

Abstract
The primary cilium has critical roles in human development and disease, but the mechanisms that regulate ciliogenesis are not understood. Here we show that Tau tubulin kinase 2 (TTBK2) is a dedicated regulator of the initiation of ciliogenesis in vivo. We identified a null allele of mouse Ttbk2 based on loss of Sonic hedgehog activity, a signaling pathway that requires the primary cilium. Despite a normal basal body template, Ttbk2 mutants lack cilia. TTBK2 acts at the distal end of the basal body, where it promotes the removal of CP110, which caps the mother centriole, and promotes recruitment of IFT proteins, which build the ciliary axoneme. Dominant truncating mutations in human TTBK2 cause Spinocerebellar Ataxia type 11 (SCA11); these mutant proteins do not promote ciliogenesis and inhibit ciliogenesis in wild-type cells. We propose that cell cycle regulators target TTBK2 to the basal body, where it modifies specific targets to initiate ciliogenesis.
Introduction
Single non-motile cilia (primary cilia) are found on the surface of most vertebrate cells, both during embryogenesis and in the adult. During embryonic development, the primary cilium is required for the activity of the Sonic hedgehog (Shh) pathway (Huangfu et al., 2003; Goetz and Anderson, 2010). Because of the critical roles of Shh signaling in embryonic patterning, disruptions in primary cilia cause lethality at mid-gestation associated with defects in neural and limb patterning. Later in development and after birth, cilia are important for the development of most organ systems and for the maintenance of stem cells of some tissues. Mutations that disrupt the assembly or function of cilia cause a set of complex human genetic syndromes, collectively termed ciliopathies, which have phenotypes as diverse as obesity and cystic kidney disease (Badano et al., 2006; Goetz and Anderson, 2010; Green and Mykytyn, 2010).
Cilia are templated by the basal body, a modified form of the more mature of the cell's two centrioles, the mother centriole. During maturation of the basal body, the mother centriole forms two sets of specialized appendages: the subdistal appendages that mediate the attachment of the basal body to cytoplasmic microtubules; and distal appendages that mediate the docking of the basal body to the plasma membrane (Ishikawa and Marshall, 2011; Pedersen et al., 2008). The ciliary axoneme is assembled by the evolutionarily conserved process of intraflagellar transport (IFT). During IFT, proteins are transported into the cilium by Kinesin-2, a dedicated anterograde motor, in conjunction with two multiprotein complexes, IFT-A and IFT-B. Ciliary proteins are recycled to the base of the cilium by a dedicated dynein motor, cytoplasmic Dynein-2 (Iomini et al., 2001; Pedersen et al., 2008).
Despite the importance of primary cilia in development and disease, it is not known what controls the initiation of ciliogenesis or how ciliogenesis is regulated by the cell cycle or by cell type. The cilium is a dynamic organelle that is rapidly assembled and disassembled during the cell cycle to enable the centrioles to become components of the spindle poles during mitosis (Seeley and Nachury, 2010). Regulation of ciliogenesis by cell cycle factors must differ between cell types, as many cultured cells ciliate only after withdrawal from the cell cycle, whereas most rapidly cycling cells in mouse embryos have cilia (Fonte et al., 1971; Ocbina et al., 2011). In addition, although most cell types have primary cilia, some differentiated cell types such as pancreatic acinar cells and hepatocytes are not ciliated (Aughsteen, 2001; Wheatley et al., 1996), and cilia are lost during development of some tumors (Seeley and Nachury, 2010).
Analysis of the factors that control the decision of a cell whether or not to make cilia has focused on a set of negative regulators. The centriolar proteins CP110 and CEP97 cap the distal centrioles: knockdown of these proteins causes inappropriate formation of cilia in cycling cultured cells, and over-expression of CP110 is sufficient to block cilia formation in quiescent cells (Spektor et al., 2007). However, the mechanism by which CP110 is displaced from the mother centriole to enable cilia formation is unknown. Several positive regulators of cilia formation, including Ran, Rab8 and Rab11 have been identified, but there is, as yet, no evidence that any of these proteins regulate ciliogenesis in the animal (Fan et al., 2011; Westlake et al., 2011).
In the course of a genetic screen for mutations that affect neural patterning in the mouse embryo, we identified a gene required for Shh signaling because it was required for formation of primary cilia. Here we show that this gene encodes a serine-threonine kinase, Tau tubulin kinase 2 (TTBK2), and that TTBK2 is a critical regulator of the initiation of ciliogenesis in vivo. TTBK2 was first identified based on its physical association with microtubules and its ability to phosphorylate microtubule-associated proteins in vitro (Takahashi et al., 1995; Tomizawa et al., 2001). We show that TTBK2 is not required for the formation of mature basal bodies, but is required to initiate assembly of the ciliary axoneme. Truncating mutations in human TTBK2 cause the dominant human neurodegenerative disorder Spinocerebellar Ataxia type 11 (SCA11) (Bauer et al., 2010; Houlden et al., 2007). We find that the disease-associated variants of TTBK2 can inhibit ciliogenesis in wild-type cells, suggesting that the mutations cause disease by interfering with the product of the wild-type allele. The findings raise the possibility that cilia may play a role in preventing neural degeneration.
Results
A null mutation in mouse Ttbk2 blocks Shh signaling and ciliogenesis
We identified an ENU-induced mutation, bartleby (bby), based on morphological defects of homozygous embryos at midgestation similar to those seen in mutants that lack cilia, including holoprosencephaly, twisted body axis, abnormal limb development and randomized laterality of heart looping; the mutants die at midgestation (~E10.5) (Figure 1A, B). Mouse embryos that lack cilia show holoprosencephaly because they lack ventral neural cell types that depend on activation of the Shh pathway (Huangfu and Anderson, 2005; Huangfu et al., 2003; Liu et al., 2005; May et al., 2005). Cilia are also required for normal proteolytic processing of Gli3, the transcriptional repressor that keeps Shh target genes off in the absence of ligand. As a result, mutants that lack cilia show an increased basal level of target gene expression that causes phenotypes such as polydactyly (Huangfu and Anderson, 2005; Liu et al., 2005; May et al., 2005).
Figure 1. Altered Shh-dependent patterning in bby mutant embryos.

(A and B) Whole wild-type (WT) (A) and bby (B) mutant embryos at E10.5. (C-J) Neural patterning in WT and bby embryos at E10.5. Sections were taken at the forelimb level; the same pattern of expression of neural markers was seen at lumbar levels. Shh (C, D), Nkx2.2 (E, F) and Isl1 (G, H) are not expressed in the neural tube of bby mutant embryos. Pax6 (I, J) is expanded ventrally in bby mutants. Dorsal is up. Nuclei are stained with DAPI (blue). Scale bar =100 μm. (K-R) Limb patterning in WT and bby mutant embryos. Ptch1 (K, L) is not expressed in limb buds of bby embryos. Fgf4 (M, N) is expanded anteriorly in E10.5 bby mutants; expression of Hoxd11 (O, P) and Hand2 (Q, R) are expanded anteriorly in E11.5 bby mutants. Anterior is up. Please see also Figure S1 for information on the bby mutation.
