

Abstract
An efficient and scalable synthesis of (−)-DAPD and (−)-APD has been developed. We discovered that t-butyl cyanoacetate can be used as a new additive for the sugar nucleoside base coupling step en route to DAPD with improved β-selectivity and an isolated yield four fold greater than the original process scale method. Using this new process, (−)-DAPD has been prepared on greater than 20 g scale. In the synthesis of (−)-APD, a key enzyme-catalyzed hydrolysis reaction afforded the water-soluble deprotected α-anomer while leaving the β-anomer completely untouched.
Keywords: Amdoxovir, DAPD, Diaminopurine, Dioxolane, Nucleoside, Glycosylation
1. Introduction
The World Health Organization (WHO) reported that there were more than 34 million individuals living with HIV and about 2 million deaths from AIDS worldwide in 2010, including 250,000 children.1 During the last two decades, many drugs and treatment regimes for this disease have been successfully discovered and developed. (−)-β-D-2,6-Diaminopurine dioxolane (Amdoxovir, DAPD), a prodrug of (−)-β-D-dioxolane guanine (DXG), was found to exhibit good anti-HIV-1 activity in both humans2 and against virus containing the M184V/I mutation,3,4 and is currently in advanced Phase 2 clinical trials5 (Figure 1). (−)-β-D-2-Aminopurine dioxolane (APD), a more water soluble prodrug of DXG, also exhibits excellent anti-HIV activity in animal models as well as an enhanced pharmacokinetic profile in rhesus monkeys.6,7 Herein, we describe improved synthetic routes for these important anti-HIV nucleoside analogs. For the synthesis of DAPD, a cyanoacetate was employed as an additive in the Vobrüggen glycosylation reaction to improve the β/α anomeric ratio and overall β anomer isolated yield. A scale-limiting chromatographic purification was eliminated through the use of an enzyme catalyzed hydrolysis in the synthesis (−)-APD.
Fig. 1.

(−)-β-D-2,6-Diaminopurine dioxolane (DAPD) and (−)-β-D-2-Aminopurine dioxolane (APD).
Since the first racemic dioxolane nucleosides were reported by Belleau8 in 1989, a number of enantiomerically pure dioxolane analogs have been synthesized and identified as potent and non-toxic anti-HIV agents. For example, the Chu/Schinazi groups9 and later Gu et al.10 reported (±)-DAPD and (±)-DXG both inhibited HIV-l in various cell systems. A process for obtaining enantiomerically pure β-D-1,3-dioxolane nucleosides via a stereospecific synthesis has also been reported (Scheme 1). The fourteen-step synthesis of (−)-DAPD from 1,6-anhydro-D-mannose11 resulted in a modest overall yield. It involved a difficult and low yielding Pb(OAc)4 oxidation to dioxolane acetate, 112 and multiple chromatography purification steps that make the process unsuitable for commercial development. It is noteworthy that reaction of intermediate 2 with ammonia gave a mixture of both 6-Cl and 2-F substitution with 2-flouro ipso displacement representing the major product occurring in the presence of the fairly reactive 6-Cl substituent.
The next significant advancement for the dioxolane nucleosides was the development of a more efficient synthesis of the dioxolane nucleoside analogs that included an enzyme driven resolution of the R/S enantiomers as well as a more scalable overall process.13 The resolution relies on a simple two-step synthesis of racemic, ester protected, dioxolane lactone from commercially available 2,2-dimethoxy or 2,2-diethoxy ethanol in an isolated 79% yield. An enzyme resolution of the racemic dioxolane lactone completes the three-step sequence to the key intermediate (R)-lactone B, 3 (Scheme 2).14 The new synthesis was scaled to produce over one thousand kilograms of (R)-lactone B, 315.
Scheme 2.

Reagents and conditions: (a) (i) LiAl(Ot-Bu)3H, THF, −20 °C, 2 h; (ii) Ac2O, DMAP, −10 °C, 2 h, rt, overnight, 60%; (b) (i) 2,6-dichloropurine, 5, HMDS, (NH4)2SO4, reflux, 2 h; (ii) TMSI, CH2Cl2, −10 °C to rt, 6 h, 9–18%; (c) (i) NaN3, DMF, rt, 1 h; (ii) n-BuBr, rt, 0.5 h; (d) H2, 5% Pd/C, DMF, rt; (e) (i) n-butyl amine, MeOH, reflux, 4h; (ii) recrystallization denatured ethanol:water 52:48 w/w 61–91% (four steps).
This more efficient dioxolane nucleosides synthesis was used to prepare kilogram quantities for the first human clinical trials.13 As shown in Scheme 2, this process started with the selective reduction of (R)-lactone B, 3 with lithium tri-tert-butoxyaluminum hydride followed by acylation with acetic anhydride to provide (2R-4R/S)-4-acetoxy-2-isobutyryloxymethyl-1,3-dioxolane, 4. In the key coupling step, 2,6-dichloropurine, 5 and dioxolane acetate, 4 were used under Vobrüggen conditions to afforded a crude mixture of nucleoside analogs as a 1:1 mixture of α- and β-anomers. A single recrystallization provided the pure β-anomer, 6. Unfortunately, the yield ranged from a disappointing 9 to 18%. A two-step conversion to the desired 2,6-diaminopurine nucleoside involved treatment of 6 with sodium azide in DMF, filtration, and direct palladium catalyzed hydrogenation. Excess NaN3 was consumed prior to filtration by the addition of n-butyl bromide. Removal of the isobutyl ester was effected with butylamine in refluxing methanol to reveal DAPD that was crystallized in denatured ethanol:water to provide active pharmaceutical ingredient (API) quality (−)-DAPD. The large scale use of sodium azide provided an additional challenge in commercial development (Scheme 2). Despite these shortcomings, over 100 kg of (−)-DAPD has been prepared utilizing this process.
