

SUMMARY
The dehydratases (DHs) of modular polyketide synthases (PKSs) catalyze dehydrations that occur frequently in the biosynthesis of complex polyketides, yet little is known about them structurally or mechanistically. Here, the structure of a DH domain, isolated from the fourth module of the erythromycin PKS, is presented at 1.85 Å resolution. As with the DH of the highly related animalian fatty acid synthase (FAS), the DH monomer possesses a double hotdog fold. Two symmetry mates within the crystal lattice make a contact that likely represents the DH dimerization interface within an intact PKS. Conserved hydrophobic residues on the DH surface indicate potential interfaces with two other PKS domains, the ketoreductase (KR) and the acyl carrier protein (ACP). Mutation of an invariant arginine at the hypothesized ACP docking site in the context of the erythromycin PKS resulted in decreased production of the erythromycin precursor 6-deoxyerythronolide B. The structure elucidates how the α-hydrogen and β-hydroxyl group of a polyketide substrate interact with the catalytic histidine and aspartic acid in the DH active site prior to dehydration.
Keywords: Polyketide, polyketide synthase, fatty acid synthase, dehydratase, erythromycin
INTRODUCTION
Polyketides are a diverse class of secondary metabolites, most of which possess biological activity and many of which have become important human pharmaceuticals. Complex polyketides, such as the antibiotic erythromycin, the immunosuppressant rapamycin, and the anticancer agent epothilone, are assembled by modular polyketide synthases (PKSs)1–4. Each module within these megasynthases adds a ketide unit to the growing polyketide chain. Thus, the six modules of the well-studied erythromycin PKS cooperate in the assembly line synthesis of the erythromycin aglycone, 6-deoxyerythronolide B (6-dEB)(Figure 1A). Modules are composed of several enzymes: an acyltransferase (AT) that selects the organic building blocks that are condensed with the growing polyketide chain, a ketosynthase (KS) that controls the condensation event, a ketoreductase (KR) that reduces the β-carbonyl formed by the condensation, a dehydratase (DH) that creates a double bond through an elimination reaction, and an enoylreductase (ER) that reduces this double bond to yield a methylene group (KR, DH, and ER are referred to as β-carbon processing enzymes). Modules also contain an acyl carrier protein (ACP) that uses an ~18 Å phosphopantetheinyl arm to covalently shuttle the growing polyketide between the enzymes.
Figure 1.
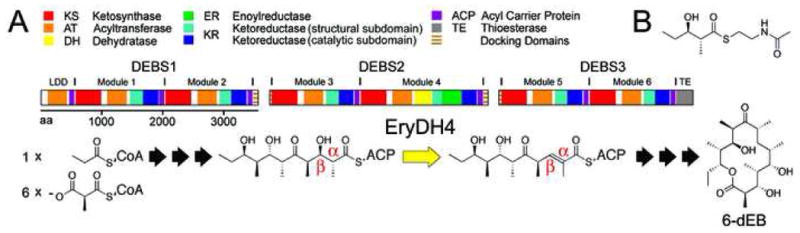
The Erythromycin PKS DH. A) Three polypeptides, DEBS1-3, contain the six modules of the erythromycin PKS, which synthesize 6-deoxyerythronolide B (6-dEB) from one molecule of propionyl-CoA and six molecules of (2S)-methylmalonyl-CoA in assembly line fashion. The dehydratase, EryDH4, catalyzes the only dehydration reaction in the synthesis to yield an intermediate containing a trans- double bond. B) Isolated from DEBS2, EryDH4 did not dehydrate the synthetic substrate mimic, (2R,3R)-2-methyl-3-hydroxypentanoate N-acetylcysteamine thioester.
Recent structural advances on PKSs and fatty acid synthases (FASs) have enabled intermediate- and high-resolution descriptions of components of these highly related megasynthases (both FASs and PKSs can be classified as type I and type II depending on whether component enzymes are fused within large polypeptides or contained within separate polypeptides, respectively; type I is implied when not explicitly stated)4. The 3.1 Å Saccharomyces cerevisiae FAS structure shows that the fungal FAS has a different overall architecture than PKSs and animalian FASs and will not be further discussed5. The 4.5 Å resolution structure of the porcine FAS reveals its head-to-head homodimeric architecture and the relative locations of its domains, except for ACP and thioesterase (TE)6. The atomic resolution details of the KS+AT didomain, KR, and ACP have been obtained from fragments of PKS modules7–11; TEs from PKSs and the human FAS have also been characterized12,13. High-resolution structures are necessary to determine the details of megasynthase architecture and the catalytic mechanisms of constituent enzymes.
The atomic details of DH, the central domain of animalian FASs and most PKS modules, are highly anticipated, as even the boundaries of animalian FAS DHs and PKS DHs have not yet been determined. What is known about DH comes from recent structural and mutagenic studies on the animalian FAS: DH possesses a double hotdog fold; DH makes homodimeric contact across the twofold axis of the synthase; DH contacts KR and possibly ER; and the segment connecting the KS+AT didomain to DH is flexible6,14.
The dehydration mechanism of animalian FAS DHs and PKS DHs is hypothesized to be similar to that of type II FAS dehydratases, such as FabA and FabZ; however, the reactions performed by PKS DHs are more mysterious15,16. In contrast to animalian FAS DHs, PKS DHs often operate on acyl chains with methyl, ethyl, or methoxy α-substituents. How PKS DHs accommodate these groups is unclear. PKS DHs can also catalyze the formation of cis- double bonds, which indicates that the dehydrating PKS DH needs to accommodate a product with a very different geometry than the more common trans- product17,18.
Here, the structure of DH from the fourth module of the erythromycin PKS (EryDH4; this nomenclature describes a fragment by its PKS and module) is presented at 1.85 Å resolution. Dimers of the EryDH4 domain are apparently formed within the crystal, although the observed conformation is ~60° straighter than the DH dimer of the porcine FAS structure. The monomers possess a double hotdog fold, in which the catalytic histidine is supplied from one of the halves and the catalytic aspartic acid is supplied from the other. The N-termini are located near the twofold axis and reveal the location of the short, unconserved linkers that connect the KS+AT and DH domains. The DH structure does not show any obvious surfaces that interface with KS or ER; however, surfaces comprised of conserved hydrophobic residues indicate potential interfaces with KR and ACP. Mutagenesis of an invariant arginine at the hypothesized ACP docking site in the context of the erythromycin PKS resulted in reduced polyketide production. The dehydration reactions that yield the common trans- double bond and the rare cis- double bond are discussed. A comparison of the PKS DH domain and the animalian FAS DH domain is also presented.
RESULTS
DH Fragment Characterization
The KS+AT and KR structures were used to predict the boundaries for DH7–10. H2362 was chosen as the N-terminus as it immediately follows the KS+AT didomain, and G2653 was chosen as the C-terminus as it immediately precedes the KR domain. The purified DH fragment migrates at ~50 kDa in gel filtration studies, between the expected monomer and dimer molecular weights (33 kDa and 66 kDa)(Supplementary Figure 1).
The natural substrate of EryDH4 is (2R,3R,4R,6R,7S,8S,9R)-3,7,9-trihydroxy-5-oxo-2,4,6,8-tetramethylundecanoate holo-EryACP4 thioester(Figure 1A). As this substrate is difficult to obtain, a simplified substrate, (2R,3R)-3-hydroxy-2-methylpentanoate N-acetylcysteamine thioester, was used to assay EryDH4 for activity (an equivalent substrate lacking the α-methyl substituent is dehydrated, albeit slowly, by the rat FAS DH)(Figure 1B)14. No dehydrated product was observed by LC/MS analysis.
Overall Structure
The EryDH4 monomer possesses a double hotdog fold(Figure 2). This fold is a variation of the hotdog fold, which derives its name from its resemblance of a bun holding a sausage (a curved β-sheet surrounding an α-helix). Many type II dehydratases, such as FabA and FabZ, possess a hotdog fold and dimerize to resemble a double hotdog – a large bun holding two sausages15,16. When an enzyme constructed from a single polypeptide folds in this manner it is said to possess a double hotdog fold, as first observed in thioesterase II from Escherichia coli19. Although EryDH4 has a double hotdog fold, structurally it is more similar to a type II dehydratase dimer than a thioesterase II monomer.
Figure 2.
