

Abstract
Background
Human parvovirus B19 (B19) is a common pathogen which causes a variety of diseases. Persistent B19 infection is related to the degree of host immunodeficiency in patients with human immunodeficiency virus (HIV) infection. However, the existence, loading, virus evolution and distribution of B19 in Chinese HIV-positive patients have not been determined.
Materials and methods.
We investigated 573 HIV-positive blood donors and AIDS patients in Sichuan, China in the last two decades. Bl9-specific serology and quantitative polymerase chain reaction were used to determine the prevalence of B19/HIV co-infection. Viral genome fragments were subjected to phylogeny and haplotype analysis.
Results
B19 genomic DNA was found in 26 of 573 (4.5%) HIV-positive individuals, a higher prevalence than in blood donors. DNA levels ranged from 5.3×102–1.1×105 copies/mL. The seroprevalence of IgG was significantly lower in HIV-positive samples than in HIV-negative blood donors, indicating deficient production of B19-specific IgG in the former. The B19 isolates were genotype-1 subtype B19-1A which formed a monophyletic group; seven distinct haplotypes were discovered with 60% of the B19/HIV co-infected variants sharing one central haplotype.
Discussion.
This study on the prevalence, phylogeny and distribution of human parvovirus B19 in Sichuan, China, demonstrates the persistence of B19 in the circulation of both immunocompetent and immunocompromised subjects, with implications for blood safety.
Keywords: human parvovirus B19, human immunodeficiency virus, co-infection, donors, AIDS patients
Introduction
The human parvovirus B19 virus is a small, non-enveloped, single-strand DNA human virus with a genome size of 5.6 kb belonging to the genus Erythrovirus of the family Parvoviridae1. The B19V genome contains three open reading frames (ORF). The first ORF encodes the non-structural protein NS1 involved in viral DNA replication and transcription while the second encodes two structural proteins, VP2 (58 kDa) and VP1 (83 kDa), from the same ORF at the right hand of the viral genome2. In addition to the three major proteins, the third ORF encodes a non-structural 11-kDa protein, which seems to have an important role in the synthesis of structural proteins3. A 7.5 kDa protein of unknown function is also encoded in the genome. Three genotypes have been found so far. Genotype 1 is the most prevalent global B19V strain and is represented by the prototype B194. Subsequently, genotype 1 was suggested to have divided into two sub-genotypes by genetic divergence: B19-1A, which contains the prototype B19, and B19-1B, found only in Vietnam5. Genotype 2 is considered to have been replaced by genotype 1 in the 1970s in Europe6, although it sporadically circulates in North America and Europe at a lower frequency than genotype 1. Genotype 2 is represented by the strains A67 and K718. Erythrovirus V9 was classified as the prototype of genotype 39 and circulates in Ghana, West Africa, France, Brazil and the United States of America (USA)4,10–12.
Parvovirus B19 exhibits a marked tropism to human bone marrow and replicates only in erythroid progenitor cells. The virus is transmissible via the respiratory route or by blood and blood products13, and its infection in humans has been associated with a wide spectrum of clinical manifestations. B19 virus is often responsible for a mild disease, erythema infectiosum (fifth disease, a common illness in children), aplastic crises, chronic pure red cell aplasia, foetal hydrops and foetal death. The virus is also associated with anaemia in immunocompromised patients, arthropathies, hepatitis and a variety of other syndromes and diseases14,15.
In immunocompetent individuals, the viral load rapidly increases during the first week after infection and may exceed 1×1012 genome equivalents/mL. The humoral immune response plays an important role in viral clearance. Within 7–14 days after infection, B19-specific immunoglobulin M (IgM) appears, with declining viraemia, and may persist for as long as 6 months. Specific IgG is detectable by the third week following infection and remains for several years16.
Parvovirus B19 has been described as a cause of chronic anaemia and a variety of other diseases in immunosuppressed patients, including those infected with human immunodeficiency virus (HIV)14,15,17,18. As indicated in several studies, B19V infection in immunocompromised patients causes severe chronic anaemia because of the host’s inability to produce neutralising antibodies, and consequent persistence of virus replication19,20; furthermore, another study demonstrated that in some cases B19V antibodies were produced but did not neutralise the virus21. The central problem in these patients is, therefore, a qualitative or quantitative defect in the production of parvovirus Bl9-specific antibody, resulting from various underlying immunological problems. The diagnosis of B19V infection is very important because B19 viraemia is a treatable cause of anaemia in patients with HIV infection22.
In this study we used parvovirus B19-specific serology and quantitative polymerase chain reaction (Q-PCR) to investigate the prevalence of human parvovirus B19 infection in HIV-infected individuals, who were identified among blood donors and AIDS patients in Sichuan, China. We also used phylogenetic and haplotype invetigations to reveal the evolutionary status of B19 distribution in this area during the last two decades.
Materials and methods
Sample collection
The study was performed on 408 first-time blood donors diagnosed by serology to be HIV-positive and on 165 patients with clinical acquired immunodeficiency syndrome (AIDS) from hospitals in the province of Sichuan, China. All these samples were collected between 1 995 and 2010. HIV-negative samples collected in 2009 from normal blood donors were used as controls. The samples collected in this study were from various districts of Chengdu city, in the province of Sichuan, except where specifically indicated. The positivity of all 573 HIV-positive blood samples was first determined by enzyme-linked immunosorbent assay (ELISA) with two different reagents in each blood centre and later confirmed by western blotting using an HIV 1/2 antibody immunoblot kit (AUSIA, Hangzhou, China) afterwards at the HIV Confirmation Laboratory of the Institute of Blood Transfusion, Chinese Academy of Medical Science. An HIV-positive sample was defined as one positive by western blotting. An HIV-negative sample was defined as one being non-reactive to both ELISA reagents. An AIDS patient was defined as having a positive western blot result and clinical symptoms of AIDS.
Serological tests
Seventy-nine HIV-positive individuals and 92 HIV-negative individuals (blood donors) were tested for B19 virus-specific antibodies using a commercial assay kit for the detection of parvovirus IgG/IgM (Virion/Serion, Friedrich-Bergius-Ring 1997076, Würzburg, Germany) according to the manufacturer’s recommendations. Single tests were performed: if a result was borderline, the assay was repeated and the unambiguous result was taken as the final one for the specimen.
Viral genome DNA extraction
Aliquots of plasma (50 μL) from four HIV-positive blood samples were pooled and subjected to nucleic acid extraction using a QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. After Q-PCR, 200 μL of plasma from each sample from B19V-positive pools underwent nucleic acid extraction independently using the same protocol as mentioned above. All of the DNA extracts were stored at −80 °C prior to PCR analysis.