As in mutants that lack cilia, a set of Shh-dependent ventral neural cell types were not specified in bby mutants, including Shh-expressing floor plate cells (Figure 1C, D), Nkx2.2-positive V3 interneuron progenitors (Figure 1E, F) and Isl1-positive motor neurons (Figure 1G, H); ventral cells in the mutant neural tube expressed Pax6, which is normally repressed by Shh in ventral neural progenitors (Figure 1I, J). Anterior-posterior patterning of the limb bud also depends on the activity of the Shh pathway (Zeller et al., 2009). Ptch1, a direct downstream target of Shh signaling, was robustly expressed in the posterior of the wild-type (WT) limbs but was absent in bby mutant embryos (Figure 1K, L), indicating that in the limb, as in the neural tube, Shh failed to activate target genes in bby embryos. Expression of Fgf4, Hand2 and Hoxd11, which are restricted to the posterior limb, was expanded anteriorly in bby mutants (Figure 1 M-R), which suggested that Gli3 repressor activity was decreased in the mutants, similar to cilia mutants (Huangfu and Anderson, 2005; Liu et al., 2005). Thus bby mutant embryos showed the same types of disruptions of Shh-dependent patterning in the neural tube and limb that are seen in mutants that lack cilia.
Because of the phenotypic similarities of bby embryos and mutants with defects in cilia, we analyzed cilia formation in bby mutants. Arl13b is a sensitive and specific marker for cilia in wild-type cells (Caspary et al., 2007), but no Arl13b-positive cilia were detected in the bby neural tube (Figure 2A, B) or in mouse embryonic fibroblasts (MEFs) derived from bby embryos (Figure 2C, D). Nearly every cell in the wild-type E10.5 neural tube had a single cilium that could be detected by scanning electron microscopy (SEM) (Figure 2E, arrowheads), but cilia were not detected by SEM in the bby neural tube (Figure 2F).
Figure 2. Ttbk2bby embryos lack neural and mesenchymal cilia.

(A and B) Arl13b (red) staining shows cilia in the WT E10.5 ventral neural tube (A) and the absence of cilia in the Ttbk2bby neural tube (B). (C and D) Arl13b (red) and γ-tubulin (green) in embryonic fibroblasts derived from WT (C) and Ttbk2bby mutant embryos (D). DAPI is in blue. Scale bar = 10 μm. (E and F) Scanning electron micrographs of the apical surface of the neural tube (viewed en face) in E10.5 WT (E) and Ttbk2bby mutant (F) embryos. Scale bar = 1 μm. (G -J) Transmission electron micrographs of cilia in the E10.5 neural tube of WT (G) and Ttbk2bby mutant (H) embryos. Arrowheads denote the distal appendages. Cross sections through the basal body in WT (I) and Ttbk2bby mutant embryos (J) show the presence of the nine triplet microtubules in the centriole of the mutant. Scale bars = 200 nm. Please see Figure S2 for additional TEM images.
We mapped the bby mutation by meiotic recombination to a 1 Mb region on chromosome 2 (Figure S1A). Analysis of the gene sequences in the interval identified an A->T nucleotide substitution in the coding region of Tau tubulin kinase 2 (Ttbk2). This nucleotide change converted lysine 142 to a stop codon, truncating the protein within the kinase domain (Figure S1B). TTBK2 is a member of the Casein Kinase 1 family of serine-threonine protein kinases (Qi et al., 2009) and was first identified based on its physical association with microtubules (Takahashi et al., 1995). Ttbk2 was also identified in siRNA-based screens for kinases that modulate Hh signaling (Evangelista et al., 2008) and for genes important in cilia formation (Kim et al., 2010), and embryos homozygous for a targeted knock-in allele of Ttbk2 die at midgestation with a morphology similar to that of bby embryos (Bouskila et al., 2011); we therefore concluded that the mutation in the Ttbk2 gene was responsible for the bby phenotype.
Ttbk2 is required to initiate ciliogenesis from the basal body
To define the cellular and molecular basis of the defect in ciliogenesis, we examined basal bodies and cilia in the E10.5 Ttbk2bby mutant neural tube by transmission electron microscopy (TEM). Although ciliary axonemes were not found in Ttbk2bby mutant tissue (Figure 2G, H; Figure S2), the centrioles in Ttbk2bby cells had the normal 9 triplets of microtubules (Figure 2 I, J). The basal bodies (modified mother centrioles) in Ttbk2bby cells were correctly apposed to the membrane, and both the subdistal appendages that anchor the centriole to microtubules and the distal appendages that help anchor the basal body to the cell surface were apparent in the micrographs (Figure 2G, H; Figure S2). Despite normal docking of the basal body to the cell surface, no cilium extending from the basal body was detected by TEM.
We then examined centrosomal and ciliary markers by immunofluorescence in MEFs derived from Ttbk2bby mutant embryos. Ninein, a marker of the subdistal appendages of the mother centriole (Delgehyr et al., 2005), was present at the mutant centrosome, as in WT (Figure 3A; Figure S3A). CEP164, a distal appendage protein that is required for ciliogenesis in cultured cells (Graser et al., 2007), was also associated with the centrosome in Ttbk2bby cells (Figure 3B; Figure S3B). Thus, markers of the subdistal and distal appendages were correctly localized to the basal body in Ttbk2bby cells, consistent with the normal appearance of the appendages seen in TEM.
Figure 3. Normal localization of appendage and transition zone markers in Ttbk2bby basal bodies.
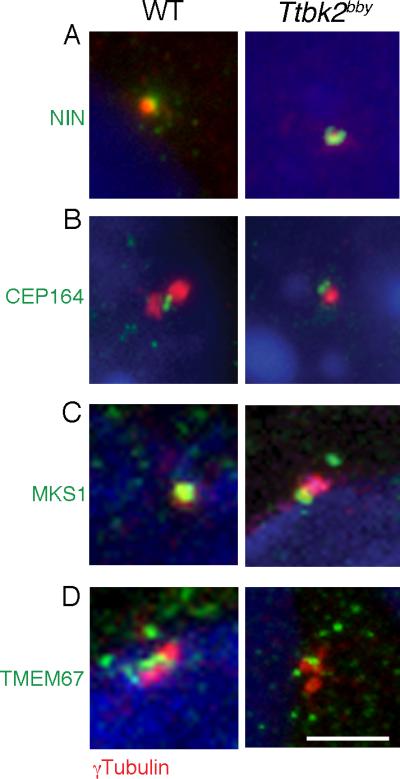
(A) The subdistal appendage marker Ninein (NIN; green) is expressed normally in Ttbk2bby MEFs. γ-tubulin (red) labels the centrioles. Images shown are representative of 14 cells imaged for Ttbk2bby and 12 for wild-type. (B) Localization of the distal appendage-associated protein CEP164 (green) distal to the centrosome, marked by γ-tubulin (red) in MEFs derived from WT and Ttbk2bby mutant embryos. 23 cells were imaged for Ttbk2bby and 20 for wild-type. (C, D) Transition zone proteins MKS1 (C) and TMEM67 (D) (green) are expressed in the transition zone of WT and at the distal basal body in Ttbk2bby cells. Centrosomes are labeled by γ-tubulin (red). Scale bar = 2 μm. Total number of cells imaged for each marker was: MKS1- 17 for Ttbk2bby; 13 for wild-type; TMEM67- 19 for Ttbk2bby; 22 for wild-type. Similar images were obtained from a minimum of 2 independent experiments. Please see Figure S3 for additional images.
The transition zone, which has been proposed to regulate the entry and exit of proteins and protein complexes into the ciliary axoneme (Craige et al., 2010; Ishikawa and Marshall, 2011), lies between the centriolar appendages and the ciliary axoneme. MKS1 and TMEM67, components of a large protein complex at the transition zone (Dowdle et al., 2011; Garcia-Gonzalo et al., 2011), were correctly localized to the mother centriole in Ttbk2bby mutant cells (Figure 3C, D; Figure S3C, D). Thus, these components of the mature basal body assembled correctly in the absence of TTBK2, despite a failure of axoneme elongation.