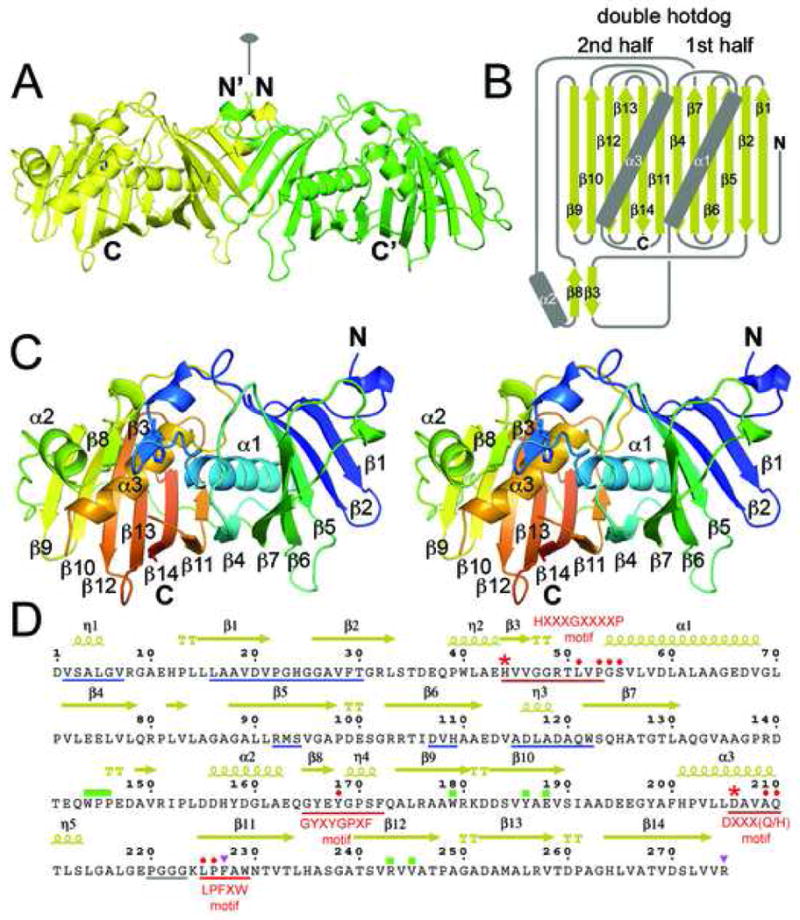
DH Structure. A) The EryDH4 dimer (one monomer is colored green for clarity) observed in the crystal lattice, showing the location the dimer interface along the twofold axis. The catalytic histidine of one monomer is represented in sticks. B) Schematic of the DH double hotdog fold (α-helices are the sausages and the β-sheet is the bun). Together, α2, β3, and β8 comprise a structural element that harbors the catalytic histidine. C) Stereodiagram of an EryDH4 monomer indicating elements of secondary structure. The protein is colored from N-terminus to C-terminus (blue to red). The catalytic histidine is represented in sticks. D) Important DH residues. Hallmark DH motifs, such as HXXXGXXXXP, are underlined in red. The catalytic histidine and aspartic acid are marked by red asterisks. Supporting active site residues are marked by red circles. Regions supporting dimerization are underlined in blue. An unobserved loop is underlined in gray. The “WPP” motif and interacting residues are marked by green rectangles. Residues hypothesized to contact ACP are marked by purple triangles. Residues indicated by “TT” help form β-turns.
The putative dimer interface observed between EryDH4 monomers in the crystal lattice buries a surface area of 1040 Å2 per monomer (9% of the monomer surface) and is formed by residues in the first half of the double hotdog: η1, a β-hairpin turn between β1 and β2, residues on the exteriors of the hotdog buns, and residues surrounding η3(Figure 3). The equivalent surface is observed to form the dimerization interface of the porcine FAS DH dimer; however, the angle between the EryDH4 monomers is ~60° greater (the angle is defined as the intersection of the lines connecting the midpoint of β1 and the C-terminal end of α3 in each monomer)(Figure 3A, 3F)6. Several small residues in η1 make hydrophobic interactions across the twofold axis(Figure 3B). These contacts are enabled through a turn made by the nearly invariant glycine, G6 (numbering is based on the first observable residue; to obtain correct numbering add 2,363). Situated at the β-hairpin turn between β1 and β2, P22 is positioned to fill a cavity in the opposite monomer formed by R92, H109, and W122(Figure 3C). Also present at the interface are two chloride ions.
Figure 3.
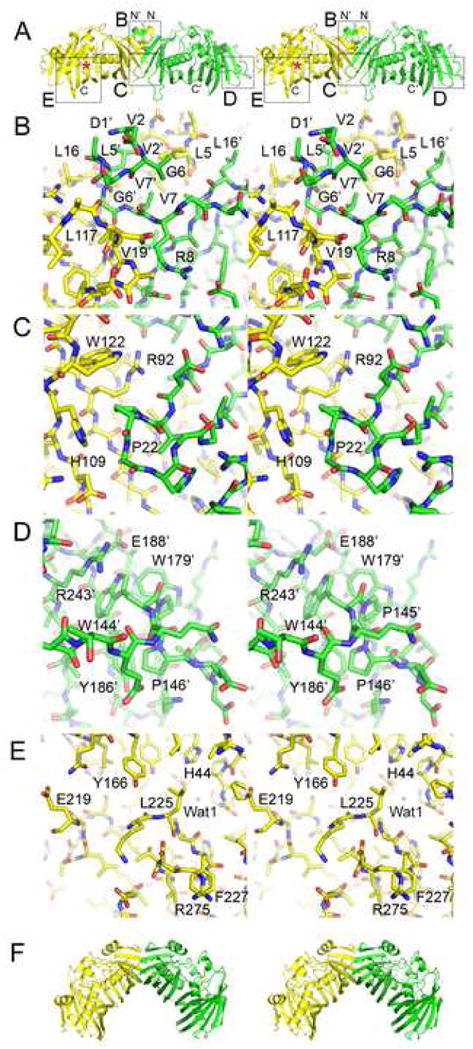
DH Features. A) Stereodiagram of the observed EryDH4 dimer. The N-termini are located adjacent to the dimer interface. The active site is marked with an asterisk. Regions highlighted in subsequent panels are indicated by boxes. B) The dimer interface near the N-termini is mediated by η1 through hydrophobic contacts across the twofold axis. G6 is nearly invariant. C) A cavity at the dimer interface is filled by P22′. D) The “WPP” motif helps guide the 25-residue loop between β7 and α2 that connects the halves of the double hotdog. E) The last observable DH residue, R275, is invariant and may help form an ACP docking site, along with the highly conserved surface residue F227. A loop adjacent to the active site (P220-G223) is invisible in the electron density maps. F) The DH dimer configuration observed in the porcine FAS structure is bent ~60° relative to the observed EryDH4 dimer.
The pseudosymmetric halves of the DH monomer are connected by a 25-residue loop similar to those observed in double hotdog enzymes thioesterase II and 2-enoyl-CoA hydratase 219,20. A “WPP” motif (W144, P145, and P146) in the middle of the loop makes a tight interaction with the body of the enzyme(Figure 3D). W144 stacks with R243, hydrogen bonds with E188, and makes hydrophobic interactions with W179, Y186, E188, R243, and V245; P145 and P156 make hydrophobic interactions with W179 and W186, respectively. Regions from both halves assemble to form a structured element (distinct from the double hotdog fold) that helps create the active site. The first half contributes β3, containing the catalytic histidine, H44, while the second half contributes α2 and β8. These elements combine with α3 to build the active site around the second half of the double hotdog(Figure 3E).
The DH domain is recognizable in PKS sequences by its signature HXXXGXXXXP motif near its N-terminal boundary, in which the catalytic histidine is invariant and both the glycine and proline are nearly invariant(Figure 2D)21. The structure reveals that the glycine helps makes a turn to enable the histidine and proline to make contact. The catalytic histidine, H44, is oriented both through van der Waals contact with P53 and through a hydrogen bond to the backbone carbonyl of L51(Figure 4A). The catalytic aspartic acid, D206, is positioned through a hydrogen bond with the sidechain NH2 of Q210. While glutamic acid serves as the general acid in type II dehydratases, this role is always assumed by an aspartic acid in type I PKS DHs. The supporting glutamine, Q210 (usually substituted by a histidine residue), is in turn anchored through a hydrogen bond to a nearly invariant tyrosine, Y158. The second tyrosine of the GYXYGPXF motif, Y168, is positioned immediately adjacent to the catalytic histidine and aspartic acid. L225, P226, and an unobservable loop (P220-G223) also surround the active site. Water Wat1 is bound by the S55 NH and the D206 side chain and may be in a special location: it is hypothesized in FabA to be where the water removed from the acyl chain remains bound to the enzyme15.
Figure 4.