More recently the glycosylation of 2,6-diaminopurines or silyl 2,6-diaminopurines with dioxolane acetates was reported to be facilitated by the presence of a 1,3-dicarbonyl compound such as t-butyl acetoacetate.16 Although it is an improvement, this process suffers in that only a 33% yield of the desired β-isomer is obtained from the critical glycosylation step (Table 1, entry 2). Interestingly, the α/β ratio was 1:1.8 whereas in the absence of t-butyl acetoacetate the α/β ratio was 1:1.2 (Table 1, entry 1) close to the roughly equal mix of anomers that had been observed with all previous dioxolane glycosylation reactions. The role played by the glycosylation additive t-butyl acetoacetate was not discussed by the authors and our studies in this regard provided no clear explanation for the improved yield and β-selectivity. To support our clinical efforts, it is advantageous for us to continue the development of a more cost-effective process scalable synthesis for preparing API quality dioxolane nucleoside derivatives such as (−)-DAPD and (−)-APD.
Table 1.
Glycosylationa results from the reaction of 2,6-diaminopurine 9 with dioxolane acetate 4.
| Entry | Additive | Solvent | Lewis Acid | Yield(%) | β/α |
|---|---|---|---|---|---|
| 1 | none | CH2Cl2 | TMSI | 41 | 1.2 |
| 2 | CH3COCH2COOtBu | CH2Cl2 | TMSI | 62b | 1.8 |
| 3 | NCCH2COOMe | CH2Cl2 | TMSI | 53 | 1.3 |
| 4 | NCCH2COOEt | CH2Cl2 | TMSI | 45 | 1.7 |
| 5 | NCCH2COOtBu | CH2Cl2 | TMSI | 72c | 2.2 |
| 6 | NCCH2COtBu | CH2Cl2 | TMSI | 54d | 2.4 |
| 7 | NCCH2COOtBu | CH2Cl2 | SnCl4 | 0 | NA |
| 8 | NCCH2COOtBu | CH2Cl2 | TiCl3(OiPr) | 34 | 0.8 |
| 9 | none | CH2Cl2 | TMSOTf | 11 | 0.8 |
| 10 | NCCH2COOtBu | CH2Cl2 | TMSOTf | 14 | 0.8 |
| 11 | NCCH2COOtBu | CH3CN | TMSI | 55 | 1.5 |
| 12 | NCCH2COOtBu | CHCl3 | TMSI | 51 | 1.8 |
| 13 | NCCHC(OTMS)tBue | CH2Cl2 | TMSI | 50f | 2.6 |
Conditions for the glycosylation reaction: (i) HMDS, (NH4)2SO4, reflux, (ii) additive, Lewis acid, −10 to −15 °C; 1 h; 4 °C, 15 h, rt for 3h.
33% isolated yield of β-isomer.
45% isolated yield of β-isomer.
33% isolated yield of β-isomer.
98:2 Z:E-enol silane based on 1H NMR.
31% isolated yield of β-isomer
2. Results and Discussion
2.1 Synthesis of (−)-DAPD
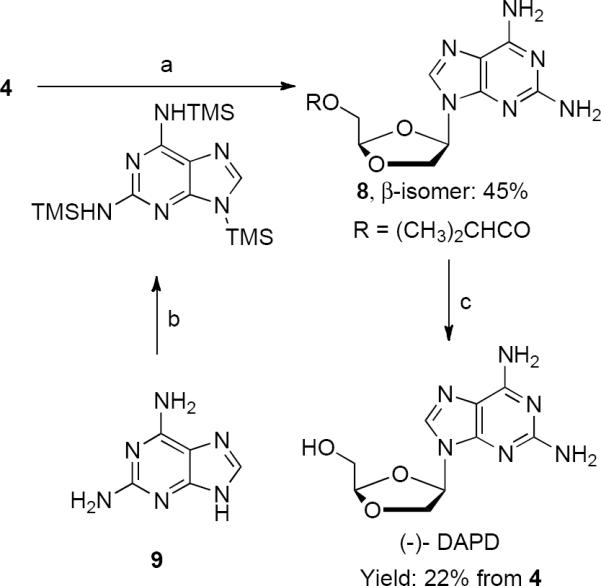
Herein we report a more robust process for the large-scale synthesis of (−)-DAPD starting from (R)-lactone B, 3. Dioxolane acetate, 4 was prepared from 3 as described in Scheme 2. Although our studies to uncover the role of t-butyl acetoacetate in the higher yielding and more facial selective dioxolane glycosylation were inconclusive we envisioned an active methylene derived complex or adduct that preferentially blocks the α-face leading to a preference for the incoming nucleoside base to attack the β-face of the dioxolane oxonium complex. The overall yield and anomeric ratios with and without t-butyl acetoacetate are shown in Table 1 entries 1 and 2. Part of our study involved investigation of α-cyano carbonyl compounds as additives along with variation of the solvent and Lewis acid in the glycosylation reaction of dioxolane acetate, 4 with silylated 2,6-diaminopurine (Table 1). We found that methyl cyano acetate (Table 1, entry 3) had very little effect on the yield or anomeric ratio versus the reaction without additives (Table 1, entry 4). Ethyl cyano acetate gave a 1:1.7 ratio of α- and β-anomers favoring the β-anomer, however, in an unimproved overall yield of 45% (Table 1, entry 4). When the dioxolane acetate, 4 was coupled with silylated 2,6-diaminopurine in the presence of the additive t-butyl cyanoacetate (Table 1, entry 5) a 1:2.2 ratio of α- and β-anomers in a much improved 72% total yield was obtained and the β-isomer 8 was obtained in an isolated 45% yield after a single recrystallization from isopropanol. The corresponding t-butyl cyanoketone (Table 1, entry 6) gave a similar yield of 70% but the 1.5:1 β/α anomeric ratio was much less favorable when compared to t-butyl cyanoacetate.