Quantification of the B19 viral genome
In order to avoid unsuccessful amplification by commercial assays23, Q-PCR was performed using a previously described in-house assay to detect B19V DNA-positive samples and to determine the viral load with consensus primers located in the NS1 region. An ABI Prism 7900 Sequence Detection System platform (Applied Biosystems, Foster City, CA, USA) was employed. The sequences of the primers are shown in Table I. Before quantifying the B19V genome in the extracted DNA, the universality of the primers and the sensitivity of the detection system were tested. The DNA extracted from three plasma samples each contained different genotypes of B19V (B19, A6, and V9 strains) (The 1st WHO International Reference Panel for parvovirus B19V genotypes for nucleic acid amplification technique [NAT]-based assays, NIBSC code: 09/110, National Institute for Biological Standards and Control, Hertfordshire, United Kingdom) were used to validate the universality of the primers. Meanwhile, a serial dilution of plasmid pYT-103 which contains genomic sequences of B19V genotype 1 was used to investigate the sensitivity of the detection system. A serial dilution (10x) of three B19V genotypes from 2 to 2×104 IU per reaction (20 μL) of the 1st WHO International Reference Panel for Parvovirus B19 Genotypes for NAT based assays (genotypes 1, 2 and 3) were tested for their lowest detection limit. Subsequently, B19V screening and quantification of the viral load in the extracted DNA were performed as described elsewhere24. Briefly, the 20 μL reaction mix consisted of 10 μL FastStart Universal SYBR Green Master (Roche Diagnostics, Indianapolis, IN, USA), 12.5 pmol of each primer, and 5 μL of extracted DNA as a template. Hot-start amplification was performed under the following conditions: 1 cycle of 95 °C for 10 min; 45 cycles of 95 °C for 15 s, 60 °C for 30 s, and 72 °C for 30 s; and a final cycle of 95 °C for 15 s, 60 °C for 15 s, and then gradually increased to 95 °C in 30 min at a ramp rate of 2%.
Table I.
Primers used in this study.
| Primer names | Forward primers’ sequence (5′-3′) | Reverse primers’ sequence (5′-3′) | Product length (bp) | Usage |
|---|---|---|---|---|
| B19_Q-PCR_F / B19_Q-PCR_R | TGCAGATGCCCTCCACCCA | GCTGCTTTCACTGAGTTCTTC | 103 | Q-PCR |
| PVB-1f-1747 / PVB-1r-1967 | CACTATGAAAACTGGGCAATAAAC | AATGATTCTCCTGAACTGGTCC | 242 | Confirmation for Q-PCR, 1st round |
| PVB-2f-1765 / PVB-2r-1965 | ATAAACTACACTTTTGATTTCCCTG | TCTCCTGAACTGGTCCCG | 218 | Confirmation for Q-PCR, 2nd round |
| b19_f_701 / b19_r_4978 | AGACACTTCTGACTGGGAACCACTAAC | AGGGCACGGTGGGGAGTGTTTA | 4,287 | Nest PCR for genome analysis/PCR 1st round |
| b19_fN_725 / b19_r_2440 | TAACTCATACTAACAGACTAATGGCAAT | ACAAAGTCCCAAGCCTCCCC | 1,736 | Nest PCR for genome analysis/PCR 2nd round / sequencing |
| b19_f_2262 / b19_r_3505 | TCAGGAGAATCATTTGTCGGAAGC | AACCTTTGCCTCYTTTCCACT | 1,264 | Nest PCR for genome analysis/PCR 2nd round / sequencing |
| b19_f_3432 / b19_rN_4883 | GTTTTTAATTCCTAATGACCCAGAGC | TGTGGTGTTGTTTTGCATCTGTAG | 1,475 | Nest PCR for genome analysis/PCR 2nd round / sequencing |
| B19_f_1453 | ACTTTGAGCAGGTTATGTG | sequencing | ||
| B19_f_2966 | TACACAAGCCTGGGCAAGT | sequencing |
Nested polymerase chain reactions
Q-PCR-detected B19V DNA-positive samples were confirmed by nested PCR using the conserved primers located in the NS1 region described previously25,26, with positions referring to the parvovirus V9 sequence (GenBank accession number AX003421). The first-round and second-round primers are shown in Table I. The first-round PCR was performed with Taq DNA Polymerase (Tiangen, PR China) in a total volume of 40 μL with the following reaction conditions: pre-denaturation at 95 °C for 5 min, followed by 35 cycles of 15 s denaturation (95 °C), 30 s annealing (50 °C), and 30 s extension (72 °C) with a final extension at 72 °C for 5 min. The cycling conditions of the second-round PCR were the same as those of the first round, and 2 μL of the products of the first-round PCR were used as the template.
Nested PCR was carried out using the primer pairs shown in Table I. The first-round PCR was performed to amplify the 4287 bp fragment with KOD FX DNA Polymerase (TOYOBO, Tokyo, Japan) in a total volume of 40 μL with the following reaction conditions: pre-denaturation at 95 °C for 5 min, followed by 35 cycles of denaturation (95 °C) at 15 s, annealing (62 °C) for 30 s, and a 6 min extension (72 °C) with a final extension at 72 °C for 10 min. The second-round PCR was performed to amplify three overlapping fragments and the cycling conditions of the second-round PCR again matched those of the first-round PCR with the exception of a shorter extension time (2 min); 2 μL of products of the first-round PCR were used as the template. All PCR cycling was performed on a Veriti Thermal Cycler (Applied Biosystems Inc., Foster City, CA, USA).
Sequencing, phylogenetic and haplotype analysis
The PCR products were purified using the NucleoSpin Extract II kit (Machery-Nagel GmbH & Co. KG, Düren, Germany) according to the manufacturer’s instructions. Purified products were used as templates in cycle-sequencing reactions. The sequencing primers were the same as the second-round primers used in the previous PCR reactions. The resulting sequences were read directly by an ABI 3730 Genetic Analyzer (Applied Biosystems Inc., Foster City, CA, USA). Sequences were determined in both directions. The 4060 bp sequences were aligned using Clustal X 1.8327 after cutting ambiguous sites off both ends. Bayesian inference (BI) and maximum likelihood (ML) analyses were employed to detect the phylogenetic position of the samples in this study using reference sequences with GenBank accession numbers shown in Figure 3a and the B19V sequences found in blood donors in our previous study24 in Luoyang and Urumqi. The best fitting model of sequence evolution for BI analyses was obtained using MrModeltest 2.228 under the Akaike Information Criterion. The BI analysis was performed with MrBayes 3. 1. 229. Posterior distributions were obtained by the Markov chain Monte Carlo method with one cold chain and three heated chains for 2,000,000 generations, with every 100th sample retained. The first 5,000 samples were discarded as burn-in and the remaining samples were used to generate a majority rule consensus tree. All Markov chain Monte Carlo runs were repeated twice to confirm consistent approximation of the posterior parameter distributions. The ML analysis was performed using RAxML Web-Servers30. RAxML searches were executed in 1000 rapid bootstrap inferences and thereafter with a thorough ML search. Free model parameters were estimated by RAxML. All of the resulting phylogenetic trees were rooted on B19 genotype 3.
Figure 3.
