

Abstract
Three types of linear thiol-functionalized siloxane oligomers and three types of ene-functionalized oligomers were synthesized and subsequently photopolymerized. Within each type of thiol-functionalized oligomer, the ratio of mercaptan repeat units to non-reactive phenyl repeat units was varied to manipulate both the crosslink density and the degree of secondary interactions through pi-pi stacking. Similarly, the repeat units of the three ene-functionalized oligomers are composed of allyl-functional monomers, benzene-functional monomers, and octyl-functional monomers in varying ratios of benzene:octyl but with a constant fraction of allyl moieties. The structural composition of the siloxane oligomers plays a pivotal role in the observed material properties of networks formed through thiol-ene photopolymerization. Networks with a high concentration of thiol functionalities exhibit higher rubbery moduli, ultimate strengths, and Young’s moduli than networks with lower thiol concentrations. Moreover, the concentration of functionalities capable of participating in secondary interactions via hydrogen bonding or pi-pi stacking directly impacts the network glass transition temperature and elasticity. The combination of low crosslink density and high secondary interactions produces networks with the greatest toughness. Finally, the fraction of octyl repeats correlates with the hydrophobic nature of the network.
Keywords: polysiloxanes, thiol-ene, networks, elastomers, structure-property relations, mechanical properties
INTRODUCTION
Polysiloxanes, particularly poly(dimethylsiloxane) or PDMS, have received extensive attention in the polymer field for their wealth of advantageous physical properties. Relative to their carbon-based counterparts, siloxanes exhibit unusually high flexibility originating from two types of mobility, torsional and bending. The Si-O-Si bond is particularly flexible and commonly reaches bond angles of 135° to 180°.1 Additionally, polysiloxanes exhibit excellent permeability as well as low surface energy.2,3 The combination of such properties makes polysiloxanes ideal candidates for high-performance elastomers, water repellents, mold release agents, adhesives, and protective coatings.4–7 Moreover, polysiloxanes have been recognized by the biomedical community as relatively inert, biocompatible platforms for designing catheters, artificial skin, contact lenses, and drug delivery systems.8,9
Further, the synthetic ease with which polysiloxanes can be generated allows for any number of functionalities to be readily incorporated into their structure. Linear, branched, and crosslinked polysiloxanes can be produced through the condensation of pendant –Cl, -OH, or –OR groups by a variety of catalytic species including acids, bases, and stannous salts.1 Careful selection of the non-condensable silane species enables the physical properties of the resulting polymer to be tailored towards a specific application. For example, incorporation of urethane functionalities can significantly improve the mechanical strength, toughness, and abrasion resistance of polysiloxane coatings, while inclusion of ethylene glycol side chains can render the hydrophobic PDMS a more hydrophilic material capable of performing as a hydrogel without altering its transparency.10,11 Siloxanes are also highly amenable to ring-opening polymerizations, which afford control over functionality ordering within a linear polysiloxane. Indeed, sequential anionic copolymerizations of cyclosiloxanes have been utilized to produce siloxane block copolymers for use as amphiphilic emulsifiers.12
While incorporation of nonreactive functional groups within a linear polysiloxane can directly contribute to the chemical and physical properties, the use of reactive moieties has the added advantage of moderating network attributes and properties. Photosensitive units such as epoxides, methacrylates, or vinyls have been used to manipulate the crosslink density of linear siloxane polymers or oligomers following their condensation. Biomedical polymers such as intraocular lenses, drug delivery devices, and dental impression materials commonly employ this strategy to yield highly crosslinked, biocompatible elastomers.13–15 However, the susceptibility of (meth)acrylate and vinyl polymerizations to oxygen inhibition greatly limits their viability in vitro, particularly when such units are incorporated into highly permeable polysiloxanes. Furthermore, the propensity of vinyls and (meth)acrylates to shrink during polymerization hinders their use in applications that demand negligible dimensional change and where the internal stresses created by the polymerization shrinkage can lead to mechanical and optical failure.16,17
Alternatively, the free radical mediated thiol-ene click reaction proceeds via a step-growth polymerization mechanism consisting of alternating propagation/chain transfer steps. As illustrated in Scheme 1, an initiating radical species abstracts a proton from a thiol monomer creating a thiyl radical.18,19 The thiyl radical propagates through a vinyl monomer and forms a carbon-centered radical. This radical then abstracts a proton from a second thiol monomer, which continues the polymerization cycle. Theoretically, termination may occur through the combination of any two radical species. The thiol-ene click reaction mechanism is well documented in the literature as a rapid reaction that produces no by-products, is not inhibited by oxygen, and accumulates less volumetric shrinkage than a vinyl homopolymerization.20–24 Furthermore, thiol and vinyl species can be readily incorporated into any number of biological or organic chemical structures, including polysiloxanes, without impairing the heat resistance, weather stability, low-temperature flexibility, or low surface tension of the crosslinked siloxane network.25
SCHEME 1.