With t-butyl cyanoacetate identified as a superior additive various Lewis acids and solvents were evaluated to further optimize the dioxolane glycosylation conditions. Although tin (IV) chloride was employed with good results as a Lewis acid in the dioxolane and oxathiolane pyrimidine nucleoside synthesis,17 no desired product was detected in the synthesis of DAPD from 2,6-diaminopurine (Table 1, entry 7). When TiCl3(OiPr)7a was used only low yields and poor β-isomer selectivity was observed (Table 1, entry 8). Trimethylsilyl trifluorosulfonate (TMSOTf) both with and without t-butyl cyanoacetate gave only low yields which surprisingly favored the α-anomer (Table 1, entries 9 and 10). Iodotrimethylsilane (TMSI) was found to be the best Lewis acid tested and it was used to test a few of the common glycosylaton solvents. Both acetonitrile and chloroform proved inferior to dichloromethane in both yield and anomer selectivity (Table 1, entries 11 and 12). In an effort to gain mechanistic insight into these glycosylation conditions we attempted to prepare the TMS ketene acetal of t-butyl cyanoacetate for use as an alternate additive. Despite the use of hexamethyldisilazane, TMSCl, and TMSOTf as silylation agents with either triethylamine or lithium diisopropyl amide as base, we were unable to isolate or detect the presence of TMS ketene acetal by 1H-NMR. Conversely the t-butyl cyanoketone was readily converted to the TMS enol ether by exposure to LDA/TMSCl and its use as an additive in the glycosylation reaction gave a 2.5:1 β/α anomeric ratio; the highest ratio observed under any conditions (Table 1, entry 13). Unfortunately the isolated yield of the pure β anomer was less than half that observed under the best conditions. By using the additive t-butyl cyanoacetate and dichloromethane as solvent the ratio of β/α was improved and the isolated yield of β-isomer, 8, obtained from the anomer mixture by a single crystallization, was increased to 45%. This represents a 37 % improvement in the isolated yield of 8 versus that obtained with t-butyl acetoacetate. (−)-DAPD was obtained in high yield after deprotection using methanol saturated with ammonia to provide an overall 22% yield of (−)-DAPD based on the starting (R)-lactone B, 3 (Scheme 3).
Scheme 3.
Reagents and conditions: (a) CNCH2COOtBu, TMSI, CH2Cl2, −10 to −15 °C; 1 h; 4 °C, 15 h, rt for 3h, 45%; (b) HMDS, (NH4)2SO4, reflux, 4 h; (c) NH3/MeOH, rt, 25 h, 83%
2.2 Synthesis of (−)-APD
An optimized TMSI mediated glycosylation reaction has been previously reported for the synthesis of APD.7 In an effort to further improve this reaction, we applied the above optimized conditions to the synthesis of (−)-APD, and surprisingly, the additives t-butyl cyanoacetate and t-butyl acetoacetate had no effect on the outcome of the anomeric ratio in the glycosylation reaction. This implicates the involvement of the purine base in the facially selective glycosylation step. Our studies also revealed that 6-chloro-2-amino-purine consistently gave significantly better yields of the desired beta isomer when compared to 2-amino purine. A simple high yielding catalytic hydrogenation of 11 in ammonium hydroxide/methanol provided simultaneous reduction and deprotection to provide the desired (−)-APD nucleoside (Scheme 4).
Scheme 4.

Reagents and conditions: (a) TMSI, CHCl3, silylated 2-amino-6-chloropurine −10 ~ −15 °C, 2 h; rt, 15h; reflux, 5 h; (b) silica gel chromatography (hexane:ethyl acetate 100:0 to 0:100) 46% from 4; (c) H2/Pd-C, NH4OH/MeOH, rt, 20 h, 84%.
In this sequence, the key difficulty for kilogram of scale synthesis (−)-APD was the separation of pure β-isomer, 11 from the β/α mixture, 10 where on small scale flash chromatography was successfully employed (Scheme 4). As the reaction was scaled to larger amounts crystallization from a large number of solvent systems was investigated but was not successful at separating the desired β-isomer, 11.
Although enzymes have been employed widely for biocatalytic resolutions,18 little information exists regarding the enantioselective enzyme-catalyzed hydrolysis of racemic nucleosides.19 Wengel and co-workers20 reported the first use of esterase catalyzed diastereoselective deacetylation to solve the problem of nucleoside anomer separation. Thus, a lipase-catalyzed deacylation of an anomeric mixture of peracetylated 2'-deoxyribofuranosyl thymines was carried out using two enzymes: wheat germ lipase (WGL) and porcine liver esterase (PLE). The β-anomer of thymidine was the only completely deprotected nucleoside product.
In order to realize a more scaleable process for the synthesis of (−)-APD we examined the potential for selective enzymatic de-esterification of the α-anomer of the β/α mixture of nucleosides, 10. If successful, the resulting alcohol containing α-anomer, 13, would be significantly more polar thus facilitating its separation from the intact β-anomer, 11. Pseudomonas fluorescens lipase (PFL), porcine liver esterase (PLE) and Candida antarctica lipase (CAL) were chosen because of their commercial availability, relative low cost, and tolerance for a wide class of substrates. The hydrolysis reactions were typically incubated at room temperature in 1 mL of pH 7.0 buffer (0.05 M potassium phosphate) and 1 mL of organic solvent with 50 mg of nucleoside 10 and enzymes PFL, CAL or PLE (25 mg, 25 mg or 100 units respectively). The hydrolysis reactions were monitored by thin layer chromatography and high pressure liquid chromatography-mass spectrometry (LC/MS). The results are summarized in Table 2. When PFL was employed no hydrolysis of 10 took place in tetrahydrofuran (THF), methanol (MeOH), or dimethyl formamide (DMF) (entries 1–3, Table 2), and only trace amounts of the α-isomer was hydrolyzed with CH3CN as co-solvent (entry 4, Table 2). No reaction was observed when PLE was used in THF, MeOH, or DMF (entries 5–7, Table 2). When the PLE reaction was conducted in CH3CN, the starting material 10 was completely but nonselectively converted to nucleosides 12 and 13. Fortunately, selective enzyme-catalyzed hydrolysis took place when CAL was used with all four solvents (entries 9–12, Table 2). The α-isomer of 10 can be selectively hydrolyzed leaving the β-isomer intact. Recovery of the desired unreacted β-anomer, ester 10, was accomplished by a simple extraction which left the undesired α-anomer in the water layer. The β-anamer 10 was obtained sufficiently pure to use in further reactions without additional purification. Simultaneous removal of the isobutyl ester and reduction of the 6-chloro group was performed by hydrogenation with Pd/C as catalyst in MeOH containing NH4OH. High purity (−)-APD was obtained in a highly scalable process by filtering the resulting material through a short silica gel column followed by crystallization in CH2Cl2/MeOH. The overall yield of APD from (R)-lactone B was 23% (Scheme 4).