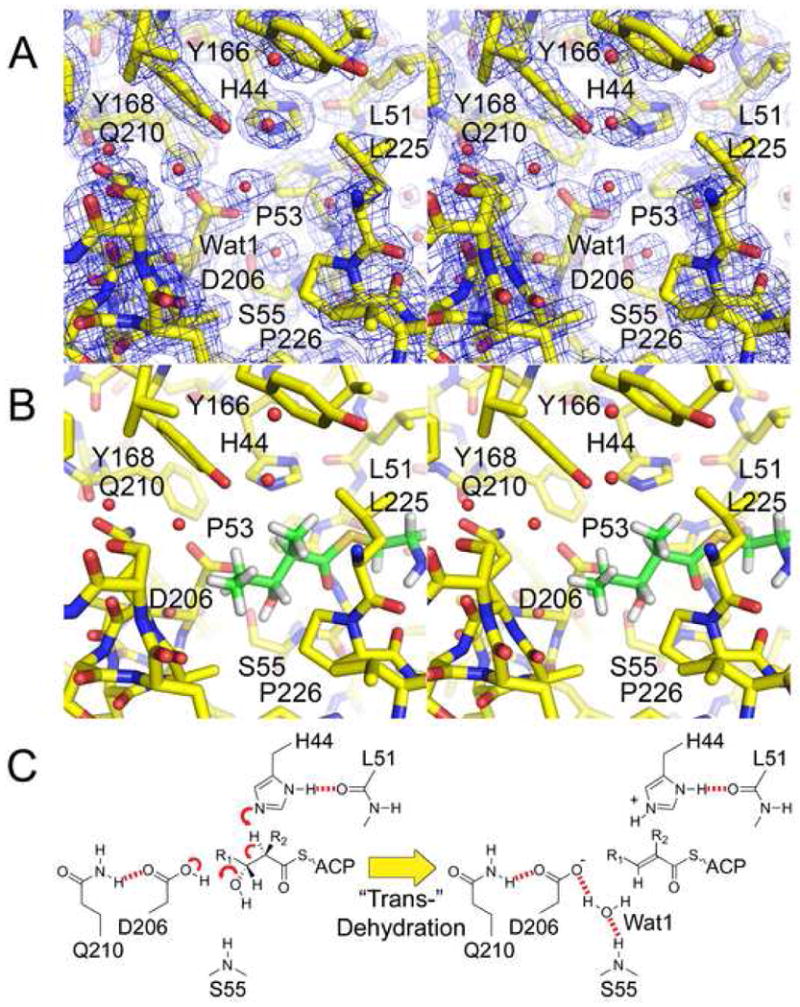
The DH Reaction. A) Stereodiagram of the 1.85 Å resolution 2Fo−Fc electron density map at the active site (contoured at 1.4 σ). The catalytic histidine, H44, and the catalytic aspartic acid, D206, are oriented by the L51 backbone carbonyl and the Q210 side chain, respectively. B) A diketide substrate mimic is modeled in the active site so that its α-hydrogen and β-hydroxyl groups are adjacent to H44 and D206, respectively. The β-hydroxyl group may partially replace Wat1 and be attracted towards the backbone NH of S55. C) Putative mechanism for the most common elimination, which yields a trans- double bond. In cis-dehydration (e.g. RifDH10) the β-hydroxyl group may remain in the same position; however, the R1 acyl chain and the β-hydrogen would be swapped.
Polyketide Substrate Modeled in the Active Site
As a mimic of polyketide substrates dehydrated by DHs, (2R,3R)-2-methyl-3-hydroxypentanoate N-acetylcysteamine thioester was modeled into the active site using the program Coot (the stereochemistries at the α- and β-positions are representative of substrates that are dehydrated to yield a trans- double bond)(Figure 1B, 4B, 4C)22,10,23,24. The α-hydrogen was positioned adjacent to the His44 Nε2, and the β-hydroxyl group was positioned adjacent to the D206 carboxylic acid, as close as possible to the location of Wat1 in the structure. The thioester carbonyl was also positioned to hydrogen bond to the backbone NH of G54. Thus, the orientation of the modeled substrate is similar to the covalently-trapped substrate in the FabA:3-decynoyl-N-acetylcysteamine complex15.
DH Interfaces and Connections
The porcine FAS structure reveals that DH is centrally located within the synthase and may contact the KS+AT didomain, KR, and ER(Figure 5A)6. The interaction between DH and KR appears to be one of the largest. To obtain a more detailed model of the potential PKS DH/KR interface, a PKS DH (the EryDH4 monomer) and a PKS KR (EryKR1; PDB Code: 2FR0) were superposed onto the equivalent domains within the porcine FAS structure using Coot(Figure 5B).
Figure 5.
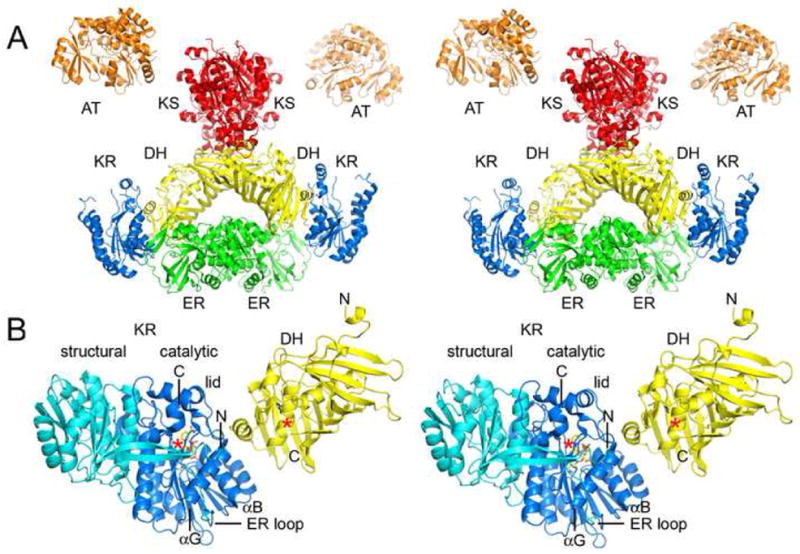
Domain Interfaces. A) Stereodiagram of the porcine FAS structure (obtained by Ban and coworkers by fitting the observed 4.5 Å resolution electron density map with solved type II enzymes)6. The structure indicates DH may contact KS, KR, and ER, and that DH monomers make contact across the twofold axis. B) PKS DH and KR domains (EryDH4 and EryKR1) were superposed on the equivalent porcine FAS domains to yield a more detailed model of the DH/KR interface. When ER is present, it inserts into the “ER loop” and makes contact across the twofold axis. The C-terminus of the DH fragment is ~10 Å from the N-terminus of the KR fragment. ACP is connected to the C-terminus of KR and would need to move to present polyketide intermediates to both active sites, which are separated by ~30 Å.
To better detail the interactions a PKS DH makes with both the KR domain and the KS+AT didomain within a module, a sequence alignment was assembled of the N- and C-terminal ends of PKS DHs(Supplementary Figure 2). To identify PKS DH residues that may interact with ER, a sequence alignment was assembled comparing DHs from modules containing an ER with DHs from modules not containing an ER(Supplementary Figure 3).
Within a PKS, DH interacts with ACP during the presentation of the polyketide substrate to the DH active site. Two surface residues, F227 and R275, are nearly invariant across all PKS DH types and are located near the active site entrance in analogous positions to residues in type II dehydratases hypothesized to contact acyl carrier proteins(Figure 3E)16. In the structure of the type II FAS acyl carrier protein synthase : acyl carrier protein complex, two of the most important contacts are made between a phenylalanine and an arginine on the surface of the acyl carrier protein synthase with a leucine and aspartate in the acyl carrier protein “DSL” motif25. Positively charged patches near active sites of type II FAS enzymes have been correlated with acyl carrier protein docking sites26. The surface near the EryDH4 active site is slightly positively charged, while the remainder of the EryDH4 dimer is negatively charged (the average isoelectric point of PKS DHs is 4.8).
Mutational Analysis of the Putative ACP-Docking Site
The importance of the nearly invariant R275 was examined by site-directed mutagenesis in the context of the erythromycin PKS. R275 was mutated to an aspartate, and the engineered erythromycin PKS was expressed in E. coli K207-3, a strain that carries the inducible genes for the surfactin phosphopantetheinyl transferase and enzymes that synthesize methylmalonyl-CoA from propionate supplied to the media27. 6-dEB was produced by the mutated PKS at reduced titer (~15%) compared to the native PKS. No hydrated 6-dEB analog was detected by LC/MS analysis(Figure 6, Supplemental Figure 4).
Figure 6.
