilize an immunocompetent mouse host that provides the advantage of mimicking the complex interactions between tumour cells and the relevant host microenvironment (Fan, 2010). While excellent for evaluating primary tumour and metastatic progression, syngeneic models lack the species-specific information that is critical to OS pathogenesis in people. As such, xenograft models of OS are commonly used for studying human OS biology and new anticancer treatments. In xenograft models, human OS cells are injected into immunodeficient mouse hosts; therefore, a weakness of the model is that it fails to mimic accurately important aspects of the host microenvironment–tumour cell interactions that participate in primary tumour development and metastatic OS progression (Fan, 2010). In mouse models of OS, tumour cells can be transplanted orthotopically into the intermedullary cavity or paraosseous structures of either the proximal tibia or distal femur or heterotopically to the subcutis or musculature. Orthotopic transplantation of OS cells more closely replicates critical host stromal cell–tumour cell interactions that likely participate in OS pathogenesis and biology (Fidler et al., 1990; Fu et al., 1991; Fan, 2010). Currently, syngeneic, orthotopic transplantation mouse models are the most common experimental system used to study OS biology (Fidler et al., 1990; Khanna et al., 2000; Hoffman, 2001). Finally, genetically engineered mouse (GEM) models provide an experimental system to model the genetics of human OS.
The most successful GEM models regulate the genetic modification (i.e. knock-in or knock-out) in a tissue-specific manner through tissue-specific promoters or tissue-specific recombination events. In OS, conditional knockout mouse models represent an experimental system that supports endogenous, spontaneous generation of tumours through knockouts of critical tumour suppressor genes. These models have proven very useful in identifying regulatory pathways in OS tumourigenesis and metastatic progression as well as providing a valuable platform for developing novel therapeutic strategies (Khanna et al., 2000; Berman et al., 2008; Walkley et al., 2008). Conditional knockout is based on a tissue-specific inactivation of the gene of interest. This can be achieved by means of Cre-lox recombination, a genetic technology that allows site-specific recombination events including deletions (Sauer and Henderson, 1988). While the aetiology of OS is unknown, it is evident that the tumour suppressor genes p53 and Rb (retinoblastoma) play a role, as children with familial mutation syndromes affecting either of these genes have higher incidences of OS (Hansen, 1991). Two GEM models lacking the p53 and Rb genes have been created using Cre-loxP recombination strategies. These models produce F1-generation mice that readily develop OS; however, while p53 loss is associated with the development of OS, the Rb gene mutation alone is not sufficient to induce osteosarcomagenesis. Instead, it must act synergistically with p53 to induce osteosarcomagenesis (Berman et al., 2008; Walkley et al., 2008). These models replicate many defining features of human OS including gene-transcription signatures; however, the observed biological behaviour of OS generated in the mice does not consistently mimic all aspects of the disease in man (Walkley et al., 2008).
Companion animals naturally develop OS and the disease is almost identical in dogs and people. As such, the dog represents a naturally occurring and spontaneous metastatic animal model of OS (Vail and MacEwen, 2000; Withrow and Khanna, 2009; Fan, 2010; Paoloni et al., 2010). The clinical presentation, biology, treatment, complications and outcomes are similar in both dogs and people, although human patients have better outcomes and the incidence of OS is much higher in dogs (Withrow and Wilkins, 2010). Another noteworthy difference in canine OS is that the median age of onset is in adulthood. In addition, physeal plates in dogs (versus children) are closed at the age of onset of OS in the breeds of dogs that are overrepresented for OS development (Fan, 2010; Withrow and Wilkins, 2010). Despite these differences, the biology and genetics of OS are highly conserved between people and dogs (Paoloni et al., 2009).
Importantly, the canine OS model includes spontaneous and natural development of the primary tumour and pulmonary metastases within an immunocompetent host. The increased risk of OS development in specific breeds of dogs has raised the possibility that the dog with OS may be useful in identification of genes linked to cancer formation. The opportunity for studies of OS biology and genetics has been enhanced through the development of the Canine Comparative Oncology and Genomics Consortium (CCOGC). The CCOGC has developed a national canine OS biospecimen repository (the Pfizer Canine Comparative Oncology and Genomics Consortium Biospecimen Repository). This US biospecimen repository seeks to collect cancer, normal tissues and biofluids from over 1,800 dogs with a variety of cancers including approximately 600 dogs with OS. Release of tissues from this repository, based on meritorious scientific proposals, is expected before the end of 2012. The translational opportunity to better develop new cancer treatments for OS patients through studies of dogs with OS, has been widely recognized (Paoloni and Khanna, 2008; Withrow and Khanna, 2009). Clinical trials conducted by single institutions and multicentre studies are conducted commonly in dogs with OS with this intent. These studies are increasingly integrated within existing drug development plans of cooperative groups and pharmaceutical companies interested in OS treatments for human patients (Khanna, 2008).
Metastasis
As suggested above, the single most significant negative prognostic factor in the treatment of cancer is the development of metastatic disease. Treating metastatic OS remains a challenge for oncologists. In part this is the result of the differences in the biological behaviour of metastatic tumours from primary tumours (Krishnan et al., 2005; Kansara and Thomas, 2007; Bacci et al., 2008; De Nigris et al., 2008; Guo et al., 2008; Posthuma-DeBoer et al., 2011). As such, the therapeutic regimens that target primary tumours may not be successful in the treatment of metastatic disease. The primary research need in the field is to understand the biology of metastasis in OS and to use this understanding to develop therapeutics that target the metastatic phenotype.
Dissemination of cancer may occur through one of three pathways: (1) direct seeding of body cavities or surfaces, (2) lymphatic spread and (3) haematogenous spread (Slauson and Cooper, 2002; Kumar et al., 2005). Haematogenous spread is typical of solid tumours such as OS. The metastatic cascade is an extremely complex multistage process. In order for a metastatic lesion to become clinically detectable, it must successfully complete a sequential series of steps of the metastatic cascade. Within this process, each step is subject to a wide variety of influences; thus, at any point in the sequence the tumour cells may not survive. Failure to complete any step results in failure to colonize a distant site. The metastatic cascade can be divided into two phases: (1) invasion of the extracellular matrix (ECM) and (2) vascular dissemination and arrest in a distant site (Kumar et al., 2005). Prior to invasion of the ECM, clonal expansion, growth, diversification and angiogenesis must occur in order to produce metastatic subclones within the primary tumour (Krishnan et al., 2006). These ‘transformed’ tumour cells must then separate from the primary tumour and interact with the ECM at several stages in the metastatic cascade. Integrins are adhesion molecules that promote stable interactions between cells and their environment. They bind to ECM proteins and send anchorage-dependent signals that can influence cell survival by suppressing apoptosis (Griffiths et al., 2011). Apoptosis results from a loss of integrin-mediated cell attachment to the ECM.