Table 2.
Enzyme-catalyzed reactions of nucleosides 10

| Entry | Enzyme | Solvent | Reaction Time (h) | Conversion (%) | |
|---|---|---|---|---|---|
| 12 | 13 | ||||
| 1 | PFL | THF | 24 | 0 | 0 |
| 2 | PFL | MeOH | 24 | 0 | 0 |
| 3 | PFL | DMF | 24 | 0 | 0 |
| 4 | PFL | CH3CN | 24 | 0 | trace |
| 5 | PLE | THF | 24 | 0 | 0 |
| 6 | PLE | MeOH | 30 | 0 | 0 |
| 7 | PLE | DMF | 24 | 0 | 0 |
| 8 | PLE | CH3CN | 15 | 100 | 100 |
| 9 | CAL | THF | 15 | 0 | 100 |
| 10 | CAL | MeOH | 18 | 0 | 100 |
| 11 | CAL | DMF | 18 | 0 | 100 |
| 12 | CAL | CH3CN | 24 | 0 | 100 |
PFL: Pseudomonas fluorescens lipase; PLE: porcine liver esterase; CAL: lipase acrylic resin from Candida antarctica.
3. Conclusions
A novel, scalable and efficient processes for the synthesis of (−)-DAPD and (−)-APD was developed. We found that t-butyl cyanoacetate can be used as an additive for the coupling step in route to (−)-DAPD and resulted in improved β-selectivity with an isolated yield four times greater than the original process scale method. The mechanistic role of this new additive and its enhancement of β-selectivity are currently under investigation. A difficult silica gel chromatography step in route to (−)-APD was replaced by an enzyme-catalyzed hydrolysis reaction that allowed the pure β-anomer, 10, to be isolated by a simple water extraction.
4. Experimental
4.1. General
Anhydrous solvents (Drisolv®) were purchased from EMD Chemicals Inc (Philadelphia, PA). Reagents were purchased from commercial sources. Unless noted otherwise, the materials used in the examples were obtained from readily available commercial suppliers or synthesized by standard methods known to one skilled in the art of chemical synthesis. Thin layer chromatography (TLC) was carried out on precoated silica gel thin layer sheets 60 F254 from EMD (E. Merck, Darmstadt, Germany) or on Silica Gel Uniplate GHLF 250 μm (Analtech, Newark DE, part number 21521). 1H-NMR spectra were taken on a Varian Unity Plus 400 spectrometer (Varian, Inc., Polo Alto, CA, USA) at room temperature and reported in parts per million downfield from internal tetramethylsilane. Signal multiplicities are represented by s (singlet), d (doublet), dd (doublet of doublets), t (triplet), q (quadruplet), br (broad), bs (broad singlet), m (multiplet). All J values are in hertz. Mass spectral analyses were performed on a Micromass TOF instrument (Hewlett-Packard HPLC driven electrospray MS instrument). Analytical HPLC analyses were performed on a Hewlett-Packard HPLC with a Phenomenex Gemini-NX column (2 mm × 50 mm, 3 μm, C18, 110 Å). Mobile phase flow was 0.7 ml/min with a 3.5 min gradient from 96% aqueous media (0.05% formic acid) to 96% CH3CN (0.05% formic acid) with a 5.5 min total acquisition time and 190–360 nm PDA detection.
4.1.1. Silylation of 2,6-diaminopurine
2,6-Diaminopurine, 9 (750 mg, 5.0 mmol), ammonium sulfate (750 mg, 5.7 mmol), and hexamethyldisilazane (20 mL) were added to a 250 mL three-neck flask. The suspension was heated to reflux with stirring at 130–135 °C (oil-bath) for 4 h. During this period the solution became homogeneous. The solution was cooled to 85 °C and the excess hexamethyldisilazane was subsequently removed by distillation at reduced pressure. After the hexamethyldisilazane was completely removed, the residue was cooled to rt under vacuum then 10 mL of anhydrous methylene chloride was added to prepare a solution of crude silylated 2,6-diamino purine of suitable purity for the preparation of dioxolane nucleosides 8. This material is highly moisture sensitive and therefore used directly in the next step.