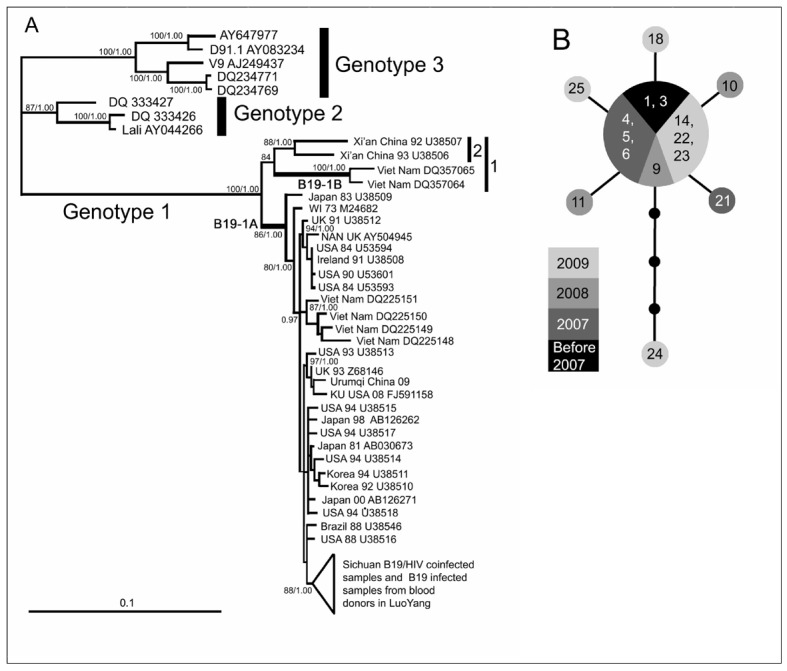
Phylogeny and haplotype analysis. (a) Bayesian inference (BI) tree estimated using an aligned 4060 bp sequence. Maximum likelihood bootstrap values >80 and BI posterior possibilities >0.95 are shown at the nodes. The GenBank accession number, collection time and locality of each reference sequence are shown at the tips. (b) Median-joining network depicting the relationships among B19/HIV co-infected samples. Grey circle: B19 DNA positive samples; Black circle: Missing intermediates. Each branch between two (sampled or missing) haplotypes indicates a single mutational step. The size of each circle is proportional to the corresponding haplotype frequency. The numbers shown in the circles correspond to the samples’ serial numbers in Table II.
All acquired 4060 bp B19V sequences from HIV-positive individuals in Sichuan were used to construct a median-jointing genetic network using NETWORK 4.5.1.6 (Fluxus-Engineering, www.fluxus-technology.com) and then modified by hand. The genetic distance was calculated by MEGA 4.031 using an uncorrected p-distance.
Statistical analysis
Statistical analyses were conducted using SPSS statistics software (SPSS, Inc., Chicago, IL). The chi-square test was applied to assess associations between categorical variants. P-values <0.05 were considered statistically significant.
Results
Socio-demographic characteristics
In this study, 573 serologically HIV-positive participants were investigated, among whom 408 were blood donors (332 male, 76 female) and 165 were clinical patients (121 male, 44 female) (Table II). The mean age of these individuals was 33.4±11.9 years (range, 15–79 years). There were 453 (79.1%) men with a median age of 33.7±12.1 years (range, 15–79 years) and 120 (20.9%) women with a median age of 32.1±11.3 years (range, 18–67 years).
Table II.
Characteristics and B19 genome prevalence of HIV-positive samples.
| Total (%) | B19-positive (%) | 95% CI | HIV-positive blood donors | AIDS patients | |||||
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| Total | B19-positive (%) | 95% CI | Total | B19-positive (%) | 95% CI | ||||
| Gender | |||||||||
| Male | 453 (79.1) | 20 (4.4) | 2.5–6.3 | 332 | 16 (4.8) | 2.5–7.1 | 121 | 4 (3.3) | 0.1–6.5 |
| Female | 120 (20.9) | 6 (5.0) | 1.2–8.8 | 76 | 3 (3.9) | 0–8.3 | 44 | 3 (6.8) | 0–14.2 |
| Total | 573 | 26 (4.5) | 2.8–6.2 | 408 | 19 (4.7) | 2.7–6.8 | 165 | 7 (4.2) | 1.1–7.3 |
| Age | |||||||||
| ≤45 | 451 (78.7) | 22 (4.9) | 2.9–6.9 | 365 | 19 (5.2) | 2.9–7.5 | 86 | 3 (3.5) | 0–7.4 |
| >45 | 71 (12.4) | 3 (4.2) | 0–8.9 | 30 | 0 | 41 | 3 (7.3) | 0–15.3 | |
| Unknown | 51 (8.9) | 1 (2.0) | 0–5.8 | 13 | 0 | 38 | 1 (2.6) | 0–7.7 | |
| Year | |||||||||
| Before 2007 | 109 (19.0) | 4 (3.7) | 0.2–7.2 | 82 | 3 (3.7) | 0–7.8 | 27 | 1 (3.7) | 0–10.8 |
| 2007 | 76 (13.2) | 5 (6.6) | 1.0–12.2 | 57 | 4 (7.0) | 0.4–13.6 | 19 | 1 (5.3) | 0–15.3 |
| 2008 | 169 (29.5) | 4 (2.4) | 0.1–4.7 | 135 | 4 (3.0) | 0.1–5.9 | 34 | 0 | |
| 2009 | 149 (26.0) | 11 (7.4) | 3.2–11.6 | 92 | 7 (7.6) | 2.2–13.0 | 57 | 4 (7.0) | 0.4–13.6 |
| 2010 | 70 (12.2) | 2 (2.9) | 0–6.8 | 42 | 1 (2.3) | 0–6.8 | 28 | 1 (3.5) | 0–10.3 |
The sensitivity and specificity of the quantitative polymerase chain reaction
Prior to determining the prevalence of B19V genome DNA by Q-PCR, the specificity and the sensitivity of the detection system were validated. A plasmid containing the entire genome of human parvovirus B19 (pYT-103) was used as a standard for amplification in a range of 5 to 5×106 copies. DNA extracted from three plasma samples containing three different genotypes of B19V (B19, A6, and V9 strains) were used to test the specificity of the method. A liner standard curve could be routinely generated from a starting copy number of five or more (Figure 1a). As few as five template copies could be distinguished from background levels (Figure 1a). To further verify the sensitivity of the assay, WHO standards including three genotypes were serially diluted (10x) from 2 to 2×104 IU per reaction (20 μL). As shown in Figure 1a, 2 IU of the three genotypes could be detected. Simultaneously, seven quantitative standards (5×106, 5×105, 5×104, 5×103, 5×102, 5×101, 5 [copies/mL]) of plasmid pYT-103 were used to generate a linear, standard curve with R2>0.99. Melting curve analysis demonstrated that this in-house PCR method amplified a single predominant product with a distinct Tm, as shown for a parvovirus B19 fragment (Figure 1b). The melting curves of the three genotype amplifications suggested that all three genotypes of human parvovirus B19 were successfully amplified (Figure 1c) albeit with slightly different melting curves.
Figure 1.

Q-PCR to determine the specificity and the sensitivity of the detection system. (a) Standard curve generated by a serial dilution (10x) of plasmid pYT-103 (■), from 5 to 5×106 copies per reaction (20 μL), and a serial dilution (10x) of three B19V genotypes from 2 to 2×104 IU per reaction (20 μL) of the 1st WHO International Reference Panel for Parvovirus B19 Genotypes for NAT-based assays [genotype 1 (●), genotype 2 (x), and genotype 3 (▲)] were tested for the lowest detection limit; (b) Melting curve of B19V-positive samples; (c) Melting curve of three genotypes of B19V with plasmid pYT-103.