In order for the neoplastic cells to breach the underlying basement membrane they must first detach themselves from one another and their surroundings. After the cells are detached, it is necessary for them to attach to matrix components (e.g. laminin, collagen and fibronectin) so that the basement membrane can be degraded enzymatically to create passageways for tumour cell migration (Slauson and Cooper, 2002). The basement membrane is degraded by serine, cysteine and matrix metalloproteinases (MMPs). Loss of either cell–cell or cell–matrix interaction activates caspase proteases, the hallmark of apoptosis (Frisch and Francis, 1994). Tumour cells resist detachment-induced apoptosis (anoikis) by establishing contacts with other tumour cells (homotypic interactions) or with host cells such as platelets and inflammatory cells (heterotypic interactions). Both types of interactions generate intracellular signals that prevent anoikis (Grossmann, 2002; Krishnan et al., 2006). Furthermore, there can be overexpression of essential proteins by cancer cells that directly inhibit anoikis. TrkB, a tyrosine kinase receptor (Douma et al., 2004), the integrin pair α2β3 (Ruoslahti and Reed, 1994), adenomatous polyposis coli (APC) gene product (Beavon, 2000), focal adhesion kinase, galectin-3 and transforming growth factor-β (Krishnan et al., 2006) are reportedly involved in resistance to anoikis.
Once tumour cells have invaded the surrounding tissues and migrated through the basement membranes into the bloodstream (lymphatics or peritoneal space), they are susceptible to destruction by innate and adaptive immune defences. In addition, destruction of intravasated tumour cells by haemodynamic forces and shearing is thought to contribute to metastatic inefficiency (Weiss et al., 1992). However, it has been demonstrated recently that some tumour cells in the bloodstream can arrest in capillary beds and extravasate with high efficiency (Luzzi et al., 1998). Neoplastic cells must strategically evade detection and destruction by the immune system and other antitumour effectors at all stages of metastatic progression (Slauson and Cooper, 2002; Hunter, 2004; Kumar et al., 2005; Krishnan et al., 2005, 2006).
Adhesion among tumour cells together with adhesion between tumour cells and platelets allows tumour cells to aggregate in clumps (tumour emboli) within the bloodstream (Kumar et al., 2005). These platelet–platelet aggregates may enhance tumour cell survival and implantation. Arrest and extravasation of tumour emboli at distant sites involves adhesion to the endothelium followed by exit through the basement membrane. Involved in this process are adhesion molecules such as integrins and laminin receptors and previously mentioned proteolytic enzymes (Slauson and Cooper, 2002). At the new site, the tumour cells need to survive, proliferate, develop a vascular supply and avoid and survive apoptotic signals and host immune responses. The site at which the circulating tumour cells leave the capillaries to form secondary deposits is defined, in part, by the anatomical location of the primary tumour, the associated venous drainage and the first capillary bed encountered by metastatic cells (Weiss et al., 1988; Kumar et al., 2005). With venous invasion, neoplastic cells follow the venous flow draining the site of the tumour; hence, the liver and lungs are most frequently involved secondarily in haematogenous dissemination (Kumar et al., 2005). Arterial dissemination may occur when tumour cells pass through the pulmonary capillary bed or when pulmonary metastases give rise to additional tumour emboli. It is important to note, however, that venous drainage and capillary bed proximity alone does not define the sites of metastasis. Currently, it is believed that the specific site for distant metastasis is not simply due to an anatomical location, the primary tumour or proximity to secondary sites, but rather involves interactions between tumour cells and the local microenvironment at the secondary site.
One theory that attempts to explain the selectivity of certain tumour types to metastasize to specific organ sites, for example OS to lung and bone, is the ‘seed and soil hypothesis’ first proposed in 1889 by the surgeon Stephen Paget and then experimentally tested by Fidler in the 1980s (Paget, 1889; Hart and Fiddler, 1980). Paget hypothesized that communication between the cancer cell (seed) and the target organ (soil) via molecular interactions resulted in non-random selection of target organs by the cancer cells. Modulation of the tumour microenvironment (soil) by the tumour cell (seed) is a critical determinant of survival of metastatic cells (Paget, 1889). Mendoza and Khanna (2009) recently suggested in their review of cancer metastasis that a critical outcome of the seed–soil interaction is resistance to the stresses tumour cells encounter that would otherwise impede metastatic progression.
The final critical step in the development of metastasis is the generation of a new blood supply (angiogenesis). Proliferation of endothelial cells is a rigidly controlled balance between proangiogenic factors (e.g. vascular endothelial growth factor [VEGF], fibroblastgrowth factor [FGF] and interleukin [IL]-8) and their antagonists (e.g., thrombospondin-1, maspin and angiostatin) (Slauson and Cooper, 2002; Kumar et al., 2005; Krishnan et al., 2005, 2006). In cancer, this balance is shifted towards proangiogenic factors.
Metastasis, Stress, and Protein Translation
As outlined above, one of the most critical determinants of a tumour cell’s success in metastasis may be its ability to resist the stresses that are associated with progression through the metastatic cascade and survival at secondary sites. Stresses encountered at the secondary site include low oxygen tension (hypoxia), reactive oxygen species, inflammation, nutrient deprivation and changes in pH. These stresses can challenge the growth and progression of a tumour cell and commonly lead to the death of the vast majority of tumour cells that reach a secondary site (Hunter et al., 2008). The ability to detect and overcome these stresses is critical to the metastatic phenotype of cancer. Additionally, it is believed that such stresses may also provide a selective pressure favouring growth of more metastatically ‘fit’ cells (Witz and Levy-Nissenbaum, 2006). The theory that metastatic cells are programmed or can adapt to the numerous stresses experienced during metastatic progression has been reviewed elsewhere (Mendoza and Khanna, 2009).