Thiol-Ene Reaction Mechanism, adapted from Kloxin et al.19 The thiol-ene polymerization proceeds through a cyclic step growth mechanism consisting of alternating propagation/chain transfer steps following initiation and prior to termination. The reaction mechanism assumes ideal conditions in which the alternating steps proceed at the same overall rate.
Schreck et al. have examined the use of thiol-ene chemistry as a means to create functionalized silsesquioxane particles, but, to date, the use of thiol-ene polymerizations in siloxane systems has focused on the coupling of nonreactive moieties onto an existing linear segment.26 Chojnowski et al. used a thermally initiated thiol-ene reaction to convert vinyl functionalities to carboxylic acids in their amphiphilic emulsifiers, and Herczynska et al. used a similar mechanism to initiate the reaction of vinyl units with thiolated pyridines in their linear polysiloxanes.12,25 However, the implementation of thiol-ene chemistry as a medium for inducing gelation and regulating crosslink density by coupling multiple linear polysiloxane chains remains unexplored. Thus, the aim of this research is to investigate the application of thiol-ene chemistry to polysiloxane systems in an attempt to combine synergistically the desirable features and characteristics of both siloxane and thiol-ene chemistries. Specifically, this work will address the synthesis and subsequent characterization of thiol- and allyl-functionalized siloxane oligomers as well as the networks formed through their copolymerization.
As previously discussed, polysiloxanes can be synthesized by a variety of methods and their functionalization manipulated in either the monomeric or polymeric state. Here, three types of thiol-functionalized siloxane oligomers and three types of allyl-functionalized siloxane oligomers were synthesized via the acid-catalyzed hydrolytic condensation of dialkoxy silanes (Scheme 2). The molecular structure of each oligomer was systematically regulated through the inclusion of varying degrees of nonreactive repeat units bearing benzene or octane moieties, while the mechanical properties of the networks to be formed were further manipulated through the incorporation of a urethane moiety within the allyl repeats. Thus, variation in the synthetic ratios of the silane monomers depicted in Scheme 3 allowed the effects of average crosslink density, extent of secondary interactions (i.e., hydrogen bonding and pi-pi stacking), and relative chain stiffness to be investigated in siloxane networks with regards to their thermomechanical properties, toughness, extensibility, and surface energy.
SCHEME 2.

Acid-Catalyzed Polycondensation Mechanism for Synthesizing Functionalized Siloxane Oligomers. Represented here is the formation of a thiol-functionalized oligomer containing diphenyl silanes within its backbone. Trimethyl silane was used as an end-capping agent to control molecular weight. The dialkoxy silanes were reacted for 7 days at 60 °C, and the resulting product was purified by vacuum distillation.
SCHEME 3.

Silane Monomers Used in This Study. a) SiNHC=C, b) SiMe, c) SiSH, d) SiDP, and e) SiDO.
