

Abstract
Persistent infections involving slow-growing or non-growing bacteria are hard to treat with antibiotics that target biosynthetic processes in growing cells. Consequently, there is a need for antimicrobials that can treat infections containing dormant bacteria. In this Review, we discuss the emerging concept that disrupting the bacterial membrane bilayer or proteins that are integral to membrane function (including membrane potential and energy metabolism) in dormant bacteria is a strategy for treating persistent infections. The clinical applicability of these approaches is exemplified by the efficacy of lipoglycopeptides that damage bacterial membranes and of the diarylquinoline TMC207, which inhibits membrane-bound ATP synthase. Despite some drawbacks, membrane-active agents form an important new means of eradicating recalcitrant, non-growing bacteria.
Antimicrobial chemotherapy seeks to eradicate the infecting pathogen from its host in the shortest possible treatment period, often using antibiotics that effectively kill or inhibit the growth of metabolically active bacteria1, 2. Many of these agents were developed during the golden era of antibiotic drug discovery (1940s–1980s), and most target five biosynthetic processes that occur in actively growing bacteria: the biosynthesis of proteins, RNA, DNA, peptidoglycan and folic acid3. However, most of these classical antimicrobial strategies are not effective for eradicating persistent infections in which bacteria are quiescent (that is, slow-growing or dormant) (Box 1). Well-known clinical examples of such infections include the staphylococcal biofilms that result in endocarditis and other medical device-related infections, Pseudomonas aeruginosa infections of the lungs of patients with cystic fibrosis, streptococcal otitis media4, ischemic osteomyelitis containing slow-growing microorganisms5, and tuberculous granulomas that contain latent Mycobacterium tuberculosis6. These cases require prolonged treatment periods to cure or alleviate the burden of disease. For example, a drug regimen lasting for at least 6 months is required to cure tuberculosis (TB)7, and for endocarditis and osteomyelitis, a minimum of 4 weeks of treatment is necessary8, 9. Alternatively, as is the case for medical device-related infections, surgical removal can often be required. The presence of different subpopulations of persisting bacteria (also known as recalcitrant bacteria) with varying antibiotic susceptibilities further challenges the overall efficacy of an antibiotic, and it may be that no single agent kills all of the subpopulations effectively (although some agents may slowly kill specific subpopulations; pyrazinamide, for example, kills M. tuberculosis). Consequently, unconventional antimicrobial strategies will be needed to cure infections that contain quiescent bacteria (Box 2), as previously described1, 6, 10. Such strategies could shorten the treatment period, reduce disease relapse and limit the emergence of antibiotic resistance arising from surviving bacteria11. Therefore, newly approved and emerging therapeutic modalities should be evaluated for their potential to fulfill unmet medical needs such as the treatment of persistent infections. Dormant bacteria have not traditionally been targeted in antibiotic development programmes, in part owing to the difficulty of discovering successful agents. As a result, the types of agents that kill dormant organisms remain poorly defined, although some established drugs, including rifampicin and moxifloxacin, are known to kill some subpopulations of dormant bacteria12–15
Box 1. Antibiotic survival and why we need new antimicrobials.
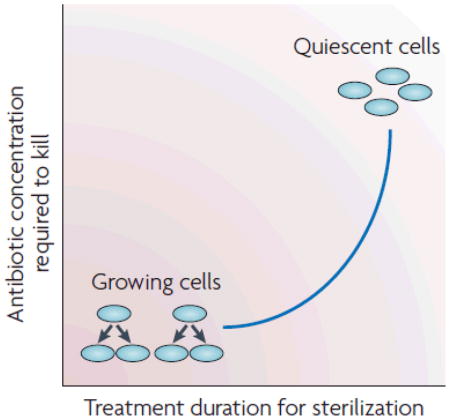
Most of the antibiotic classes that are currently in clinical use were discovered during the ‘golden era’ of antibiotic discovery, which lasted from the 1940s to 1980s. These antibiotics were discovered by screening for the ability to inhibit or kill logarithmically growing bacteria. Therefore, most developed antibiotics target processes required for growth - that is, the biosynthesis of proteins, peptidoglycan, folic acid, DNA and RNA. In response to the bacterial acquisition of antibiotic resistance, chemical modification of the existing drug classes was carried out to obtain analogues with improved activity. Like their precursor drugs, these analogues are efficacious against metabolically active, rapidly growing bacteria (see the figure). However, in several types of infections, bacteria encounter unfavourable conditions that cause cells to enter into a quiescent state of slow growth or even no growth. These metabolically inactive organisms (see the figure) can survive high concentrations of antibiotics, and extended treatment is therefore required for drug efficacy. When the drug concentration falls below levels that kill or inhibit the growing cells, dormant bacteria can reactivate to become growing cells, and the host begins to show symptoms of disease again. Providing that these reactivated cells have not evolved genetic resistance to the drug, they may be killed on subsequent rounds of treatment. The reason why dividing cells become dormant is multifactorial, as this phenomenon occurs in response to environmental factors such as the depletion of nutrients and oxygen, and an acidic pH, all of which decrease and eventually stop bacterial growth. For an extensive review of this phenomenon, see Refs1,2,10,24.
Biofilm-mediated infections and tuberculosis (TB) provide two major examples of unmet clinical need owing to bacteria that are either slow growing or dormant and therefore hard to treat. Since the mid-1990s, the recognition of biofilm diseases has greatly increased, with estimates by the US CDC suggesting that up to 65% of all infections in developed countries involve biofilms. Biofilm-mediated infections range from those involving medical-device implants, including bloodline catheters and heart implants, to those associated with cystic fibrosis, wounds and superficial skin infections4. The unmet medical need for a TB treatment also came to light in the mid-1990s, which saw outbreaks of drug-resistant bacterial strains and the lethal synergy of the disease with HIV. TB is the most devastating bacterial infection of humans, causing ~2 million deaths per year. It is estimated that one-third of the world’s population is infected with asymptomatic, dormant M. tuberculosis, from which new cases of active infection arise (~8 million annually)21.
Now, more than a decade on, and with much intense research effort, it is emerging that antimicrobials that damage the membrane structure to perturb its function may provide a novel avenue for treating persisting infections. Similarly, organisms that can become dormant, such as M. tuberculosis and biofilm bacteria (for example, Staphylococcus aureus and Pseudomonas aeruginosa) may use anaerobic metabolism (for example, substrate level phosphorylation and anaerobic respiration) to sustain viability in the absence of growth. Because of this, enzymes involved in these processes are also being explored as targets for drug development.
Box 2. Targeting persistence.
Targeting of proteins involved in the formation of quiescent cells is being considered as a co-therapy approach to treat persistent infections152. Although this concept is plausible, genetic mechanisms that c confer bacterial persistence are currently not well understood and may involve multiple genes and redundant pathways46, which would complicate target selection for drug discovery. Indeed, a screen of a well-defined transposon library in Escherichia coli (the Keio collection) failed to identify a single mutant unable form persisters, supporting the hypothesis that bacteria use multiple pathways to reduce growth and form persister cells46, 153, 154. Hence, an alternative strategy could be to target processes that are required to maintain bacterial viability in already formed quiescent cells. These cell types are often present in an infection before therapy is administered1, 10.
In our opinion, antimicrobials that target either the organization of the bacterial membrane bilayer (in bacterial biofilms) or the functions of membrane-associated respiratory enzymes (in latent TB) are promising therapeutic approaches for treating slow-growing or dormant bacterial infections. The general actions of these types of agents are shown in Fig. 1. Inhibitors of energy metabolism bind directly to target enzymes in the membrane that are involved in energy generation and the redox balancing of NAD+/NADH (Fig. 1a). By contrast, membrane-damaging antimicrobials are generally lipophilic in nature and directly interact with the bacterial membrane bilayer, disrupting its function (or functions) and its physical integrity (Fig. 1b). Although some membrane-damaging agents may also disrupt the function of membrane-bound proteins, such as the enzymes involved in energy production, this action is indirect. The clinical effectiveness of damaging the bacterial membrane is demonstrated by the recently approved agents daptomycin and telavancin, which disrupt the membrane bilayer and are in clinical use for treating Staphylococcus aureus infections (Fig. 2; Table 1). Furthermore, the drug TMC207 disrupts energy production in M. tuberculosis by targeting its ATP synthase, and it is presently in Phase II clinical trials for TB treatment (Fig. 3; Table 1). Several other membrane-active agents and inhibitors of energy metabolism are at various stages of pre-clinical and clinical development (Table 1). The potential therapeutic benefits of such agents arises from the fact that the membrane is vital to both active and metabolically inactive pathogens, and quiescent bacteria, like all living cells, require some form of cellular energy and redox homeostasis to maintain cell viability, even in the absence of growth16. Although these emerging paradigms have arisen from seemingly separate research areas, they represent a change from traditional discovery efforts. In this Review, we discuss whether antimicrobial strategies can be devised to treat infections of dormant and slow-growing bacteria, and whether lessons learned from the development of new therapeutics for one indication can be applied to another. We focus in particular on studies concerning the potential use of membrane-active antibiotics against biofilms and of agents that attack the membrane-associated anaerobic respiratory system of dormant M. tuberculosis. Accordingly, we stress that the concept of using membrane disruption to counter bacterial biofilms should be applicable to killing dormant M. tuberculosis to treat chronic TB; similarly, as membrane-mediated anaerobic metabolism is increasingly being recognized as essential for the maintenance of biofilms17, 18, 19, 20, corrupting this function may also counter biofilm diseases. As the clinical relevance of biofilm formation by M. tuberculosis is unknown21, 22, this Review does not address the action of antimicrobials against M. tuberculosis biofilms.
Figure 1.

Figure 2.

Table 1.
| Antibiotic | Active against | Mode of action | Antibiotic status | Refs |
|---|---|---|---|---|
| Daptomycin | Gram-positive bacteria (for example, Staphylococcus aureus and Enterococcus spp.); active against biofilms | Membrane permeabilization and depolarization; disruption of multiple cellular processes | Approved (2003) (or cSSSI; S. aureus bacteremia and right-side endocarditis | 160 |
| Telavancin | Gram-positive bacteria (for example, S. aureus, including VISA); active against biofilms | Inhibition of peptidoglycan biosynthesis by binding to the D-Ala-D-Ala termini; membrane permeabilization and depolarization; disruption of multiple cellular processes | Approved (2009) for cSSSI; completed Phase III for pneumonia | 63 |
| Oritavancin | Gram-positive bacteria (S. aureus, including VISA, and Enterococcus spp.); active against biofilms and stationary phase cells | Inhibition of peptidoglycan biosynthesis by binding to the D-Ala–D-Ala termini; membrane permeabilization and depolarization; disruption of multiple cellular processes | Completed Phase III trials for cSSSI and Phase II trials for S. aureus bacteremia | 63 |
| Dalbavancin | Gram-positive bacteria (for example, S. aureus, including VISA); prevents biofilm formation in vivo | Inhibition of peptidoglycan biosynthesis by binding to the D-Ala-D-Ala termini; alternative modes of action are presumed to involve membrane disruption | Completed Phase III trials for cSSSI | 53.63 |
| Reutericyclin (Lee-867) | Gram-positive bacteria (for example, S. aureus); active against biofilms | Membrane depolarization; disruption of multiple cellular processes | Discovery stage for topical use against S. aureus | 56 |
| XF-73 | Gram-positive bacteria (for example, S. aureus) | Membrane permeabilization and depolarization; disruption of multiple cellular processes | Discovery stage for topical use against S. aureus | 50.54 |
| CSA-13 | Broad spectrum drug, mainly active against Gram-positive bacteria (for example, S. aureus); active against biofilms | Membrane permeabilization and depolarization: disruption of multiple cellular processes | Pre-clinical stage for topical use and biofilm prevention on medical devices | 158.161.162 |
| HT61 | Gram-positive bacteria (for example, S. aureus) | Membrane permeabilization and depolarization; disruption of multiple cellular processes | Clinical trials for nasal decolonization of S. aureus | 163 |
| Clofazimine derivatives | Mycobacterium tuberculosis and S. aureus; active against NRP cells | Complex action, including membrane depolarization | Discovery optimization stage | TB Alliance* |
| TMC207 | M. tuberculosis; active against NRP cells | Inhibition of mycobacterial ATP synthetase | Phase II trials for TB disease | TB Alliance* |
| Nitroimidazoles (PA-824 and OPC67683) | M. tuberculosis; active against NRP cells | Activated by the Rv3547–Fgd1 pathway to Form reactive free radical antimicrobial species | Phase II trials for TB disease | TB Alliance* and Otsuka pharmaceuticals‡ |
| Nitrofurans (such as Lee-1106) | M. tuberculosis; active against NRP cells | Activated by an unknown mechanism to form reactive free antimicrobial species | Discovery optimization stage | 164.165 |
| Nitazoxanide | Broad spectrum drug, active against Escherichia coli and S. aureus biofilms and NRP M. tuberculosis | Inhibition of PFOR; undergoes reduction by other redox enzymes to form reactive free radical antimicrobial species | Approved for anaerobic intenstinal parasites (for example, Giadria lamblia and Cryptosporidium parvum) | 147.166 |
| Ro48-8071 | M. tuberculosis | Inhibition of menaquinone biosynthesis, leading to respiratory chain arrest | Discovery optimization stage | 115 and TB Alliance* |
cSSSL, complicated skin and skin structure infection; NRPcells, non-replicating persistent cells; PFOR, pyruvate–ferredoxin oxidoreductase; Fgd1.F420-dependent glucose-6-phosphate dehydrogenase; TB, tuberculosis; VISA, vancomycin-intermediate S. aureus.