Within a dynamic process like metastasis, it is attractive to hypothesize that the ability of a metastatic cancer cell to respond and manage these numerous stresses could be accomplished through an efficient mechanism to deliver critical proteins to specific locations in a cell in a rapid manner. Since it is energetically impossible to express all proteins at all times, a premium would be afforded to cells with such protein translational machinery. More generally, there is a growing body of evidence that suggests links between dysregulation of protein synthesis and malignant progression of cancer. Indeed, the development of rapamycin and its structural analogues as inhibitors of the mTOR pathway (and cap-dependent translation) as anticancer drugs is evidence of the potential clinical value of blocking the protein translation machinery.
From the perspective of this review, activity and target modulation of these inhibitors of translation initiation in OS models has been reported (Wan et al., 2005; Paoloni et al., 2010). Clemens and Bommer (1999) have discussed the many ways in which translational control is relevant to the topic of cancer, including how the efficient expression of important proteins involved in cell growth regulation, proliferation or cell death may be controlled at the translational level by changes in the activity of components of the protein synthesis machinery. Furthermore, mutations that lead to changes in the structure of individual mRNAs may alter the rates at which the proteins encoded by these mRNAs are produced. Finally, disruptions in the regulation of signalling pathways that result in a loss of control of the synthesis of growth-promoting proteins (or impair the synthesis of growth-inhibitory or pro-apoptotic proteins) may alter the balance of production of important cellular components (Clemens and Bommer, 1999).
Translation is the second process of protein biosynthesis, which is part of the overall process of gene expression. In translation, mRNA is decoded to produce a polypeptide based on the genetic code. There are three distinct phases of translation including initiation, elongation and termination. In order for translation to proceed there must first be activation. In activation, the correct amino acid is joined to the correct transfer RNA (tRNA). When the tRNA has an amino acid linked to it, it is termed ‘charged’. Once the tRNA has been charged, initiation can begin (Lehninger et al., 1993; Pain, 1996). Initiation involves the small subunit of the ribosome binding to the 5′ end of mRNA with the help of other proteins that assist the process (initiation factors [IFs]) (Pain, 1996). Elongation takes place when the next charged tRNA in line binds to the ribosome along with GTP and an elongation factor (EF). Termination of the polypeptide happens when the A site of the ribosome faces a stop codon (Pain, 1996). Of the three phases, initiation is the most important as it is considered to be rate-limiting in the translation process. Cap-dependent translation initiation is the major translation initiation pathway in eukaryotes. All eukaryotic mRNAs present a 5′ terminal nuclear modification, the cap structure (Gingras et al., 1999). In translation, the cap structure marks the spot where the small ribosomal subunit (40S) is to be recruited.
Important in this recruitment process is the eukaryotic initiation factor 4F (eIF4F) complex. eIF4F is a 3-subunit complex composed of eIF4E, eIF4A and eIF4G (Gingras et al., 1999; Mamane et al., 2004). eIF4G is a scaffolding protein that brings together eIF4F, as it has two binding sites for eIF4A and one binding site for eIF4E, but more importantly, it bridges the mRNA cap (via eIF4E) and the 40S ribosomal subunit (via eIF3) (Gingras et al., 1999; Mamane et al., 2004). eIF3 is associated with the small ribosomal subunit and plays a role in preventing the large ribosomal subunit from binding prematurely (Fig. 1). eIF4A is an ATP-dependent RNA helicase. Finally, eIF4E is the cap-binding protein, which is a critical node in RNA regulation impacting nearly every stage of cell cycle progression (Mamane et al., 2004) Specifically, eIF4E coordinately promotes the mRNA export of several genes involved in the cell cycle.
Fig. 1.
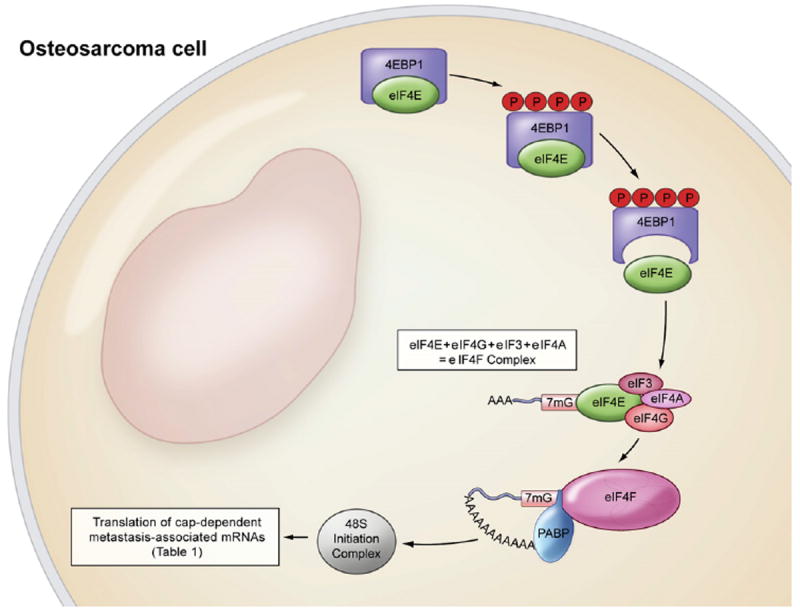
Translational machinery. Under physiological conditions, eIF4E is bound to its inhibitory binding protein 4EBP1. Upon phosphorylation (P) of 4EBP1, eIF4E is liberated from 4EBP1 and able to bind with capped mRNA (7mG) and eIF4G-eIF3-eIF4A subunits, forming the eIF4F complex. eIF4E is the rate-limiting component of the eIF4F complex. eIF4F is a necessary component of the 48S initiation complex. PABP, poly(A) binding protein.
A model describing a hierarchy of weakly and strongly translated proteins in cells has emerged (Baserga, 1990; Graff and Zimmer, 2003). These proteins are differentially regulated by translation. The ‘strongly translated’ proteins are a stable set of ‘less regulated’ proteins with short, unstructured 5′ untranslated regions (UTRs) that are expressed at a basal rate with limited input from external stimuli. In contrast, ‘weakly translated’ proteins typically have lengthy, G–C rich, highly structured 5′UTRs and are maintained as stable mRNA transcripts. These so-called weakly translated proteins are not translated unless the translational machinery is up-regulated by stimuli such as growth factors or cellular stress (Sonenberg and Dever, 2003; Bjornsti and Houghton, 2004; De Benedetti and Graff, 2004). This hierarchy of proteins provides efficiency in biological systems by eliminating the need for indiscriminate, large-scale translation of all proteins at all times and locations in the cell.