IFT is required to assemble the microtubule doublets of the ciliary axoneme from the basal body template (Pedersen et al., 2008). Both IFT140, a component of the IFT-A complex, and IFT88, a component of the IFT-B complex, localized to the transition zone and the ciliary axoneme in wild-type cells (Figure 4A, B; Figure S3E, F). In Kif3a-/- mutants, which lack the anterograde kinesin-II IFT motor, both IFT140 and IFT88 localized to the basal body (Figure 4A, B; Figure S3E, F), although no axoneme was extended. IFT140 also localized to the basal body in the IFT-B mutants Ift88-/- and Ift172wim/wim, which lack cilia, and IFT88 was present at the basal body of Ift172wim/wim cells (Figure S4A, B, D). Thus, IFT proteins can be recruited to the basal body under conditions where IFT is not actively promoting cilia formation. In contrast, Ttbk2bby mutant cells lacked detectable IFT88 or IFT140 at the basal body (Figure 4A, B; Figure S3E, F). We conclude, therefore, that TTBK2 acts upstream of IFT and is required for the recruitment or retention of IFT complexes at the basal body prior to cilia assembly.
Figure 4. TTBK2 acts upstream of IFT and CP110.

(A) IFT140 (green), an IFT-A complex protein, localizes primarily to the transition zone in WT and can be seen at lower levels in the axoneme. IFT140 is present at the mother centriole (red, γ-tubulin) in Kif3a-/- MEFs, but is not detectable at the Ttbk2bby centrosomes. 56 Ttbk2bby, 17 Kif3a-/- and 27 WT cells were examined. (B) The IFT-B complex component IFT88 (green) localizes to transition zone and ciliary axoneme in WT MEFS and to the mother centriole (red) in Kif3a-/- MEFs, but is not associated with the centrosomes of Ttbk2bby cells. Centrosomes are labeled with γ-tubulin (red). 52 Ttbk2bby cells, 21 Kif3a-/-- and 31 WT cells were examined. (C) CP110 (green) is present on the distal end of the WT daughter centriole (red), the centriole that does not template the ciliary axoneme (purple). CP110 is detected on one of the two centrosomes in Kif3a-/- MEFs. In Ttbk2bby cells, CP110 is present on both centrosomes in most cells. Scale bar is 2 μm. (D) The percentage of cells with CP110 seen on both centrosomes in WT (white bar, 16.0+/-5.2) and Ttbk2bby (black bar, 88.8+/-3.6) MEFs. Error bars represent the SEM. p < 0.0001, 5 fields of cells were counted for a total of 114 Ttbk2bby and 116 WT cells. Please see Figures S3 and S4 for additional images.
CP110 is a centriolar protein that caps the distal end of centrioles (Chen et al., 2002), and removal of CP110 correlates with the ability to initiate ciliogenesis in cultured cells (Spektor et al., 2007). As expected, we observed that CP110 was localized only to the daughter centriole (the centriole not associated with a cilium) in the vast majority of wild-type MEFs that had been serum-starved to induce cilia formation. Similarly, CP110 was restricted to one of the two centrosomes in Kif3a-/- and Ift88-/- cells, (Figure 4C; Figure S3G; Figure S4C). In contrast, CP110 was present on both centrosomes in 88.8% of Ttbk2bby cells (Figure 4C, D; Figure S3G, Figure S4F,G). Thus TTBK2 is required for removal of CP110 from the mother centriole to allow ciliogenesis.
Ttbk2 localizes to the transition zone in response to cell cycle signals that promote ciliogenesis
To determine whether TTBK2 could directly control cilia formation at the basal body, we expressed tagged forms of mouse TTBK2 in wild-type MEFs and assessed their localization within the cell. TTBK2 tagged at the N-terminus with eGFP (mTTBK2::eGFP ) localized to the transition zone of ciliated cells, between the axoneme and the basal body (Figure 5A). In cells where the ciliary axoneme had not yet extended, tagged TTBK2 was seen on one of the two centrosomes (Figure 5B). TTBK2 tagged at the C-terminus with V5 also localized to the transition zone of ciliated cells (Figure 5C), where it co-localized with IFT88 (Figure 5D), consistent with a role for TTBK2 in mediating the recruitment or retention of IFT complexes to this compartment. In contrast, we did not detect TTBK2 at the spindle poles of dividing cells (data not shown). An antibody to human TTBK2 confirmed that the protein localized to the ciliary transition zone of human RPE cells (Figure 5E). Thus, the TTBK2 protein was present at the basal body and transition zone, where it could directly regulate the initiation and maintenance of ciliogenesis.
Figure 5. TTBK2 localizes to the basal body in response to serum withdrawal.

(A-D) Localization of TTBK2 fusion protein constructs expressed in mouse cells. (A, B) WT MEFs were infected with a mouse N-terminal-eGFP-TTBK2 construct. TTBK2-eGFP (green) is present in the transition zone between the mother centriole, labeled with γ-tubulin (purple), and the axoneme, labeled by acetylated α-tubulin (red), in ciliated cells (A), and is localized to one of the two centrosomes in non-ciliated cells (B). (C, D) WT MEFs infected with mouse TTBK2 with a C-terminal V5 tag (green); TTBK2-V5 also localizes to the transition zone (C). TTBK2-V5 co-localizes with IFT88 (red) in the transition zone (D). Insets show high magnification images that highlight the overlap of IFT88 and TTBK2 at the base of the cilium. (E) An antibody to human TTBK2 (green) shows that the endogenous protein localizes to the transition zone of human RPE cells. Scale bar = 2 μm. (F-K) WT MEFs stably expressing mTTBK2::eGFP after shift to serum-free medium. 48 hours of serum withdrawal, cells were shifted back to growth media induce re-entry into the cell cycle (K). Localization of mTTBK2::eGFP was assessed using an antibody against GFP (green), and cells were counterstained with γ-tubulin (purple) to label the centrosome and acetylated α-tubulin (red) to label the ciliary axoneme. (L) Line graph shows the mean percentage of cells with centrosomal localization of mTTBK2::eGFP (green) the mean percentage of ciliated cells (red), and the mean percentage of cells with CP110 on both centrioles (black) at each time point from 4-6 randomly selected fields of cells from at least two different slides; see Supplemental Methods. Error bars indicate SEM. See also Figure S5.
The initiation of cilia formation is coupled to the cell cycle, as cilia are not found on most cells undergoing mitosis (Rieder et al., 1979; Seeley and Nachury, 2010; Wheatley et al., 1996), and it is known that a key negative regulator of cilia assembly, CP110, is removed from the mother centriole in a cell-cycle dependent manner (Spektor et al., 2007). However, no pathway that links cell cycle signals to ciliogenesis has been defined. In fibroblasts, efficient cilia formation is triggered by serum starvation, high cell density and withdrawal of cells from the cell cycle (Seeley and Nachury, 2010). We therefore compared the kinetics of cilia induction with the localization of TTBK2 to the centriole in serum-starved MEFs. In asynchronously dividing fibroblasts in growth media, we observed that approximately 6% of cells were ciliated (Figure 5L), as assayed by presence of an acetylated α-tubulin-positive axoneme adjacent to a γ-tubulin-positive centrosome. mTTBK2::eGFP was diffusely cytoplasmic in most of these asynchronous cells, and was enriched at one of the two centrosomes in about a quarter (23.1%) of the cells (Figure 5F, L). Similar low frequencies of both centrosomal TTBK2 localization and ciliation were observed after 6 hours of serum starvation (Figure 5G, L). At 12 hours of serum starvation, TTBK2::eGFP had accumulated at one of the two centrosomes of most (71.9%) cells (Figure 5H, I, L), whereas only 13.2% of cells were ciliated at that time point. After 48 hours of serum starvation, nearly half of the cells were ciliated (43.7%), and the fraction of cells with TTBK2 at the basal body remained high (64.4%) (Figure 5J, L). A similar time course of ciliation was seen using Arl13b as a cilia marker instead of acetylated α-tubulin (Figure S5A). Recruitment of TTBK2 to the basal body therefore preceded the induction of detectable cilia formation in quiescent fibroblasts.