EXPERIMENTAL
Materials
3-(mercaptopropylmethyl) dimethoxysilane (SiSH), 3-(aminopropylmethyl) diethoxysilane, diphenyl dimethoxysilane (SiDP), di-n-ocytyl dimethoxysilane (SiDO), and trimethyl methoxysilane (SiMe) were purchased from Gelest (Morrisville, PA). Allylchloroformate (97 %) and triethylamine (≥99 %) were purchased from Sigma Aldrich (St. Louis, MO). Irgacure 184 (1-hydroxy-cyclohexyl-phenyl-ketone) was purchased from Ciba Specialty Chemicals and used without further purification.
Synthesis of allyl(3-(diethoxy(methyl)silyl) propyl)carbamate (SiNHC=C, Scheme 3a)
3-(aminopropylmethyl)diethoxy silane (90.71 g, 0.47 mol) and triethylamine (75.45 g, 0.75 mol) were loaded into a sealed 1000 mL three-neck round-bottomed flask and purged with nitrogen for 1 h at 0 °C while stirring. Allylchloroformate (80 g, 0.66 mol) was added dropwise, and HCl gas evolved followed by a white precipitate. Upon complete addition of the allylchloroformate, the reaction mixture was allowed to mix for 2 h as the contents were slowly warmed to room temperature. The product was filtered and washed with an equal volume of 2M HCl followed by an equal volume of brine. The product was then dried over sodium sulfate and purified under vacuum to give 78.4 g (82%) of clear, yellow liquid. 1H NMR (500 MHz, CDCl3, δ): 5.88 (ddd, J = 5.4, 10.8, 16.2, 1H;-CH=), 5.27 (d, J = 17.1, 1H; =CH2), 5.17 (d, J = 10.4, 1H; =CH2), 4.95 (s, 1H, -NH-), 4.52 (d, J = 5.4, 2H;-CH2-), 3.76 – 3.68 (m, 4H, -CH2-), 3.14 (dt, J = 6.3, 12.5, 2H;-CH2-), 1.60 – 1.50 (m, 2H, -CH2-), 1.18 (td, J = 0.6, 7.0, 6H; CH3-), 0.61 – 0.55 (m, 2H, -CH2-), 0.13 – 0.03 (m, 3H, CH3-).
Synthesis of SiNHC=C DP DO Oligomers
Allyl (3-(diethoxy(methyl)silyl)propyl) carbamate was co-condensed with diphenyldimethoxy silane and di-n-octyldimethoxy silane in three ratios and the polymer chain lengths controlled through the addition of trimethylmethoxy silane. In the first ratio, SiNHC=C (54.4 g, 0.20 mol), SiDP (38.6 g, 0.16 mol), SiDO (12.5 g, 0.040 mol), and SiMe (8.23 g, 0.079 mol) were loaded into a round-bottomed flask with 1M HCl (15.4 g, 0.85 mol) and heated to 60 °C. After stirring for 7 days, the reaction by-products (methanol, ethanol and water) were evaporated, and 87.2 g (99 %) a clear, viscous liquid with a dark yellow color was obtained. The remaining two SiNHC=C DP DO oligomers were synthesized by an identical method but with lower starting fractions of SiDP and higher fractions of SiDO. Specifically, the second SiNHC=C DP DO oligomer was synthesized from SiNHC=C (54.4 g, 0.20 mol), SiDP (28.9 g, 0.12 mol), SiDO (25.0 g, 0.079 mol), and SiMe (8.23 g, 0.079 mol) and purified to 89.5 g (98 %) of product. The final SiNHC=C DP DO oligomer was synthesized from SiNHC=C (43.5 g, 0.16 mol), SiDP (15.4 g, 0.063 mol), SiDO (30.0 g, 0.095 mol), and SiMe (6.58 g, 0.063 mol) and purified to 73.8 g (98 %) of product. The purity of all three SiNHC=C DP DO oligomers was determined by 1H NMR (500 MHz, CDCl3) through the disappearance of peaks corresponding to ethanol, methanol, and water protons. Final spectra of the three oligomers, collected at equivalent concentrations, are presented in Figure 1.
FIGURE 1.

1H NMRs of the SiNHC=C DP DO oligomers. From spectra a) to spectra c), the SiDP content increases incrementally while the SiDO content decreases and the SiNHC=C content remains constant.