The model of weakly and strongly translated proteins suggests that, under specific signals and requirements, specific proteins can be translated and delivered to specialized locations in the cell (Richter and Sonenberg, 2005). For example, when the cell is exposed to stress or growth factors, there is an increased concentration of unbound (free) eIF4E available to bind with the eIF4F complex creating the specifically enhanced translation initiation that the weakly translated proteins require for efficient translation. The end result is disproportionately enhanced translation of weak proteins (De Benedetti and Graff, 2004). Interestingly, several mRNAs with complexity (lengthy, G–C rich, highly structured) in the 5′ UTR (weakly translated proteins) have been associated with cancer (Table 1), including FGF-2 (Nathan et al., 1997a), VEGF (Scott et al., 1998), c-myc (Saito et al., 1983) and cyclin-D1 (Stacey, 2003). Additionally, there are many proto-oncogenes that are regulated at the translational level including c-myc, c-fos and c-jun (De Benedetti and Graff, 2004; Mamane et al., 2004). Such weakly translated and regulated proteins may be ideally suited for rapid expression and delivery to a metastatic cancer cell that is facing a novel stress during metastatic progression.
Table 1.
Cap-dependent metastasis-associated mRNAs
| Function | Metastasis-related gene |
|---|---|
| Cell proliferation | c-Myc |
| CDK2 | |
| Cyclin-D1 | |
| ODC | |
| Angiogenesis | VEGF |
| FGF-2 | |
| PDGF | |
| Anti-apoptotic | Mcl-1 |
| Bcl-2 | |
| Bcl-xL | |
| Survivin | |
| Invasion | MMP-9 |
| Heparanase |
CDK2, cyclin-dependent kinase 2; ODC, ornithine decarboxylase; PDGF, platelet-derived growth factor; Mcl-1, induced myeloid leukemia cell differentiation protein; Bcl-2, B-cell lymphoma 2; Bcl-xL, B-lymphoma × isoform long.
eIF4E is a 25 kDa mRNA cap-binding phosphoprotein (Rhoads et al., 1993; Sonenburg and Gingras, 1998). eIF4E is an important modulator of cell growth and proliferation. It is the least abundant component of the translation initiation machinery (Rhoads et al., 1993). Within translation initiation, the abundance and activation of eIF4E is considered both rate and process limiting (Rhoads et al., 1993; Sonenburg and Gingras, 1998). Numerous studies have now implicated eIF4E in tumour formation and, potentially, in metastatic progression. Overexpression of eIF4E in the cell lines, NIH3T3, CREF and MM3MG has resulted in cellular transformation and tumourigenesis (De Benedetti and Rhoads, 1990; Lazaris-Karatzas et al., 1990; De Benedetti et al., 1994; Li et al., 2001). Antisense RNA-mediated suppression of eIF4E suppressed proliferation and changed cell morphology in HeLa cells (De Benedetti and Rhoads, 1990) and suppressed soft-agar colonization as well as tumour formation and growth in ras-transformed CREF cells (Rinker-Schaeffer et al., 1993). Furthermore, the ability of the ras-transformed CREF cells to invade surrounding normal tissues and metastasize was also markedly reduced (Graff et al., 1995). Expression of antisense RNA to eIF4E in human breast, head and neck cancer cell lines suppressed tumour formation and angiogenesis (Nathan et al., 1997a, b; DeFatta et al., 2000). Finally, functional blockage of eIF4E by expressing 4EBP1 can cause reversion of the transformed and tumourigenic phenotype (Rousseau et al., 1996). Overexpression of eIF4E has been documented in human carcinomas of the breast (Kerekatte et al., 1995; Scott et al., 1998; De Benedetti and Graff, 2004), head and neck (Nathan et al., 1997b; Franklin et al., 1999), bladder (Crew et al., 2000), cervix (Matthews-Greer et al., 2005), lung (Rosenwald et al., 2001; Seki et al., 2002), prostate (Graff et al., 2009) and colon and rectum (Rosenwald et al., 1999; Berkel et al., 2001), as well as in non-Hodgkin’s lymphomas (Wang et al., 1999) when compared with normal tissues and benign lesions.
Collectively, these data suggest that eIF4E may play a key role in both tumour formation and metastatic progression by specifically enhancing the translation of a subset of key genes (weakly translated proteins) necessary for overriding normal growth constraints (c-myc, cyclin-D1), inducing angiogenesis (VEGF, FGF-2) and facilitating tumour invasion and metastasis (MMP-9, heparanase) (Zimmer et al., 2000; Jiang and Muschel, 2002; Yang et al., 2003). eIF4E enables cells to coordinate efficiently the translation of these needed transcripts during metastatic progression, thus increasing success in the demanding process of metastasis. While there has been a wealth of evidence in both experimental cancer models and in human cancer tissues implicating eIF4E in tumour development and progression, the majority of this work has been conducted in epithelial tumours. Expression and activity of eIF4E in mesenchymal tumours, particularly OS, requires further investigation. We have recently reported on the consistent expression of eIF4E in the majority of human OS primary tumours and metastases (Osborne et al., 2011). Ongoing studies have demonstrated the modulation of the metastatic phenotype in OS model systems following interference of eIF4E expression by shRNA targeting. Based on the multiplicity of stresses faced by metastatic cells, the targeting of a common mechanism by which cells can manage these stresses may be a useful novel therapy. Novel therapies that target the translational machinery or novel approaches to the use of approved pharmaceuticals that interfere with protein translation (i.e. rapamycin) should be considered within a metastasis-specific drug development path for OS. Comparative studies of such novel therapies in a number of model systems may assist in the development of such therapies.
Footnotes
Conflict of Interest
The authors of the above manuscript have not declared any conflict of interest that may arise from being named as an author on the manuscript.