Re-addition of growth media to quiescent serum-starved cells causes rapid disassembly of cilia, a process that requires the Aurora A centrosomal kinase (Pugacheva et al., 2007). Six hours after serum re-addition to cells that had been starved for 48 hours, the majority of cells lacked cilia. In these cells, TTBK2 was no longer enriched at the mother centriole and was instead again diffusely cytoplasmic (Figure 5K, L). Thus, removal of TTBK2 from the basal body accompanied cilia disassembly.
We also followed the localization of CP110, a negative regulator of ciliogenesis, in this serum-starvation time course (Figure 5L, Figure S5B-G). Prior to serum withdrawal, CP110 was found on both centrosomes in 56.4% of asynchronously growing fibroblasts. Six hours after serum withdrawal, CP110 was detected on both centrosomes in a slightly smaller fraction of cells (49.0%). At 12 hours of serum starvation, the time point when TTBK2 was found at the mother centriole in the majority of cells, the fraction of cells with CP110 at both centrosomes dropped to 24.8%. By 48 hours, when most cells were ciliated, only 11.4% of cells had CP110 on both centrosomes. Upon re-addition of serum for 6 hours, when nearly all cilia had been disassembled, the percentage of cells with CP110 on both centrosomes increased to 38.5% Thus in response to serum withdrawal, the reduction of CP110 at the mother centriole paralleled the enrichment of TTBK2 at this structure prior to the initiation of ciliogenesis. The results suggest that TTBK2 activity at the centrosome may lead to the removal of CP110, either directly or indirectly, in order to prime the basal body for cilia formation.
Rescue of the Ttbk2bby ciliogenesis defect by wild-type and mutant constructs
To establish an assay to define important structural features of the TTBK2 protein, we tested whether expression of tagged forms of TTBK2 (Figure 6A) could rescue the defects in ciliogenesis in Ttbk2bby mutant MEFs. Cilia are induced in wild-type MEFs after removal of serum, but no cilia were ever observed in uninfected Ttbk2bby MEFs under the same conditions. In contrast, 52.7 +/- 3.5% of Ttbk2bby cells infected with mTTBK2::eGFP formed cilia of normal length (Figure 6B, F), which was not significantly different from the fraction of ciliated cells seen in control wild-type MEFs (57.3 +/- 3.6%). This rescue experiment confirmed that the ciliogenesis defect seen in Ttbk2bby cells was due to the absence of functional TTBK2 and provided a quantitative assay for structure-function studies.
Figure 6. TTBK2 kinase activity is required for ciliogenesis and the C-terminal domain is required for localization to the centriole.

(A) Schematics of the mouse Ttbk2 truncations used in rescue experiments. (B-E) Cilia are marked by acetylated α-tubulin (red); centrosomes by γ-tubulin (magenta); GFP-tagged TTBK2 is in green. (B) WT mouse TTBK2::eGFP localizes to the transition zone and rescues the Ttbk2bby ciliogenesis defect. (C) The kinase-dead mTTBK2D163A::eGFP construct localizes to the centrosome but does not rescue ciliogenesis. (D) The kinase domain alone (mTTBK213-306::eGFP) localized inefficiently to the centrosome and rescued ciliogenesis weakly. (E) The C-terminal tail (mTTBK2307-1243::eGFP) localizes to the centrosome but does not rescue ciliogenesis. Scale bar = 2 μm. (F) Graph shows the percentage of ciliated cells observed in Ttbk2bby (black bars) or WT (white bars) MEFs infected with the indicated construct. (G) Graph showing the percentage of cells with centrosomal localization of GFP for the indicated construct. For F and G, bars represent the mean percentage of ciliated cells (F), or cells with centriolar GFP (G) from 4-8 randomly selected fields of cells per condition, and fields were selected from at least two different slides and had a minimum of 25 cells per field; see Supplemental Methods. Error bars indicate the SEM. Please see also Figure S6.
We used the rescue assay to test the importance of the kinase activity of TTBK2. Mutation of a conserved aspartic acid residue within the catalytic domain to alanine has been shown to block TTBK2 activity in in vitro kinase assays (TTBK2D163A) (Bouskila et al., 2011). This kinase-dead form of mouse TTBK2 generated a stable protein (Figure S5A) and localized correctly to the mother centriole (Figure 6C, G), but failed to rescue cilia in Ttbk2bby mutant cells (Figure 6F). Thus, the kinase activity of TTBK2 was required to promote ciliogenesis.
TTBK2 has a long (936AA) conserved tail C-terminal to the kinase domain. A construct encoding only the kinase domain of TTBK2 and lacking the C-terminus (TTBK213-306) had a dramatically decreased ability to rescue cilia formation: only 2.2% of Ttbk2bby cells infected with this construct were ciliated. This truncated protein was detected at centrioles only at very low frequency (1.9%; Figure 6D, G), although a stable protein was detected on Western blots (Figure S6A). A construct that expressed the C-terminal 936 amino acids of the protein, without the kinase domain (TTBK2307-1243), localized to the mother centriole at approximately half the efficiency of the wild-type protein (Figure 6E, G), but did not rescue cilia formation (Figure 6F). Thus, the C-terminal tail was required to recruit TTBK2 to the centriole, and the kinase domain was required to promote ciliogenesis.
Truncating mutations associated with SCA11 inhibit ciliogenesis
Mutations in human TTBK2 appear to be the cause of the neurodegenerative disorder SCA11 (Bauer et al., 2010; Houlden et al., 2007). The three familial mutations associated with SCA11 are predicted to produce very similar truncated proteins that include the kinase domain and 122-137 amino acid residues C-terminal to the kinase domain, but lack the remainder of the long C-terminus of the protein. SCA11, like other forms of Spinocerebellar Ataxia, is a dominant disorder, although it is not clear whether the disease is caused by haploinsufficiency of TTBK2 or by abnormal activity of the mutant protein. We therefore generated human TTBK2::eGFP constructs harboring two of the familial mutations described previously (Houlden et al., 2007) (hTTBK2Fam1 and hTTBK2Fam2, truncating the protein after 443 and 428 AA, respectively; Figure 7A) and compared the ability of the wild-type hTTBK2 (human TTBK2) and the two human mutants to localize to the mother centriole and to rescue the cilia defect in Ttbk2bby MEFs. Like mouse TTBK2, wild-type human TTBK2::eGFP localized to the mother centriole and rescued ciliogenesis in mouse Ttbk2bby MEFs (Figure 7B, E), although the heterologous hTTBK2::eGFP rescued cilia in fewer mouse cells than did the wild-type mouse construct (20.9% vs. 52.7%) (Figures 6F, 7E). The localization of hTTBK2::eGFP to the mother centriole of mouse cells was also less efficient than that of the mouse construct (20.5% vs. 57%) (Figures 6G, 7F).
Figure 7. SCA11-associated variants of hTTBK2 do not rescue cilia formation in Ttbk2bby cells and interfere with ciliogenesis in WT cells.