TB Alliance (The Global Alliance for TB Drug Development) can be Found at www.tballiance.org
Otsuka Pharmaceuticals can be found at http://www.otsuka-us.com.
Figure 3.

The problem of antibiotic recalcitrance
Resistance to antibiotics is widely associated with treatment failure. In addition to genetic resistance, pathogenic bacteria commonly undergo physiological adaptations that render cells slow-growing or non-growing and the bacteria thereby become recalcitrant to killing by bactericidal antibiotics (Box 1) without becoming genetically resistant; they regain sensitivity to antibiotics on the resumption of growth. This recalcitrance is due to two phenomena: ‘antibiotic persistence’6,23, in which a fraction of non-growing specialized persister cells arises during stationary phase and can survive antibiotic exposure, and ‘antibiotic indifference’2,24 in which a bacterial subpopulation becomes slow-growing or non-growing in response to unfavourable conditions (such as host responses, low pH, nutrient deprivation and oxygen deprivation) and is therefore not susceptible to killing by antibiotics. ‘Antibiotic survival’, a more comprehensive term encompassing both phenomena, indicates that bacteria have multiple ways of avoiding being killed by bactericidal antibiotics11. As a result of several studies, a consensus is emerging that persisting bacteria can evade antibiotic killing by substantially downregulating the biosynthetic processes that are targeted by most antibiotics without affecting bacterial survival in themetabolically inactive state1,2,6,10,11,24 (Box 1). For example, β-lactam antibiotics kill by activating autolysins and this requires active peptidoglycan synthesis in cells undergoing division. Below, we explain why agents that disorganize the structure of the membrane or that inhibit respiratory enzymes involved in energy production and establishing the membrane’s proton motive force are valid approaches for treating infections containing quiescent bacteria.
A case for targeting the cell membrane
Antimicrobial properties that are applicable to the discovery and therapeutic use of membrane-active agents include the essentiality of the bacterial membrane target, as well as the ability of some agents to kill target pathogens selectively or to disrupt multiple parts of the cell; together, these are potent bactericidal properties that result in low prospects for the emergence of resistance.
Essentiality and selectivity
The bacterial membrane is essential, irrespective of the metabolic status of the cell, as it provides selective permeability for cellular homeostasis and metabolic energy-transduction. In addition, the membrane contains about one third of the proteins in a cell and is the site for crucial processes, such as active transport of nutrients and wastes, bacterial respiration, establishment of the proton motive force in association with respiratory enzymes (Fig. 1a), ATP generation and cell-cell communication in biofilms25. Antimicrobial peptides (AMPs) made by the host and several bioactive molecules that act on the membrane26,27 validate its significance as an antibacterial target site. In spite of this, traditional discovery efforts have not focused on the membrane as a target for antibiotic development. Several synthetic and natural-product chemotypes that damage the bacterial membrane, and could possibly kill dormant bacteria, are under-explored as chemotherapeutics, possibly owing to concern over the potential for these agents to disrupt the mammalian plasma membrane as well27 and the lack of knowledge regarding chemical optimization of such molecules to attain pathogen selectivity (Box 3). However, the successful medical use of the membrane-active antibiotics daptomycin (a cyclic lipopeptide) and telavancin, oritavancin and dalbavancin (lipoglycopeptides) indicate that bacterial specificity is achievable. These agents preferentially bind to bacterial membranes owing to the predominance of negatively charged phospholipids (phosphatidylglycerol and cardiolipin) and zwitterionic phosphatidylethanolamine, which are rare in the outer leaflets of mammalian cells28, and the absence of cholesterol. For example, daptomycin oligomerizes in the presence of calcium ions (Ca2+) to form a micelle-like amphipathic structure, with its hydrophobic decanoyl side chain facing inwards29 and this provides the drug with a pseudo-positively charged surface that increases its affinity for the negatively charged bacterial membranes29. The micelle is disrupted by interaction with the membrane, allowing the insertion of the hydrophobic tail of daptomycin, similar to the action of cationic antimicrobial peptides29. Similarly, oritavancin carries a net positive charge and destabilizes model membranes that contain large amounts of bacterial phosphatidylglycerol and cardiolipin30,31. In addition, binding to membrane-located peptidoglycan precursors and bacterial membrane proteins may contribute to the specificity of daptomycin and lipoglycopeptides.
Box 3. Challenges and opportunities for membrane-active agents.
Disruption of the membrane may offer a superior approach for treating dormant infections, but several obstacles need to be considered during the discovery and assessment of these agents. The most obvious challenges and opportunities are outlined below.
Selective toxicity should, ideally, be achieved against the target pathogen. There is a wealth of potential compounds in chemical-product and natural-product screening libraries, many of which have been shown to kill pathogens, but these candidates often also perturb mammalian membranes27. Therefore, structure–activity relationships need to be established to understand how to create species-specific molecules. A starting point would be to apply the design strategies from antimicrobial peptides and peptidomimetics to small molecules.
During antibacterial whole-cell screening, the red blood cell haemolysis assay provides a simple approach for eliminating molecules that damage mammalian membranes. Rather than discarding these molecules, counter-selective screening in bacterial membrane-damage assays155 should be carried out to catalogue molecules with some selectivity for bacteria. These compounds can then be subjected to structure–activity relationship medicinal chemistry efforts.
The physicochemical properties of membrane-active agents may not be best suited for oral administration. Therefore, these parameters need to be considered during optimization and may require suitable formulations for oral and parenteral delivery, including specialized dosing regimens and formulations. This is exemplified by the antifungal amphothericin B, which damages the membrane of Candida albicans: its toxicity can be mitigated by specialized liposomal formulations, allowing this drug to be used effectively at higher doses to treat systemic mycoses156.
Not all antibacterial drugs kill in the same manner. The determination of bactericidal kinetics for membrane-active agents, to determine whether they are concentration dependent or time dependent, may be used to develop optimum dosing regimens that limit the development of resistance and maximize efficacy and safety. Furthermore, not all membrane-active agents will be bactericidal against all forms of persisting infections. Hence, the spectrum of diseases that can be covered by agents should be defined early during the discovery stages. This will enable further clinical development or chemical optimization to expand the potential for activity against bacteria in different quiescent states.
The complex lipid-rich cell wall of Mycobacterium tuberculosis contributes to its resilience against chemical challenges and may restrict molecules from reaching the plasma membrane target. The outer-membrane of Gram-negative bacteria will impose similar effects. Therefore, the amphipathic properties of molecules must be taken into account during the development of agents for use against these organisms.
As is the case for pyrazinamide, agents showing high minimum inhibitory concentration values in vitro might still eradicate dormant cells from an infection by synergizing with the host immune system. But the discovery of such molecules would require suitable animal models or in vitro systems that can reproduce the complex conditions found in the host.
In addition, minimum inhibitory concentration values are not indicative of antimicrobial potency against non-growing bacteria; therefore, more representative parameters such the minimum stationary-cidal concentration1, 37, minimum dormicidal concentration1 or minimum biofilm eradication concentration50 should be determined during discovery of compounds for treating persistent infections.
As membrane-active agents do not rely on intracellular targets for their activities, they may be immobilized to the surface of medical devices as part of a ‘smart surface approach’ to prevent bacterial contamination and biofilm formation. This is demonstrated by the agent chitosan157 and by a new class of antimicrobial peptidomimics known as ceragenins158.
A complex multitarget mode of action
Several membrane-damaging agents interfere with multiple targets29, 31, 32 through the interaction of a lipophilic moiety with the bacterial membrane (causing disruption of membrane architecture and functional integrity), through steric inhibition of membrane-embedded proteins and/or through alteration of the proton motive force, which may lead to leakage of cytosolic content and eventual cell death (Fig. 1). Agents causing damage to the membrane may lead to lethal pleiotropic effects in quiescent bacteria, but dissipation of the proton motive force alone is not bactericidal in all species (although this property may constrain energy supply in already metabolically inactive cells). By contrast, M. tuberculosis, unlike other pathogens33, 34, seems to require a fully energized membrane for survival under both aerobic and hypoxic conditions16, 35, and therefore dissipation of the proton motive force by the ionophores nigericin or valinomycin is highly bactericidal to active and dormant M. tuberculosis35. Furthermore, increasing the proton permeability of the membrane makes mycobacteria more sensitive to the effect of reactive free radicals (nitric oxide and superoxide) in macrophages, as this mechanism is enhanced at acidic pHs36.
A low potential for the development of resistance
As membrane-active agents may interact with multiple targets in the membrane, the ability of bacteria to acquire resistance to these agents is limited. In vitro studies have been carried out with lipopeptides22, lipoglycopeptides (I.C. and A.J.O., unpublished observations), a range of cationic antimicrobial peptides13 and HT61, a recently described quinolone-derived membrane-active compound37, and these studies suggest that de novo mutations conferring resistance to membrane-active antibiotics do not readily arise. The induction of lipopolysaccharide modification systems (such as those regulated by the PmrA–PmrB two-component system; also known as the BasR–BasS system) and the expression of efflux pumps (including MexAB–OprM) allow Gram-negative organisms such as P. aeruginosa to avert killing by cationic peptides38, 39. However, the rapid elimination of pathogens that are inherently susceptible to membrane-active drugs should reduce the likelihood of resistance emerging, as long as drug concentrations remain bactericidal at the site of infection.