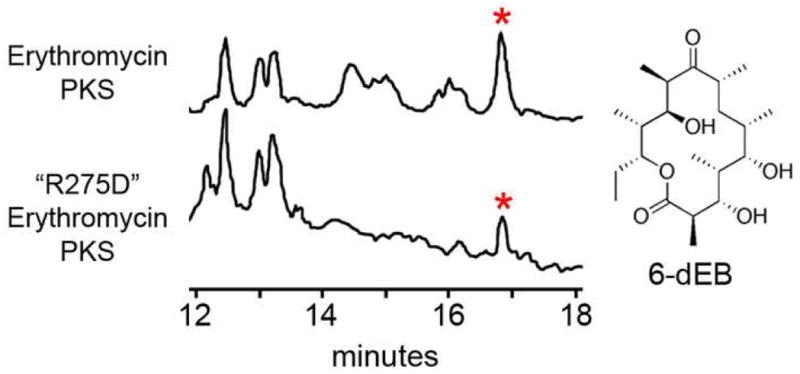
Decreased 6-dEB Production by “R275D” Erythromycin PKS. LC/MS traces of media extracts from E. coli K207-3 expressing either the erythromycin PKS or the erythromycin PKS containing a point mutation at the hypothesized EryDH4 ACP-docking site. Polyketides are detected via ion count mode. Peaks representing 6-dEB (marked by asterisks) yield the anticipated 6-dEB fragmentation pattern (Supplementary Figure 4).
Comparing DHs
Sequence alignments were constructed of PKS DHs comparing DHs that dehydrate substrates containing various α-substituents, DHs that are hypothesized to form a cis-double bond, and DHs that are apparently nonfunctional(Supplementary Figure 5, 6). Significant differences in active site residues are only observed in the nonfunctional DHs. Each of the nonfunctional DHs possesses significant alterations to one or more of the HXXXGXXXXP, GYXYGPXF, LPFXW, and DXXX(Q/H) motifs.
To determine what differences exist between animalian FAS DHs and PKS DHs, sequences from representative domains were compared (Supplementary Figure 7). A satisfactory alignment could not be made between these DHs. Instead, they were aligned separately and then compared. The regions between the N-terminal boundary and the DXXX(Q/H) motif are similar; however, the FAS DH C-terminal boundary is difficult to identify. Based on the anticipated C-terminal boundary of DH (from a size comparison with the PKS DH) and the most likely location of the N-terminal boundary of the KR structural subdomain (β1), there are ~150 unassigned residues between DH and the KR structural subdomain in mammalian FASs.
DISCUSSION
Structure
In contrast to homodimeric type II dehydratases, which have two equivalent active sites, type I DHs only possess one active site per double hotdog. As type I enzymes are hypothesized to have evolved from type II enzymes, type I DHs may have arisen from an ancestral gene duplication of a type II dehydratase with one of the active sites subsequently being lost, a recurring theme in PKS enzymes (i.e. ketosynthase and chain length factor in type II PKSs; the catalytic and structural subdomains of KR)9,28. The DH dimerization interface is formed exclusively by the first half of the double hotdog fold, while the active site is formed by both halves. Dimerization may not be necessary for catalysis as DHs in FAS monomers are functional29.
The EryDH4 dimer observed in the crystal lattice is ~60° straighter than the porcine FAS DH dimer(Figure 3A, 3F, 5A)6. Given their evolutionary relationship, it is unlikely that PKS and animalian FAS β-carbon processing enzymes make fundamentally different interactions across the twofold axis of these synthases3. The PKS DH dimer may bend similar to the animalian FAS DH dimer when all β-carbon processing enzymes are present within a module. If the modeled PKS DH/KR interface is accurate, the straight DH dimer conformation would prevent ER dimerization. Thus, the DH dimer may be capable of a hinge motion, and the bent conformation may be stabilized through dimerization of ER. The DH dimer in PKS modules containing only DH and KR may not be as conformationally restricted.
While it is challenging to compare animalian FAS DHs with PKS DHs, it does appear that mammalian FASs possess an additional ~150 residue region between DH and the KR structural subdomain. Intriguingly, this region is predicted by the program Phyre to be primarily α-helical and contains two invariant cysteines30. Insect FASs apparently do not share this region and may be more similar to PKSs, although it is difficult to identify β1 of the KR structural subdomain within these FASs.
Domain Interactions
The N-termini of the observed EryDH4 dimer help reveal how DHs connect to the KS+AT didomain(Figure 5, Supplementary Figure 2). They are adjacent to the dimer interface, and a mere four residues in EryMod4 connect them to the KS+AT didomain (i.e. the post-AT segment). These residues are unconserved and often include prolines, possibly indicating they are unstructured. In the porcine FAS structure this connection (the “waist”) was observed to be flexible and may provide degrees of freedom for the β-carbon processing enzymes and ACP relative to the KS+AT didomain so ACP can access the active sites of the synthase6. The C-termini of the Ery(KS+AT)5 dimer are separated by ~15 Å7, while the N-termini in the EryDH4 dimer are separated by ~10 Å. If the C-termini of the KS+AT didomain and the N-termini of the DH dimer are located approximately where they are located in the crystal structures, the connection between the KS+AT didomain and the β-carbon processing enzymes may resemble a “loose swing” more than a “swivel.” It remains to be determined how restricted the β-carbon processing enzymes are relative to the KS+AT didomain; however, the orientation of the KS+AT didomain relative to the DH dimer in the porcine FAS structure is similar to the orientation obtained by modeling the C-termini of the PKS KS+AT didomain adjacent to the N-termini of the PKS DH dimer.
Nearly all modules that contain DH also contain KR. The animalian FAS DH/KR interaction has been observed at 4.5 Å resolution in the porcine FAS and is likely to be similar in PKSs (Figure 5B)6. KRs from modules containing DHs possess several conserved residues that may contribute to the PKS DH/KR interface9. A conserved αB histidine and a conserved αG aspartate (KR nomenclature9) may be well-positioned on the KR surface to contact the 10–15 residue segment connecting DH to KR. The KR lid also possesses several conserved residues that may contact DH. The DH surface expected to contact KR based on the porcine FAS structure is concave and exposes hydrophobic residues such as I152 and L214. The ~15 residues spanning from R275 to β1 of KR may be structured in the presence of KR and also contribute to the PKS DH/KR interface. The EryDH4 dimer does not reveal any obvious surfaces that might interface with ER. A sequence comparison of DHs from modules containing ER with DHs from modules not containing ER also does not reveal any conserved residues that may participate in an ER interaction(Supplementary Figure 3).
A few surface residues are conserved between DHs from all types of modules and are clustered in the analogous location to residues in type II dehydratases hypothesized to interact with acyl carrier protein(Supplementary Figure 5)26,16. In particular, a highly conserved phenylalanine (F227) is located near the active site entrance adjacent to several smaller, semi-conserved hydrophobic residues(Figure 3E). A nearly invariant arginine (R275) is also adjacent to the active site and may form a salt-bridge with the conserved aspartate of the ACP “DSL” motif. Interestingly, the phenylalanine and arginine are not conserved in nonfunctional DHs, which suggests that ACP does not dock on them(Supplementary Figure 6).
Mutagenesis
That the isolated EryDH4 domain did not dehydrate (2R,3R)-3-hydroxy-2-methylpentanoate N-acetylcysteamine thioester at millimolar concentrations raised the question whether EryDH4 isolated from the erythromycin PKS is functional. As DH from monomeric FAS and the truncated FAS construct KS+AT+DH were shown to be functional, it is anticipated that isolated EryDH4 display some level of activity. Perhaps the isolated EryDH4 domain does not bind (2R,3R)-3-hydroxy-2-methylpentanoate N-acetylcysteamine thioester with sufficient affinity for the dehydration reaction to proceed(Figure 1B). A large component of the binding energy of the natural substrate, (2R,3R,4R,6R,7S,8S,9R)-3,7,9-trihydroxy-5-oxo-2,4,6,8-tetramethylundecanoate holo-EryACP4 thioester, may come from EryACP4. Indeed, in the related FAS, ACP-bound acyl chains proved better substrates for the FAS DH than the corresponding pantetheinyl-bound acyl chains14. If EryACP4 docks to EryDH4 through conserved surfaces residues such as F227 and R275, then mutating these residues could compromise the dehydration reaction. The “R275D” erythromycin PKS produced significantly less 6-dEB than the unmutated erythromycin PKS(Figure 6, Supplementary Figure 4). Thus, R275 may help build the EryACP4 docking site. Other residues that might be involved in ACP-docking, such as F227, should also be assayed.
Dehydration Mechanism
The catalytic residues of PKS DHs are similar to those of related dehydratases. The type II FAS dehydratase FabZ is hypothesized to catalyze dehydration via the catalytic histidine abstracting the α-proton and the catalytic glutamic acid donating a proton to the β-hydroxyl group16. Hydrogen and water are anticipated to leave from Cα and Cβ, respectively (from the same side of the bound acyl chain), as electrons from the Cα-H bond migrate to form the Cα-Cβ trans- double bond. H44 and D206 likely catalyze a similar reaction in EryDH4 by directly interacting with the α-hydrogen and β-hydroxyl group, to form a trans- double bond in the polyketide intermediate(Figure 1, 4C). The stereochemistries of the α-proton and β-hydroxyl group that are normally removed from the polyketide intermediate are equivalent to the analogous intermediate in fatty acid biosynthesis(Figure 4E)23.