(A) Schematic of human TTBK2, which is 89% identical to mouse TTBK2; and two familial SCA11 mutations that truncate the protein at 443 or 428 AA. (B-D) Localization of WT hTTBK2::eGFP (A) and SCA11-associated Fam1 and Fam2 (C and D) variants expressed in Ttbk2bby MEFs. GFP is in green, γ-tubulin is in purple, and acetylated α-tubulin is in red. The Fam1 and Fam2 fusion proteins generally localized diffusely in the cytoplasm; C show a rare cell in which GFP is localized to centrosome. Scale bar = 2 μm. (E) Graph showing the percentage of ciliated cells observed in Ttbk2bby mutant (black bars) or WT (white bars) MEFs infected with the indicated construct. (F) Graph showing the percentage of cells with centrosomal localization of GFP for the indicated construct. For E and F, bars represent the mean percentage of ciliated cells (E) or cells with centriolar GFP (F) from 4-8 randomly selected fields of cells per condition. Fields were selected from at least two different slides and had a minimum of 25 cells per field; see Supplemental Methods. Error bars indicate the SEM. Please see also Figure S6.
We then examined the localization and activity of the SCA11-associated forms of the human protein. Both mutant human proteins localized to the mother centriole, indicating that these truncated proteins retain sequences sufficient for centrosomal localization, although the efficiency of localization to the centrosome was reduced approximately 7-fold compared to the wild-type human protein (Figure 7C, D, F). Both SCA11-associated forms produced a stable protein product (Figure S6B), however neither showed any rescue of the ciliogenesis defect of Ttbk2bby cells (Figure 7E), suggesting that the protein was inactive even when it was localized to the centriole. Because of the dominant inheritance of SCA11, we also measured the percentage of ciliated cells in wild-type MEFs that stably expressed these constructs. There was a modest, but significant (p= 0.017), decrease in the proportion of ciliated cells infected with hTTBK2Fam1 and hTTBK2Fam2 constructs (45.8% and 44.0%), compared to those infected with the wild-type hTTBK2 construct (59.4%; Figure 7E). These data suggest that, in addition to being unable to promote cilia formation, the proteins encoded by human SCA11-associated TTBK2 alleles may interfere with the function of the wild-type gene product. Thus, the disease-causing SCA11 mutations could act as antimorphic alleles, accounting for the dominant inheritance of SCA11.
Discussion
A TTBK2 pathway for the initiation of ciliogenesis
The data presented here define the specific role of the serine-threonine kinase TTBK2 in mouse development: it acts at the mature basal body to promote the initiation of ciliogenesis. The embryonic phenotypes seen in Ttbk2bby mutant embryos can all be traced to defects in ciliogenesis, as Ttbk2bby embryos arrests at mid-gestation with defects in Shh signaling, the same phenotype caused by the absence of primary cilia.
In the absence of TTBK2, a mature basal body docks at the apical membrane, but no ciliary axoneme extends from the cell surface. TTBK2 is localized to the distal end of the mother centriole, where it promotes ciliogenesis through the displacement of the negative regulator CP110 and the enrichment of IFT complex components at the base of the cilium. This set of defects appears to be unique to Ttbk2bby mutant cells, as some IFT proteins can localize normally in Kif3a-/-, Ift88-/- and Ift172wim/wim mutants that lack other components of the IFT machinery, despite the failure of all of these mutants to extend an axoneme. The OFD1 protein is also required for the recruitment of the IFT complexes to the basal body, but the OFD1 mutant basal body is elongated and fails to recruit appendage proteins (Singla et al., 2010), whereas the mother centrioles in Ttbk2bby mutant cells have the normal structure and normal appendage proteins. Thus, TTBK2 promotes the initial extension of the ciliary axoneme at a step upstream of the recruitment of the IFT machinery to the mature basal body.
The data show that TTBK2 is required for the displacement CP110 from the basal body. Cell-based studies have indicated that CP110 is a negative regulator of cilia formation that, together with CEP97 (Spektor et al., 2007), CEP290 (Tsang et al., 2008), and KIF24 (Kobayashi et al., 2011), prevents formation of cilia at inappropriate stages of the cell cycle. Displacement of CP110 from the mother centriole appears to be required for the initiation of ciliogenesis, as CP110 is never associated with a basal body connected to a cilium, and over-expression of CP110 suppresses cilia formation in quiescent cells (Spektor et al., 2007). CP110 is confined to one of the two centrioles in serum-starved Kif3a-/- and Ift88-/- mutant cells, implying that retention of CP110 on both centrioles is not a general hallmark of non-ciliated cells, and instead reflects a specific defect in Ttbk2bby mutant cells. After removal of serum, CP110 is lost from the mother centriole with the same kinetics as TTBK2 is recruited at the mother centriole, prior to cilia formation. Thus it appears that recruitment of TTBK2 to the mother centriole in response to cell cycle signals immediately precedes the removal of CP110 from the mother centriole, which suggests that TTBK2 may initiate ciliogenesis by phosphorylating one or more of the proteins in the CP110/CEP97/CEP290/KIF24 cilia-suppression pathway.
Although a small number of other kinases, including NEK1, NEK8, Aurora A, CCRK and MAK regulate aspects of cilia structure and function (Ko et al., 2010; Omori et al., 2010; Otto et al., 2008; Pugacheva et al., 2007; Thiel et al., 2011), TTBK2 is the only kinase identified to date that is specifically required for the initiation of ciliogenesis. Identification of TTBK2 substrates at the basal body should define the events that initiate ciliogenesis, and identification of upstream regulators that control the activity of TTBK2 may define both cell cycle and cell type-specific mechanisms that regulate cilia formation. The position of TTBK2 at the nexus of cell cycle regulators and the initiation of ciliogenesis suggest that TTBK2 may provide an attractive target for small molecule manipulation of cilia formation.
TTBK2 and SCA11
To date, 27 genes associated with different forms of SCA have been identified, but the cellular and molecular bases for this pathology remain poorly defined (Matilla-Duenas, 2012). SCA11 has been linked to three different familial mutations that are all predicted to produce nearly identical proteins that truncate 122-137 amino acids C-terminal to the kinase domain. SCA11, like other SCA subtypes, is an autosomal dominant disorder; however, the molecular basis by which these mutations in TTBK2 result in a dominant neurological disorder is not clear (Bauer et al., 2010; Bouskila et al., 2011; Houlden et al., 2007). We find that the SCA11-associated variants of human TTBK2 can make stable proteins, but we find that these proteins do not rescue cilia formation when expressed in Ttbk2bby mutant cells.
The SCA11-associated forms of the TTBK2 protein can localize to the basal body, albeit inefficiently, which is consistent with our observation that expression of the SCA11 mutants in wild-type cells caused a reduction in the percentage of ciliated cells. This suggests that the mutant alleles not only attenuate TTBK2 function, but also interfere with the function of wild-type TTBK2. In contrast, the mouse kinase-domain only (TTBK213-306) construct, which localized to the centriole at about the same frequency as the mutant human proteins, did not interfere with wild-type ciliogenesis. The first biochemical studies of TTBK2 showed that a truncated form of the protein that terminated immediately after the kinase domain, similar to TTBK213-306, was an active kinase (Tomizawa et al., 2001). Thus, it is likely that the 122-137 amino acids C-terminal to the kinase domain mediate association with another protein that inhibits activity of the kinase domain. We hypothesize that the proteins that inhibit the SCA11 forms of TTBK2 reside at the centriole and that the SCA11 proteins inhibit the activity of wild-type TTBK2 at the basal body in trans. Because our experiments are based on over-expression of mutant TTBK2 in wild-type cells, the disruption of ciliogenesis caused by these constructs may be due to higher levels of the mutant protein than would be present in individuals with SCA11, who are heterozygous for these mutations. It will therefore be important to examine cilia formation in mouse models in which the human SCA11-associated mutations are present at the endogenous Ttbk2 locus.