Membrane-damaging agents against biofilms
Several agents that disrupt the bacterial cytoplasmic membrane possess activity against biofilms in vitro (Fig. 2; Table 1), and one of these, daptomycin, has been used clinically since 2003. Telavancin was approved for use in the United States and Canada in 2009, and other lipoglycopeptides are in late-stage development40. Daptomycin kills staphylococcal cells quicker than most other antibacterials and can, in some cases, completely eradicate S. aureus biofilms41, 42. Indeed, clinical observations indicate that it is useful for treating biofilm-mediated staphylococcal and enterococcal endocarditis43. However, daptomycin may not be able to eradicate all types of staphylococcal biofilm infections, as in a mouse model it cleared less than 7% of catheter-associated biofilms after 7 days of treatment44. Thus, even a dormant bacterial population may contain different subpopulations of persistent cells, depending on the existing environmental or host conditions6, 17, 45, 46; similarly, the composition of membrane lipids may vary between different subpopulations of persistent cells47, 48, thereby affecting the interactions of the drug with the membrane11. Combination therapies may be required to eradicate particular biofilm diseases, as the clinical efficacy of daptomycin, like that of all other antimicrobials, will depend on the pharmacokinetics and pharmacodynamics of the drug at the site of infection. Furthermore, the finding that daptomycin is less effective against stationary phase bacteria than against cells in logarithmic phase exemplifies the fact that not all membrane-damaging agents will be effective in certain slow-growing or dormant infections49. This does not mean, however, that other, structurally distinct membrane-disrupting agents will lose their bactericidal activities against the different forms of slow-growing or dormant bacteria50, 51. For example, telavancin can eradicate staphylococcal and enterococcal biofilms at concentrations close to those that inhibit the growth of planktonic cells52. Similarly, the related lipoglycopeptide oritavancin is equally potent for killing staphylococcal biofilms and stationary phase cells51. Although telavancin and oritavancin have multiple modes of action, including inhibition of peptidoglycan biosynthesis, their anti-biofilm activity probably results primarily from their ability to permeabilize the bacterial membrane51, 52. Corroborating this idea, the prototypical glycopeptide vancomycin, which inhibits peptidoglycan crossbridge formation but lacks activity against the staphylococcal membrane, is inactive against staphylococcal biofilms51. These membrane-active lipoglycopeptides may have an important role in combating osteomyelitis, endocarditis and catheter-related infections, as suggested by experimental animal models, as long as they can reach the site of infection40. Dalbavancin, the lipophilic derivative of the glycopeptide teicoplanin, may also have membrane-damaging properties, but its mechanism of action and potential activity against quiescent bacteria, including S. aureus biofilms, is unknown; however, a study examining its efficacy in preventing S. aureus colonization of catheters in rabbits indicates some improvement over the related agent vancomycin53.
Several pre-clinical or experimental membrane-active agents also display potent activity against biofilms (Fig. 2; Table 1). The novel anti-staphylococcal porphyrin agents XF70 and XF73 perturb the bacterial membrane54 and eradicate both S. aureus biofilms and stationary phase S. aureus cells at concentrations close to the minimum inhibitory concentration (MIC) for planktonic cultures, similarly to lipoglycopeptides50. Anti-biofilm activity has also been noted for derivatives of the tetramic acid natural product reutericyclin (Fig. 2) (obtained from Lactobacillus reuteri55), which depolarize staphylococcal cells and therefore have potential for treating persistent topical infections56. Interestingly, reutericyclin derivatives also kill stationary phase Clostridium difficile cells that are insensitive to vancomycin and metronidazole (J.G.H. and R.E.L., unpublished observations). Antimicrobial peptides such as LL-37 (Ref. 57) and synthetic peptidomimetics58 (Fig. 2), which cause permeabilization of the inner and outer membranes in Gram-negative bacteria, can kill bacterial biofilms.
The small quinolone-derived compound HT61 was discovered in a screen for novel antimicrobials that kill non-growing bacteria37. In this unorthodox but highly commendable approach, compounds were prioritized according to their killing of non-growing cells rather than on conventional MIC testing against growing bacteria (Box 3). HT61 rapidly and preferentially kills non-growing staphylococci through multiple mechanisms involving rapid permeabilization of the bacterial membrane (Table 1), and the drug is currently in clinical trials as a topical agent for nasal decolonization of methicillin-resistant S. aureus. No anti-biofilm activity has been reported for HT61, but its method of discovery could prove useful for guiding other approaches to obtain a new generation of antimicrobials that kill persisting cells37.
Challenges for membrane-active agents
Despite these encouraging examples, there are problems associated with the current anti-biofilm agents and their evaluation.
Test methods
A standardized methodology for evaluating anti-biofilm activity is lacking, and there is substantial variation in the experimental approaches that are used by different laboratories1. Existing methods include the use of static biofilm devices (such as minimum biofilm eradication concentration (MBEC) assays and colorimetric microtitre systems), in which biofilms are cultured without a continuous flow of fresh nutrients, and flow biofilm models (including the CDC biofilm reactor and the BioFlux system), in which biofilms are cultured with a continuous supply of fresh nutrients59. Consequently, the activity of agents tested in unrelated studies cannot be directly compared, and the relative activities of these agents against biofilms is therefore unknown.
The spectrum of activity
Most approaches do not consider that medical biofilms often have a polymicrobial composition60 and that an effective anti-biofilm strategy must therefore eliminate multiple bacterial species or at least synergize with other antibiotics to achieve complete sterilization11. Furthermore, the use of membrane-active antimicrobials that also target Gram-negative biofilms needs to be expanded, but this requires agents that either damage the outer membrane (as has been shown for the anti-biofilm action of polymyxin38) or traverse this layer and damage the cytoplasmic membrane. Furthermore, little is known about the structure–activity relationships that are required for small membrane-active molecules to display specificity for bacterial membranes11, 61. However, we believe that this gap can be filled by applying concepts from antimicrobial peptides or peptidomimetics to the design of amphipathic molecules that selectively target bacterial membranes (Box 3).
Pharmacology
Commonly associated toxicities (such as nephrotoxicity) and factors that affect drug disposition and efficacy (such as serum binding and tissue penetration) are also problems intrinsic to the design and development of membrane-active agents. For instance, the key to the clinical development of daptomycin was the choice of dosing regimen — it is dosed once per day at a high concentration — to maximize its concentration-dependent killing mechanism and to avoid an associated time-dependent myotoxicity43. Other membrane-active drugs will probably have similar physiochemical properties and will thus perhaps also exhibit pharmacological liabilities. Such drugs will tend to be partly lipophilic in nature, and they will therefore become bound to protein and may not distribute well into all tissues. For example, daptomycin is inactive against respiratory infections, and this is thought to be due to high levels of protein binding, as occurs in epithelial lining fluid (ELF) obtained by bronchoalveolar lavage62. Telavancin is also highly protein bound (90–93%) compared with vancomycin (10–55%), but its efficacy is not affected by ELF, as the concentration of unbound telavancin remains above the MIC against methicillin-resistant S. aureus63, 64. Dalbavancin and oritavancin are also highly protein bound (93–98% and 86–90%, respectively), with prolonged terminal half-lives (195 hours and 257 hours, respectively) and high retention times in mammalian cells and tissues48, which could extend the duration of potential side effects40. In culture models, accumulation of oritavancin, a drug that is important for killing intracellular S. aureus, induces lysosomal lipid storage disorder, but its corresponding toxic effects in vivo are unknown65, 66. These factors, along with a narrower safety margin, have discouraged the development of membrane-active agents in the past. However, owing to the urgent need for antibiotics that decrease the time required to effectively treat biofilm infections and other persistent infections, such agents may become important parts of future antibacterial treatment strategies.
Resistance
Laboratory studies indicate that the development of resistance is less frequent for several membrane-active agents than for other drugs; nonetheless, resistance (including cross-resistance between different structural classes of membrane-active agents with similar mechanisms of action) could emerge following widespread clinical use of these agents. For example, resistance to daptomycin was detected shortly after its clinical introduction67, 68, but the resulting daptomycin-resistant strains are exceedingly rare. This resistance is multifactorial and partially mediated by overproduction of phosphatidylglycerol lysyltransferase (MprF)69, 70, which adds L-lysine to phosphatidylglycerol, thereby increasing the net positive charge of the bacterial surface and decreasing the binding of daptomycin and some cationic antimicrobial peptides70, 71. However, telavancin is still active against daptomycin-resistant mutants of S. aureus containing mutations in MprF72, suggesting that MprF does not affect telavancin activity. Expression of the dltABCD operon, which encodes the enzymes that mediate the addition of alanine to lipoteichoic acid, also confers daptomycin non-susceptibility by increasing the net positive charge of the bacterial surface73, but its effects on other membrane-active agents is unknown. Lipoglycopeptide resistance can also result from expression of the vanA gene clusters; this expression leads to the production of peptidoglycan precursors ending in D-Ala-D-lactate instead of D-Ala-D-Ala, reducing the binding of glycopeptides to peptidoglycan by up to 1,000-fold74. Vancomycin-resistant S. aureus and vancomycin-resistant enterococci expressing vanA are resistant to dalbavancin (MIC > 32 μg ml 1) and only partially susceptible to telavancin (MIC = 2–16 μg ml 1) compared with wild-type strains (MIC ≤ 4 μg ml 1 for both drugs)63, 72. However, these resistant strains remain sensitive to oritavancin owing to an enhanced ability of this drug to form homodimers to improve its interaction with peptidoglycan precursors; this interaction is facilitated by the chlorobiphenyl side chain of oritavancin, which ensures that the drug is anchored to the membrane75, 76. Despite this, constitutive expression of vanA can lead to decreased susceptibility to oritavancin77. The clinical prevalence of vanA-mediated resistance in S. aureus is low, as expression of the vanA cluster is associated with a fitness cost74, 78; this may enable long-term and widespread use of lipoglycopeptides to treat staphylococcal infections. As current and future membrane-active antimicrobials progress, examination of the associated mechanisms of resistance, the ease with which resistance can occur and the potential for cross-resistance will be important parameters for the clinical development of different chemical classes of these agents.
Membrane-damaging agents against tuberculosis
The concept of damaging the structure of the mycobacterial membrane bilayer as a therapeutic strategy has received little attention. The development of membrane-active drugs with activity against M. tuberculosis could prove valuable in the endeavour to shorten the treatment period for TB, as is evident from the anti-TB activities of ionophores and the third-line anti-TB drug clofazimine. The mode of action of clofazimine is not well defined, but it may affect membrane architecture79, 80, with subsequent accumulation of lysophospholipids and depletion of K+ (Ref. 81). These effects are associated with membrane depolarization. Not surprisingly, clofazimine shows potent in vitro bactericidal activity against dormant M. tuberculosis under hypoxia82 and near-sterilizing activity in mice83. Although clofazimine causes reversible skin discoloration, this molecule is a chemotype for the development of less toxic derivatives84.
Several host-derived peptides are known to be active against many important bacterial pathogens, but only recently have these antimicrobials been reported to kill M. tuberculosis85. For example, ubiquitin-derived peptides kill persister cells that are tolerant to rifampicin (G. E. Purdy, personal communication). Although the therapeutic development of antimicrobial peptides is challenged by several factors, including the high costs of production, peptidomimetics that also cause membrane damage86 may provide another way forward for obtaining novel anti-TB drugs.
Damaging membranes in atypical bacteria
Most bacterial pathogens can form persistent infections that thwart antibiotic efficacy87, 88. Therefore, as our understanding of various types of bacterial persistent infections improves, it will become possible to define other bacterial pathogens to which the concepts about membrane-damaging agents may be applied. In particular, studies will be required to determine whether this approach is applicable to atypical microorganisms that have both extracellular and intracellular niches during dormant infections; such microorganisms include Mycoplasma spp.89, 90 and Chlamydia spp.91. Several studies report that antimicrobial peptides92–95 and analogues of host-derived membrane-active antimicrobial lipids96 kill metabolically inactive extracellular elementary bodies of Chlamydia trachomatis91 effectively. By contrast, few of these studies describe the killing of the metabolically active intracellular reticulate bodies of Chlamydia spp., which may indicate that poor cell penetration by these peptides is an issue97. Indeed, the intracellular expression of the peptide melittin on a tetracycline-inducible plasmid in mammalian cells achieved a 75% reduction in reticulate bodies98. Similarly, Pep-1, a synthetic transport carrier peptide in eukaryotes, is active against reticulate bodies, although it lacks activity against elementary bodies97. If cell penetration can be achieved by antimicrobial peptides, their peptidomimetics or other membrane-active agents, activity against reticulate bodies may be achieved to provide a comprehensive therapeutic strategy. Cell culture and animal models also indicate that some membrane-active peptides are bactericidal against persistent infections of Mycoplasma spp.99–101. These examples suggest that the future could see the application of membrane-damaging agents to atypical pathogens, although variations in membrane lipid composition (stemming from biphasic lifestyles and the ability of these pathogens to display eukaryote-like lipids in their membrane102–104) may alter drug–target interactions and the activities of membrane-active agents.