4.1.2. ((2R)-4-acetoxy-1,3-dioxolan-2-yl)methyl isobutyrate (4)
To a well stirred solution of LiAl(OtBu)3H (25.4 g, 100.0 mmol) in dry THF (150 mL) at −10 to −20 °C was added a precooled (R)-(4-oxo-1,3-dioxolan-2-yl)methyl isobutyrate (12.5 g, 66.4 mmol) over a period of 10 min under N2 atmosphere. The reaction mixture was allowed to stir for 2 h at −10 to −20 °C. To this solution DMAP (7.0 g, 57 mmol) was added in one portion and stirred for 30 min followed by dropwise addition of Ac2O (46.0 mL, 443 mmol). After stirring the bright yellow solution for 2 h at −10 °C, the reaction was allowed to slowly raise to room temperature and stirred overnight. The dark brown solution was poured into saturated aqueous NH4Cl (180 mL), stirred for 30 min, filtered (to remove Li salts), concentrated in vacuo and extracted with ethyl acetate (3 × 60 mL). The combined organic solutions were washed with saturated aqueous NaHCO3 (2 × 50 mL), brine (1 × 25 mL), dried over anhydrous Na2SO4, filtered, and evaporated under reduced pressure to afford crude 4 (red syrup). This crude material is suitable for immediate use in glycosylation reactions. Alternatively, it can be stored for extended periods at −80 °C protected from air and moisture as a 1 M solution in CHCl3. Rf: 0.45 (ethyl acetate:hexanes 1:4). 1H-NMR analysis showed a 1:1 mixture of α- and β–isomers. 1H-NMR (400 MHz, CDCl3) δ: 1.18 (d, 6H, J=7.2 Hz, 2 × CH3); 1.19 (d, 6H, J=6.8 Hz, 2 × CH3); 2.09 (s, 3H, CH3); 2.10 (s, 3H, CH3); 2.58–2.61 (m, 2H, 2 × CH); 4.20–4.42 (m, 8H, 4 × CH2); 5.32 (t, 1H, J=4.4 Hz, CH); 5.42 (t, 1H, J=4.4 Hz, CH); 6.35 (d, 1H, J=4.0 Hz, CH); 6.41 (dd, 1H, J=4.0 Hz, J=1.6 Hz, CH).
Anhydrous CHCl3 was added to a total volume of 40 mL to prepare a 1 M solution (based on a 60% yield)
4.1.3. (2R,4R)-2-Isobutyryloxymethyl-4-(2,6-diaminopurin-9-yl)-[1,3]-dioxolane (8)
To the solution of silylated 2,6-diaminopurine in dry methylene chloride from above was added 4 mL of 1 M ((2R, 4R/S)-4-acetoxy-1,3-dioxolan-2-yl)methyl isobutyrate, 4 in chloroform and 0.75 mL of t-butyl cyanoacetate under a nitrogen atmosphere. The resulting solution was cooled to between −10 and −15 °C then a solution of iodotrimethylsilane (1.5 mL) in 2 mL of dry methylene chloride was added dropwise over the course of 2–3 min. The mixture was then stirred between −10 and −15 °C for 1 h, 4 °C for 15h and rt for 3h. The reaction mixture was added dropwise to 0.5 M hydrochloric acid (18 mL) at 0 °C. The mixture was warmed with stirring to 25 °C and stirred for another 20 min. The phases were separated and the organic phase was back-extracted once with 0.5 M hydrochloric acid (18 mL). The combined aqueous phases were washed twice with 25 mL of methylene chloride. Then, after addition of a further 50 mL of methylene chloride, the pH was adjusted to 9.0 with approximately 40 mL of 10% sodium carbonate solution. The mixture was stirred at 25 °C for 1 h and the phases were separated. The aqueous phase was back-extracted twice with 30 mL of methylene chloride. The combined organic phases were washed once with 25 mL of water. The solvent was removed under vacuum and the resulting residue was passed through a short flash silica gel column, eluting with 0 to 15% methanol in methylene chloride to yield 940 mg of yellowish solid. LC/MS analysis showed the isomer ratio (β/α ratio = 2.2:1). The crude product was crystallized from isopropanol and 690 mg (45% yield) of colorless β-isomer crystals were obtained. NMR analysis revealed one mol equivalent of isopropanol in addition to (2R, 4R)-2-isobutyryloxymethyl-4-(2,6-diaminopurin-9-yl)-1,3-dioxolane, 813. 1H-NMR (400 MHz, DMSO-d6) δ: 0.95–1.04 (m, 12H, 4 × CH3); 2.44–2.40 (m, 1H, CH); 3.71–3.75 (m, 1H, CH), 4.17–4.52 (m, 5H, CH, 2 × CH2); 5.20 (t, 1H, J=3.2 Hz, CH); 5.80 (brs, 2H, NH2) 6.17 (dd, 1H, J=1.6 Hz, J=5.6 Hz, CH); 6.71 (brs, 2H, NH2); 7.74 (s, 1H, ArH).