Based on our data, we suggest the hypothesis that ciliogenesis may be perturbed in individuals with SCA11. However, we cannot distinguish whether the pathology of SCA11 is due to interference with ciliogenesis or to other targets of TTBK2 in the nervous system. TTBK2 can phosphorylate β-tubulin and the microtubule-associated proteins Tau and MAP2 in vitro (Takahashi et al., 1995; Tomizawa et al., 2001), and it has been hypothesized that SCA11 is caused by misregulation of Tau (Houlden et al., 2007). However, the finding that TTBK2 is essential for ciliogenesis and the specificity of the Ttbk2 mutant phenotype in the animal raises the possibility that cilia are important in the adult CNS to prevent neural degeneration.
There are a number of indications that cilia have important functions in the adult brain. Shh and cilia are required for maintenance of adult neural stem cells (Ahn and Joyner, 2005; Han et al., 2008). The SCA10-associated protein ATXN10 (Matsuura et al., 2000) has been shown to be part of a protein complex localized to the basal body and is mutated in several cases of nephronophthisis, a ciliopathy (Sang et al., 2011). A number of transmembrane proteins known to be important in complex neurological functions such as somatostatin receptor 3 (SSTR3), serotonin receptor 6 (Berbari et al., 2008), a subset of dopamine receptors (DR) (Domire et al., 2010; Marley and von Zastrow, 2010), and the p75 neurotrophin receptor (Chakravarthy et al., 2010) have all been found to localize to cilia. The localization of SSTR3 and DR1 to cilia is disrupted in individuals with the ciliopathy Bardet Biedl syndrome (BBS) (Berbari et al., 2008; Domire et al., 2010) and may contribute to the disease phenotypes. In the future, genetic removal of TTBK2 in the adult brain should make it possible to test whether TTBK2 prevents neural degeneration through cilia or through other mechanisms.
Experimental Procedures
Imaging
For SEM, E10.5 embryos were dissected in PBS at room temperature and immediately fixed in 2.5% glutaraldehyde and 2% PFA in 0.1 M sodium cacodylate buffer, pH 7.4 (Electron Microscopy Sciences) overnight at 4°C. The neural tube of the embryo at the level of the forelimbs and cut open to allow en face imaging of the apical surface of the neural epithelium. This tissue was processed as described (Huangfu et al., 2003). Scanning electron micrograph images were taken on a Zeiss SUPRA 25 FESEM. Samples for TEM were processed as described (Liem et al., 2012) and viewed on a JEOL 1200EX transmission electron microscope at 80kV.
MEFs were isolated from E10.5 embryos and prepared, fixed and stained as described (Ocbina and Anderson, 2008; Ocbina et al., 2011).Confocal microscopy was performed using an upright Leica TCS SP5 AOBS laser scanning microscope. Images were taken with a 63X water objective and 1.7X zoom. Z-stacks were taken at 0.20 μm intervals. Extended views of the confocal datasets were processed using the Volocity software package (Improvision). The percentage of ciliated cells was measured as the proportion of cells with Arl13b localized to the cilium in 4-8 randomly selected fields of cells per condition. Fields were selected from at least two different slides and had a minimum of 25 cells per field. Slides were imaged as described above, except that they were imaged using a 40X water objective with 1X zoom to increase the number of cells counted in each field. Cells were counted using ImageJ. In situ hybridization was performed on E10.5 and E11.5 embryos as previously described (Weatherbee et al., 2009).
TTBK2::GFP and ::V5 Constructs
Mouse and human Ttbk2 Gateway-ready clones were obtained Open Biosystems. Truncations were generated by PCR and cloned into the Gateway pENTR vector (Invitrogen). The mTtbk2D163A, hTtbk2Fam1 and hTtbk2Fam2 mutations were generated by site directed mutagenesis using the Quick Change Mutagenesis Kit (Agilent) according to manufacturer's instructions. These clones were then transferred into Gateway Destination vectors by recombination using Gateway LR Clonase (Invitrogen) according to manufacturer's instructions. For C-terminal V5 tagged constructs, Gateway pcDNA-DEST40 was used, for N-terminal GFP tagged constructs, a retroviral vector expression vector modified to contain eGFP and FLAG was used.
Retroviral Transduction
Inserts were cloned into a pQCXIN retroviral expression vector (Clontech) modified to contain FLAG and eGFP upstream of the Gateway cloning elements (Burkard et al., 2007; Terret et al., 2009) (gift of P. Jallepalli) and co-infected with the pVSV-G pantropic envelope vector into Phoenix cells using Lipofectamine 2000 (Invitrogen). Retrovirus-containing supernatants were collected after 48 hours and diluted 1:1 in complete growth medium. Virus-containing medium was then added to target cells (wild-type or Ttbk2bby mutant MEFs immortalized by serial passaging) and incubated overnight. The medium was replaced by complete growth medium; after 24 hours, the cells were passaged into complete growth medium containing 500 μg/mL G418 for selection. The stability of each construct was assessed by infection of 293T cells. For each construct, one 10 cm plate of infected cells was analyzed by Western blotting with an antibody against GFP to detect the fusion proteins.
Supplementary Material
Acknowledgments
We are grateful to P. Jallepalli and B. Tsou and their laboratories for reagents and advice on retroviral transduction. We thank James Sillibourne and Michel Bornens (Institut Curie), Brian Dynlacht (NYU), and Greg Pazour (U. Massachusetts Medical School) for antibodies. We thank the MSKCC Molecular Cytology Core Facility and the Bio-Imaging Resource Center at Rockefeller University for help with imaging; the MSKCC Electron Microscopy Core Facility for SEM; Peter Satir and Leslie Gunther at the Analytical Imaging Center of Albert Einstein College of Medicine for assistance with TEM. We thank A. Parrish, H. Bazzi and F. Bangs for comments on the manuscript. This work was funded by NIH grant NS044385 to KVA and an American Cancer Society Postdoctoral Fellowship to SCG.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahn S, Joyner AL. In vivo analysis of quiescent adult neural stem cells responding to Sonic hedgehog. Nature. 2005;437:894–897. doi: 10.1038/nature03994. [DOI] [PubMed] [Google Scholar]
- Aughsteen AA. The ultrastructure of primary cilia in the endocrine and excretory duct cells of the pancreas of mice and rats. Eur J Morphol. 2001;39:277–283. doi: 10.1076/ejom.39.5.277.7380. [DOI] [PubMed] [Google Scholar]
- Badano JL, Mitsuma N, Beales PL, Katsanis N. The ciliopathies: an emerging class of human genetic disorders. Annu Rev Genomics Hum Genet. 2006;7:125–148. doi: 10.1146/annurev.genom.7.080505.115610. [DOI] [PubMed] [Google Scholar]
- Bauer P, Stevanin G, Beetz C, Synofzik M, Schmitz-Hubsch T, Wullner U, Berthier E, Ollagnon-Roman E, Riess O, Forlani S, et al. Spinocerebellar ataxia type 11 (SCA11) is an uncommon cause of dominant ataxia among French and German kindreds. J Neurol Neurosurg Ps. 2010;81:1229–1232. doi: 10.1136/jnnp.2009.202150. [DOI] [PubMed] [Google Scholar]