When the polyketide substrate has entered the active site so that the α-hydrogen is adjacent to the H44 Nε2, the β-hydroxyl group may be able to eject Wat1 from its position at the N-terminal end of α1, but due to geometric constraints it cannot occupy the former position of Wat1(Figure 4B). The positive dipole of α1 may attract the oxygen of the β-hydroxyl group and polarize the Cβ-O bond. This may help the β-hydroxyl group to leave the polyketide during the formation of the double bond.
The highly conserved leucine L225 is well-positioned in the active site to clamp the substrate through hydrophobic interactions during catalysis(Figure 4B). This residue may contact the α-substituent to some extent - when the native polyketide substrate contains an α-ethyl substituent, it is sometimes replaced by a methionine(Supplementary Figure 5). The nearly invariant P226 is also positioned to make hydrophobic interactions with the polyketide substrate. The second tyrosine of the GYXYGPXF motif, Y168, is rarely substituted by phenylalanine, indicating that the tyrosine hydroxyl group is serving a function. In the structure, it is bound by a water molecule that is also coordinated by the catalytic aspartic acid, D206(Figure 4A). Perhaps this interaction helps order the active site in the absence of a polyketide substrate.
Where the equilibrium lies between hydrated and dehydrated polyketide intermediates is unclear; however, hydrated fatty acid intermediates have been demonstrated to be thermodynamically favored over dehydrated fatty acid intermediates31,32. Dehydration is the final β-carbon processing event in many modules before the polyketide is accepted by the downstream KS or TE. Thus, ER is not required by PKSs to pull the reaction forward, as is apparently the case in FASs. In fact, when EryER4 was knocked out through site-directed mutagenesis in the erythromycin PKS, it was the dehydrated intermediate from EryMod4 that transferred to EryKS533. Perhaps the rate of transesterification to the active site of a downstream KS or TE can be correlated with the hydrophobicity of the polyketide intermediate proximal to the thioester bond. Thus, the equilibrium may be towards the hydrated polyketide, analogous to fatty acid synthesis, and a downstream KS or TE may pull the dehydrated polyketide forward.
PKS DHs, unlike FAS DHs, often dehydrate acyl chains possessing methyl, ethyl, or methoxy α-substituents. These polyketide intermediates are reduced by B1-type KRs and, less frequently, by A1-type KRs10. In both cases, the α-substituent of the resulting intermediate possesses “R” stereochemistry; if the α-substituent possessed “S” stereochemistry, the α-hydrogen would be inaccessible to the catalytic histidine (quotes are placed around R and S, as the convention used to label chiral centers in polyketides can deviate from the RS system: when discussing chirality at the α-position, the α-substituent is given the lowest priority after the hydrogen: when discussing chirality at the β-position, the γ-position is given the lowest priority after the hydrogen)(Figure 4B). The active site residues of DHs that dehydrate α-substituent-containing substrates are not significantly different from DHs that dehydrate substrates without α-substituents(Supplementary Figure 5). Thus, α-substituents may not need to be recognized by DHs.
Cis- Dehydration
The stereochemistry of the β-hydroxyl group of a polyketide substrate may be the primary determinant for whether a cis- or a trans- double bond will form34. When an A-type KR accompanies DH, a polyketide containing a β-hydroxyl group possessing “S” stereochemistry is presented to DH. For the “S” hydroxyl group to be bound by D206, the chain must rotate ~120° about the Cα-Cβ bond relative to the more common substrate orientation(Figure 4). Due to the chain configuration, an elimination reaction would then result in the formation of a cis- double bond. Adequate space for this conformation of the substrate, as well as the cis-dehydrated product, needs to exist within DHs that catalyze cis- dehydration.
Few PKS DHs hypothesized to catalyze the formation of a cis- double bond have been sequenced (PhoDH1, PhoDH2, and RifDH10 are the best examples)17,18. The polyketide vacidin A contains two adjacent cis- double bonds; however, the sequence of its PKS has not been published35. Candicidin may possess analogous adjacent cis- double bonds (the modules that would create these double bonds contain A-type KRs)36,37. A comparison of these “cis-” DHs to “trans-” DHs does not reveal any obvious differences in the active sites (Supplemental Figure 5).
Some PKS DHs may be epimerases (e.g. AscDH2, AscDH4, MyxDH2, RapDH3, RapDH6, RifDH6, and RifDH7). These DHs could use the conserved catalytic histidine to abstract the α-proton of an α-substituted, β-keto intermediate to form an enol that can tautomerize to the epimerized polyketide, similar to the mechanism proposed for epimerizing KRs10. Six out of seven of these DHs have a mutation to the leucine or proline of the LPFXW motif at the active site (the exception is AscDH4, in which the catalytic aspartic acid is replaced by a valine)(Supplementary Figure 5).
Polyketide Structure Prediction and Engineering
Predicting the structure of a polyketide based on the sequence of its PKS has traditionally been complicated by a lack of knowledge of DHs. Differences in DH activity can now be related through the EryDH4 structure to differences in DH sequences detected in sequence alignments. To predict whether a DH is nonfunctional, the catalytic motifs that build the active site should be analyzed for alterations (made apparent by a sequence alignment with functional DHs as in Supplementary Figure 6). To predict if a DH might epimerize, the LPFXW motif should be examined for mutations. To predict rare cis-double bond formation, the KR with which the DH cooperates should be checked for the absence of the “LDD” motif34,38,10.
One of the most direct ways of engineering new polyketides is through knocking out the function of enzymes within a PKS33,39. The only reported success in knocking out a DH while retaining a functional PKS was accomplished in PikDH2 by mutating the catalytic histidine to a phenylalanine40. The equivalent mutation was attempted within EryDH4 in the context of the erythromycin PKS; however, no polyketide was produced41. This has been attributed to the specificity of EryKS5 for its natural substrate. In general, the substrate specificities of KSs are quite broad and knocking out DHs within a PKS is a promising strategy to generate libraries of novel polyketides.
MATERIALS AND METHODS
Cloning, Expression, and Purification
The DNA encoding the DH fragment was cloned from a synthetic version of eryAII (Kosan Biosciences) using primers 5′-GCAGATATACATATGCACCGCCCAGCAGAT GTTAGC-3′ and 5′-GTGGTGCTCGAGTCACCCGCGCGGTTCCGGTTGTTC-3′42. The DNA was digested with NdeI and XhoI and ligated into pET28b (Novagen). E. coli BL21(DE3) was transformed with this plasmid and grown in LB at 37°C until A600=0.4 at which time protein expression was induced with 1 mM IPTG and the temperature was decreased to 15°C. After 16 h the cells were collected and resuspended in lysis buffer (30 mM Tris pH 7.4, 500 mM NaCl) and sonicated. The lysate was collected and poured through a column filled with Ni-NTA resin (Qiagen). After washing the resin twice with 15 mM imidazole in lysis buffer, the DH fragment was eluted with 150 mM imidazole in lysis buffer. The protein was concentrated and injected onto a Superdex 200 gel filtration column equilibrated with 10 mM Tris pH 7.4, 150 mM NaCl. The protein (yield >20 mg/L media, purity >99% by SDS-PAGE/Coomassie) was collected and concentrated to 10 mg/mL.
Gel Filtration
A Superdex 200 gel filtration column was equilibrated with 10 mM Tris pH 7.4, 150 mM NaCl and injected with 0.2 mL samples. The DH fragment was compared to known standards (Gel Filtration Standard; Biorad Laboratories). Dextran blue 2000 and tyrosine were used to determine Vo and Vt, respectively. Kav = (Ve−Vo)/(Vt−Vo), where Ve is the volume at which the protein elutes.
Assay for EryDH4 Activity
Milligram quantities of (2R,3R)-3-hydroxy-2-methylpentanoate N-acetylcysteamine thioester were produced as described previously through a preparative scale synthesis using (2RS)-3-oxo-2-methylpentanoate N-acetylcysteamine thioester, NADPH, and TylKR110. (2R,3R)-3-hydroxy-2-methylpentanoate N-acetylcysteamine thioester (0.1, 1, or 10 mM) was incubated with EryDH4 (1, 10, or 100 μM) in 10 mM Tris (pH 6.5 or 7.5), NaCl (10, 100, or 200 mM) at either 21 or 37°C for one day. The reactions were extracted with ethyl acetate, and the dried extract was analyzed by LC/MS using a C18 reverse phase column with a 0–30% acetonitrile/water (0.1% TFA) gradient.