Synthesis of SiSH DP Oligomers
3-(mercaptopropylmethyl)dimethoxy silane was co-condensed in three ratios with diphenyldimethoxy silane and capped with trimethylmethoxy silane following the procedure described above. Briefly, the first oligomer was synthesized with equal molar quantities of SiSH (37.1 g, 0.21 mol) and SiDP (50.2 g, 0.21 mol) with SiMe (8.58 g, 0.082 mol) added to control polymer chain lengths. The second oligomer was synthesized with a 2X molar excess of SiSH (93.3 g, 0.52 mol) relative to SiDP (63.2 g, 0.26 mol) in the initial mixture with SiMe (16.2 g, 0.16 mol), while the synthesis of the third oligomer utilized a 3X molar excess of SiSH (105 g, 0.58 mol) to SiDP (47.4 g, 0.19 mol) with SiMe (16.2 g, 0.16 mol) again used to control polymer chain length. Purity of the SiSH DP oligomers was determined by 1H NMR (500 MHz, CDCl3) through the disappearance of peaks corresponding to methanol and water protons. The final spectra of the three SiSH DP oligomers are presented in Figure 2.
FIGURE 2.

1H NMRs of the SiSH DP oligomers. The SiDP fraction rises from spectra a) to spectra c) while the SiSH fraction lessens.
Characterization
MALDI-TOF mass spectrometry was performed on each polysiloxane (Voyager-DE STR Biospectrometry Workstation). The analysis followed a previously reported method in which individual solutions of the trans-3-indoleacrylic acid matrix (10 mg/mL), sample (2 mg/mL), and sodium trifluoroacetate (1 mg/mL) were prepared using a 7/3, v/v mixture of tetrahydrofuran/methanol.27 Equal volumes of the three solutions were hand-spotted onto the MALDI plate.
Polymerization conversion studies were performed on formulated thiol-ene mixtures in the near IR (Nicolet Magna-IR 750 series II FTIR spectrometer) using glass slides separated by a 300 μm spacer as the sample holders. Following an initial scan, each specimen was irradiated with a high-pressure mercury vapor short arc lamp (EXFO Acticure 4000) equipped with a 365 nm narrow bandpass filter. Light intensity was measured with a radiometer equipped with a GaAsP detector (International Light IL1400A, model SEL005), a wide bandpass filter (WBS320), and a quartz diffuser (model W). The final conversions of thiol and allyl functionalities were calculated as one minus the ratio of final to initial peak areas centered at 2570 cm−1 (SiSH DP, S-H stretch) and 4490 cm−1 (SiNHC=C DP DO, C=C stretch), respectively. The impact of network formation on secondary interactions was quantified by the change in hydrogen bonds from the peak centered at 3330 cm−1 (O-H stretch) in the mid IR.
Dynamic mechanical analysis (DMA) was performed in triplicate on each crosslinked thiol-ene siloxane networks in tension using a TA Instruments Q800 scanning at 1 °C/min from −40 to 40 °C at a frequency of 1 Hz and a strain of 0.1%. The crosslink density was estimated from the elastic modulus (E′) in the rubbery regime according to rubber elasticity theory.28 The glass transition temperature (Tg) was defined as the temperature corresponding to the maximum in the tan δ curve.
Toughness and extensibility of each crosslinked formulation was obtained using a mechanical tester (MTS 858 Mini Bionix II). Five dogbone-shaped specimen (ASTM D638, Type IV) of each formulation were extended at 10 mm/min until rupture. Specimen toughness (TEB) was calculated as the area under the resulting stress vs. strain curve, while Young’s modulus (E) was determined from the initial slope. The stress and strain (%) at rupture were taken to be the ultimate strength (σU) and elongation at break (%El), respectively. Samples were prepared as for testing as follows. An uncured formulation was injected between two glass slides (7” × 5”) separated by an 0.04” thick spacer, and the filled sample holder was run through a UV processor (Fusion UV, model DRS-10/12) operating at 100% light intensity and a belt speed of 9 rpm. The crosslinked, cured samples were then removed from the glass slides, and five specimen were extracted using a custom metal dye (Global Dyes, ASTM D638, Type IV).