Figure 2.
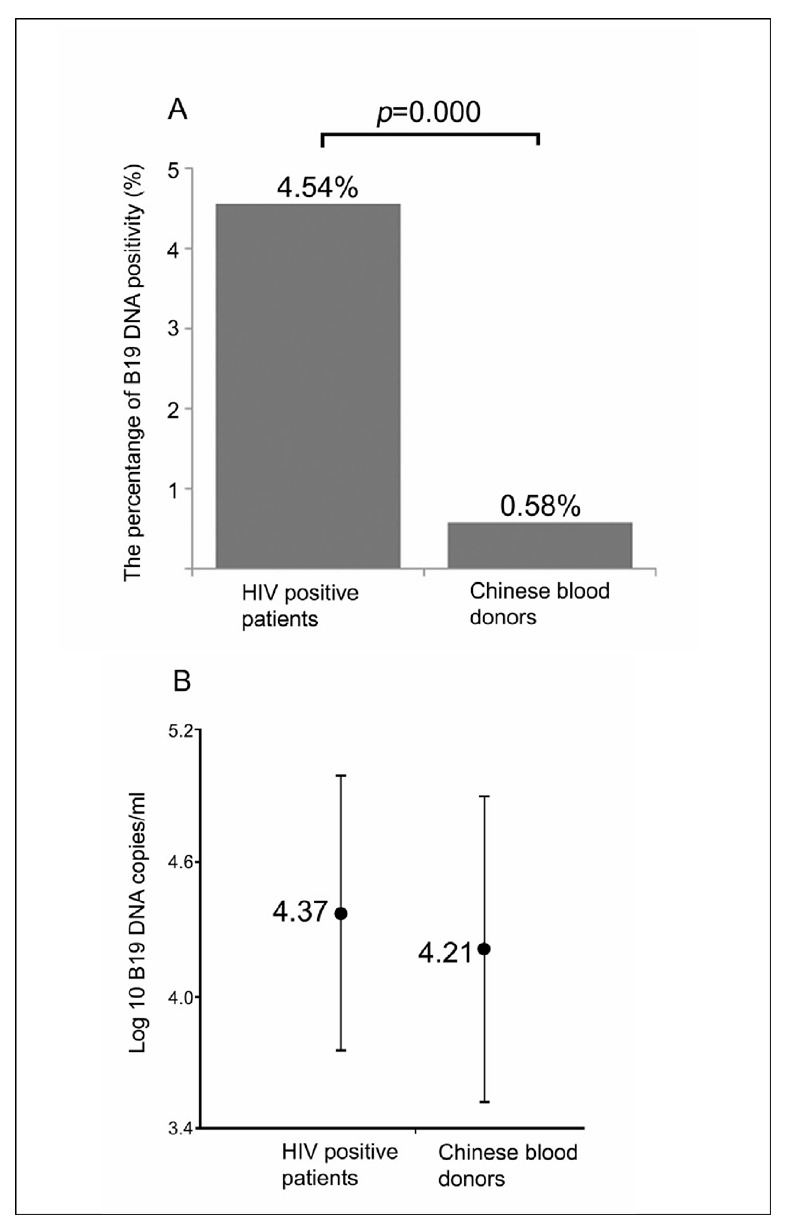
The prevalences of B19 viral genome (a) and viral load (b) in HIV-positive samples compared with those in the normal Chinese blood donor samples.
B19V genome DNA prevalence, viral load and sero-status in HIV- positive individuals
As shown in Table II, 26 of 573 of the HIV-positive individuals tested positive for B19V genome DNA, with an overall prevalence of B19V genome DNA of 4.54% (95% CI: 2.8% –6.2%). We previously demonstrated24 that the prevalence of B19V genome DNA in normal blood donors was 0.58%. The prevalence of the B19V genome DNA was significantly higher in HIV-positive subjects than in normal Chinese blood donors (Figure 2a). The HIV-positive individuals were divided into two groups: blood donors and clinical AIDS patients (Table II). The prevalence of B19 virus genome DNA in HIV-positive blood donors was 4.7% (19 out of 408; 95% CI: 2.7%–6.8%) while that in clinical AIDS patients was 4.2% (7 out of 165; 95% CI: 1.1%–7.3%). There was no significant difference in B19V genome DNA prevalence between these two groups. Additionally, univariate analysis showed no significant difference of B19V genome DNA prevalence by gender in both groups (data not shown). By contrast, among clinical AIDS patients there was a higher prevalence in females (6.8%) than in males (3.3%) although the difference was not statistically significant.
In order to determine the association of B19V genome prevalence with age, the HIV-positive populations from blood banks and hospitals were divided into two age groups: <45 years (young) and ≥45 years (middle-aged). As shown in Table II, the prevalence of B19V genome DNA in the younger age group was not significantly different from that in the older age group.
We also determined the prevalence of B19V/HIV co-infected samples over time: the prevalence in samples from 1995 to 200 6 was 3.7% (4 out of 109; 95% CI: 0.2%–7.2%); the prevalence in 2007 was 6.6% (5 out of 76; 95% CI: 1%–12.2%); in 2008 it was 2.4% (4 out of 169; 95% CI: 0.1%–4.7%); and in 2010 it was 2.9% (2 out of 70; 95% CI: 0%–6.8%). The B19V genome DNA prevalence in samples taken in 2009 was 7.4% (11 out of 149; 95% CI: 3.2%–11.6%) which was significantly higher than the mean level and the levels in other years except for 2007, both in HIV-positive blood donors and clinical AIDS patients (p<0.05) (Table II), which suggests that 2009 was a B19 epidemic year.
Parvovirus B19 DNA levels and the characteristics of the B19V genome DNA-positive samples, including the providers’ age, gender, status of B19V-specific antibodies (IgM/IgG), geographic location, sample source (blood bank or hospital) and collection dates, are shown in Table III. Overall, the DNA levels ranged from 5.3×102–1.1×105 copies/mL and there was no statistically significant difference in viral load between samples from hospitals and blood banks. Furthermore, the mean viral load between HIV-infected individuals and the regular Chinese blood donor population was not statistically different (Figure 2b).
Table III.