Targeting energy metabolism
Persistent colonization of the host requires that bacteria adopt various mechanisms of energy production to survive the scarcity of nutrients and oxygen that may occur in these infections. Dormant M. tuberculosis and organisms in biofilms survive these conditions and maintain redox homeostasis by using fermentation and anaerobic respiration to fulfil the lower energy demands of these persisting bacteria16, 19, 105. Anaerobic respiration is driven by the proton motive force, yielding a lower, but adequate, amount of ATP energy that sustains the viability of dormant cells. As dormant bacteria require ATP and redox balancing to survive, the inhibition of membrane-bound proteins that facilitate anaerobic metabolism is increasingly being proposed as a strategy to limit the survival of persisting bacteria16, 105–107. Although this strategy is a new direction for antibiotic discovery, there are several challenges. First, bacteria possess multiple ways of recycling or generating energy to compensate for the loss of their favoured energy-producing mechanisms. For example, staphylococci can forgo their respiratory chain to form small-colony variants that survive solely by fermentation and that persist in the host for prolonged periods108, 109. Second, as persisting infections consist of metabolically heterogeneous populations, a failure to kill all populations will necessitate the use of additional agents to sterilize the infection. Nevertheless, as our understanding of bacterial metabolism in dormant bacteria increases, novel targets will be identified that may be suitable for therapeutic intervention. Notable examples illustrating the potential value of this strategy are described here for P. aeruginosa and M. tuberculosis, and these examples reveal that targeting energy metabolism is likely to be a pathogen-specific approach.
Inactivation of cytochrome cbb3 oxidase 1, cytochrome cbb3 oxidase 2 and the cyanide-insensitive oxidase in P. aeruginosa results in impaired growth and poor biofilm formation under aerobic and microaerophilic conditions110. Similarly, deletion of the rhlRI quorum sensing system (which acts as a transcriptional regulator of anaerobic respiration) kills P. aeruginosa in biofilms owing to intoxication by intracellular nitric oxide18, which is generated as a by-product of denitrification. Biofilm formation in staphylococci is also disrupted by nitric oxide111. Therefore, using prodrug nitroheterocyclic antibiotics to generate nitric oxide in bacteria, a strategy currently in development for the treatment of latent TB, may be a viable strategy for the treatment of other diseases.
Dormant M. tuberculosis is exceptionally susceptible to agents that inhibit its respiratory chain (Figs 1, 3) and therefore cause membrane depolarization, ATP depletion and changes in the cellular redox state of the bacterium35, 112. This high sensitivity seems to correlate with an unusual need for M. tuberculosis to maintain a fully energized membrane under both aerobic and anaerobic conditions, which could reflect the inability of the species to survive by fermentation alone. Therefore, ATP synthase, which couples the proton motive force to energy transduction, is essential for the survival of active and dormant M. tuberculosis cells, even though this enzyme is downregulated during dormancy113. ATP synthase is also important in Streptococcus pneumoniae27, but it can be deleted in other bacteria, including Escherichia coli and Salmonella enterica, although this leads to virulence attentuation114. Thus, inhibition of ATP synthase may be only an adjunctive approach in some organisms.
In mycobacteria, enzymes that initiate the transfer of electrons into the respiratory chain, enzymes that are involved in menaquinone biosynthesis, and enzymes that maintain NAD+/NADH redox pools are further points of vulnerability. For example, the inhibition of menaquinone production kills M. tuberculosis115, and the NADH type II dehydrogenase (NDH2; present in two copies encoded by ndh and ndhA), a nonproton-pumping enzyme that transfers electrons to the respiratory chain, is essential in active and dormant M. tuberculosis35, 107 and is important for recycling NAD+, the main cellular oxidant and an important cofactor in cells35, 112. Hence, the inhibition of NDH2 deprives M. tuberculosis of both energy production and maintenance of its redox pools, which leads to cell death. As NAD+ recycling is also important in biofilms116, 117 and under anaerobic conditions118, 119, this enzyme could be a promising target for drugs. This will require studies to probe the antimicrobial potential of inhibiting NADH dehydrogenases and other enzymes that affect redox homeostasis in biofilms, as the effects are still, for the most part, unknown120.
On the basis of studies in E. coli, it has been proposed that several key bactericidal agents (that is, β-lactams, aminoglycosides and fluoroquinolones) cause cell death through a common mechanism of disturbing bacterial metabolism and the respiratory chain, leading to the formation of lethal reactive oxygen species121. However, it remains to be seen whether this concept is applicable to other classes of bactericidal antibiotics and other bacteria. Nevertheless, by directly perturbing the membrane and essential respiratory components, it may be possible to generate reactive oxygen species to impair numerous cellular processes and, eventually, cause cell death. Other respiratory and metabolic enzymes have been proposed as targets for anti-infective approaches in P. aeruginosa105, 106, S. aureus120, 122 and M. tuberculosis107, 123, but this is beyond the scope of this article.
Inhibiting energy metabolism in tuberculosis
With the renewed interest in drug discovery for TB treatment, energy metabolism in M. tuberculosis has re-emerged as an important drug target. Pyrazinamide (PZA) provided the first indication that energy metabolism is vulnerable in dormant TB, as this drug reduced the period of TB treatment from 9 months to 6 months. PZA disrupts the intracellular pH, an important component of the proton motive force, and only kills metabolically dormant cells at acidic pH and under oxygen-limiting conditions124, 125. Paradoxically, PZA has poor in vitro activity (MIC ≈ 60 mg l−1), even under acidic conditions, which is not predictive of its potent sterilizing properties. It is possible that by reducing the intracellular pH, PZA synergizes with host-derived reactive oxygen and nitrogen species36, thus reducing the MIC in vivo. Agents possessing this activity would thus be expected to kill more efficiently in acidified phagosomes in the host. This demonstrates the potential technical challenges when developing agents that specifically target latent or slow-growing subpopulations: namely, that such agents will probably have poor activity in standard MIC assays against dividing bacteria, and that the in vivo conditions that favour their synergistic activity are difficult to reproduce in vitro.
Among the anti-TB agents discovered in the past decade, the diarylquinoline TMC207, which is active against metabolizing and non-growing cells, seems to be the leading candidate126. TMC207 specifically binds to subunit C of the mycobacterial F0F1-ATPase, sterically blocking the enzyme’s rotational motor and thereby inhibiting proton translocation and coupled ATP biosynthesis127. Mutations in subunit C confer resistance to TMC207 (Ref. 128). Combinations of PZA and TMC207 are highly synergistic in mice (compared with other anti-TB combinations)129, due to their inhibition of sequential processes: the uncoupling of the proton gradient by PZA and specific inhibition of the ATP synthase by TMC207. Further studies to explore this observation are needed, as synergy with PZA was not reported in clinical trials with TMC207 (Ref. 130). Like PZA, TMC207 displays slow-onset killing131, which might point to the partial use of alternative energy systems by the bacteria or the presence of a pool of ATP in the bacteria that needs to be depleted before the bactericidal effects of inhibiting ATP synthase can be detected. However, in one study using a guinea pig model of TB infection, TMC207 was unable to kill a fraction of M. tuberculosis132. These findings illustrate the complexity of using a single antibiotic to sterilize the different physiological states of persisting bacteria, and they also highlight the need for standardized test models as well as for models that evaluate the effects of antibiotics on dormant cells exposed to multiple stresses (for example, simultaneous nutrient and oxygen starvation)133, which may be more representative of in vivo conditions. Efforts are already underway to find other ATP synthase inhibitors using structure-aided technologies134.
Although there are currently no drugs approved for TB treatment that target mycobacterial NDH2, experimental observations with the NDH2 inhibitors phenothiazines, chlorpromazine and thioridazine (which are antipsychotic drugs) provide a case for targeting NDH2 (Refs 135, 136). Chlorpromazine and thioridazine are both bactericidal and reduce the pulmonary mycobacterial load in mice137. However, phenothiazines are present at subinhibitory levels at the site of infection, and toxic side effects occur following routine administration, two factors that slow their clinical development138. Nevertheless, these agents demonstrate that NDH2 inhibition kills both nutrient-starved and hypoxic M. tuberculosis at concentrations similar to those that kill cells in the logarithmic phase35, 139. The lethality of NDH2 inhibition, resulting from disruption of both ATP synthesis and NAD+/NADH recycling, demonstrates that antibiotics acting at different points of the M. tuberculosis respiratory chain may display enhanced sterilizing activity by disrupting redox homeostasis. As the development of non-phenothiazine inhibitors of NDH2 progresses140, 141, attention must be paid to potential cross-resistance with isoniazid-resistant mutants that contain mutations in NDH2 (Ref. 142).
Prodrug nitroheterocyclic antibiotics are used to treat infections with parasites and anaerobic bacteria, and derivatives of these drugs with the potential to sterilize the lungs have emerged as good candidates for TB therapy. These agents include two nitroimidazoles (PA-824 and OPC-67683) that are in current clinical trials143, nitazoxanide144, and nitrofurans that are still in the early discovery stage145. These agents are activated in the bacterium by reduction of the precursor, resulting in the formation of reactive oxygen or nitrogen intermediates that damage multiple targets, including cell envelope lipids, DNA and proteins. Furthermore, the nitric oxide that is generated is predicted to poison the respiratory chain of dormant cells by inhibiting cytochrome oxidases112, thereby changing the redox status of cells and dramatically reducing the amount of intracellular ATP146. Cross-resistance between nitroheterocyclic antibiotics is a concern, particularly if they require the same nitroreductase or biochemical pathway for activation. However, the potential for cross-resistance can be reduced by optimizing the electrochemical potential of agents such that different nitroreductases and pathways are used to activate the prodrugs147. Indeed, changing the head group of OPC-67683 to a nitrofuran (Fig. 3) maintained the efficacy of the drug against M. tuberculosis mutants resistant to PA-824 and OPC-67683 (J.G.H., R. B. Lee, R.E.L., M. S. Scherman and M. R. McNeil, unpublished observations). Nitroheterocyclic antibiotics could potentially also be used as anti-biofilm agents, given their multitarget mode of action and good chemical tractability, and indeed nitazoxanide inhibits the formation of biofilms for E. coli and S. aureus148, 149. Similarly, ranbezolid, which is active against M. tuberculosis and contains a nitrofuran ring fused to an oxazolidone scaffold, exhibits good anti-biofilm properties against staphylococci compared with linezolid150. Ranbezolid is also reported to damage the membrane of Staphylcoccus epidermidis, probably owing to nitroheterocyclic action151. However, the use of nitroheterocyclic antibiotics for anti-biofilm control will require the presence of a nitroreductase that can activate the prodrug, and for that nitroreductase to be expressed in active and quiescent subpopulations. In addition, these molecules would have to be non-mutagenic, so they would have to avoid reduction by the host redox enzymes.