Lastly, surface energy was quantified by measuring the static contact angle (DROPimage Advanced, v.2.0.10) of DI water atop a film of each crosslinked network with a goniometer (Ramé-Hart Instruments, Model 500 Advanced). Samples were again prepared by injecting uncured formulations between two glass slides separated by glass spacers (1mm thick). Samples were photopolymerized following the UV processor method described above. Complete conversion of each sample was confirmed by FTIR.
RESULTS AND DISCUSSION
The molecular weights (Mn and Mw) as well as the polydispersity (PDI) of the three SiNHC=C DP DO oligomers and the three SiSH DP oligomers were determined by MALDI-TOF MS (Figure 3). From this data, the average chain compositions (x:y:z) were calculated from the synthetic silane mole fractions assuming a polymer composition that reflects the relative initial monomer ratios given the consistency between the synthetic monomer ratios and the 1H NMR peak ratios of each component (Table 1). The molecular weights of the polysiloxanes typically follow a Poisson distribution and exhibit rather low polydispersities, a feature representative of their chain-length dependent nature of condensation.1 However, for molecular weights greater than 1000, the Poisson distribution approximates the Gaussian distribution, which implicates an equal probability of condensation for all monomer species. Theoretically, silanes with pendant functional groups that concentrate electron density around the central silicon atom, such as benzene, promote condensation.29 Thus, the phenyl-functional silane monomers would be expected to exhibit greater reactivity than the other silane monomers used in this study, yet the low PDI values (1.06 – 1.16 by MALDI and 1.08 – 1.25 by GPC) of the allyl- and thiol-functionalized siloxane oligomers suggest a consistency among all chain structures of a single synthetic ratio, which further validates the assumption of equal reactivity of all silane monomers in approximating polysiloxane chain compositions.
FIGURE 3.

Representative MALDI plot of a functionalized siloxane polymer distribution. The 5:5 SiSH DP oligomer is shown. The peak intensities are plotted as a fraction of the maximum peak height. Mass/charge is corrected for mass of the ionizing agent, sodium trifluoroacetate, by subtracting the molecular weight of sodium (23 g/mol).
TABLE 1.
Average molecular weights (Mn and Mw) and repeat compositions of a) the three SiNHC=C DP DO oligomers and b) the three SiSH DP oligomers. Repeat compositions (x:y:z) were calculated from the synthetic silane mole fractions under the assumption of equal monomer reactivity.
| Siloxane Structure | x:y:z | Mn (g/mol) | Mw (g/mol) | Mw/Mn |
|---|---|---|---|---|
|
|
||||
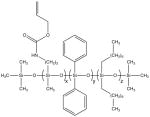
|
5:4:1 | 2130 | 2360 | 1.11 |
|
| ||||
| 5:3:2 | 2240 | 2370 | 1.06 | |
|
| ||||
| 5:2:3 | 2390 | 2570 | 1.08 | |
|
| ||||
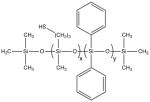
|
5:5 | 1820 | 2120 | 1.16 |
|
| ||||
| 7:3 | 1700 | 1940 | 1.14 | |
|
| ||||
| 8:3 | 1680 | 1940 | 1.15 | |
The compositions given in Table 1 afford the ability to control the material properties of the siloxane networks formed by polymerizing combinations of thiol- and allyl-functionalized oligomers. For the SiNHC=C DP DO oligomers, the ratio of allyl- (A) to phenyl- (P) to octyl-(O) functional monomer was varied from 5:4:1 to 5:2:3 A:P:O in an effort to achieve similar crosslink densities while altering the chain stiffness and degree of pi-pi stacking. Similarly, within the SiSH DP oligomers, the ratio of thiol-functional monomer to phenyl-functional monomer was increased from 1:1 to 3:1 in an attempt to alter the ultimate crosslink density, chain stiffness, and degree of pi-pi stacking. Secondary interactions offered by the urethane and phenyl functionalities (i.e., hydrogen bonding and pi-pi stacking) are known to increase the glass transition temperature and elastic modulus. Conversely, chain flexibility and high molecular weights between crosslinks (Mc), which increase with increasing octyl-functional monomer content, reduce the glass transition temperature and elastic modulus, respectively.28