Viral load, antibody status and information on the subjects from whom B19 DNA-positive samples were collected.
| Samples | Viral load | Gender | Age | IgM | IgG | Collection date | Source | Location |
|---|---|---|---|---|---|---|---|---|
| 1 | 8.2×103 | M | 24 | − | + | 17/05/1995 | Blood bank | Santai |
| 2 | 1.8×103 | F | 18 | − | − | 11/11/2004 | Blood bank | Zigong |
| 3 | 5.8×102 | M | 28 | − | − | 24/05/2005 | Blood bank | Yingshan |
| 4 | 2.3×104 | M | 47 | + | + | 11/10/2007 | Blood bank | Deyang |
| 5 | 1.0×104 | M | 32 | − | − | 17/10/2007 | Blood bank | Ya’an |
| 6 | 3.7×104 | M | 35 | NA | NA | 01/11/2007 | Blood bank | Mianyang |
| 7 | 4.8×104 | M | 26 | − | − | 01/11/2007 | Blood bank | Qionglai |
| 8 | 9.3×102 | M | 24 | − | − | 25/03/2008 | Blood bank | Dazhou |
| 9 | 5.5×104 | F | 21 | − | − | 24/04/2008 | Blood bank | Chengdu |
| 10 | 5.0×103 | M | 15 | − | − | 08/12/2008 | Blood bank | NA |
| 11 | 2.8×104 | M | 21 | NA | NA | 10/12/2008 | Blood bank | Chengdu |
| 12 | 4.6×104 | M | 19 | − | − | 13/01/2009 | Blood bank | Guangyuan |
| 13 | 1.6×104 | M | 22 | + | − | 20/01/2009 | Blood bank | Chengdu |
| 14 | 8.9×103 | F | 25 | − | − | 29/04/2009 | Blood bank | Zizhong |
| 15 | 1.2×104 | M | 40 | − | − | 07/07/2009 | Blood bank | Chengdu |
| 16 | 2.9×104 | M | 37 | − | − | 23/07/2009 | Blood bank | Mianyang |
| 17 | 4.6×104 | M | 26 | − | − | 16/09/2009 | Blood bank | Chengdu |
| 18 | 2.7×104 | M | 19 | − | − | 16/09/2009 | Blood bank | Pixian |
| 19 | 5.0×103 | M | 36 | NA | NA | 24/02/2010 | Blood bank | Chengdu |
| 20 | 5.3×102 | F | 58 | + | + | 28/06/2005 | Hospital | Chengdu |
| 21 | 2.2×104 | M | 33 | − | + | 14/11/2007 | Hospital | Dujiangyan |
| 22 | 3.6×104 | F | 27 | NA | NA | 09/06/2009 | Hospital | Deyang |
| 23 | 4.0×103 | M | N/A | NA | NA | 10/07/2009 | Hospital | NA |
| 24 | 1.1×105 | F | 52 | − | + | 19/08/2009 | Hospital | Zigong |
| 25 | 2.5×104 | M | 50 | − | + | 20/08/2009 | Hospital | Chengdu |
| 26 | 5.1×103 | M | 21 | NA | NA | 24/02/2010 | Hospital | Langzhong |
NA: not available
Among the 26 B19V/HIV co-infected samples, six were positive for IgG, while two were positive for both IgM and IgG. However, the B19V genome level in these two samples was not demonstrated to be significant differently from that in other B19V genome-positive samples (Table III); we assume that these subjects were in the late phase of an acute infection.
Seroprevalence of parvovirus B19 antibodies in HIV-positive samples
In order to determine the seroprevalence of parvovirus B19 antibody in HIV-positive samples, B19V-specific antibodies (IgG/IgM) were investigated among 79 HIV-positive samples (45 from HIV-positive blood donors and 34 from patients with AIDS) and the results compared to those in 92 HIV-negative blood bank samples collected in 2009 in the Chengdu blood centre. As shown in Table IV, among the 79 HIV-positive samples tested for B19V-specific immunoglobulins, 12 (15.2%, 95% CI, 7.3–23.1%) were positive for IgG and four (5.1%, 95% CI, 0.2–9.9%) were positive for IgM. No samples were found to be positive for both subtypes of antibody, but 63 (79.7%, 95% CI, 70.8–88.6%) were shown to be negative for both antibodies. The prevalences of B19V IgG and IgM in HIV-positive blood donors were 22.2% (10 of 45) and 4.4% (2 of 45), respectively, and those in AIDS patients were 5.9% (2 of 34) and 5.9% (2 of 34), respectively. The seroprevalence of B19V IgG in HIV-negative blood donors (32.6%, 95% CI, 23.0–42.2%, p<0.05) was significantly higher than that in HIV-positive samples, while the seroprevalence of B19V IgM was not statistically different. The IgG prevalence in HIV-positive blood donors was lower than in normal control blood donors, but the difference was not statistically significant. The prevalence of B19V IgG in AIDS patients was significantly lower than that in control blood donors. Interestingly, the ratio of both antibody-negative samples in HIV-positive individuals (79.7%, 95% CI, 70.8–88.6%) was statistically higher than that in regular blood donors (51.1%, 95% CI, 40.9–61.3%) (p<0.05). The presence of the B19 genome in these 79 HIV-positive samples was also determined. There were four B19 DNA-positive samples, of which three were from HIV-positive blood donors (corresponding to cases n. 13, 15, and 17 in Table III) and one was from a patient with AIDS (corresponding to case n. 25 in Table III). All the HIV-negative controls were also negative for B19 DNA.
Table IV.
The seroprevalence of B19 antibody in HIV-positive patients and Chinese blood donors.
| Number of samples | IgG-positive (%) (95%CI) | IgM-positive (%) (95%CI) | IgG & IgM positive (95%CI) | IgG & IgM negative (95%CI) | |
|---|---|---|---|---|---|
| HIV-positive blood donors | 45 | 10 (22.2), (10.1–34.3) | 2 (4.4), (0–10.4) | 0 | 33 (73.3), (60.4–86.2) |
| AIDS patients | 34 | 2 (5.9), (0–13.8)* | 2 (5.9), (0–13.8) | 0 | 30 (88.2), (77.4–99.0)* |
| HIV-negative blood donors** | 92 | 30 (32.6), (23.0–42.2) | 7 (7.6), (2.2–13.0) | 2 (2.2), (0–5.2) | 47 (51.1), (40.9–61.3) |
Legend:
P<0.05;
10 samples tested gave borderline results.
Phylogeny and haplotype analysis
The aligned sequences of 4060 bp in length were employed to reconstruct the phylogeny using BI and ML methods. The best fitting model for the data was GTR+I+G. Both BI and ML estimations provided identical and well-supported tree topologies; most branches were strongly supported with over 80% bootstrap support (BS) values and 0.9 Bayesian posterior probabilities (PP) (Figure 3a). The BI tree is shown in Figure 3a with BS/PP values on the corresponding branches (only BS>80 and PP>0.9 were considered to be credible and are shown).
The three distinct clades in Figure 3a correspond to the three genotypes of human parvovirus B19. Within genotype 1, two main clades appeared as two monophyletic groups. Clade B19-1A contained most samples, which belonged to genotype 1 sub-genotype B19-1A that were used in the phylogenetic study. Meanwhile, clade 1 also consisted of two solid monophyletic groups. Clade B19-1B was determined as being the genotype 1 subtype B19-1B found in Vietnam around 20005; simultaneously, clade 2 was formed by two sequences sampled in Xi’an, China, in 1992 and 199332.
Our samples fell in B19-1A (Figure 3a). The B19V/HIV co-infected samples from Sichuan formed a monophyletic group (BS=88, PP=1.0) together with the sequences from Luoyang detected in our previous study. The genetic distance within this clade was very short (p-distance=0.00038), and this clade had a close relationship with the B19V from the USA and Brazil (Figure 3a). The B19V from Urumqi was clustered with the sequence from the USA and the United Kingdom (BS=97, PP=1.0). Furthermore, the U38509 sequence was a sister to other B19-1A sequences (BS=86, PP=1.0).