References
- Bacci G, Rocca M, Salone M, Balladelli A, Ferrari S, et al. High grade osteosarcoma of the extremities with lung metastases at presentation: treatment with neoadjuvant chemotherapy and simultaneous resection of primary and metastatic lesions. Journal of Surgical Oncology. 2008;98:415–420. doi: 10.1002/jso.21140. [DOI] [PubMed] [Google Scholar]
- Baserga R. The cell cycle: myths and realities. Cancer Research. 1990;50:6769–6771. [PubMed] [Google Scholar]
- Beavon IR. The E-cadherine–catenin complex in tumour metastasis: structure, function and regulation. European Journal of Cancer. 2000;36:1607–1620. doi: 10.1016/s0959-8049(00)00158-1. [DOI] [PubMed] [Google Scholar]
- Berkel H, Turbat-Herrera E, Shi R, De Benedetti A. Expression of the translation initiation factor eIF4E in the polyp-cancer sequence in the colon. Cancer Epidemiology, Biomarkers and Prevalence. 2001;10:663–666. [PubMed] [Google Scholar]
- Berman SD, Calo E, Landman AS, Danielian PS, Miller ES, et al. Metastatic osteosarcoma induced by inactivation of Rb and p53 in the osteoblast lineage. Proceedings of the National Academy of Sciences USA. 2008;105:11851–11856. doi: 10.1073/pnas.0805462105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielack SS, Kempf-Bielack B, Delling G, Exner GU, Flege S, et al. Prognostic factors in high-grade osteosarcoma of the extremities or trunk: an analysis of 1,702 patients treated on neoadjuvant cooperative osteosarcoma study group protocols. Journal of Clinical Oncology. 2002;20:776–790. doi: 10.1200/JCO.2002.20.3.776. [DOI] [PubMed] [Google Scholar]
- Bjornsti MA, Houghton PJ. The TOR pathway:a target for cancer therapy. Nature Reviews Cancer. 2004;4:335–348. doi: 10.1038/nrc1362. [DOI] [PubMed] [Google Scholar]
- Blouin S, Basle MF, Chappard D. Rat models of bone metastases. Clinical and Experimental Metastasis. 2005;22:605–614. doi: 10.1007/s10585-006-9002-5. [DOI] [PubMed] [Google Scholar]
- Clemens MJ, Bommer UA. Translational control: the cancer connection. International Journal of Biochemistry and Cell Biology. 1999;31:1–23. doi: 10.1016/s1357-2725(98)00127-7. [DOI] [PubMed] [Google Scholar]
- Crew J, Fuggle S, Bicknell R, Cranston D, De Benedetti A, et al. Eukaryotic initiation factor-4E in superficial and muscle invasive bladder cancer and its correlation with vascular endothelial growth factor expression and tumour progression. British Journal of Cancer. 2000;82:161–166. doi: 10.1054/bjoc.1999.0894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlin D, Coventry M. Osteogenic osteosarcoma. A study of six hundred cases. Journal of Bone and Joint Surgery. 1967;49:101–110. [PubMed] [Google Scholar]
- De Benedetti A, Graff JR. eIF-4E expression and its role in malignancies and metastases. Oncogene. 2004;23:3189–3199. doi: 10.1038/sj.onc.1207545. [DOI] [PubMed] [Google Scholar]
- De Benedetti A, Joshi B, Graff J, Zimmer S. CHO cells transformed by the translation factor eIF-4E display increased c-myc expression but require overexpression for Max tumorigenicity. Molecular and Cellular Differentiation. 1994;2:347–371. [Google Scholar]
- De Benedetti A, Rhoads RE. Overexpression of eukaryotic protein synthesis initiation factor 4E in HeLa cells results in aberrant growth and morphology. Proceedings of the National Academy of Sciences USA. 1990;87:8212–8216. doi: 10.1073/pnas.87.21.8212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Nigris F, Rossiello R, Schiano C, Arra C, Williams-Ignarro S, et al. Deletion of Yin Yang 1 protein in osteosarcoma cells on cell invasion and CXCR4/angiogenesisand metastasis. Cancer Research. 2008;68:1797–1808. doi: 10.1158/0008-5472.CAN-07-5582. [DOI] [PubMed] [Google Scholar]
- DeFatta RJ, Nathan CO, De Benedetti A. Antisense RNA to eIF4E suppresses oncogenic properties of a head and neck squamous cell carcinoma cell line. Laryngoscope. 2000;110:928–933. doi: 10.1097/00005537-200006000-00007. [DOI] [PubMed] [Google Scholar]
- Douma S, Van Laar T, Zevenhoven J, Meuwissen R, Van Garderen E, et al. Suppression of anoikis and induction of metastasis by the neurotrophic receptor TrkB. Nature. 2004;430:1034–1039. doi: 10.1038/nature02765. [DOI] [PubMed] [Google Scholar]
- Fan TM. Animal models of osteosarcoma. Expert Reviews of Anticancer Therapy. 2010;10:1327–1338. doi: 10.1586/era.10.107. [DOI] [PubMed] [Google Scholar]
- Fidler IJ, Naito S, Pathak S. Orthotopic implantation is essential for the selection, growth and metastasis of human renal cell cancer in nude mice [corrected] Cancer Metastasis Reviews. 1990;9:149–165. doi: 10.1007/BF00046341. [DOI] [PubMed] [Google Scholar]
- Franklin S, Pho T, Abreo F, Nassar R, De Benedetti A, et al. Detection of the proto-oncogene eIF4E in larynx and hypopharynx cancers. Archives of Otolaryngology and Head and Neck Surgery. 1999;125:177–182. doi: 10.1001/archotol.125.2.177. [DOI] [PubMed] [Google Scholar]
- Frisch SM, Francis H. Disruption of epithelial cell–matrix interactions induces apoptosis. Journal of Cell Biology. 1994;124:619–626. doi: 10.1083/jcb.124.4.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu XY, Besterman JM, Monosov A, Hoffman RM. Models of human metastatic colon cancer in nude mice orthotopically constructed by using histologically intact patient specimens. Proceedings of the National Academy of Sciences USA. 1991;88:9345–9349. doi: 10.1073/pnas.88.20.9345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingras A, Raught B, Sonenberg N. eIF4 initiation factors: effectors of mRNA recruitment to ribosomes and regulators of translation. Annual Reviews of Biochemistry. 1999;68:913–963. doi: 10.1146/annurev.biochem.68.1.913. [DOI] [PubMed] [Google Scholar]