- Berbari NF, Lewis JS, Bishop GA, Askwith CC, Mykytyn K. Bardet-Biedl syndrome proteins are required for the localization of G protein-coupled receptors to primary cilia. Proc Natl Acad Sci USA. 2008;105:4242–4246. doi: 10.1073/pnas.0711027105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouskila M, Esoof N, Gay L, Fang EH, Deak M, Begley M, Cantley LC, Prescott A, Storey KG, Alessi DR. TTBK2 kinase substrate specificity and the impact of spinocerebellar ataxia-causing mutations on expression, activity, localisation and development. Biochem J. 2011;427:157–167. doi: 10.1042/BJ20110276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkard ME, Randall CL, Larochelle S, Zhang C, Shokat KM, Fisher RP, Jallepalli PV. Chemical genetics reveals the requirement for Polo-like kinase 1 activity in positioning RhoA and triggering cytokinesis in human cells. Proc Natl Acad Sci s USA. 2007;104:4383–4388. doi: 10.1073/pnas.0701140104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspary T, Larkins CE, Anderson KV. The graded response to Sonic Hedgehog depends on cilia architecture. Dev Cell. 2007;12:767–778. doi: 10.1016/j.devcel.2007.03.004. [DOI] [PubMed] [Google Scholar]
- Chakravarthy B, Gaudet C, Menard M, Atkinson T, Chiarini A, Dal Pra I, Whitfield J. The p75 neurotrophin receptor is localized to primary cilia in adult murine hippocampal dentate gyrus granule cells. Biochem Biophys Res Commun. 2010;401:458–462. doi: 10.1016/j.bbrc.2010.09.081. [DOI] [PubMed] [Google Scholar]
- Chen Z, Indjeian VB, McManus M, Wang L, Dynlacht BD. CP110, a cell cycle-dependent CDK substrate, regulates centrosome duplication in human cells. Dev Cell. 2002;3:339–350. doi: 10.1016/s1534-5807(02)00258-7. [DOI] [PubMed] [Google Scholar]
- Craige B, Tsao CC, Diener DR, Hou Y, Lechtreck KF, Rosenbaum JL, Witman GB. CEP290 tethers flagellar transition zone microtubules to the membrane and regulates flagellar protein content. J Cell Biol. 2010;190:927–940. doi: 10.1083/jcb.201006105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgehyr N, Sillibourne J, Bornens M. Microtubule nucleation and anchoring at the centrosome are independent processes linked by ninein function. J Cell Sci. 2005;118:1565–1575. doi: 10.1242/jcs.02302. [DOI] [PubMed] [Google Scholar]
- Domire JS, Green JA, Lee KG, Johnson AD, Askwith CC, Mykytyn K. Dopamine receptor 1 localizes to neuronal cilia in a dynamic process that requires the Bardet-Biedl syndrome proteins. Cell Mol Life Sci. 2010;68:2951–2960. doi: 10.1007/s00018-010-0603-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowdle WE, Robinson JF, Kneist A, Sirerol-Piquer MS, Frints SG, Corbit KC, Zaghloul NA, van Lijnschoten G, Mulders L, Verver DE, et al. Disruption of a ciliary B9 protein complex causes Meckel syndrome. Am J Hum Genet. 2011;89:94–110. doi: 10.1016/j.ajhg.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evangelista M, Lim T, Lee J, Parker L, Ashique A, Peterson A, Ye W, Davis D, De Sauvage F. Kinome siRNA Screen Identifies Regulators of Ciliogenesis and Hedgehog Signal Transduction. Science signaling. 2008;1:ra7. doi: 10.1126/scisignal.1162925. [DOI] [PubMed] [Google Scholar]
- Fan S, Whiteman EL, Hurd TW, McIntyre JC, Dishinger JF, Liu CJ, Martens JR, Verhey KJ, Sajjan U, Margolis B. Induction of Ran GTP drives ciliogenesis. Mol Biol Cell. 2011;22:4539–4548. doi: 10.1091/mbc.E11-03-0267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonte VG, Searls RL, Hilfer SR. The relationship of cilia with cell division and differentiation. J Cell Biol. 1971;49:226–229. doi: 10.1083/jcb.49.1.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Garcia MJ, Eggenschwiler JT, Caspary T, Alcorn HL, Wyler MR, Huangfu D, Rakeman AS, Lee JD, Feinberg EH, Timmer JR, et al. Analysis of mouse embryonic patterning and morphogenesis by forward genetics. Proc Natl Acad Sci USA. 2005;102:5913–5919. doi: 10.1073/pnas.0501071102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Gonzalo FR, Corbit KC, Sirerol-Piquer MS, Ramaswami G, Otto EA, Noriega TR, Seol AD, Robinson JF, Bennett CL, Josifova DJ, et al. A transition zone complex regulates mammalian ciliogenesis and ciliary membrane composition. Nat Genet. 2011;43:776–784. doi: 10.1038/ng.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetz SC, Anderson KV. The primary cilium: a signalling centre during vertebrate development. Nat Rev Genet. 2010;11:331–344. doi: 10.1038/nrg2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graser S, Stierhof YD, Lavoie SB, Gassner OS, Lamla S, Le Clech M, Nigg EA. Cep164, a novel centriole appendage protein required for primary cilium formation. J Cell Biol. 2007;179:321–330. doi: 10.1083/jcb.200707181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green JA, Mykytyn K. Neuronal ciliary signaling in homeostasis and disease. Cell Mol Life Sci. 2010;67:3287–3297. doi: 10.1007/s00018-010-0425-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y, Spassky N, Romaguera-Ros M, Garcia-Verdugo J, Aguilar A, Schneider-Maunoury S, Alvarez-Buylla A. Hedgehog signaling and primary cilia are required for the formation of adult neural stem cells. Nat Neurosci. 2008;11:277–284. doi: 10.1038/nn2059. [DOI] [PubMed] [Google Scholar]
- Houlden H, Johnson J, Gardner-Thorpe C, Lashley T, Hernandez D, Worth P, Singleton A, Hilton D, Holton J, Revesz T, et al. Mutations in TTBK2, encoding a kinase implicated in tau phosphorylation, segregate with spinocerebellar ataxia type 11. Nat Genet. 2007;39:1434–1436. doi: 10.1038/ng.2007.43. [DOI] [PubMed] [Google Scholar]
- Huangfu D, Anderson KV. Cilia and Hedgehog responsiveness in the mouse. Proc Natl Acad Sci USA. 2005;102:11325–11330. doi: 10.1073/pnas.0505328102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huangfu D, Liu A, Rakeman AS, Murcia NS, Niswander L, Anderson KV. Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature. 2003;426:83–87. doi: 10.1038/nature02061. [DOI] [PubMed] [Google Scholar]
- Iomini C, Babaev-Khaimov V, Sassaroli M, Piperno G. Protein particles in Chlamydomonas flagella undergo a transport cycle consisting of four phases. J Cell Biol. 2001;153:13–24. doi: 10.1083/jcb.153.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, Marshall WF. Ciliogenesis: building the cell's antenna. Nat Rev Mol Cell Biol. 2011;12:222–234. doi: 10.1038/nrm3085. [DOI] [PubMed] [Google Scholar]
- Kim J, Lee JE, Heynen-Genel S, Suyama E, Ono K, Lee K, Ideker T, Aza-Blanc P, Gleeson JG. Functional genomic screen for modulators of ciliogenesis and cilium length. Nature. 2010;464:1048–1051. doi: 10.1038/nature08895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko HW, Norman RX, Tran J, Fuller KP, Fukuda M, Eggenschwiler JT. Broad-minded links cell cycle-related kinase to cilia assembly and hedgehog signal transduction. Dev Cell. 2010;18:237–247. doi: 10.1016/j.devcel.2009.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Tsang WY, Li J, Lane W, Dynlacht BD. Centriolar Kinesin Kif24 Interacts with CP110 to Remodel Microtubules and Regulate Ciliogenesis. Cell. 2011;145:914–925. doi: 10.1016/j.cell.2011.04.028. [DOI] [PubMed] [Google Scholar]