Concluding remarks
Owing to the paucity of treatment options for infections involving quiescent bacteria, we have begun to explore the premise that the bacterial membrane and enzymes involved in anaerobic respiration might be potential drug targets for controlling such infections, as exemplified by TB and biofilm diseases. Although these strategies are potentially advantageous, the main challenge will be obtaining molecules that are selective for bacteria. Although the fortuitous discovery of TMC207 reveals that this can be achieved for respiratory enzymes, an understanding of which genetic determinants are essential for anaerobic metabolism is needed for several bacterial pathogens. If these targets are sufficiently structurally distinct from human counterparts, this could then allow established drug discovery methods to be used. These efforts will probably result in pathogen-specific molecules owing to there being different mechanisms for energy production in bacteria. However, we believe that membrane-active agents could emerge as a superior therapeutic approach, with the advantages that they have multitarget effects, a potentially rapid bactericidal action, activity against growing and dormant subpopulations and low resistance prospects — providing that such agents can be developed with an acceptable selectivity verses human membranes. Moreover, disruption of membrane structure is also likely to affect membrane-embedded enzymes that carry out anaerobic metabolism and redox reactions. As most of the recently described membrane-active agents act on Gram-positive pathogens, there is a real need to extend this paradigm to Gram-negative biofilms and dormant M. tuberculosis. We anticipate that different forms of persistent bacterial infections, not covered in detail here, would be susceptible to killing by agents that can disrupt the membrane integrity of the pathogen. As this exciting field progresses, an understanding of the structure–activity relationships needed to obtain pathogen-selective molecules will prove crucial for guiding the discovery and development of novel agents that could shorten treatment periods and improve clinical outcomes for persistent infections.
Supplementary Material
Acknowledgments
We thank Drs Elaine Tuomanen and Engy Mahrous (St Jude Children’s Research Hospital) for critical reading of this manuscript. Funding for this research was provided by National Institutes of Health grants R01AI062415 and ARRA 1 R01AI079653, and the American Lebanese Syrian Associated Charities (ALSAC).
References
- 1.Coates AR, Hu Y. Targeting non-multiplying organisms as a way to develop novel antimicrobials. Trends Pharmacol Sci. 2008;29:143–150. doi: 10.1016/j.tips.2007.12.001. This review provides an excellent description of why antibiotics are needed against slow and non-growing organisms. [DOI] [PubMed] [Google Scholar]
- 2.Levin BR, Rozen DE. Non-inherited antibiotic resistance. Nat Rev Microbiol. 2006;4:556–562. doi: 10.1038/nrmicro1445. Through mathematical modeling and computer simulations, this paper describes how non-inherited resistance could extend antibiotic treatment duration, cause treatment failure and lead to the emergence of genetic resistance. [DOI] [PubMed] [Google Scholar]
- 3.Chopra I, Hesse L, O’Neill AJ. Exploiting current understanding of antibiotic action for discovery of new drugs. J Appl Microbiol. 2002;92 (Suppl):4S–15S. [PubMed] [Google Scholar]
- 4.Costerton JW, Stewart PS, Greenberg EP. Bacterial biofilms: a common cause of persistent infections. Science. 1999;284:1318–1322. doi: 10.1126/science.284.5418.1318. [DOI] [PubMed] [Google Scholar]
- 5.Wright JA, Nair SP. Interaction of staphylococci with bone. Int J Med Microbiol. 2010;300:193–204. doi: 10.1016/j.ijmm.2009.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lewis K. Persister cells, dormancy and infectious disease. Nat Rev Microbiol. 2007;5:48–56. doi: 10.1038/nrmicro1557. This article presents an excellent description of different types of bacterial mechanisms of persistence, their clinical importance and plausible counter approaches. [DOI] [PubMed] [Google Scholar]
- 7.Stewart GR, Robertson BD, Young DB. Tuberculosis: a problem with persistence. Nat Rev Microbiol. 2003;1:97–105. doi: 10.1038/nrmicro749. [DOI] [PubMed] [Google Scholar]
- 8.Mader JT, Shirtliff ME, Bergquist SC, Calhoun J. Antimicrobial treatment of chronic osteomyelitis. Clin Orthop Relat Res. 1999;360:47–65. doi: 10.1097/00003086-199903000-00008. [DOI] [PubMed] [Google Scholar]
- 9.Baddour LM, et al. Infective endocarditis: diagnosis, antimicrobial therapy, and management of complications: a statement for healthcare professionals from the Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease, Council on Cardiovascular Disease in the Young, and the Councils on Clinical Cardiology, Stroke, and Cardiovascular Surgery and Anesthesia, American Heart Association: endorsed by the Infectious Diseases Society of America. Circulation. 2005;111:e394–434. doi: 10.1161/CIRCULATIONAHA.105.165564. [DOI] [PubMed] [Google Scholar]
- 10.Coates A, Hu Y, Bax R, Page C. The future challenges facing the development of new antimicrobial drugs. Nat Rev Drug Discov. 2002;1:895–910. doi: 10.1038/nrd940. This is an excellent discussion on new directions for drug discovery against non-growing bacteria, indicating how it limit emergence of drug resistance. [DOI] [PubMed] [Google Scholar]
- 11.O’Neill AJ. Bacterial phenotypes refractory to antibiotic-mediated killing: mechanisms and mitigation. Emerging Trends in Antibacterial Discovery. 2010 This review details various theories regarding bacterial persistence and develops the term antibiotic survival as a new description. [Google Scholar]
- 12.Roveta S, Schito AM, Marchese A, Schito GC. Activity of moxifloxacin on biofilms produced in vitro by bacterial pathogens involved in acute exacerbations of chronic bronchitis. Int J Antimicrob Agents. 2007;30:415–421. doi: 10.1016/j.ijantimicag.2007.06.029. [DOI] [PubMed] [Google Scholar]
- 13.Hu Y, Coates AR, Mitchison DA. Comparison of the sterilising activities of the nitroimidazopyran PA-824 and moxifloxacin against persisting Mycobacterium tuberculosis. Int J Tuberc Lung Dis. 2008;12:69–73. [PubMed] [Google Scholar]
- 14.Lenaerts AJ, et al. Preclinical testing of the nitroimidazopyran PA-824 for activity against Mycobacterium tuberculosis in a series of in vitro and in vivo models. Antimicrob Agents Chemother. 2005;49:2294–2301. doi: 10.1128/AAC.49.6.2294-2301.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rose WE, Poppens PT. Impact of biofilm on the in vitro activity of vancomycin alone and in combination with tigecycline and rifampicin against Staphylococcus aureus. J Antimicrob Chemother. 2009;63:485–488. doi: 10.1093/jac/dkn513. [DOI] [PubMed] [Google Scholar]
- 16.Boshoff HI, III, Barry CE. Tuberculosis — metabolism and respiration in the absence of growth. Nat Rev Microbiol. 2005;3:70–80. doi: 10.1038/nrmicro1065. This article provides an understanding of how tuberculosis adapts and survives during long term persistence. [DOI] [PubMed] [Google Scholar]
- 17.Stewart PS, Franklin MJ. Physiological heterogeneity in biofilms. Nat Rev Microbiol. 2008;6:199–210. doi: 10.1038/nrmicro1838. [DOI] [PubMed] [Google Scholar]
- 18.Yoon SS, et al. Pseudomonas aeruginosa anaerobic respiration in biofilms: relationships to cystic fibrosis pathogenesis. Dev Cell. 2002;3:593–603. doi: 10.1016/s1534-5807(02)00295-2. [DOI] [PubMed] [Google Scholar]
- 19.Resch A, Rosenstein R, Nerz C, Gotz F. Differential gene expression profiling of Staphylococcus aureus cultivated under biofilm and planktonic conditions. Appl Environ Microbiol. 2005;71:2663–2676. doi: 10.1128/AEM.71.5.2663-2676.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Falsetta ML, McEwan AG, Jennings MP, Apicella MA. Anaerobic metabolism occurs in the substratum of gonococcal biofilms and may be sustained in part by nitric oxide. Infect Immun. 2010;78:2320–2328. doi: 10.1128/IAI.01312-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barry CE, 3rd, et al. The spectrum of latent tuberculosis: rethinking the biology and intervention strategies. Nat Rev Microbiol. 2009;7:845–855. doi: 10.1038/nrmicro2236. Provides new insights on the biology of TB disease from non-human primates and ways to combat this organism. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ojha AK, et al. Growth of Mycobacterium tuberculosis biofilms containing free mycolic acids and harbouring drug-tolerant bacteria. Mol Microbiol. 2008;69:164–174. doi: 10.1111/j.1365-2958.2008.06274.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bigger JW. Treatment of staphylococcal infections with penicillin. Lancet. 1944;244:497–500. [Google Scholar]
- 24.Tuomanen E, Durack DT, Tomasz A. Antibiotic tolerance among clinical isolates of bacteria. Antimicrob Agents Chemother. 1986;30:521–527. doi: 10.1128/aac.30.4.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang YM, Rock CO. Transcriptional regulation in bacterial membrane lipid synthesis. J Lipid Res. 2009;50 (Suppl):S115–119. doi: 10.1194/jlr.R800046-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nolan EM, Walsh CT. How nature morphs peptide scaffolds into antibiotics. Chembiochem. 2009;10:34–53. doi: 10.1002/cbic.200800438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat Rev Drug Discov. 2007;6:29–40. doi: 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- 28.Verkleij AJ, et al. The asymmetric distribution of phospholipids in the human red cell membrane. A combined study using phospholipases and freeze-etch electron microscopy. Biochim Biophys Acta. 1973;323:178–193. doi: 10.1016/0005-2736(73)90143-0. [DOI] [PubMed] [Google Scholar]
- 29.Straus SK, Hancock RE. Mode of action of the new antibiotic for Gram-positive pathogens daptomycin: comparison with cationic antimicrobial peptides and lipopeptides. Biochim Biophys Acta. 2006;1758:1215–1223. doi: 10.1016/j.bbamem.2006.02.009. This article presents a nice summary of daptomycin’s mode of action. [DOI] [PubMed] [Google Scholar]
- 30.Domenech O, Dufrene YF, Van Bambeke F, Tukens PM, Mingeot-Leclercq MP. Interactions of oritavancin, a new semi-synthetic lipoglycopeptide, with lipids extracted from Staphylococcus aureus. Biochim Biophys Acta. 2010 doi: 10.1016/j.bbamem.2010.06.011. [DOI] [PubMed] [Google Scholar]
- 31.Domenech O, et al. Interactions of oritavancin, a new lipoglycopeptide derived from vancomycin, with phospholipid bilayers: Effect on membrane permeability and nanoscale lipid membrane organization. Biochim Biophys Acta. 2009;1788:1832–1840. doi: 10.1016/j.bbamem.2009.05.003. [DOI] [PubMed] [Google Scholar]