Seven haplotypes were found in the successfully sequenced 15 B19V/HIV co-infected samples from Sichuan (the other B19V genome-positive samples which were detected by Q-PCR and confirmatory nested-PCR failed to amplify the long DNA segment). The haplotype network is shown in Figure 3b. The frequency of central haplotypes, including nine B19V/HIV co-infected samples, was 60% and the frequency of the other six haplotypes, consisting of one sample each, was 6.7%. Overall, there were ten mutations in the network and the most diverged haplotype with the highest viral load in this study was that with the four mutations from the central haplotype and was collected in 2009 (Figure 3b). The central haplotype was composed of one sample collected in 1995, one sample collected in 2005, three samples collected in 2007, one sample collected in 2008, and three samples collected in 2009 (Figure 3b).
Discussion
In the current study, we used ELISA and Q-PCR to investigate the prevalence of serological evidence of parvovirus B19 and its genomic DNA in 573 HIV-infected individuals. As indicated in Figure 2a and 2b, although the prevalence of genomic DNA was significantly different, it was very interesting to find that there was no statistical difference between the viral loads. High B19V viral loads, which are common in western countries33,34, have not emerged yet. At the same time, the seroprevalence of IgG against B19V was 15.2% in HIV-infected individuals, which was significantly lower than that in the normal blood donor controls (p<0.05). This is consistent with the results of a previous study that indicated the level to be 7.6%21 and suggests that chronic parvovirus Bl9 infection occurred in HIV-infected individuals whose immune systems were not capable of mounting an adequate response. Considering the relatively low seroprevalence of B19 in the normal blood donor population (Table III), this is a convincing argument. However, the seroprevalence of parvovirus B19-specific IgG showed large variations. One study conducted in 1996 also indicated that there was no statistically significant difference in seroprevalence of parvovirus B19-specific IgG between HIV-infected patients and male blood donors35. Other studies involving small numbers of patients indicated a higher prevalence of IgG antibodies to B19V36–38. Our results demonstrated that the proportion of samples negative for both IgG and IgM antibodies was statistically higher among the HIV-positive patients (79.7%, 95% CI, 70.8–88.6%) than among the blood donors (51.1%, 95% CI, 40.9–61.3%) (p<0.05). The discrepancy between B19 antibody prevalences in HIV-positive samples in different reports35,39 and in our study, is intriguing and needs further investigation. As demonstrated in Table IV, the prevalence of IgG in HIV-positive blood donors and AIDS patients was 22.2% and 5.9%, respectively. Both these values are statistically lower than the prevalence in normal HIV-negative blood donors. It seems that the value is inversely correlated with the degree of immunodeficiency. However, we could not determine the CD4 T-cell count in order to elucidate the subjects’ immune status because blood cells were not available. To our surprise, 100% of HIV-positive/B19 DNA-positive AIDS patients tested (4/4) were IgG-positive and three samples were IgM-positive (two from HIV-positive blood donors, one from an AIDS patient) (Table III). This seems to contradict the data presented in Table IV. The concentration of B19 genome DNA ranged from 5.3×102 to 1.1×105 copies/mL in these B19 DNA-positive samples. It, therefore, seems that none of these samples was from people with B19 acute infection because in the early phase of acute infection the B19 genome load can reach 1013 copies/mL or even higher. There are several possible reasons why 100% of HIV+/B19 DNA+ AIDS patients tested were IgG-positive (4/4). It could be a non-specific cross-reactivity or perhaps, the result of a coincidence in a sample size that is too small for statistically meaningful conclusions. We also assume that these were persistent B19 infections, which are usually associated with anomalies of immune status or specificities of the immune response. One study found that an underlying cause of persistent infection might be quantitative or qualitative defects in the humoral response to B1919 because some persistently infected individuals have no production of antibodies to B19 (a quantitative defect), whereas others produce B19 antibodies that cannot neutralise the virus (a qualitative defect). Similar results were obtained with genotype 3 B19, where persistence was presented in the form of the viral particles being complexed to specific antibodies40. However, mechanisms other than the immune response could cause persistence41. One of the alternative hypothesised mechanisms for persistence is that the virus exists in the body without replication following the acute phase of infection26,42. One study proposed the concept of a “Bioportfolio”, which was defined as the capacity of human tissues to possess a lifelong storage mechanism of B19 viruses41.
As demonstrated in Figure 3a, B19 virus distribution in China could be classified into three groups: the Xi’an group (clade 2) which was a sister to B19-1B, the Urumqi group which clustered with sequences from the United States of America and United Kingdom, and the Sichuan and Luoyang group which was closely related to sequences from the United States of America and Brazil. Thus, the B19 virus distribution in China was polyphyletic which indicates the possibility of multiple origins of the virus in this country; two B19-1A strains could have spread independently into China from western countries. On the other hand, the Xi’an group did not belong to either of the known two sub-genotypes of B19V genotype 1. The origin of this diverged group remain a mystery.
Until recently, mutation rates of B19V genome were considered to be around 1×10−4 substitutions per site per year43,44. Further studies concluded that the substitution rate was reduced by relatively invariant regions in VP1/2, and the plasma-derived B19V genome evolutionary rate was corrected after eliminating the tissue-derived samples6; it is now being considered to be around 4×10−4 substitutions per site per year in ORF2. Nevertheless, the Sichuan B19V/HIV co-infected samples had only seven haplotypes and the sample collected in 1995 shared a haplotype with the samples collected in the late 2000s (Figure 3b). This phenomenon might suggest a relatively low substitution rate in the B19 viruses from Sichuan (and Luoyang, which share the same central haplotype, data not shown); however, it is also possible that this was a result of limited sampling. Consequently, thorough investigation is needed to uncover the B19 virus evolutionary pattern in south-western and central China where substitution rates might differ from those in other places investigated previously6,43,44.
Three genotypes of human parvovirus B19 were defined by Servants et al.4, with genotype 1 being considered the predominant genotype worldwide. Soon afterwards, Toan et al.5 divided B19 genotype 1 into two sub-genotypes as a result of a molecular phylogenetic study to determine the reason for a diverged monophyletic group within B19 genotype 1 in B19V/HBV co-infected patients in Vietnam. That monophyletic group was then defined as B19 -1B, which is a sister to the B19-1A lineage that includes the prototype of human parvovirus B19. However, in this study, both the BI and ML tree showed that, except for the two subgroups defined by Toan et al.5, the sequences from Xi’an collected in 1992 and 1993 formed another monophyletic group which appeared to be a sister group to B19 -1B (Figure 3a). The uncorrected p-distances among these three groups are as follows: the Xi’an group to B19-1A=0.043; the Xi’ans group to B19-1B=0.058; and B19-1A to B19-1B=0.053. These relatively large differences among three groups in the B19 genotype 1 make it hard to determine into which sub-genotype the Xi’an group should be placed, or whether the Xi’an group is another new sub-genotype of B19 genotype 1. Furthermore, the huge difference between the Xi’an group and B19-1A had already been discovered and, therefore, excluded from the calculation of B19 genotype 1 virus evolutionary rate in the previous study43. However, we believe that inferences regarding the subdivision of B19 genotype 1 should be made carefully because the limitation of sampling could influence the relationship among B19-1A, B19-1B and the Xi’an group. We, therefore, do not nominate the monophyletic Xi’an group as a new subgenotype of B19 genotype 1, and we suggest that a vast investigation of B19 virus distribution in China, focused on the Xi’an area and south-western China up to the Vietnamese border, should be launched in order to discover more samples of B19 virus which belong to clade 1 in Figure 3a and to reveal the origin, divergence as well as evolutionary history of B19 virus genotype 1.