- Graff JR, Boghaert ER, De Benedetti A, Tudor DL, Zimmer CC, et al. Reduction of translation initiation factor 4E decreases the malignancy of rastransformed cloned rat embryo fibroblasts. International Journal of Cancer. 1995;60:255–263. doi: 10.1002/ijc.2910600221. [DOI] [PubMed] [Google Scholar]
- Graff JR, Konicek BW, Lynch RL, Dumstorf CA, Dowless MS, et al. eIF4E activation is commonly elevated in advanced human prostate cancers and significantly related to reduced patient survival. Cancer Research. 2009;69:3866–3873. doi: 10.1158/0008-5472.CAN-08-3472. [DOI] [PubMed] [Google Scholar]
- Graff JR, Zimmer SG. Translational control and metastatic progression: enhanced activity of the mRNA cap-binding protein eIF-4E selectively enhances translation of metastasis-related mRNAs. Clinical and Experimental Metastasis. 2003;20:265–273. doi: 10.1023/a:1022943419011. [DOI] [PubMed] [Google Scholar]
- Griffiths G, Grundl M, Leychenko A, Reiter S, Young-Robbins S, et al. Bit-1 mediates integrin-dependent cell survival through activation of the NFkappaB pathway. Journal of Biological Chemistry. 2011;286:14713–14723. doi: 10.1074/jbc.M111.228387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossmann J. Molecular mechanisms of ‘detachment-induced apoptosis–Anoikis’. Apoptosis. 2002;7:247–260. doi: 10.1023/a:1015312119693. [DOI] [PubMed] [Google Scholar]
- Guo Y, Rubin E, Xie J, Zi X, Hoang B. Dominant negative LRP5 decreases tumorigenicity and metastasis of osteosarcoma in an animal model. Clinical Orthopaedic Related Research. 2008;466:2039–2045. doi: 10.1007/s11999-008-0344-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurney JG, Swensen AR, Bulterys M. Malignant Bone Tumors. Bethesda: 1999. pp. 99–110. NIH Publication Number 99-4649. [Google Scholar]
- Hansen MF. Molecular genetic considerations in osteosarcoma. Clinical Orthopaedic Related Research. 1991;270:237–246. [PubMed] [Google Scholar]
- Hart I, Fiddler I. Role of organ selectivity in the determination of metastatic patterns of B16 melanoma. Cancer Research. 1980;40:2281–2287. [PubMed] [Google Scholar]
- Hattinger C, Pasello M, Ferrari S, Picci P, Serra M. Emerging drugs for high-grade osteosarcoma. Expert Opinion in Emerging Drugs. 2010;15:615–634. doi: 10.1517/14728214.2010.505603. [DOI] [PubMed] [Google Scholar]
- Hayden JB, Hoang BH. Osteosarcoma: basic science and clinical implications. Orthopaedic Clinics of North America. 2006;37:1–7. doi: 10.1016/j.ocl.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Hoffman RM. Clinically accurate orthotopic mouse models of cancer. Methods in Molecular Medicine. 2001;58:251–275. doi: 10.1385/1-59259-137-X:251. [DOI] [PubMed] [Google Scholar]
- Hunter KW. Ezrin, a key component in tumor metastasis. Trends in Molecular Medicine. 2004;10:201–204. doi: 10.1016/j.molmed.2004.03.001. [DOI] [PubMed] [Google Scholar]
- Hunter KW, Crawford NP, Alsarraj J. Mechanisms of metastasis. Breast Cancer Research. 2008;10(Suppl. 11):S12. doi: 10.1186/bcr1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huvos AG, Rosen G, Marcove RC. Primary osteogenic osteosarcoma: pathogenic aspects in 20 patients after treatment with chemotherapy, en bloc resection, and prosthetic bone replacement. Archives in Pathology and Laboratory Medicine. 1977;101:14–18. [PubMed] [Google Scholar]
- Jiang Y, Muschel RJ. Regulation of matrix metalloproteinase-9 (MMP-9) by translational efficiency in murine prostate carcinoma cells. Cancer Research. 2002;62:1910–1914. [PubMed] [Google Scholar]
- Kansara M, Thomas DM. Molecular pathogenesis of osteosarcoma. DNA and Cell Biology. 2007;26:1–18. doi: 10.1089/dna.2006.0505. [DOI] [PubMed] [Google Scholar]
- Kerekatte V, Smiley K, Hu B, Smith A, Gelder F, et al. The proto-oncogene/translation factor eIF4E: a survey of its expression in breast carcinomas. International Journal of Cancer. 1995;64:27–31. doi: 10.1002/ijc.2910640107. [DOI] [PubMed] [Google Scholar]
- Khanna C. Novel targets with potential therapeutic applications in osteosarcoma. Current Oncology Reports. 2008;10:350–358. doi: 10.1007/s11912-008-0054-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna C, Prehn J, Yeung C, Caylor J, Tsokos M, et al. An orthotopic model of murine osteosarcoma with clonally related variants differing in pulmonary metastatic potential. Clinical and Experimental Metastasis. 2000;18:261–271. doi: 10.1023/a:1006767007547. [DOI] [PubMed] [Google Scholar]
- Krishnan K, Bruce B, Hewitt S, Thomas D, Khanna C, et al. Ezrin mediates growth and survival in Ewing’s sarcoma through the AKT/mTOR, but not the MAPK, signaling pathway. Clinical and Experimental Metastasis. 2006;23:227–236. doi: 10.1007/s10585-006-9033-y. [DOI] [PubMed] [Google Scholar]
- Krishnan K, Khanna C, Helman LJ. The biology of metastases in pediatric sarcomas. Cancer Journal. 2005;11:306–313. doi: 10.1097/00130404-200507000-00006. [DOI] [PubMed] [Google Scholar]
- Kumar V, Abbas A, Fausto N. Robbins’ and Cotran’s Pathologic Basis of Disease. Elsevier Saunders; Philadelphia: 2005. pp. 309–313. [Google Scholar]