- Liem KF, Jr., Ashe A, He M, Satir P, Moran J, Beier D, Wicking C, Anderson KV. The IFT-A Complex Regulates Shh Signaling Through Cilia Structure and Membrane Protein Trafficking. J Cell Biol. 2012;197:789–800. doi: 10.1083/jcb.201110049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu A, Wang B, Niswander LA. Mouse intraflagellar transport proteins regulate both the activator and repressor functions of Gli transcription factors. Development. 2005;132:3103–3111. doi: 10.1242/dev.01894. [DOI] [PubMed] [Google Scholar]
- Marley A, von Zastrow M. DISC1 regulates primary cilia that display specific dopamine receptors. PLoS One. 2010;5:e10902. doi: 10.1371/journal.pone.0010902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matilla-Duenas A. The ever expanding spinocerebellar ataxias. Cerebellum. 2012 Mar 24; doi: 10.1007/s12311-012-0376-4. 2012, Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Matsuura T, Yamagata T, Burgess DL, Rasmussen A, Grewal RP, Watase K, Khajavi M, McCall AE, Davis CF, Zu L, et al. Large expansion of the ATTCT pentanucleotide repeat in spinocerebellar ataxia type 10. Nat Genet. 2000;26:191–194. doi: 10.1038/79911. [DOI] [PubMed] [Google Scholar]
- May SR, Ashique AM, Karlen M, Wang B, Shen Y, Zarbalis K, Reiter J, Ericson J, Peterson AS. Loss of the retrograde motor for IFT disrupts localization of Smo to cilia and prevents the expression of both activator and repressor functions of Gli. Dev Biol. 2005;287:378–389. doi: 10.1016/j.ydbio.2005.08.050. [DOI] [PubMed] [Google Scholar]
- Ocbina PJ, Anderson KV. Intraflagellar transport, cilia, and mammalian Hedgehog signaling: analysis in mouse embryonic fibroblasts. Dev Dyn. 2008;237:2030–2038. doi: 10.1002/dvdy.21551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ocbina PJ, Eggenschwiler JT, Moskowitz I, Anderson KV. Complex interactions between genes controlling trafficking in primary cilia. Nat Genet. 2011;43:547–553. doi: 10.1038/ng.832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omori Y, Chaya T, Katoh K, Kajimura N, Sato S, Muraoka K, Ueno S, Koyasu T, Kondo M, Furukawa T. Negative regulation of ciliary length by ciliary male germ cell-associated kinase (Mak) is required for retinal photoreceptor survival. Proc Natl Acad Sci USA. 2010;107:22671–22676. doi: 10.1073/pnas.1009437108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto EA, Trapp ML, Schultheiss UT, Helou J, Quarmby LM, Hildebrandt F. NEK8 mutations affect ciliary and centrosomal localization and may cause nephronophthisis. J Am Soc Nephrol. 2008;19:587–592. doi: 10.1681/ASN.2007040490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen LB, Veland IR, Schroder JM, Christensen ST. Assembly of primary cilia. Dev Dyn. 2008;237:1993–2006. doi: 10.1002/dvdy.21521. [DOI] [PubMed] [Google Scholar]
- Pugacheva EN, Jablonski SA, Hartman TR, Henske EP, Golemis EA. HEF1-dependent Aurora A activation induces disassembly of the primary cilium. Cell. 2007;129:1351–1363. doi: 10.1016/j.cell.2007.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi H, Yao C, Cai W, Girton J, Johansen KM, Johansen J. Asator, a tau-tubulin kinase homolog in Drosophila localizes to the mitotic spindle. Dev Dyn. 2009;238:3248–3256. doi: 10.1002/dvdy.22150. [DOI] [PubMed] [Google Scholar]
- Rieder CL, Jensen CG, Jensen LC. The resorption of primary cilia during mitosis in a vertebrate (PtK1) cell line. J Ultrastruct Res. 1979;68:173–185. doi: 10.1016/s0022-5320(79)90152-7. [DOI] [PubMed] [Google Scholar]
- Sang L, Miller JJ, Corbit KC, Giles RH, Brauer MJ, Otto EA, Baye LM, Wen X, Scales SJ, Kwong M, et al. Mapping the NPHP-JBTS-MKS Protein Network Reveals Ciliopathy Disease Genes and Pathways. Cell. 2011;145:513–528. doi: 10.1016/j.cell.2011.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeley ES, Nachury MV. The perennial organelle: assembly and disassembly of the primary cilium. J Cell Sci. 2010;123:511–518. doi: 10.1242/jcs.061093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singla V, Romaguera-Ros M, Garcia-Verdugo JM, Reiter JF. Ofd1, a human disease gene, regulates the length and distal structure of centrioles. Dev Cell. 2010;18:410–424. doi: 10.1016/j.devcel.2009.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spektor A, Tsang W, Khoo D, Dynlacht B. Cep97 and CP110 Suppress a Cilia Assembly Program. Cell. 2007;130:678–690. doi: 10.1016/j.cell.2007.06.027. [DOI] [PubMed] [Google Scholar]
- Takahashi M, Tomizawa K, Sato K, Ohtake A, Omori A. A novel tau-tubulin kinase from bovine brain. FEBS Lett. 1995;372:59–64. doi: 10.1016/0014-5793(95)00955-9. [DOI] [PubMed] [Google Scholar]
- Terret ME, Sherwood R, Rahman S, Qin J, Jallepalli PV. Cohesin acetylation speeds the replication fork. Nature. 2009;462:231–234. doi: 10.1038/nature08550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel C, Kessler K, Giessl A, Dimmler A, Shalev SA, von der Haar S, Zenker M, Zahnleiter D, Stoss H, Beinder E, et al. NEK1 mutations cause short-rib polydactyly syndrome type majewski. Am J Hum Genet. 2011;88:106–114. doi: 10.1016/j.ajhg.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomizawa K, Omori A, Ohtake A, Sato K, Takahashi M. Tau-tubulin kinase phosphorylates tau at Ser-208 and Ser-210, sites found in paired helical filament-tau. FEBS Lett. 2001;492:221–227. doi: 10.1016/s0014-5793(01)02256-6. [DOI] [PubMed] [Google Scholar]
- Tsang W, Bossard C, Khanna H, Peranen J, Swaroop A, Malhotra V, Dynlacht B. CP110 Suppresses Primary Cilia Formation through Its Interaction with CEP290, a Protein Deficient in Human Ciliary Disease. Dev Cell. 2008;15:187–197. doi: 10.1016/j.devcel.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weatherbee SD, Niswander LA, Anderson KV. A mouse model for Meckel syndrome reveals Mks1 is required for ciliogenesis and Hedgehog signaling. Hum Mol Genet. 2009;18:4565–4575. doi: 10.1093/hmg/ddp422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westlake CJ, Baye LM, Nachury MV, Wright KJ, Ervin KE, Phu L, Chalouni C, Beck JS, Kirkpatrick DS, Slusarski DC, et al. Primary cilia membrane assembly is initiated by Rab11 and transport protein particle II (TRAPPII) complex-dependent trafficking of Rabin8 to the centrosome. Proc Natl Acad Sci USA. 2011;108:2759–2764. doi: 10.1073/pnas.1018823108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheatley DN, Wang AM, Strugnell GE. Expression of primary cilia in mammalian cells. Cell Biol Int. 1996;20:73–81. doi: 10.1006/cbir.1996.0011. [DOI] [PubMed] [Google Scholar]
- Zeller R, Lopez-Rios J, Zuniga A. Vertebrate limb bud development: moving towards integrative analysis of organogenesis. Nat Rev Genet. 2009;10:845–858. doi: 10.1038/nrg2681. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.