As for the B19-1A sub-genotype, this sub-genotype originated around the 1950s in Europe and, by the early 1970s, replaced B19 genotype 2 in large parts of the world6. In Figure 3a, the most basal sequence of B19 -1a sub-genotype was U3 8509 which was collected in Japan in 198332. The uncorrected p-distance between U38509 and the other B19-1a samples was 0.0048. As a result, we also placed the U38509 sequence in the B19-1A sub-genotype. The globally-distributed B19-1A sub-genotype could have spread into Asia after its origin in Europe or have originated somewhere in Asia and then spread into Europe to replace the predominant genotype at that time, i.e. B19 genotype 2, by the early 1970s6.
This study has several limitations. First, some demographic data were not recorded when the samples were collected which made the investigation of some factors impossible. Second, the data collected in this study were not population-based. The prevalence estimation derived from this population might, therefore, not be representative of or generalizable to all B19V/HIV co-infected people in Sichuan, China and, in particular, individuals residing in rural areas and those with no access to HIV care or blood donation facilities. Third, our definition of chronic B19V co-infection was based on the presence of a single positive B19V DNA result; we might, therefore, have misclassified some individuals with acute B19V infection as being persistently co-infected and, consequently, overestimated the prevalence of B19V. As shown in Table IV, there were four IgM-positive samples among 79 HIV-positive samples, probably reflecting acute infections. Fourth, information on whether these subjects were anaemic was not available, making it impossible to investigate the role of B19V in this context. Despite these limitations, this was the first time that human parvovirus B19 in HIV-positive individuals in Sichuan, China had been investigated. This study elucidated the prevalence, phylogeny and distribution of human parvovirus B19 in Sichuan, China and raises a potential concern for blood safety related to persistent parvovirus B19 in the circulation of not only immunocompromised individuals, but also in immunocompetent ones.
Acknowledgments
This work was supported by a grant from the Institute of Pathogen Biology, Chinese Academy of Medical Sciences (grant n. 2009IPB203). We thank Edward A. Myers and Alexander D. McKelvy for helpful discussion and for reviewing the manuscript.
Footnotes
The Authors declare no conflicts of interest.
References
- 1.Corcoran A, Doyle S. Advances in the biology, diagnosis and host-pathogen interactions of parvovirus B19. J Med Microbiol. 2004;53:459–75. doi: 10.1099/jmm.0.05485-0. [DOI] [PubMed] [Google Scholar]
- 2.Shade RO, Blundell MC, Cotmore SF, et al. Nucleotide sequence and genome organization of human parvovirus B19 isolated from the serum of a child during aplastic crisis. J Virol. 1986;58:921–36. doi: 10.1128/jvi.58.3.921-936.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhi N, Mills IP, Lu J, et al. Molecular and functional analyses of a human parvovirus B19 infectious clone demonstrates essential roles for NS1, VP1, and the 11-kilodalton protein in virus replication and infectivity. J Virol. 2006;80:5941–50. doi: 10.1128/JVI.02430-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Servant A, Laperche S, Lallemand F, et al. Genetic diversity within human erythroviruses: identification of three genotypes. J Virol. 2002;76:9124–34. doi: 10.1128/JVI.76.18.9124-9134.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Toan NL, Duechting A, Kremsner PG, et al. Phylogenetic analysis of human parvovirus B19, indicating two subgroups of genotype 1 in Vietnamese patients. J Gen Virol. 2006;87:2941–9. doi: 10.1099/vir.0.82037-0. [DOI] [PubMed] [Google Scholar]
- 6.Norja P, Eis-Hubinger AM, Soderlund-Venermo M, et al. Rapid sequence change and geographical spread of human parvovirus B19: comparison of B19 virus evolution in acute and persistent infections. J Virol. 2008;82:6427–33. doi: 10.1128/JVI.00471-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nguyen QT, Wong S, Heegaard ED, Brown KE. Identification and characterization of a second novel human erythrovirus variant, A6. Virology. 2002;301:374–80. doi: 10.1006/viro.2002.1585. [DOI] [PubMed] [Google Scholar]
- 8.Hokynar K, Soderlund-Venermo M, Pesonen M, et al. A new parvovirus genotype persistent in human skin. Virology. 2002;302:224–8. doi: 10.1006/viro.2002.1673. [DOI] [PubMed] [Google Scholar]
- 9.Nguyen QT, Sifer C, Schneider V, et al. Detection of an erythrovirus sequence distinct from B19 in a child with acute anaemia. Lancet. 1998;352:1524. doi: 10.1016/S0140-6736(05)60330-3. [DOI] [PubMed] [Google Scholar]
- 10.Parsyan A, Addo-Yobo E, Owusu-Ofori S, et al. Effects of transfusion on human erythrovirus B19-susceptible or -infected pediatric recipients in a genotype 3-endemic area. Transfusion. 2006;46:1593–600. doi: 10.1111/j.1537-2995.2006.00952.x. [DOI] [PubMed] [Google Scholar]
- 11.Rinckel LA, Buno BR, Gierman TM, Lee DC. Discovery and analysis of a novel parvovirus B19 genotype 3 isolate in the United States. Transfusion. 2009;49:1488–92. doi: 10.1111/j.1537-2995.2009.02160.x. [DOI] [PubMed] [Google Scholar]
- 12.Sanabani S, Neto WK, Pereira J, Sabino EC. Sequence variability of human erythroviruses present in bone marrow of Brazilian patients with various parvovirus B19-related hematological symptoms. J Clin Microbiol. 2006;44:604–6. doi: 10.1128/JCM.44.2.604-606.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Young NS, Brown KE. Parvovirus B19. N Engl J Med. 2004;350:586–97. doi: 10.1056/NEJMra030840. [DOI] [PubMed] [Google Scholar]
- 14.Heegaard ED, Brown KE. Human parvovirus B19. Clin Microbiol Rev. 2002;15:485–505. doi: 10.1128/CMR.15.3.485-505.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Buyukkose M, Kozanoglu E, Basaran S, et al. Seroprevalence of parvovirus B19 in fibromyalgia syndrome. Clin Rheumatol. 2009;28:305–9. doi: 10.1007/s10067-008-1044-4. [DOI] [PubMed] [Google Scholar]
- 16.Parsyan A, Candotti D. Human erythrovirus B19 and blood transfusion - an update. Transfus Med. 2007;17:263–78. doi: 10.1111/j.1365-3148.2007.00765.x. [DOI] [PubMed] [Google Scholar]