Crystallization and Structure Determination
Crystals were obtained in one week by hanging drop vapor diffusion at 21°C with a 2.15 M ammonium sulfate, 100 mM sodium cacodylate pH 6.4 crystallization condition using 2 μL protein : 1 μL crystallization condition. The concentration of ammonium sulfate was high enough that no cryoprotectant was required.
Data were collected at the Advanced Light Source Beamline 8.3.1 and reduced with HKL200043. Every protein with a hotdog or double hotdog fold in the RCSB Database was used as a molecular replacement search model using Phaser; however, no solution was obtained44. Thus, crystals were soaked for 1 minute in 2.15 M ammonium sulfate, 1 M sodium bromide, 100 mM sodium cacodylate prior to collecting data at the bromine K edge45. The program PHENIX located 9 bromine sites and automatically built ~90% of the protein model46. The final model was constructed using a native dataset through several rounds of refinement using Coot and CNS(Table 1)24,47.
Table 1.
Crystallographic Data and Refinement Statistics. Statistics in parentheses refer to the highest resolution shell (1.85–1.92 Å).
| Native | Bromide Derivative | |
|---|---|---|
| Data collection | ||
| Wavelength (Å) | 1.1159 | 0.9200 |
| Space group | P43212 | P43212 |
| Cell dimensions | ||
| a, b, c (Å) | 67.0, 67.0, 186.2 | 67.5, 67.5, 186.3 |
| Resolution (Å) | 50–1.85 | 50–2.60 |
| Rmerge | 0.076 (0.659) | 0.131(0.459) |
| I/σ(I) | 42.5 (2.5) | 31.8 (4.7) |
| Completeness (%) | 100 (100) | 99.9 (99.9) |
| Redundancy | 14.0 (14.2) | 10.8 (9.9) |
| Refinement | ||
| Resolution (Å) | 50–1.85 | |
| No. reflections | 37,089 | |
| Rwork/Rfree | 0.197/0.227 | |
| No. atoms | ||
| Protein | 1,990 | |
| Water | 247 | |
| Chloride | 1 | |
| B-factors (Å2) | ||
| Protein | 31 | |
| Water | 42 | |
| Chloride | 30 | |
| R.m.s. deviations | ||
| Bond lengths (Å) | 0.019 | |
| Bond angles (°) | 1.8 | |
The twofold axis of the apparent DH dimer is equivalent to the twofold axis of the spacegroup (P43212). Three regions of the constructed DH fragment were unobservable in the electron density maps: 1) the N-terminal histidine tag and H2362-A2365, (2) the C-terminal residues S2641-G2653, and 3) the loop P2582-G2585.
Sequence Alignments
Most PKS sequences were obtained from the PKSDB database48. Sequence alignments were assembled with the program ClustalX using default parameters49.
Engineering the “R275D” Erythromycin PKS Mutant
The SacI-NotI fragment from KOS207-4 (a plasmid containing a synthetic version of DEBS2 obtained from Kosan Biosciences) was ligated into pUC18. QuickChange was performed using primers 5′-GTTGATTCTCTTGTGGTCGACTCAACTGGTG AGAAATG-3′ and 5′-CATTTCTCACCAGTTGAGTCGACCACAAGAGAATCAAC-3′50. The NdeI-EcoRI fragment encoding DEBS2 from KOS207-4 was ligated into pUC18. The SacI-NotI fragment from the site-directed mutagenesis was ligated into this plasmid, which was also digested with SacI and NotI. Finally, the NdeI-EcoRI fragment was ligated with the NdeI-EcoRI backbone of KOS207-4.
Erythomycin PKS Assay
K207-3 cells transformed with plasmids encoding natural versions of DEBS1 and DEBS3 were transformed with plasmids encoding either DEBS2 (KOS207-4) or engineered “R275D” DEBS2 (plasmids were obtained from Kosan Biosciences)50. These cells were grown in 500 mL M9 minimal media (4 g/L glucose, 6 g/L Na2HPO4, 3 g/L KH2PO4, 1 g/L NH4Cl, 0.5 g/L NaCl, 1 mM MgSO4, 0.1 mM CaCl2) containing 20 mg/L kanamycin, 10 mg/L tetracycline, 20 mg/L streptomycin, and 1 mM β-alanine at 37 °C until A600= 0.4 when 50 mM glutamate, 50 mM succinate, and 5 mM propionate (pH 7.0) and 1 mM IPTG were supplied to the media and the temperature was reduced to 21 °C. Cells were spun down after 2 days. The supernatant was acidified to pH 1.5 with sulfuric acid, and polyketides were extracted with ethyl acetate, dried with MgSO4, and concentrated. The extract was resuspended in 50% ACN (0.1% TFA) and analyzed via LC/MS using a C18 reverse phase column with a 0–100% acetonitrile/water (0.1% TFA) gradient. Side-by-side (unmutated and mutated PKS) assays were repeated in triplicate; representative traces are displayed in Figure 6.
Supplementary Material
Gel Filtration. EryDH4 migrates at ~50 kDa compared to standard proteins. The expected monomer and dimer molecular weights are 33 kDa and 66 kDa, respectively.
{kind=link}
Residues Connecting Domains. The unconserved segment that often contains prolines between the post-AT segment and η1 of DH may be unstructured. The segment between DH and the structural subdomain of KR contains semi-conserved hydrophobic residues, indicating some level of structure between these domains.
{kind=link}
Investigating the DH/ER Interface. This sequence alignment shows there are no consistent differences between DHs from modules containing ER and DHs from modules not containing ER, indicating that there is not much contact between DH and ER.
{kind=link}
The “R275D” Erythromycin PKS Produces 6-dEB. The mass fragmentation patterns from the 6-dEB peaks of the LC/MS traces presented in Figure 6 show that 6-dEB (386 Da) is produced by the engineered “R275D” erythromycin PKS.
{kind=link}
Investigating Residues that may Regulate Selectivity and Reactivity. A sequence alignment of PKS DHs compares DHs that form trans- double bonds, DHs hypothesized to form cis-double bonds, and DHs hypothesized to catalyze epimerization of α-substituents in β-keto intermediates. Active site residues are indicated by red rectangles. DHs that form trans- double bonds are separated by the α-substituent of the polyketide they dehydrate. There may be an increased propensity for a methionine to replace L225 in DHs that dehydrate α-ethyl substituted polyketides; however, most DHs appear not to make specific interactions with α-substituents. The putative epimerases lack conserved residues particularly at the LPFXW motif; in six out of seven cases, either the leucine is mutated to methionine or the proline is mutated to alanine.
{kind=link}
Detecting Nonfunctional PKS DHs. A sequence alignment of EryDH4 to DHs hypothesized to be nonfunctional reveals obvious alterations to multiple catalytic motifs. AmpDH15 would be the most challenging to detect; however, its invariant catalytic aspartic acid is replaced by a histidine. Interestingly, the phenylalanine of the LPFXW and terminal arginine hypothesized to contact ACP are not conserved in these DHs, indicating that ACP does not dock to nonfunctional DHs.
{kind=link}
PKS and FAS Domain Boundaries Surrounding DH. The known PKS domain boundaries may help to determine the location of the FAS domain boundaries. The PKS and FAS sequences are aligned separately as they do not align well together. While the HXXXGXXXXP, GYXYGPXF, and DXXX(Q/H) motifs can be identified in FAS DHs, it remains challenging to pinpoint the boundary between DH and the structural subdomain of KR. Mammalian FASs may possess a ~150 residue domain between DH and the KR structural subdomain. Insect FASs may not possess this domain; however, the DH/KR boundary is still unclear as β1 of the KR structural subdomain is difficult to identify. Man, Homo sapiens; Rat, Rattus norvegicus; Pig, Sus scrofa; Bee, Apis mellifera; Wasp, Nasonia vitripennis; Fly, Drosophila melanogaster; Mosq., Anopheles gambiae.
{kind=link}
Acknowledgments
Ralph Reid, Chris Reeves, and Hugo Menzella shared their knowledge of PKS enzymology. Janet Finer-Moore and Robert Stroud helped interpret experimental results. Research was supported by National Cancer Institute, National Institutes of Health grant CA63081 (to Robert Stroud).