- 32.Zhang L, Dhillon P, Yan H, Farmer S, Hancock RE. Interactions of bacterial cationic peptide antibiotics with outer and cytoplasmic membranes of Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2000;44:3317–3321. doi: 10.1128/aac.44.12.3317-3321.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kashket ER. Proton motive force in growing Streptococcus lactis and Staphylococcus aureus cells under aerobic and anaerobic conditions. J Bacteriol. 1981;146:369–376. doi: 10.1128/jb.146.1.369-376.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kashket ER. Effects of aerobiosis and nitrogen source on the proton motive force in growing Escherichia coli and Klebsiella pneumoniae cells. J Bacteriol. 1981;146:377–384. doi: 10.1128/jb.146.1.377-384.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rao SP, Alonso S, Rand L, Dick T, Pethe K. The protonmotive force is required for maintaining ATP homeostasis and viability of hypoxic, nonreplicating Mycobacterium tuberculosis. Proc Nat Acad Sci USA. 2008;105:11945–11950. doi: 10.1073/pnas.0711697105. This research can be considered a landmark achievement, demonstrating the importance of the proton motive force in M. tuberculosis, showing why dormant TB cells are killed by agents that affect this property. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vandal OH, Nathan CF, Ehrt S. Acid resistance in Mycobacterium tuberculosis. J Bacteriol. 2009;191:4714–4721. doi: 10.1128/JB.00305-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hu Y, Shamaei-Tousi A, Liu Y, Coates A. A new approach for the discovery of antibiotics by targeting non-multiplying bacteria: a novel topical antibiotic for staphylococcal infections. PLoS One. 2010;5:e11818. doi: 10.1371/journal.pone.0011818. This paper reports a novel approach of discovering antibiotics targeting non-growing bacteria and the prioritization of molecules that kill non-growing cells over those with good MICs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pamp SJ, Gjermansen M, Johansen HK, Tolker-Nielsen T. Tolerance to the antimicrobial peptide colistin in Pseudomonas aeruginosa biofilms is linked to metabolically active cells, and depends on the pmr and mexAB-oprM genes. Mol Microbiol. 2008;68:223–240. doi: 10.1111/j.1365-2958.2008.06152.x. [DOI] [PubMed] [Google Scholar]
- 39.McPhee JB, Lewenza S, Hancock RE. Cationic antimicrobial peptides activate a two-component regulatory system, PmrA-PmrB, that regulates resistance to polymyxin B and cationic antimicrobial peptides in Pseudomonas aeruginosa. Mol Microbiol. 2003;50:205–217. doi: 10.1046/j.1365-2958.2003.03673.x. [DOI] [PubMed] [Google Scholar]
- 40.Guskey MT, Tsuji BT. A comparative review of the lipoglycopeptides: oritavancin, dalbavancin, and telavancin. Pharmacotherapy. 2010;30:80–94. doi: 10.1592/phco.30.1.80. [DOI] [PubMed] [Google Scholar]
- 41.Raad I, et al. Comparative activities of daptomycin, linezolid, and tigecycline against catheter-related methicillin-resistant staphylococcus bacteremic isolates embedded in biofilm. Antimicrob Agents Chemother. 2007;51:1656–1660. doi: 10.1128/AAC.00350-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.LaPlante KL, Mermel LA. In vitro activity of daptomycin and vancomycin lock solutions on staphylococcal biofilms in a central venous catheter model. Nephrol Dial Transplant. 2007;22:2239–2246. doi: 10.1093/ndt/gfm141. [DOI] [PubMed] [Google Scholar]
- 43.Warren RE. Daptomycin in endocarditis and bacteraemia: a British perspective. J Antimicrob Chemother. 2008;62(Suppl 3):iii25–33. doi: 10.1093/jac/dkn370. [DOI] [PubMed] [Google Scholar]
- 44.Weiss EC, et al. Impact of sarA on daptomycin susceptibility of Staphylococcus aureus biofilms in vivo. Antimicrob Agents Chemother. 2009;53:4096–4102. doi: 10.1128/AAC.00484-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mikkelsen H, Duck Z, Lilley KS, Welch M. Interrelationships between colonies, biofilms, and planktonic cells of Pseudomonas aeruginosa. J Bacteriol. 2007;189:2411–2416. doi: 10.1128/JB.01687-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lewis K. Persister Cells. Annu Rev Microbiol. 2010;64:357–372. doi: 10.1146/annurev.micro.112408.134306. This article describes the search for persister genes and findings indicating that multiple genes/pathways are involved. [DOI] [PubMed] [Google Scholar]
- 47.Chiu TH, Hung SA. Effect of age on the membrane lipid composition of Streptococcus sanguis. Biochim Biophys Acta. 1979;558:267–272. doi: 10.1016/0005-2736(79)90261-x. [DOI] [PubMed] [Google Scholar]
- 48.Zhang YM, Rock CO. Membrane lipid homeostasis in bacteria. Nat Rev Microbiol. 2008;6:222–233. doi: 10.1038/nrmicro1839. [DOI] [PubMed] [Google Scholar]
- 49.Mascio CT, Alder JD, Silverman JA. Bactericidal action of daptomycin against stationary-phase and nondividing Staphylococcus aureus cells. Antimicrobial agents and chemotherapy. 2007;51:4255–4260. doi: 10.1128/AAC.00824-07. This papers shows that daptomycin is less active against stationary phase cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ooi N, et al. XF-70 and XF-73, novel antibacterial agents active against slow-growing and non-dividing cultures of Staphylococcus aureus including biofilms. J Antimicrob Chemother. 2010;65:72–78. doi: 10.1093/jac/dkp409. [DOI] [PubMed] [Google Scholar]
- 51.Belley A, et al. Oritavancin kills stationary-phase and biofilm Staphylococcus aureus cells in vitro. Antimicrob Agents Chemother. 2009;53:918–925. doi: 10.1128/AAC.00766-08. This paper describes the killing of various persistent cells by oritavancin with good potency. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.LaPlante KL, Mermel LA. In vitro activities of telavancin and vancomycin against biofilm-producing Staphylococcus aureus, S. epidermidis, and Enterococcus faecalis strains. Antimicrob Agents Chemother. 2009;53:3166–3169. doi: 10.1128/AAC.01642-08. This paper describes telavancin’s activity against biofilm cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Darouiche RO, Mansouri MD. Dalbavancin compared with vancomycin for prevention of Staphylococcus aureus colonization of devices in vivo. J of Infect. 2005;50:206–209. doi: 10.1016/j.jinf.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 54.Ooi N, et al. XF-73, a novel antistaphylococcal membrane-active agent with rapid bactericidal activity. J Antimicrob Chemother. 2009;64:735–740. doi: 10.1093/jac/dkp299. [DOI] [PubMed] [Google Scholar]
- 55.Ganzle MG. Reutericyclin: biological activity, mode of action, and potential applications. Appl Microbiol Biotechnol. 2004;64:326–332. doi: 10.1007/s00253-003-1536-8. [DOI] [PubMed] [Google Scholar]
- 56.Hurdle JG, Yendapally R, Sun D, Lee RE. Evaluation of analogs of reutericyclin as prospective candidates for treatment of staphylococcal skin infections. Antimicrob Agents Chemother. 2009;53:4028–4031. doi: 10.1128/AAC.00457-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Overhage J, et al. Human host defense peptide LL-37 prevents bacterial biofilm formation. Infect Immun. 2008;76:4176–4182. doi: 10.1128/IAI.00318-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Flemming K, et al. High in vitro antimicrobial activity of synthetic antimicrobial peptidomimetics against staphylococcal biofilms. J Antimicrob Chemother. 2009;63:136–145. doi: 10.1093/jac/dkn464. [DOI] [PubMed] [Google Scholar]
- 59.Benoit MR, Conant CG, Lonescu-Zanetti C, Schwartz M, Matin A. New device for high-throughput viability screening of flow biofilms. Appl Environ Microbiol. 2010;76:4136–4142. doi: 10.1128/AEM.03065-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dowd SE, et al. Survey of bacterial diversity in chronic wounds using pyrosequencing, DGGE, and full ribosome shotgun sequencing. BMC Microbiol. 2008;8:43. doi: 10.1186/1471-2180-8-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yendapally R, Hurdle JG, Carson EI, Lee RB, Lee RE. N-substituted 3-acetyltetramic acid derivatives as antibacterial agents. J Med Chem. 2008;51:1487–1491. doi: 10.1021/jm701356q. [DOI] [PubMed] [Google Scholar]
- 62.Silverman JA, Mortin LI, Vanpraagh AD, Li T, Alder J. Inhibition of daptomycin by pulmonary surfactant: in vitro modeling and clinical impact. J Infect Dis. 2005;191:2149–2152. doi: 10.1086/430352. [DOI] [PubMed] [Google Scholar]
- 63.Zhanel GG, et al. New lipoglycopeptides: a comparative review of dalbavancin, oritavancin and telavancin. Drugs. 2010;70:859–886. doi: 10.2165/11534440-000000000-00000. This paper provides a summary of microbiological and pharmacological aspects of lipoglycopeptides mentioned in title. [DOI] [PubMed] [Google Scholar]
- 64.Gotfried MH, et al. Intrapulmonary distribution of intravenous telavancin in healthy subjects and effect of pulmonary surfactant on in vitro activities of telavancin and other antibiotics. Antimicrob Agents Chemother. 2008;52:92–97. doi: 10.1128/AAC.00875-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Van Bambeke F, Saffran J, Mingeot-Leclercq MP, Tulkens PM. Mixed-lipid storage disorder induced in macrophages and fibroblasts by oritavancin (LY333328), a new glycopeptide antibiotic with exceptional cellular accumulation. Antimicrob Agents Chemother. 2005;49:1695–1700. doi: 10.1128/AAC.49.5.1695-1700.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Van Bambeke F, Mingeot-Leclercq MP, Struelens MJ, Tulkens PM. The bacterial envelope as a target for novel anti-MRSA antibiotics. Trends Pharmacol Sci. 2008;29:124–134. doi: 10.1016/j.tips.2007.12.004. This review provides a nice summary of membrane active antibiotics against MRSA. [DOI] [PubMed] [Google Scholar]
- 67.Mangili A, Bica I, Snydman DR, Hamer DH. Daptomycin-resistant, methicillin-resistant Staphylococcus aureus bacteremia. Clin Infect Dis. 2005;40:1058–1060. doi: 10.1086/428616. [DOI] [PubMed] [Google Scholar]
- 68.Hayden MK, et al. Development of daptomycin resistance in vivo in methicillin-resistant Staphylococcus aureus. J Clin Microbiol. 2005;43:5285–5287. doi: 10.1128/JCM.43.10.5285-5287.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mishra NN, et al. Analysis of cell membrane characteristics of in vitro-selected daptomycin-resistant strains of methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother. 2009;53:2312–2318. doi: 10.1128/AAC.01682-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jones T, et al. Failures in clinical treatment of Staphylococcus aureus infection with daptomycin are associated with alterations in surface charge, membrane phospholipid asymmetry, and drug binding. Antimicrob Agents Chemother. 2008;52:269–278. doi: 10.1128/AAC.00719-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ernst CM, et al. The bacterial defensin resistance protein MprF consists of separable domains for lipid lysinylation and antimicrobial peptide repulsion. PLoS Pathog. 2009;5:e1000660. doi: 10.1371/journal.ppat.1000660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kosowska-Shick K, et al. Activity of telavancin against staphylococci and enterococci determined by MIC and resistance selection studies. Antimicrob Agents Chemother. 2009;53:4217–4224. doi: 10.1128/AAC.00742-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yang SJ, et al. Enhanced expression of dltABCD is associated with the development of daptomycin nonsusceptibility in a clinical endocarditis isolate of Staphylococcus aureus. J Infect Dis. 2009;200:1916–1920. doi: 10.1086/648473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Courvalin P. Vancomycin resistance in gram-positive cocci. Clin Infect Dis. 2006;42 (Suppl 1):S25–34. doi: 10.1086/491711. [DOI] [PubMed] [Google Scholar]
- 75.Allen NE, LeTourneau DL, Hobbs JN., Jr Molecular interactions of a semisynthetic glycopeptide antibiotic with D-alanyl-D-alanine and D-alanyl-D-lactate residues. Antimicrob Agents Chemother. 1997;41:66–71. doi: 10.1128/aac.41.1.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Allen NE, Nicas TI. Mechanism of action of oritavancin and related glycopeptide antibiotics. FEMS Microbiol Rev. 2003;26:511–532. doi: 10.1111/j.1574-6976.2003.tb00628.x. [DOI] [PubMed] [Google Scholar]