4.1.4. (Z)-4,4-dimethyl-3-((trimethylsilyl)oxy)pent-2-enenitrile
A solution of diisopropylamine (1.80 mL, 13.0 mmol) in THF (20 mL) cooled to 0 °C. n-BuLi (5.20 mL of a 2.5 M solution in hexanes, 13.0 mmol) was added dropwise and the reaction was stirred a further 30 min at 0 °C. The LDA solution was cooled to −78 °C and a solution of 4,4-dimethyl-3-oxopentanenitrile (1.25 g, 10.0 mmol) in THF (5 mL) was added dropwise. After stirring at −78 °C for an additional 30 min, TMSCl (2.50 mL, 20.0 mmol) was added. The reaction was allowed to slowly warm to rt and stirred an additional 3 h. The reaction was concentrated under reduced pressure and the residue was taken up in hexanes, filtered through celite eluting with hexanes (100 mL), and solvents were removed under reduced pressure. The orange oil was distilled under reduced pressure to yield 1.14 g (58%, 102–103 °C at 13 mbar) of the titled compound as a clear oil (93:7 ratio of enol silane:ketone). This material is highly moisture sensitive and must be stored at ≤ 4 °C under an anhydrous atmosphere of nitrogen. 1H-NMR (400 MHz, CDCl3) δ 4.64 (1H, s) 1.09 (9H, s) 0.38 (9H, s). 13C-NMR (100 MHz, CDCl3) δ: 181.1, 118.5, 74.8, 38.3, 27.7, 1.2
4.1.5. [(2R,4R)-[4-(2,6-diamino-9H-purin-9-yl)-1,3-dioxolan-2-yl]methanol [(−)-DAPD]
(2R,4R)-2-Isobutyryloxymethyl-4-(2,6-diaminopurin-9-yl)-1,3-dioxolane • 2-propanol, 8 (31.1 g, 81.2 mmol) was dissolved in 310 mL of NH3-saturated methanol. The solution was stirred at 25 °C for 15 h and the solvent was removed under reduced pressure. The residue was crystallized from ethanol/water resulting in 17.1 g (83%) of (−)-DAPD as colorless crystals, mp: 236 – 238 °C (lit.21 mp: 236 – 237 °C). 1H-NMR (400 MHz, DMSO-d6) δ: 3.58 (dd, 2H, J=6.0 Hz, J=3.2 Hz); 4.17 (dd, 1H, J= 9.5 Hz, J= 5.5 Hz); 4.42 (dd, 1H, J= 9.5 Hz, J=1.8 Hz); 5.02 (t, J=3.2 Hz); 5.18 (t, J=6.0 Hz); 5.88 (brs, 2H); 6.18 (dd, 1H, J=5.5 Hz, J=1.8 Hz); 6.79 (brs, 2H); 7.86 (s, 1H). 13C-NMR (100 MHz, DMSO-d6) δ: 61.9, 71.0, 79.3, 105.9, 113.3, 135.4, 152.0, 156.7, 161.0. Calculated for C9H12N6O3: C, 42.86; H, 4.80; N, 33.32. Found: C, 42.57; H, 4.69; N, 33.02.
4.1.6. Silylation of 2-amino-6-chloropurine
2-Amino-6-chloropurine (0.85 g, 5.0 mmol), ammonium sulfate (0.75 g, 5.7 mmol), and 1,1,1,3,3,3-hexamethyl disilazane (HMDS) (3 mL) were heated to reflux under a N2 atmosphere. A clear solution was obtained after 2 h of reflux. The reaction mixture was cooled to rt and used directly in the next step.
4.1.7. Isobutyricacid-4-(R)-(2-amino-6-choloropurin-9-yl)-[1,3]-dioxolan-2-(R)-yl methyl ester (11) via chromatography
Isobutyricacid-4-acetoxy-[1,3]-dioxolan-2-(R)-yl methyl ester, 4 (3.77 mL of 1M solution in step 1, 3.77 mmol) was dissolved in CHCl3 (10 mL) and cooled to −10 to −20 °C. Iodotrimethylsilane (0.65 mL, 4.8 mmol) was slowly added by syringe over a period of 1 h and stirred for 2 h at −10 to −20 °C. To this solution was added persilylated 2-amino-6-choloro purine, dissolved in CHCl3 (15 mL) and the reaction was stirred at −10 to −20 °C for 2 h. The reaction mixture was allowed to warm to room temperature, stirred an additional 2 h at rt and then heated to gentle reflux for 4 h. The resulting mixture was cooled to rt, quenched with water, washed with saturated aqueous NaHCO3 (2 × 20mL), saturated aqueous sodium thiosulfate (20 mL), water (2 × 20 mL), and brine (2 × 20 mL). The organic layer was dried over Na2SO4, filtered and evaporated under reduced pressure to yield a yellow solid consisting of the α/β anomer mixture, 10. The residue was purified by silica gel flash column chromatography, eluting with 0 to 100% ethyl acetate in hexane to yield β-isomer, 117 as a white foam (590 mg, 45.8%). 1H-NMR (400 MHz, CDCl3) δ: 1.12 and 1.18 (2d, J=7.2 Hz, 6H, 2 × CH3); 2.60 (d, J=7.2 Hz 1H, CH); 4.28 (dd, J= 5.2 Hz, J=10.0 Hz 1H, CH2); 4.37 (m, 2H, CH2); 4.54 (dd, J=0.8 Hz, J=10.0 Hz, 1H, CH2); 5.21 (bs, 2H, NH2); 5.31 (t, J=3.2, 1H, CH); 6.37 (dd, J=1.2 Hz, J=5.2 Hz, 1H, CH) and 8.12 (s, ArH). 13C-NMR (100 MHz, CDCl3) δ: 19.15, 19.19, 33.95, 62.39, 71.41, 79.82, 103.65, 125.37, 140.05, 151.79, 153.47, 159.37, 176.85.
4.1.8. CAL catalyzed isolation of isobutyricacid-4-(R)-(2-amino-6-choloropurin-9-yl)-[1,3]-dioxolan-2-(R)-yl methyl ester (11)
The α/β anomer mixture, 10 from the above coupling step was hydrolyzed in 10 mL of pH 7.0 buffer (0.05 M potassium phosphate) and 10 mL of THF and 300 mg of lipase acrylic resin from candida antarctica (CAL). The mixture was stirred at room temperature for 20 h. After which the α-isomer was hydrolyzed completely and 100 mL of CH2Cl2 was added. The organic phase was collected and the aqueous layer was extracted with CH2Cl2 (2 × 100 mL). The combined organic layer was washed with water (50 mL), brine (50 mL) and dried over Na2SO4. After removal of the solvent under reduced pressure, 11 was obtained in 46% yield and used directly in the next step.