Abbreviations
- ACP
acyl carrier protein
- AT
acyltransferase
- CoA
coenzyme A
- DH
dehydratase
- ER
enoylreductase
- FAS
fatty acid synthase
- KR
ketoreductase
- KS
ketosynthase
- PKS
polyketide synthase
- TE
thioesterase
- Amp
Amphotericin
- Asc
Ascomycin
- Ave
Avermectin
- Can
Candicidin
- Con
Concanamycin
- Epo
Epothilone
- Ery
Erythromycin
- Mer
Meridamycin
- Myx
Myxalamide
- Nid
Niddamycin
- Nys
Nystatin
- Pim
Pimaricin
- Plm
Phoslactomycin
- Rap
Rapamycin
- Rif
Rifamycin
- Sor
Soraphen
- Spi
Spinosad
- Tyl
Tylosin
Footnotes
Protein Data Bank Accession Code
The atomic coordinates and structure factors for EryDH4 have been deposited in the RCSB Protein Data Bank as entry 3EL6.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Staunton J, Wilkinson B. Biosynthesis of Erythromycin and Rapamycin. Chem Rev. 1997;97:2611–2630. doi: 10.1021/cr9600316. [DOI] [PubMed] [Google Scholar]
- 2.Khosla C, Tang Y, Chen AY, Schnarr NA, Cane DE. Structure and mechanism of the 6-deoxyerythronolide B synthase. Annu Rev Biochem. 2007;76:195–221. doi: 10.1146/annurev.biochem.76.053105.093515. [DOI] [PubMed] [Google Scholar]
- 3.Sherman DH, Smith JL. Clearing the skies over modular polyketide synthases. ACS Chem Biol. 2006;1:505–9. doi: 10.1021/cb600376r. [DOI] [PubMed] [Google Scholar]
- 4.Smith S, Tsai SC. The type I fatty acid and polyketide synthases: a tale of two megasynthases. Nat Prod Rep. 2007;24:1041–72. doi: 10.1039/b603600g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leibundgut M, Jenni S, Frick C, Ban N. Structural basis for substrate delivery by acyl carrier protein in the yeast fatty acid synthase. Science. 2007;316:288–90. doi: 10.1126/science.1138249. [DOI] [PubMed] [Google Scholar]
- 6.Maier T, Jenni S, Ban N. Architecture of mammalian fatty acid synthase at 4.5 A resolution. Science. 2006;311:1258–62. doi: 10.1126/science.1123248. [DOI] [PubMed] [Google Scholar]
- 7.Tang Y, Kim CY, Mathews II, Cane DE, Khosla C. The 2.7-Angstrom crystal structure of a 194-kDa homodimeric fragment of the 6-deoxyerythronolide B synthase. Proc Natl Acad Sci U S A. 2006;103:11124–9. doi: 10.1073/pnas.0601924103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tang Y, Chen AY, Kim CY, Cane DE, Khosla C. Structural and mechanistic analysis of protein interactions in module 3 of the 6-deoxyerythronolide B synthase. Chem Biol. 2007;14:931–43. doi: 10.1016/j.chembiol.2007.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Keatinge-Clay AT, Stroud RM. The structure of a ketoreductase determines the organization of the beta-carbon processing enzymes of modular polyketide synthases. Structure. 2006;14:737–48. doi: 10.1016/j.str.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 10.Keatinge-Clay AT. A tylosin ketoreductase reveals how chirality is determined in polyketides. Chem Biol. 2007;14:898–908. doi: 10.1016/j.chembiol.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 11.Alekseyev VY, Liu CW, Cane DE, Puglisi JD, Khosla C. Solution structure and proposed domain domain recognition interface of an acyl carrier protein domain from a modular polyketide synthase. Protein Sci. 2007;16:2093–107. doi: 10.1110/ps.073011407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tsai SC, Lu H, Cane DE, Khosla C, Stroud RM. Insights into channel architecture and substrate specificity from crystal structures of two macrocycle-forming thioesterases of modular polyketide synthases. Biochemistry. 2002;41:12598–606. doi: 10.1021/bi0260177. [DOI] [PubMed] [Google Scholar]
- 13.Chakravarty B, Gu Z, Chirala SS, Wakil SJ, Quiocho FA. Human fatty acid synthase: structure and substrate selectivity of the thioesterase domain. Proc Natl Acad Sci U S A. 2004;101:15567–72. doi: 10.1073/pnas.0406901101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pasta S, Witkowski A, Joshi AK, Smith S. Catalytic residues are shared between two pseudosubunits of the dehydratase domain of the animal fatty acid synthase. Chem Biol. 2007;14:1377–85. doi: 10.1016/j.chembiol.2007.11.007. [DOI] [PubMed] [Google Scholar]
- 15.Leesong M, Henderson BS, Gillig JR, Schwab JM, Smith JL. Structure of a dehydratase-isomerase from the bacterial pathway for biosynthesis of unsaturated fatty acids: two catalytic activities in one active site. Structure. 1996;4:253–64. doi: 10.1016/s0969-2126(96)00030-5. [DOI] [PubMed] [Google Scholar]
- 16.Kimber MS, Martin F, Lu Y, Houston S, Vedadi M, Dharamsi A, Fiebig KM, Schmid M, Rock CO. The structure of (3R)-hydroxyacyl-acyl carrier protein dehydratase (FabZ) from Pseudomonas aeruginosa. J Biol Chem. 2004;279:52593–602. doi: 10.1074/jbc.M408105200. [DOI] [PubMed] [Google Scholar]
- 17.Alhamadsheh MM, Palaniappan N, Daschouduri S, Reynolds KA. Modular polyketide synthases and cis double bond formation: establishment of activated cis-3-cyclohexylpropenoic acid as the diketide intermediate in phoslactomycin biosynthesis. J Am Chem Soc. 2007;129:1910–1. doi: 10.1021/ja068818t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schupp T, Toupet C, Engel N, Goff S. Cloning and sequence analysis of the putative rifamycin polyketide synthase gene cluster from Amycolatopsis mediterranei. FEMS Microbiol Lett. 1998;159:201–7. doi: 10.1111/j.1574-6968.1998.tb12861.x. [DOI] [PubMed] [Google Scholar]
- 19.Li J, Derewenda U, Dauter Z, Smith S, Derewenda ZS. Crystal structure of the Escherichia coli thioesterase II, a homolog of the human Nef binding enzyme. Nat Struct Biol. 2000;7:555–9. doi: 10.1038/76776. [DOI] [PubMed] [Google Scholar]
- 20.Koski KM, Haapalainen AM, Hiltunen JK, Glumoff T. Crystal structure of 2-enoyl-CoA hydratase 2 from human peroxisomal multifunctional enzyme type 2. J Mol Biol. 2005;345:1157–69. doi: 10.1016/j.jmb.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 21.Bevitt DJ, Cortes J, Haydock SF, Leadlay PF. 6-Deoxyerythronolide-B synthase 2 from Saccharopolyspora erythraea. Cloning of the structural gene, sequence analysis and inferred domain structure of the multifunctional enzyme. Eur J Biochem. 1992;204:39–49. doi: 10.1111/j.1432-1033.1992.tb16603.x. [DOI] [PubMed] [Google Scholar]
- 22.Kao CM, McPherson M, McDaniel RN, Fu H, Cane DE, Khosla C. Alcohol Stereochemistry in Polyketide Backbones Is Controlled by the beta-Ketoreductase Domains of Modular Polyketide Synthases. J Am Chem Soc. 1998;120:2478–9. [Google Scholar]
- 23.Castonguay R, He W, Chen AY, Khosla C, Cane DE. Stereospecificity of ketoreductase domains of the 6-deoxyerythronolide B synthase. J Am Chem Soc. 2007;129:13758–69. doi: 10.1021/ja0753290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–32. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 25.Parris KD, Lin L, Tam A, Mathew R, Hixon J, Stahl M, Fritz CC, Seehra J, Somers WS. Crystal structures of substrate binding to Bacillus subtilis holo-(acyl carrier protein) synthase reveal a novel trimeric arrangement of molecules resulting in three active sites. Structure Fold Des. 2000;8:883–895. doi: 10.1016/s0969-2126(00)00178-7. [DOI] [PubMed] [Google Scholar]
- 26.Zhang YM, Rao MS, Heath RJ, Price AC, Olson AJ, Rock CO, White SW. Identification and analysis of the acyl carrier protein (ACP) docking site on beta-ketoacyl-ACP synthase III. J Biol Chem. 2001;276:8231–8. doi: 10.1074/jbc.M008042200. [DOI] [PubMed] [Google Scholar]
- 27.Murli S, Kennedy J, Dayem LC, Carney JR, Kealey JT. Metabolic engineering of Escherichia coli for improved 6-deoxyerythronolide B production. J Ind Microbiol Biotechnol. 2003;30:500–509. doi: 10.1007/s10295-003-0073-x. [DOI] [PubMed] [Google Scholar]
- 28.Keatinge-Clay AT, Maltby DA, Medzihradszky KF, Khosla C, Stroud RM. An antibiotic factory caught in action. Nat Struct Mol Biol. 2004;11:888–93. doi: 10.1038/nsmb808. [DOI] [PubMed] [Google Scholar]
- 29.Smith S, Witkowski A, Joshi AK. Structural and functional organization of the animal fatty acid synthase. Prog Lipid Res. 2003;42:289–317. doi: 10.1016/s0163-7827(02)00067-x. [DOI] [PubMed] [Google Scholar]