- Lazaris-Karatzas A, Montine KS, Sonenberg N. Malignant transformation by a eukaryotic initiation factor subunit that binds to mRNA 5′ cap. Nature. 1990;345:544–547. doi: 10.1038/345544a0. [DOI] [PubMed] [Google Scholar]
- Lehninger A, Nelson D, Cox M. Principles of Biochemistry. Worth Publishers; New York: 1993. pp. 905–923. [Google Scholar]
- Li Y, DeFatta R, Anthony C, Sunavala G, De Benedetti A. A translationally regulated tousled kinase phosphorylates histone H3 and confers radioresistance when overexpressed. Oncogene. 2001;20:726–738. doi: 10.1038/sj.onc.1204147. [DOI] [PubMed] [Google Scholar]
- Luzzi KJ, MacDonald IC, Schmidt EE, Kerkvliet N, Morris VL, et al. Multistep nature of metastatic inefficiency: dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. American Journal of Pathology. 1998;153:865–873. doi: 10.1016/S0002-9440(10)65628-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malawar M, Helman L, O’Sullivan B. In: Cancer Principles and Practice of Oncology. De Vita V, Hellman S, Rosenburg SA, editors. Lippincott Williams & Wilkins; Philadelphia: 2005. pp. 1638–1686. [Google Scholar]
- Mamane Y, Petroulakis E, Rong L, Yoshida K, Ler LW, et al. eIF4E efrom translation to transformation. Oncogene. 2004;23:3172–3179. doi: 10.1038/sj.onc.1207549. [DOI] [PubMed] [Google Scholar]
- Matthews-Greer J, Caldito G, De Benedetti A, Herrera G, Dominguez-Malagon H, et al. eIF4E as a marker for cervical neoplasia. Applied Immunohistochemistry and Molecular Morphology. 2005;13:367–370. doi: 10.1097/01.pai.0000170625.98446.3e. [DOI] [PubMed] [Google Scholar]
- Mendoza M, Khanna C. Revisiting the seed and soil in cancer metastasis. International Journal of Biochemistry and Cell Biology. 2009;41:1452–1462. doi: 10.1016/j.biocel.2009.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers PA. Muramyl tripeptide (mifamurtide) for the treatment of osteosarcoma. Expert Reviews of Anticancer Therapy. 2009;9:1035–1049. doi: 10.1586/era.09.69. [DOI] [PubMed] [Google Scholar]
- Nathan CA, Carter P, Liu L, Li BD, Abreo F, et al. Elevated expression of eIF4E and FGF-2 isoforms during vascularization of breast carcinomas. Oncogene. 1997a;15:1087–1094. doi: 10.1038/sj.onc.1201272. [DOI] [PubMed] [Google Scholar]
- Nathan CA, Liu L, Li BD, Abreo FW, Nandy I, et al. Detection of the proto-oncogene eIF4E in surgical margins may predict recurrence in head and neck cancer. Oncogene. 1997b;15:579–584. doi: 10.1038/sj.onc.1201216. [DOI] [PubMed] [Google Scholar]
- Osborne T, Ren L, Healey JH, Shapiro LQ, Chou AJ, et al. Evaluation of eIF4E expression in an osteosarcoma-specific tissue microarray. Journal of Pediatric Hematology and Oncology. 2011;33:524–528. doi: 10.1097/MPH.0b013e318223d0c1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paget S. The distribution of secondary growths in cancer of the breast. Lancet. 1889;1:571–573. [PubMed] [Google Scholar]
- Pain VM. Initiation of protein synthesis in eukaryotic cells. European Journal of Biochemistry. 1996;236:747–771. doi: 10.1111/j.1432-1033.1996.00747.x. [DOI] [PubMed] [Google Scholar]
- Paoloni M, Davis S, Lana S, Withrow S, Sangiorgi L, et al. Canine tumor cross-species genomics uncovers targets linked to osteosarcoma progression. BMC Genomics. 2009;10:625. doi: 10.1186/1471-2164-10-625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoloni M, Khanna C. Translation of new cancer treatments from pet dogs to humans. Nature Reviews Cancer. 2008;8:147–156. doi: 10.1038/nrc2273. [DOI] [PubMed] [Google Scholar]
- Paoloni MC, Mazcko C, Fox E, Fan T, Lana S, et al. Rapamycin pharmacokinetic and pharmacodynamic relationships in osteosarcoma: a comparative oncology study in dogs. PLoS One. 2010;5:e11013. doi: 10.1371/journal.pone.0011013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posthuma-DeBoer J, Witlox M, Kaspers G, van Royen B. Molecular alterations as target for therapy in metastatic osteosarcoma: a review of literature. Clinical and Experimental Metastasis. 2011;28:493–503. doi: 10.1007/s10585-011-9384-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhoads RE, Joshi-Barve S, Rinker-Schaeffer C. Mechanism of action and regulation of protein synthesis initiation factor 4E: effects on mRNA discrimination, cellular growth rate, and oncogenesis. Progress in Nucleic Acid Research and Molecular Biology. 1993;46:183–219. doi: 10.1016/s0079-6603(08)61022-3. [DOI] [PubMed] [Google Scholar]
- Richter JD, Sonenberg N. Regulation of cap-dependent translation by eIF4E inhibitory proteins. Nature. 2005;433:477–480. doi: 10.1038/nature03205. [DOI] [PubMed] [Google Scholar]
- Rinker-Schaeffer CW, Graff JR, De Benedetti A, Zimmer SG, Rhoads RE. Decreasing the level of translation initiation factor 4E with antisense RNA causes reversal of ras-mediated transformation and tumorigenesis of cloned rat embryo fibroblasts. International Journal of Cancer. 1993;55:841–847. doi: 10.1002/ijc.2910550525. [DOI] [PubMed] [Google Scholar]
- Rosenwald I, Chen J, Wang S, Savas L, London I, et al. Upregulation of protein synthesis initiation factor eIF-4E is an early event during colon carcinogenesis. Oncogene. 1999;18:2507–2517. doi: 10.1038/sj.onc.1202563. [DOI] [PubMed] [Google Scholar]