- 77.Arthur M, Depardieu F, Reynolds P, Courvalin P. Moderate-level resistance to glycopeptide LY333328 mediated by genes of the vanA and vanB clusters in enterococci. Antimicrob Agents Chemother. 1999;43:1875–1880. doi: 10.1128/aac.43.8.1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Perichon B, Courvalin P. VanA-type vancomycin-resistant Staphylococcus aureus. Antimicrob Agents Chemother. 2009;53:4580–4587. doi: 10.1128/AAC.00346-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cholo MC, van Rensburg E, RA Potassium uptake systems of Mycobacterium tuberculosis:genomic and protein organisation and potential roles in microbial pathogenesis and chemotherap. South Afr J Epidemiol Infect. 2008;23:13–16. [Google Scholar]
- 80.Oliva B, O’Neill AJ, Miller K, Stubbings W, Chopra I. Anti-staphylococcal activity and mode of action of clofazimine. J Antimicrob Chemother. 2004;53:435–440. doi: 10.1093/jac/dkh114. [DOI] [PubMed] [Google Scholar]
- 81.Steel HC, Matlola NM, Anderson R. Inhibition of potassium transport and growth of mycobacteria exposed to clofazimine and B669 is associated with a calcium-independent increase in microbial phospholipase A2 activity. J Antimicrob Chemother. 1999;44:209–216. doi: 10.1093/jac/44.2.209. [DOI] [PubMed] [Google Scholar]
- 82.Cho SH, et al. Low-oxygen-recovery assay for high-throughput screening of compounds against nonreplicating Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2007;51:1380–1385. doi: 10.1128/AAC.00055-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Adams LB, Sinha I, Franzblau SG, Krahenbuhl JL, Mehta RT. Effective treatment of acute and chronic murine tuberculosis with liposome-encapsulated clofazimine. Antimicrob Agents Chemother. 1999;43:1638–1643. doi: 10.1128/aac.43.7.1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Reddy VM, O’Sullivan JF, Gangadharam PR. Antimycobacterial activities of riminophenazines. J Antimicrob Chemother. 1999;43:615–623. doi: 10.1093/jac/43.5.615. [DOI] [PubMed] [Google Scholar]
- 85.Purdy GE, Niederweis M, Russell DG. Decreased outer membrane permeability protects mycobacteria from killing by ubiquitin-derived peptides. Mol Microbiol. 2009;73:844–857. doi: 10.1111/j.1365-2958.2009.06801.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hancock RE, Sahl HG. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat Biotechnol. 2006;24:1551–1557. doi: 10.1038/nbt1267. [DOI] [PubMed] [Google Scholar]
- 87.Blaser MJ, Kirschner D. The equilibria that allow bacterial persistence in human hosts. Nature. 2007;449:843–849. doi: 10.1038/nature06198. [DOI] [PubMed] [Google Scholar]
- 88.Furukawa S, Kuchma SL, O’Toole GA. Keeping their options open: acute versus persistent infections. J Bacteriol. 2006;188:1211–1217. doi: 10.1128/JB.188.4.1211-1217.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bradshaw CS, Chen MY, Fairley CK. Persistence of Mycoplasma genitalium following azithromycin therapy. PLoS One. 2008;3:e3618. doi: 10.1371/journal.pone.0003618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Taylor-Robinson D, Webster AD, Furr PM, Asherson GL. Prolonged persistence of Mycoplasma pneumoniae in a patient with hypogammaglobulinaemia. J Infect. 1980;2:171–175. doi: 10.1016/s0163-4453(80)91284-0. [DOI] [PubMed] [Google Scholar]
- 91.Abdelrahman YM, Belland RJ. The chlamydial developmental cycle. FEMS Microbiol Rev. 2005;29:949–959. doi: 10.1016/j.femsre.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 92.Donati M, et al. Activity of cathelicidin peptides against Chlamydia spp. Antimicrob Agents Chemother. 2005;49:1201–1202. doi: 10.1128/AAC.49.3.1201-1202.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yasin B, Harwig SS, Lehrer RI, Wagar EA. Susceptibility of Chlamydia trachomatis to protegrins and defensins. Infection and immunity. 1996;64:709–713. doi: 10.1128/iai.64.3.709-713.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yasin B, Pang M, Lehrer RI, Wagar EA. Activity of Novispirin G-10, a novel antimicrobial peptide against Chlamydia trachomatis and vaginosis-associated bacteria. Exp Mol Pathol. 2003;74:190–195. doi: 10.1016/s0014-4800(02)00022-9. [DOI] [PubMed] [Google Scholar]
- 95.Yasin B, Pang M, Wagar EA. A cumulative experience examining the effect of natural and synthetic antimicrobial peptides vs. Chlamydia trachomatis J Pept Res. 2004;64:65–71. doi: 10.1111/j.1399-3011.2004.00172.x. [DOI] [PubMed] [Google Scholar]
- 96.Lampe MF, Ballweber LM, Isaacs CE, Patton DL, Stamm WE. Killing of Chlamydia trachomatis by novel antimicrobial lipids adapted from compounds in human breast milk. Antimicrob Agents Chemother. 1998;42:1239–1244. doi: 10.1128/aac.42.5.1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Park N, et al. The cell-penetrating peptide, Pep-1, has activity against intracellular chlamydial growth but not extracellular forms of Chlamydia trachomatis. J Antimicrob Chemother. 2009;63:115–123. doi: 10.1093/jac/dkn436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lazarev VN, et al. Induced expression of melittin, an antimicrobial peptide, inhibits infection by Chlamydia trachomatis and Mycoplasma hominis in a HeLa cell line. Int J Antimicrob Agents. 2002;19:133–137. doi: 10.1016/s0924-8579(01)00479-4. [DOI] [PubMed] [Google Scholar]
- 99.Lazarev VN, et al. Effect of induced expression of an antimicrobial peptide melittin on Chlamydia trachomatis and Mycoplasma hominis infections in vivo. Biochem Biophys Res Commun. 2005;338:946–950. doi: 10.1016/j.bbrc.2005.10.028. [DOI] [PubMed] [Google Scholar]
- 100.Nir-Paz R, Prevost MC, Nicolas P, Blanchard A, Wroblewski H. Susceptibilities of Mycoplasma fermentans and Mycoplasma hyorhinis to membrane-active peptides and enrofloxacin in human tissue cell cultures. Antimicrob Agents Chemother. 2002;46:1218–1225. doi: 10.1128/AAC.46.5.1218-1225.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Beven L, Wroblewski H. Effect of natural amphipathic peptides on viability, membrane potential, cell shape and motility of mollicutes. Res Microbiol. 1997;148:163–175. doi: 10.1016/S0923-2508(97)87647-4. [DOI] [PubMed] [Google Scholar]
- 102.Hatch GM, McClarty G. Phospholipid composition of purified Chlamydia trachomatis mimics that of the eucaryotic host cell. Infect Immun. 1998;66:3727–3735. doi: 10.1128/iai.66.8.3727-3735.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wylie JL, Hatch GM, McClarty G. Host cell phospholipids are trafficked to and then modified by Chlamydia trachomatis. J Bacteriol. 1997;179:7233–7242. doi: 10.1128/jb.179.23.7233-7242.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Rotem S, Mor A. Antimicrobial peptide mimics for improved therapeutic properties. Biochim Biophys Acta. 2009;1788:1582–1592. doi: 10.1016/j.bbamem.2008.10.020. [DOI] [PubMed] [Google Scholar]
- 105.Schobert M, Tielen P. Contribution of oxygen-limiting conditions to persistent infection of Pseudomonas aeruginosa. Future Microbiol. 2010;5:603–621. doi: 10.2217/fmb.10.16. [DOI] [PubMed] [Google Scholar]
- 106.Hassett DJ, et al. Anaerobic metabolism and quorum sensing by Pseudomonas aeruginosa biofilms in chronically infected cystic fibrosis airways: rethinking antibiotic treatment strategies and drug targets. Adv Drug Deliv Rev. 2002;54:1425–1443. doi: 10.1016/s0169-409x(02)00152-7. [DOI] [PubMed] [Google Scholar]
- 107.Kana BD, et al. Electron transport and respiration in Mycobacteria. In: Parish T, Brown A, editors. Mycobacterium: Genomics and Molecular Biology. Caister Academic Press; 2010. This book chapter provides a very well detailed analysis of the mycobacterial respiratory chain and enzymes involved. [Google Scholar]
- 108.Biswas L, Biswas R, Schlag M, Bertram R, Gotz F. Small-colony variant selection as a survival strategy for Staphylococcus aureus in the presence of Pseudomonas aeruginosa. Appl Environ Microbiol. 2009;75:6910–6912. doi: 10.1128/AEM.01211-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Besier S, et al. Prevalence and clinical significance of Staphylococcus aureus small-colony variants in cystic fibrosis lung disease. J Clin Microbiol. 2007;45:168–172. doi: 10.1128/JCM.01510-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Alvarez-Ortega C, Harwood CS. Responses of Pseudomonas aeruginosa to low oxygen indicate that growth in the cystic fibrosis lung is by aerobic respiration. Mol Microbiol. 2007;65:153–165. doi: 10.1111/j.1365-2958.2007.05772.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Schlag S, Nerz C, Birkenstock TA, Altenberend F, Gotz F. Inhibition of staphylococcal biofilm formation by nitrite. J Bacteriol. 2007;189:7911–7919. doi: 10.1128/JB.00598-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Boshoff HI, et al. The transcriptional responses of Mycobacterium tuberculosis to inhibitors of metabolism: novel insights into drug mechanisms of action. J Biol Chem. 2004;279:40174–40184. doi: 10.1074/jbc.M406796200. This research examines transcriptional changes that TB cells undergo in response to various metabolic inhibitors. [DOI] [PubMed] [Google Scholar]
- 113.Karakousis PC, et al. Dormancy phenotype displayed by extracellular Mycobacterium tuberculosis within artificial granulomas in mice. J Exp Med. 2004;200:647–657. doi: 10.1084/jem.20040646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Northen H, et al. Salmonella enterica serovar Typhimurium mutants completely lacking the F(0)F(1) ATPase are novel live attenuated vaccine strains. Vaccine. 2010;28:940–949. doi: 10.1016/j.vaccine.2009.10.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Dhiman RK, et al. Menaquinone synthesis is critical for maintaining mycobacterial viability during exponential growth and recovery from non-replicating persistence. Mol Microbiol. 2009;72:85–97. doi: 10.1111/j.1365-2958.2009.06625.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Dekkers LC, et al. Role of the O-antigen of lipopolysaccharide, and possible roles of growth rate and of NADH:ubiquinone oxidoreductase (nuo) in competitive tomato root-tip colonization by Pseudomonas fluorescens WCS365. Mol Plant Microbe Interact. 1998;11:763–771. doi: 10.1094/MPMI.1998.11.8.763. [DOI] [PubMed] [Google Scholar]
- 117.Zhu L, et al. Characterization of mRNA interferases from Mycobacterium tuberculosis. J Biol Chem. 2006;281:18638–18643. doi: 10.1074/jbc.M512693200. [DOI] [PubMed] [Google Scholar]
- 118.Melo AM, Bandeiras TM, Teixeira M. New insights into type II NAD(P)H:quinone oxidoreductases. Microbiol Mol Biol Rev. 2004;68:603–616. doi: 10.1128/MMBR.68.4.603-616.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Richardson AR, Libby SJ, Fang FC. A nitric oxide-inducible lactate dehydrogenase enables Staphylococcus aureus to resist innate immunity. Science. 2008;319:1672–1676. doi: 10.1126/science.1155207. [DOI] [PubMed] [Google Scholar]