- 17.Bizjak G, Blondin D, Hammer R, et al. Acute infection with parvovirus B19 in early pregnancy. Ultrasound Obstet Gynecol. 2009;34:234–5. doi: 10.1002/uog.6454. [DOI] [PubMed] [Google Scholar]
- 18.Bremner JA, Cohen BJ. Parvovirus B19 as a cause of anemia in human immunodeficiency virus-infected patients. J Infect Dis. 1994;169:938–40. doi: 10.1093/infdis/169.4.938a. [DOI] [PubMed] [Google Scholar]
- 19.Kurtzman GJ, Cohen BJ, Field AM, et al. Immune response to B19 parvovirus and an antibody defect in persistent viral infection. J Clin Invest. 1989;84:1114–23. doi: 10.1172/JCI114274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frickhofen N, Young NS. Persistent parvovirus B19 infections in humans. Microb Pathog. 1989;7:319–27. doi: 10.1016/0882-4010(89)90035-1. [DOI] [PubMed] [Google Scholar]
- 21.Chernak E, Dubin G, Henry D, et al. Infection due to parvovirus B19 in patients infected with human immunodeficiency virus. Clin Infect Dis. 1995;20:170–3. doi: 10.1093/clinids/20.1.170. [DOI] [PubMed] [Google Scholar]
- 22.Frickhofen N, Abkowitz JL, Safford M, et al. Persistent B19 parvovirus infection in patients infected with human immunodeficiency virus type 1 (HIV-1): a treatable cause of anemia in AIDS. Ann Intern Med. 1990;113:926–33. doi: 10.7326/0003-4819-113-12-926. [DOI] [PubMed] [Google Scholar]
- 23.Koppelman MH, Rood IG, Fryer JF, et al. Parvovirus B19 genotypes 1 and 2 detection with real-time polymerase chain reaction assays. Vox Sang. 2007;93:208–15. doi: 10.1111/j.1423-0410.2007.00957.x. [DOI] [PubMed] [Google Scholar]
- 24.Ke L, He M, Li C, et al. The prevalence of human parvovirus B19 DNA and antibodies in blood donors from four Chinese blood centers. Transfusion. 2011;51:1909–18. doi: 10.1111/j.1537-2995.2011.03067.x. [DOI] [PubMed] [Google Scholar]
- 25.Corcoran C, Hardie D, Yeats J, Smuts H. Genetic variants of human parvovirus B19 in South Africa: cocirculation of three genotypes and identification of a novel subtype of genotype 1. J Clin Microbiol. 2010;48:137–42. doi: 10.1128/JCM.00610-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cassinotti P, Siegl G. Quantitative evidence for persistence of human parvovirus B19 DNA in an immunocompetent individual. Eur J Clin Microbiol Infect Dis. 2000;19:886–7. doi: 10.1007/s100960000384. [DOI] [PubMed] [Google Scholar]
- 27.Thompson JD, Gibson TJ, Plewniak F, et al. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25:4876–82. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nylander JAA. 2004. MrModeltest v2 Program distributed by the author: Evolutionary Biology Centre, Uppsala University. [Google Scholar]
- 29.Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–4. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- 30.Stamatakis A, Hoover P, Rougemont J. A rapid bootstrap algorithm for the RAxML Web servers. Syst Biol. 2008;57:758–71. doi: 10.1080/10635150802429642. [DOI] [PubMed] [Google Scholar]
- 31.Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–9. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- 32.Erdman DD, Durigon EL, Wang QY, Anderson LJ. Genetic diversity of human parvovirus B19: sequence analysis of the VP1/VP2 gene from multiple isolates. J Gen Virol. 1996;77:2767–74. doi: 10.1099/0022-1317-77-11-2767. [DOI] [PubMed] [Google Scholar]
- 33.Thomas I, Di Giambattista M, Gerard C, et al. Prevalence of human erythrovirus B19 DNA in healthy Belgian blood donors and correlation with specific antibodies against structural and non-structural viral proteins. Vox Sang. 2003;84:300–7. doi: 10.1046/j.1423-0410.2003.00299.x. [DOI] [PubMed] [Google Scholar]
- 34.Kooistra K, Mesman HJ, de Waal M, et al. Epidemiology of high-level parvovirus B19 viraemia among Dutch blood donors, 2003–2009. Vox Sang. 2011;100:261–6. doi: 10.1111/j.1423-0410.2010.01423.x. [DOI] [PubMed] [Google Scholar]
- 35.van Elsacker-Neile AM, Kroon FP, van der Ende ME, et al. Prevalence of parvovirus B19 infection in patients infected with human immunodeficiency virus. Clin Infect Dis. 1996;23:1255–60. doi: 10.1093/clinids/23.6.1255. [DOI] [PubMed] [Google Scholar]
- 36.Bremner JA, Beard S, Cohen BJ, et al. Secondary infection with parvovirus B19 in an HIV-positive patient. AIDS. 1993;7:1131–2. doi: 10.1097/00002030-199308000-00021. [DOI] [PubMed] [Google Scholar]
- 37.Naides SJ, Howard EJ, Swack NS, et al. Parvovirus B19 infection in human immunodeficiency virus type 1-infected persons failing or intolerant to zidovudine therapy. J Infect Dis. 1993;168:101–5. doi: 10.1093/infdis/168.1.101. [DOI] [PubMed] [Google Scholar]
- 38.Zuckerman MA, Williams I, Bremner J, et al. Persistent anaemia in HIV-infected individuals due to parvovirus B19 infection. AIDS. 1994;8:1191–2. doi: 10.1097/00002030-199408000-00029. [DOI] [PubMed] [Google Scholar]
- 39.Azevedo KM, Setubal S, Camacho LA, et al. Seroepidemiological study of human parvovirus B19 among human immunodeficiency virus-infected patients in a medium-sized city in Rio de Janeiro, Brazil. Mem Inst Oswaldo Cruz. 2009;104:901–4. doi: 10.1590/s0074-02762009000600014. [DOI] [PubMed] [Google Scholar]
- 40.Candotti D, Etiz N, Parsyan A, Allain JP. Identification and characterization of persistent human erythrovirus infection in blood donor samples. J Virol. 2004;78(22):12169–78. doi: 10.1128/JVI.78.22.12169-12178.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Norja P, Hokynar K, Aaltonen LM, et al. Bioportfolio: lifelong persistence of variant and prototypic erythrovirus DNA genomes in human tissue. Proc Natl Acad Sci USA. 2006;103:7450–3. doi: 10.1073/pnas.0602259103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Woolf AD, Cohen BJ. Parvovirus B19 and chronic arthritis–causal or casual association? Ann Rheum Dis. 1995;54:535–6. doi: 10.1136/ard.54.7.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Parsyan A, Szmaragd C, Allain JP, Candotti D. Identification and genetic diversity of two human parvovirus B19 genotype 3 subtypes. J Gen Virol. 2007;88(Pt 2):428–31. doi: 10.1099/vir.0.82496-0. [DOI] [PubMed] [Google Scholar]
- 44.Shackelton LA, Holmes EC. Phylogenetic evidence for the rapid evolution of human B19 erythrovirus. J Virol. 2006;80:3666–9. doi: 10.1128/JVI.80.7.3666-3669.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]