- Rosenwald I, Hutzler M, Wang S, Savas L, Fraire A. Expression of eukaryotic translation initiation factors 4E and 2alpha is increased frequently in bronchioloalveolar but not in squamous cell carcinomas of the lung. Cancer. 2001;92:2164–2171. doi: 10.1002/1097-0142(20011015)92:8<2164::aid-cncr1559>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- Rousseau D, Kaspar R, Rosenwald I, Gehrke L, Sonenberg N. Translation initiation of ornithine decarboxylase and nucleocytoplasmic transport of cyclin D1 mRNA are increased in cells overexpressing eukaryotic initiation factor 4E. Proceedings of the National Academy of Sciences USA. 1996;93:1065–1070. doi: 10.1073/pnas.93.3.1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruoslahti E, Reed JC. Anchorage dependence, integrins, and apoptosis. Cell. 1994;77:477–478. doi: 10.1016/0092-8674(94)90209-7. [DOI] [PubMed] [Google Scholar]
- Saito H, Hayday AC, Wiman K, Hayward WS, Tonegawa S. Activation of the c-myc gene by translocation: a model for translational control. Proceedings of the National Academy of Sciences USA. 1983;80:7476–7480. doi: 10.1073/pnas.80.24.7476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer B, Henderson N. The cyclization of linear DNA in Escherichia coli by site-specific recombination. Gene. 1988;70:331–341. doi: 10.1016/0378-1119(88)90205-3. [DOI] [PubMed] [Google Scholar]
- Scott P, Smith K, Poulsom R, De Benedetti A, Bicknell R, et al. Differential expression of vascular endothelial growth factor mRNA vs protein isoform expression in human breast cancer and relationship to eIF-4E. British Journal of Cancer. 1998;77:2120–2128. doi: 10.1038/bjc.1998.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki N, Takasu T, Mandai K, Nakata M, Saeki H, et al. Expression of eukaryotic initiation factor 4E in atypical adenomatous hyperplasia and adenocarcinoma of the human peripheral lung. Clinical Cancer Research. 2002;8:3046–3053. [PubMed] [Google Scholar]
- Slauson D, Cooper B. Mechanisms of Disease: a Textbook of Comparative General Pathology. Mosby; St Louis: 2002. pp. 341–353. [Google Scholar]
- Sonenberg N, Dever T. Eukaryotic translation initiation factors and regulators. Current Opinions in Structural Biology. 2003;13:56–63. doi: 10.1016/s0959-440x(03)00009-5. [DOI] [PubMed] [Google Scholar]
- Sonenburg N, Gingras A. The mRNA 5′ cap-binding protein eIF4E and control of cell growth. Current Opinions in Cell Biology. 1998;10:268–275. doi: 10.1016/s0955-0674(98)80150-6. [DOI] [PubMed] [Google Scholar]
- Stacey D. Cyclin D1 serves as a cell cycle regulatory switch in actively proliferating cells. Current Opinions in Cell Biology. 2003;15:158–163. doi: 10.1016/s0955-0674(03)00008-5. [DOI] [PubMed] [Google Scholar]
- Vail DM, MacEwen EG. Spontaneously occurring tumors of companion animals as models for human cancer. Cancer Investigations. 2000;18:781–792. doi: 10.3109/07357900009012210. [DOI] [PubMed] [Google Scholar]
- Walkley CR, Qudsi R, Sankaran VG, Perry JA, Gostissa M, et al. Conditional mouse osteosarcoma, dependent on p53 loss and potentiated by loss of Rb, mimics the human disease. Genes and Development. 2008;22:1662–1676. doi: 10.1101/gad.1656808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan X, Mendoza A, Khanna C, Helman LJ. Rapamycin inhibits ezrin-mediated metastatic behavior in a murine model of osteosarcoma. Cancer Research. 2005;65:2406–2411. doi: 10.1158/0008-5472.CAN-04-3135. [DOI] [PubMed] [Google Scholar]
- Wang LL. Biology of osteogenic sarcoma. Cancer Journal. 2005;11:294–305. doi: 10.1097/00130404-200507000-00005. [DOI] [PubMed] [Google Scholar]
- Wang S, Rosenwald I, Hutzler M, Pihan G, Savas L, et al. Expression of the eukaryotic translation initiation factors 4E and 2alpha in non-Hodgkin’s lymphomas. American Journal of Pathology. 1999;155:247–255. doi: 10.1016/s0002-9440(10)65118-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss L, Nannmark U, Johansson BR, Bagge U. Lethal deformation of cancer cells in the microcirculation: a potential rate regulator of hematogenous metastasis. International Journal of Cancer. 1992;50:103–107. doi: 10.1002/ijc.2910500121. [DOI] [PubMed] [Google Scholar]
- Weiss L, Orr F, Honn K. Interactions of cancer cells with the microvasculature during metastasis. FASEB Journal. 1988;2:12–21. doi: 10.1096/fasebj.2.1.3275560. [DOI] [PubMed] [Google Scholar]
- Whelan J, Weeden S, Uscinska B, McTiernan A. Localized extremity osteosarcoma: mature survival data from two European osteosarcoma intergroup randomised clinical trials. Proceedings of the American Society of Clinical Oncology. 2000;19:1281a. [Google Scholar]
- Withrow SJ, Khanna C. Bridging the gap between experimental animals and humans in osteosarcoma. Cancer Treatment and Research. 2009;152:439–446. doi: 10.1007/978-1-4419-0284-9_24. [DOI] [PubMed] [Google Scholar]
- Withrow SJ, Wilkins RM. Cross talk from pets to people: translational osteosarcoma treatments. ILAR Journal. 2010;51:208–213. doi: 10.1093/ilar.51.3.208. [DOI] [PubMed] [Google Scholar]
- Witz IP, Levy-Nissenbaum O. The tumor microenvironment in the post-PAGET era. Cancer Letters. 2006;242:1–10. doi: 10.1016/j.canlet.2005.12.005. [DOI] [PubMed] [Google Scholar]
- Yang L, Kuang LG, Zheng HC, Li JY, Wu DY, et al. PTEN encoding product: a marker for tumorigenesis and progression of gastric carcinoma. World Journal of Gastroenterology. 2003;9:35–39. doi: 10.3748/wjg.v9.i1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer SG, DeBenedetti A, Graff JR. Translational control of malignancy: the mRNA cap-binding protein, eIF-4E, as a central regulator of tumor formation, growth, invasion and metastasis. Anticancer Research. 2000;20:1343–1351. [PubMed] [Google Scholar]