- 120.Somerville GA, Proctor RA. At the crossroads of bacterial metabolism and virulence factor synthesis in Staphylococci. Microbiol Mol Biol Rev. 2009;73:233–248. doi: 10.1128/MMBR.00005-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kohanski MA, Dwyer DJ, Hayete B, Lawrence CA, Collins JJ. A common mechanism of cellular death induced by bactericidal antibiotics. Cell. 2007;130:797–810. doi: 10.1016/j.cell.2007.06.049. This paper and others by the author (J.J Collins) explores the cellular mechanisms/responses that occur during the killing of cells by bactericidal antibiotics. [DOI] [PubMed] [Google Scholar]
- 122.Fuchs S, Pane-Farre J, Kohler C, Hecker M, Engelmann S. Anaerobic gene expression in Staphylococcus aureus. J Bacteriol. 2007;189:4275–4289. doi: 10.1128/JB.00081-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bald D, Koul A. Respiratory ATP synthesis: the new generation of mycobacterial drug targets? FEMS Microbiol Lett. 2010;308:1–7. doi: 10.1111/j.1574-6968.2010.01959.x. [DOI] [PubMed] [Google Scholar]
- 124.Wade MM, Zhang Y. Anaerobic incubation conditions enhance pyrazinamide activity against Mycobacterium tuberculosis. J Med Microbiol. 2004;53:769–773. doi: 10.1099/jmm.0.45639-0. [DOI] [PubMed] [Google Scholar]
- 125.Zhang Y, Wade MM, Scorpio A, Zhang H, Sun Z. Mode of action of pyrazinamide: disruption of Mycobacterium tuberculosis membrane transport and energetics by pyrazinoic acid. J Antimicrob Chemother. 2003;52:790–795. doi: 10.1093/jac/dkg446. [DOI] [PubMed] [Google Scholar]
- 126.Koul A, et al. Diarylquinolines are bactericidal for dormant mycobacteria as a result of disturbed ATP homeostasis. J Biol Chem. 2008;283:25273–25280. doi: 10.1074/jbc.M803899200. [DOI] [PubMed] [Google Scholar]
- 127.Andries K, et al. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science. 2005;307:223–227. doi: 10.1126/science.1106753. This landmark work describes the discovery of the novel drug TMC-207. [DOI] [PubMed] [Google Scholar]
- 128.Petrella S, et al. Genetic basis for natural and acquired resistance to the diarylquinoline R207910 in mycobacteria. Antimicrob Agents Chemother. 2006;50:2853–2856. doi: 10.1128/AAC.00244-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ibrahim M, et al. Synergistic activity of R207910 combined with pyrazinamide against murine tuberculosis. Antimicrob Agents Chemother. 2007;51:1011–1015. doi: 10.1128/AAC.00898-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Diacon AH, et al. The diarylquinoline TMC207 for multidrug-resistant tuberculosis. N Engl J Med. 2009;360:2397–2405. doi: 10.1056/NEJMoa0808427. [DOI] [PubMed] [Google Scholar]
- 131.Rustomjee R, et al. Early bactericidal activity and pharmacokinetics of the diarylquinoline TMC207 in treatment of pulmonary tuberculosis. Antimicrob Agents Chemother. 2008;52:2831–2835. doi: 10.1128/AAC.01204-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Lenaerts AJ, et al. Location of persisting mycobacteria in the guinea pig model of tuberculosis revealed by R207910. Antimicrob Agents Chemother. 2007 doi: 10.1128/AAC.00276-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Deb C, et al. A novel in vitro multiple-stress dormancy model for Mycobacterium tuberculosis generates a lipid-loaded, drug-tolerant, dormant pathogen. PLoS One. 2009;4:e6077. doi: 10.1371/journal.pone.0006077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Upadhayaya RS, et al. Design, synthesis, biological evaluation and molecular modelling studies of novel quinoline derivatives against Mycobacterium tuberculosis. Bioorganic & medicinal chemistry. 2009;17:2830–2841. doi: 10.1016/j.bmc.2009.02.026. [DOI] [PubMed] [Google Scholar]
- 135.Yano T, Li LS, Weinstein E, Teh JS, Rubin H. Steady-state kinetics and inhibitory action of antitubercular phenothiazines on Mycobacterium tuberculosis type-II NADH-menaquinone oxidoreductase (NDH-2) J Biol Chem. 2006;281:11456–11463. doi: 10.1074/jbc.M508844200. [DOI] [PubMed] [Google Scholar]
- 136.Weinstein EA, et al. Inhibitors of type II NADH:menaquinone oxidoreductase represent a class of antitubercular drugs. Proc Nat Acad Sci USA. 2005;102:4548–4553. doi: 10.1073/pnas.0500469102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Amaral L, Boeree MJ, Gillespie SH, Udwadia ZF, van Soolingen D. Thioridazine cures extensively drug-resistant tuberculosis (XDR-TB) and the need for global trials is now! Int J Antimicrob Agents. 2010 doi: 10.1016/j.ijantimicag.2009.12.019. [DOI] [PubMed] [Google Scholar]
- 138.Ordway D, Viveiros M, Leandro C, Arroz MJ, Amaral L. Intracellular activity of clinical concentrations of phenothiazines including thioridiazine against phagocytosed Staphylococcus aureus. Int J Antimicrob Agents. 2002;20:34–43. doi: 10.1016/s0924-8579(02)00110-3. [DOI] [PubMed] [Google Scholar]
- 139.Xie Z, Siddiqi N, Rubin EJ. Differential antibiotic susceptibilities of starved Mycobacterium tuberculosis isolates. Antimicrob Agents Chemother. 2005;49:4778–4780. doi: 10.1128/AAC.49.11.4778-4780.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Mogi T, et al. Identification of new inhibitors for alternative NADH dehydrogenase (NDH-II) FEMS Microbiol Lett. 2009;291:157–161. doi: 10.1111/j.1574-6968.2008.01451.x. [DOI] [PubMed] [Google Scholar]
- 141.Mogi T, et al. Antibiotics LL-Z1272 identified as novel inhibitors discriminating bacterial and mitochondrial quinol oxidases. Biochim Biophys Acta. 2009;1787:129–133. doi: 10.1016/j.bbabio.2008.11.016. [DOI] [PubMed] [Google Scholar]
- 142.Cardoso RF, et al. Characterization of ndh gene of isoniazid resistant and susceptible Mycobacterium tuberculosis isolates from Brazil. Mem Inst Oswaldo Cruz. 2007;102:59–61. doi: 10.1590/s0074-02762007000100009. [DOI] [PubMed] [Google Scholar]
- 143.van den Boogaard J, Kibiki GS, Kisanga ER, Boeree MJ, Aarnoutse RE. New drugs against tuberculosis: problems, progress, and evaluation of agents in clinical development. Antimicrob Agents Chemother. 2009;53:849–862. doi: 10.1128/AAC.00749-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.de Carvalho LP, Lin G, Jiang X, Nathan C. Nitazoxanide kills replicating and nonreplicating Mycobacterium tuberculosis and evades resistance. J Med Chem. 2009;52:5789–5792. doi: 10.1021/jm9010719. [DOI] [PubMed] [Google Scholar]
- 145.Hurdle JG, et al. A microbiological assessment of novel nitrofuranylamides as anti-tuberculosis agents. J Antimicrob Chemother. 2008;62:1037–1045. doi: 10.1093/jac/dkn307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Manjunatha U, Boshoff HI, Barry CE. The mechanism of action of PA-824: Novel insights from transcriptional profiling. Commun Integr Biol. 2009;2:215–218. doi: 10.4161/cib.2.3.7926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Sisson G, et al. Enzymes associated with reductive activation and action of nitazoxanide, nitrofurans, and metronidazole in Helicobacter pylori. Antimicrob Agents Chemother. 2002;46:2116–2123. doi: 10.1128/AAC.46.7.2116-2123.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Shamir ER, et al. Nitazoxanide inhibits biofilm production and hemagglutination by enteroaggregative Escherichia coli strains by blocking assembly of AafA fimbriae. Antimicrob Agents Chemother. 2010;54:1526–1533. doi: 10.1128/AAC.01279-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Tchouaffi-Nana F, et al. Nitazoxanide inhibits biofilm formation by Staphylococcus epidermidis by blocking accumulation on surfaces. Antimicrob Agents Chemother. 2010;54:2767–2774. doi: 10.1128/AAC.00901-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Mathur T, Bhateja P, Pandya M, Fatma T, Rattan A. In vitro activity of RBx 7644 (ranbezolid) on biofilm producing bacteria. Int J Antimicrob Agents. 2004;24:369–373. doi: 10.1016/j.ijantimicag.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 151.Kalia V, et al. Mode of action of ranbezolid against staphylococci and structural modeling studies of its interaction with ribosomes. Antimicrob Agents Chemother. 2009;53:1427–1433. doi: 10.1128/AAC.00887-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Smith PA, Romesberg FE. Combating bacteria and drug resistance by inhibiting mechanisms of persistence and adaptation. Nat Chem Biol. 2007;3:549–556. doi: 10.1038/nchembio.2007.27. [DOI] [PubMed] [Google Scholar]
- 153.Hansen S, Lewis K, Vulic M. Role of global regulators and nucleotide metabolism in antibiotic tolerance in Escherichia coli. Antimicrob Agents Chemother. 2008;52:2718–2726. doi: 10.1128/AAC.00144-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Vazquez-Laslop N, Lee H, Neyfakh AA. Increased persistence in Escherichia coli caused by controlled expression of toxins or other unrelated proteins. J Bacteriol. 2006;188:3494–3497. doi: 10.1128/JB.188.10.3494-3497.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.O’Neill AJ, Miller K, Oliva B, Chopra I. Comparison of assays for detection of agents causing membrane damage in Staphylococcus aureus. J Antimicrob Chemother. 2004;54:1127–1129. doi: 10.1093/jac/dkh476. [DOI] [PubMed] [Google Scholar]
- 156.Walsh TJ, et al. Safety, tolerance, and pharmacokinetics of a small unilamellar liposomal formulation of amphotericin B (AmBisome) in neutropenic patients. Antimicrob Agents Chemother. 1998;42:2391–2398. doi: 10.1128/aac.42.9.2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Carlson RP, Taffs R, Davison WM, Stewart PS. Anti-biofilm properties of chitosan-coated surfaces. J Biomater Sci Polym Ed. 2008;19:1035–1046. doi: 10.1163/156856208784909372. [DOI] [PubMed] [Google Scholar]
- 158.Savage PB, et al. Antibacterial activities of thin films containing ceragenins. In: Camesano TA, Mello CM, editors. Microbial Surfaces: Structure, Interactions, and Reactivity. American Chemical Society; 2008. pp. 65–78. [Google Scholar]
- 159.Brogden KA. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nature Rev Microbiol. 2005;3:238–250. doi: 10.1038/nrmicro1098. [DOI] [PubMed] [Google Scholar]
- 160.Hawkey PM. Pre-clinical experience with daptomycin. J Antimicrob Chemother. 2008;62(Suppl 3):iii7–14. doi: 10.1093/jac/dkn367. [DOI] [PubMed] [Google Scholar]
- 161.Epand RF, Pollard JE, Wright J, Savage PB, Epand RM. Depolarization, bacterial membrane composition and the antimicrobial action of ceragenins. Antimicrob Agents Chemother. 2010 doi: 10.1128/AAC.00380-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Chin JN, Rybak MJ, Cheung CM, Savage PB. Antimicrobial activities of ceragenins against clinical isolates of resistant Staphylococcus aureus. Antimicrob Agents Chemother. 2007;51:1268–1273. doi: 10.1128/AAC.01325-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Coates T, Bax R, Coates A. Nasal decolonization of Staphylococcus aureus with mupirocin: strengths, weaknesses and future prospects. J Antimicrob Chemother. 2009;64:9–15. doi: 10.1093/jac/dkp159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Budha N, et al. Pharmacokinetically-guided lead optimization of nitrofuranylamide anti-tuberculosis agents. AAPS J. 2008;10:157–165. doi: 10.1208/s12248-008-9017-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Tangallapally RP, Yendapally R, Daniels AJ, Lee RE, Lee RE. Nitrofurans as novel anti-tuberculosis agents: identification, development and evaluation. Curr Top Med Chem. 2007;7:509–526. doi: 10.2174/156802607780059772. [DOI] [PubMed] [Google Scholar]
- 166.Ballard TE, et al. Biological activity of modified and exchanged 2-amino-5-nitrothiazole amide analogues of nitazoxanide. Bioorg Med Chem Lett. 2010;20:3537–3539. doi: 10.1016/j.bmcl.2010.04.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.