- 30.Bennett-Lovsey RM, Herbert AD, Sternberg MJ, Kelley LA. Exploring the extremes of sequence/structure space with ensemble fold recognition in the program Phyre. Proteins. 2008;70:611–25. doi: 10.1002/prot.21688. [DOI] [PubMed] [Google Scholar]
- 31.Heath RJ, Rock CO. Enoyl-acyl carrier protein reductase (fabI) plays a determinant role in completing cycles of fatty acid elongation in Escherichia coli. J Biol Chem. 1995;270:26538–42. doi: 10.1074/jbc.270.44.26538. [DOI] [PubMed] [Google Scholar]
- 32.Witkowski A, Joshi AK, Smith S. Characterization of the beta-carbon processing reactions of the mammalian cytosolic fatty acid synthase: role of the central core. Biochemistry. 2004;43:10458–66. doi: 10.1021/bi048988n. [DOI] [PubMed] [Google Scholar]
- 33.Donadio S, McAlpine JB, Sheldon PJ, Jackson M, Katz L. An erythromycin analog produced by reprogramming of polyketide synthesis. Proc Natl Acad Sci U S A. 1993;90:7119–23. doi: 10.1073/pnas.90.15.7119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reid R, Piagentini M, Rodriguez E, Ashley G, Viswanathan N, Carney J, Santi DV, Hutchinson CR, McDaniel R. A model of structure and catalysis for ketoreductase domains in modular polyketide synthases. Biochemistry. 2003;42:72–9. doi: 10.1021/bi0268706. [DOI] [PubMed] [Google Scholar]
- 35.Sowinski P, Gariboldi P, Czerwinski A, Borowski E. The structure of vacidin A, an aromatic heptaene macrolide antibiotic. I. Complete assignment of the 1H NMR spectrum and geometry of the polyene chromophore. J Antibiot (Tokyo) 1989;42:1631–8. doi: 10.7164/antibiotics.42.1631. [DOI] [PubMed] [Google Scholar]
- 36.Cybulska B, Borowski E, Prigent Y, Gary-Bobo CM. Cation permeability induced by two aromatic heptaenes, vacidin A and candicidin D on phospholipid unilamellar vesicles. J Antibiot (Tokyo) 1981;34:884–91. doi: 10.7164/antibiotics.34.884. [DOI] [PubMed] [Google Scholar]
- 37.Campelo AB, Gil JA. The candicidin gene cluster from Streptomyces griseus IMRU 3570. Microbiology. 2002;148:51–9. doi: 10.1099/00221287-148-1-51. [DOI] [PubMed] [Google Scholar]
- 38.Caffrey P. Conserved amino acid residues correlating with ketoreductase stereospecificity in modular polyketide synthases. Chembiochem. 2003;4:645–657. doi: 10.1002/cbic.200300581. [DOI] [PubMed] [Google Scholar]
- 39.Power P, Dunne T, Murphy B, Lochlainn LN, Rai D, Borissow C, Rawlings B, Caffrey P. Engineered synthesis of 7-oxo- and 15-deoxy-15-oxo-amphotericins: insights into structure-activity relationships in polyene antibiotics. Chem Biol. 2008;15:78–86. doi: 10.1016/j.chembiol.2007.11.008. [DOI] [PubMed] [Google Scholar]
- 40.Wu J, Zaleski TJ, Valenzano C, Khosla C, Cane DE. Polyketide deouble bond biosynthesis. Mechanistic analysis of the dehydratase-containing module 2 of the picromycin/methymycin polyketide synthase. J Am Chem Soc. 2005;127:17393–404. doi: 10.1021/ja055672+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Leadlay PF, Staunton J, Aparicio JF, Bevitt DJ, Caffrey P, Marsden A, Roberts GA. The erythromycin-producing polyketide synthase. Biochem Soc Trans. 1993;21:218–22. doi: 10.1042/bst0210218. [DOI] [PubMed] [Google Scholar]
- 42.Kodumal SJ, Patel KG, Reid R, Menzella HG, Welch M, Santi DV. Total synthesis of long DNA sequences: synthesis of a contiguous 32-kb polyketide synthase gene cluster. Proc Natl Acad Sci U S A. 2004;101:15573–8. doi: 10.1073/pnas.0406911101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. In: Carter CW Jr, Sweet RM, editors. Methods in Enzymology, Volume 276: Macromolecular Crystallography Part A. New York: Academic Press; 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 44.CCP4 (Collaborative Computational Project, N. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 45.Dauter Z, Dauter M, Rajashankar KR. Novel approach to phasing proteins: derivatization by short cryo-soaking with halides. Acta Crystallogr D Biol Crystallogr. 2000;56:232–7. doi: 10.1107/s0907444999016352. [DOI] [PubMed] [Google Scholar]
- 46.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr. 2002;58:1948–54. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 47.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54 (Pt 5):905–21. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 48.Yadav G, Gokhale RS, Mohanty D. Computational approach for prediction of domain organization and substrate specificity of modular polyketide synthases. J Mol Biol. 2003;328:335–63. doi: 10.1016/s0022-2836(03)00232-8. [DOI] [PubMed] [Google Scholar]
- 49.Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25:4876–82. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Menzella HG, Reisinger SJ, Welch M, Kealey JT, Kennedy J, Reid R, Tran CQ, Santi DV. Redesign, synthesis and functional expression of the 6-deoxyerythronolide B polyketide synthase gene cluster. J Ind Microbiol Biotechnol. 2006;33:22–8. doi: 10.1007/s10295-005-0038-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Gel Filtration. EryDH4 migrates at ~50 kDa compared to standard proteins. The expected monomer and dimer molecular weights are 33 kDa and 66 kDa, respectively.
Residues Connecting Domains. The unconserved segment that often contains prolines between the post-AT segment and η1 of DH may be unstructured. The segment between DH and the structural subdomain of KR contains semi-conserved hydrophobic residues, indicating some level of structure between these domains.
Investigating the DH/ER Interface. This sequence alignment shows there are no consistent differences between DHs from modules containing ER and DHs from modules not containing ER, indicating that there is not much contact between DH and ER.
The “R275D” Erythromycin PKS Produces 6-dEB. The mass fragmentation patterns from the 6-dEB peaks of the LC/MS traces presented in Figure 6 show that 6-dEB (386 Da) is produced by the engineered “R275D” erythromycin PKS.
Investigating Residues that may Regulate Selectivity and Reactivity. A sequence alignment of PKS DHs compares DHs that form trans- double bonds, DHs hypothesized to form cis-double bonds, and DHs hypothesized to catalyze epimerization of α-substituents in β-keto intermediates. Active site residues are indicated by red rectangles. DHs that form trans- double bonds are separated by the α-substituent of the polyketide they dehydrate. There may be an increased propensity for a methionine to replace L225 in DHs that dehydrate α-ethyl substituted polyketides; however, most DHs appear not to make specific interactions with α-substituents. The putative epimerases lack conserved residues particularly at the LPFXW motif; in six out of seven cases, either the leucine is mutated to methionine or the proline is mutated to alanine.
Detecting Nonfunctional PKS DHs. A sequence alignment of EryDH4 to DHs hypothesized to be nonfunctional reveals obvious alterations to multiple catalytic motifs. AmpDH15 would be the most challenging to detect; however, its invariant catalytic aspartic acid is replaced by a histidine. Interestingly, the phenylalanine of the LPFXW and terminal arginine hypothesized to contact ACP are not conserved in these DHs, indicating that ACP does not dock to nonfunctional DHs.
PKS and FAS Domain Boundaries Surrounding DH. The known PKS domain boundaries may help to determine the location of the FAS domain boundaries. The PKS and FAS sequences are aligned separately as they do not align well together. While the HXXXGXXXXP, GYXYGPXF, and DXXX(Q/H) motifs can be identified in FAS DHs, it remains challenging to pinpoint the boundary between DH and the structural subdomain of KR. Mammalian FASs may possess a ~150 residue domain between DH and the KR structural subdomain. Insect FASs may not possess this domain; however, the DH/KR boundary is still unclear as β1 of the KR structural subdomain is difficult to identify. Man, Homo sapiens; Rat, Rattus norvegicus; Pig, Sus scrofa; Bee, Apis mellifera; Wasp, Nasonia vitripennis; Fly, Drosophila melanogaster; Mosq., Anopheles gambiae.