4.1.9. Preparation of (−)-D-2-Amino purine dioxolane [(−)-APD]
A solution of isobutyric acid 4-(R)(2-amino-6-chloro-purin-9-yl)-[1,3]-dioxolan-2-(R)-yl methyl ester 11 (500 mg, 1.40 mmol), and 10% Pd/C (100 mg) in MeOH (50 mL) and NH4OH (5 mL) was kept under hydrogen (1 atm) until the TLC showed disappearance of the starting material (~ 24 h). The reaction mixture was degassed with nitrogen, filtered through celite and the solvent was removed under reduced pressure. The residue was passed through a short flash silica gel column (hexanes: ethyl acetate, 9:1 to 3:7 gradient) to give the title compound (285 mg, 84%). Pure material was obtained by crystallization in MeOH/CH2Cl2: mp: 155–156 °C. LCMS (EI): 238 (M + 1). 1H-NMR (400 MHz, DMSO-d6) δ: 3.62 (dd, J=6.0 Hz, J=3.0 Hz, 2H, CH2); 4.23 (dd, J=10.0 Hz, J=5.5 Hz, 1H, CH); 4.54 (d, J=10.0 Hz, J=1.5 Hz, 1H, CH); 5.07 (t, J=3.0 Hz, 1H, OH); 5.15 (d, J=6.0 Hz, 1H, CH); 6.33 (dd, J=5.5 Hz, J=1.5 Hz, 1H, CH); 6.62 (bs, 2H, NH2); 8.23 (s, 1H, CH); 8.60 (s, 1H, CH). 13C-NMR (100 MHz, DMSO-d6) δ: 61.0, 70.4, 78.5, 105.5, 126.5, 140.0, 149.3, 152.6, 160.6. Calculated for C9H11N5O3·0.5H2O: C, 43.90; H, 4.91; N, 28.44. Found: C, 42.91; H, 4.80; N, 28.40.
Scheme 1.

Early synthesis of (−)-DAPD.
Acknowledgements
Supported in part by NIH CFAR grant 2P30-AI-050409 and by the Department of Veterans Affairs. Emory received no funding from RFS Pharma, LLC to perform this work and vice versa.
COI: Dr. R.F. Schinazi is the Founder of RFS Pharma, LLC, a major shareholder of RFS Pharma, LIC and an inventor of DAPD/APD and may receive royalties from the future sales of these drugs.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Global summary of the AIDS epidemic, database of WHO. 2010 http://www.who.int/hiv/data/2011_epi_core_en.png.
- 2.(a) Thompson MA, Kessler HA, Eron JJ, Jr., Jacobson JM, Adda N, Shen G, Zong J, Harris J, Moxham C, Rousseau FS. AIDS. 2005;19:1607. doi: 10.1097/01.aids.0000186822.68606.05. [DOI] [PubMed] [Google Scholar]; (b) Gripshover BM, Ribaudo H, Santana J, Gerber JG, Campbell TB, Hogg E, Jarocki B, Hammer SM, Kuritzkes DR, A5118 Team Antivir Ther. 2006;11:619. [PubMed] [Google Scholar]; (c) Margolis DM, Mukherjee AL, Fletcher CV, Hogg E, Ogata-Arakaki D, Petersen T, Rusin D, Martinez A, Mellors JW. AIDS. 2007;21:2025. doi: 10.1097/QAD.0b013e3282364381. [DOI] [PubMed] [Google Scholar]
- 3.(a) Gu Z, Wainberg MA, Nguyen-Ba N, L'Heureux L, de Muys JM, Bowlin TL, Rando RF. Antimicrob Agents Chemother. 1999;43:2376. doi: 10.1128/aac.43.10.2376. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gu Z, Wainberg MA, Nguyen-Ba P, L'Heureux L, de Muys JM, Rando RF. Nucleosides Nucleotides. 1999;18:891. doi: 10.1080/15257779908041594. [DOI] [PubMed] [Google Scholar]; (c) Mewshaw JP, Myrick FT, Wakefield DA, Hooper BJ, Harris JL, McCreedy B, Borroto-Esoda K. J Acquir Immune Defic Syndr. 2002;29:11. doi: 10.1097/00042560-200201010-00002. [DOI] [PubMed] [Google Scholar]
- 4.Bazmi HZ, Hammond JL, Cavalcanti SCH, Chu CK, Schinazi RF, Mellors JW. Antimicrob. Agents Chemother. 2000;44:1783. doi: 10.1128/aac.44.7.1783-1788.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Murphy RL, Kivel NM, Zala C, Ochoa C, Tharnish P, Mathew J, Pascual ML, Schinazi RF. Antivir. Ther. 2010;15:185. doi: 10.3851/IMP1514. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hurwitz SJ, Asif G, Fromentin E, Tharnish PM, Schinazi RF. Antimicrob. Agents Chemother. 2010;54:1248. doi: 10.1128/AAC.01209-09. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Won SY, Kessler HA. In: Kucers' The Use of Antibiotics Sixth Edition. Grayson ML, editor. Vol 1. Hodder Arnold; 2010. p. 2627. [Google Scholar]
- 6.(a) Chen H, Manouilov KK, Chu CK, Schinazi RF, McClure HM, Boudinot FD. Journal of Chromatography, B: Biomedical Sciences and Applications. 1997;691:425. doi: 10.1016/s0378-4347(96)00473-2. [DOI] [PubMed] [Google Scholar]; (b) Chen H, Boudinot FD, Chu CK, McClure HM, Schinazi RF. Antimicrob. Agents Chemother. 1996;40:2332. doi: 10.1128/aac.40.10.2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Narayanasamy J, Pullagurla MR, Sharon A, Wang J, Schinazi RF, Chu CK. Antiviral Research. 2007;75:198. doi: 10.1016/j.antiviral.2007.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Belleau B. EP 0337713. 1989. [Google Scholar]
- 9.(a) Chu CK, Schinazi RF. U.S. Patent 5179104. 1990; (b) Kim HO, Schinazi RF, Nampalli S, Shanmuganathan K, Cannon DL, Alves AJ, Jeong SJ, Beach JW, Chu CK. J. Med. Chem. 1993;36:30. doi: 10.1021/jm00053a004. [DOI] [PubMed] [Google Scholar]
- 10.(a) Gu Z, Wainberg MA, Nguyen-Ba P, L'Heureux L, de Muys JM, Bowlin TL, Rando RF. Antimicrob. Agents Chemother. 1999;43:2376. doi: 10.1128/aac.43.10.2376. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gu Z, Wainberg MA, Nguyen-Ba P, L'Heureux L, de Muys JM, Bowlin TL, Rando RF. Nucleosides Nucleic Acids. 1999;18:891. doi: 10.1080/15257779908041594. [DOI] [PubMed] [Google Scholar]
- 11.Zottola MA, Alonso R, Vite GD, Fraser-Reid B. J. Org. Chem. 1989;54:6123. [Google Scholar]
- 12.(a) Chu CK, Ahn SK, Kim HO, Beach JW, Alves AJ, Jeong LS, Islam Q, Van Roey P, Schinazi RF. Tetrahedron Lett. 1991;32:3791. [Google Scholar]; (b) Kim HO, Ahn SK, Alves AJ, Beach JW, Jeong LS, Choi BG, Van, Roey P, Schinazi RF, Chu CK. J. Med. Chem. 1992;35:1987. doi: 10.1021/jm00089a007. [DOI] [PubMed] [Google Scholar]
- 13.Sznaidman ML, Du J, Pesyan A, Cleary DJ, Hurley KP, Waligora F, Almond MR. Nucleoside, Nucleotides & Nucleic Acids. 2004;23:1875. doi: 10.1081/NCN-200040643. [DOI] [PubMed] [Google Scholar]
- 14.Sznaidman M, Painter GR, Almond MR, Cleary DG, Pesyan A. 2005 WO2005074654. [Google Scholar]
- 15.Popp A, Gilch A, Mersier A-L, Petersen H, Rockinger-Mechlem J, Stohrer J. Adv. Synth. Catal. 2004;346:682. [Google Scholar]
- 16.(a) Döring W, Petersen H. U.S. Patent 0211855. 2006; (b) Döring W, Petersen H. 2005. WO2005012302. [Google Scholar]
- 17.(a) Choi W-B, Wilson LJ, Yeola S, Liotta DC, Schinazi RF. J. Am. Chem. Soc. 1991;113:9377. [Google Scholar]; (b) Jin H, Tse HLA, Evans CA, Mansour TS, Beels CM, Ravenscroft P, Humber DC, Jones MF, Payne JJ, Ramsay MVJ. Tetrahedron: Asymmetry. 1993;4:211. [Google Scholar]; (c) Wilson LJ, Choi W-B, Spurling T, Liotta DC, Schinazi RF, Cannon D, Painter GR, Clair M, Furman PA. Bioorg. & Med. Chem. Lett. 1993;3:169. [Google Scholar]
- 18.(a) García-Urdiales E, Alfonso I, Gotor V. Chem. Rev. 2005;105:313. doi: 10.1021/cr040640a. [DOI] [PubMed] [Google Scholar]; (b) Pàmies O, Bäckvall J-E. Chem. Rev. 2003;103:3247. doi: 10.1021/cr020029g. [DOI] [PubMed] [Google Scholar]; (c) Ferrero M, Gotor V. Chem. Rev. 2000;100:4319. doi: 10.1021/cr000446y. [DOI] [PubMed] [Google Scholar]; (d) Santaniello E, Ferraboschi P, Grisenti P, Manzocchi A. Chem Rev. 1992;92:1071. [Google Scholar]; (e) Klibanov AM. Arc. Chem. Res. 1990;23:114. [Google Scholar]; (f) Wong C-H. Chemtracts. 1990;3:91. [Google Scholar]; (g) Zhu L, Tedford MC. Tetrahedron. 1990;46:6587. [Google Scholar]; (h) Whitesides GM, Wong C. Aldrichim. Acta. 1983;16:27. [Google Scholar]; (i) Turner NJ. Nat. Prod. Rep. 1989;6:625. doi: 10.1039/np9890600625. [DOI] [PubMed] [Google Scholar]; (j) Sih CJ, Wu S. In: Topics in Stereochemistry. Eliel EL, Wilen SH, editors. Vol. 19. John Wiley and Sons; New York: 1989. p. 63. [Google Scholar]; (k) Walpole CSJ, Wrigglesworth R. Nat. Prod. Rep. 1989;6:311. doi: 10.1039/np9890600311. [DOI] [PubMed] [Google Scholar]; (l) Butt S, Roberts SM. Chem. Britain. 1987;127 [Google Scholar]; (m) Jones JB. Tetrahedron. 1986;42:3351. [Google Scholar]; (n) Mulzer J. In: Organic Synthesis Highlights. Mulzer J, Altenbach H, Braun M, Krohn K, Reissig H, editors. Vol. 216. VCH; New York: 1991. [Google Scholar]; (o) Mulzer J. In: Organic Synthesis Highlights. Mulzer J, Altenbach H, Braun M, Krohn K, Reissig H, editors. VCH; New York: 1991. p. 207. [Google Scholar]
- 19.(a) Yoshimura Y, Moon HR, Choi Y, Marquez VE. J. Org. Chem. 2002;67:5938. doi: 10.1021/jo020249u. [DOI] [PubMed] [Google Scholar]; (b) Hoong LK, Strange LE, Liotta DC, Koszalka GW, Burns CL, Schinazi RF. J. Org. Chem. 1992;57:5563. [Google Scholar]; (c) Hultin PG, Mueseler F-J, Jones JB. J. Org. Chem. 1991;56:5375. [Google Scholar]; (d) Secrist JA, III, Montgomery JA, Shealy YF, O'Dell CA, Clayton SJ. J. Med. Chem. 1987;30:746. doi: 10.1021/jm00387a032. [DOI] [PubMed] [Google Scholar]
- 20.Damkjær DL, Petersen M, Wengel J. Nucleosides Nucleotides. 1994;13:1801. [Google Scholar]
- 21.Furman PA, Cleary D, Trost LC, Bigley JW, Painter GR. Drugs Future. 2000;25:454. [Google Scholar]