Thus, networks formed from SiNHC=C DP DO oligomers with high phenyl compositions should display greater Tg and E′ values than those formed from oligomers with high dioctyl compositions when reacted with thiolated oligomers of equal degrees of functionalization. Such a trend is apparent in Figure 4a. As the diphenyl content increases from 2:3 to 3:2, and again from 3:2 to 4:1, a notable increase in Tg is detected. Furthermore, a slight increase in E′ occurs when increasing the diphenyl content from 2:3 to 3:2 in the oligomer, but no such rise is evident when the ratio is increased further to 4:1. Likely the equivalent number of allyl functional groups per chain for the three types of SiNHC=C DP DO oligomers is the dominant factor in crosslink density trends, which explains the lack of any significant rise in E′ with increasing phenyl content.
FIGURE 4.

Storage modulus versus temperature plots for networks formed under two scenarios. a) A single thiol oligomer (1:1 SiSH DP) polymerized with the three ene oligomers, and b) a single ene oligomer (5:3:2 SiNHC=C DP DO) polymerized with the three thiol oligomers. All samples were formulated with 0.5 wt% IR184 and irradiated for 10 min at an intensity of 25 mW/cm2. Testing was performed in triplicates; for clarity, not all data points are shown.
Although high thiol-functional compositions produce elevated crosslink densities that should increase Tg and E′, the extent of secondary interactions in the form of pi-pi stacking is diminished as the thiol functionality increases in the SiSH DP oligomers, limiting the evolution in their network properties. The crosslink density and modulus increase significantly as the thiol content increases from 1:1 to 2:1; however, the increasing thiol content in the 3:1 thiol-functional oligomer does not result in a significant increase in either the glass transition temperature or crosslink density, as indicated by the elastic modulus in Figure 4b. From the elastic modulus vs. temperature plots, the Tg depends most strongly on the allyl-functional siloxane while E′ is more closely impacted by the thiol-functional siloxane. The complete set of thermomechanical data collected for all thiol-ene polymer networks, in addition to their final fractional conversions, is provided in Table 2. Clearly, the thiol-ene reaction maintains its speed and highly quantitative conversions in these siloxane systems. Moreover, less than a 10% decrease in hydrogen bonding is noted by FTIR evaluation of networks upon photopolymerization.
TABLE 2.
Comprehensive results (avg±st dev) of the thermomechanical testing conducted in this study. The systems for DMA testing were formulated with 0.5 wt% of IR184 and irradiated for 10 min with 25 mW/cm2 of UV light (365 nm). Sample thickness was 300 μm. All tests were performed in triplicate.
| Ene/Thiol (x:y:z/x:y)a | %C=C Conversion | Tg (°C)b | E′ (MPa)c | XLD (mol/m3) |
|---|---|---|---|---|
| 5:4:1/5:5 | 82 ± 2 | 1.8 ± 1 | 1.4 ± 0.2 | 180 ± 30 |
| 5:4:1/7:3 | 86 ± 3 | 3.2 ± 1 | 2.8 ± 0.1 | 370 ± 20 |
| 5:4:1/8:3 | 85 ± 5 | 2.8 ± 1 | 2.3 ± 0.8 | 300 ± 100 |
| 5:3:2/5:5 | 83 ± 2 | −5.8 ± 1 | 1.3 ± 0.1 | 170 ± 20 |
| 5:3:2/7:3 | 90 ± 1 | −4.3 ± 1 | 3.1 ± 0.7 | 400 ± 100 |
| 5:3:2/8:3 | 92 ± 0 | −2.7 ± 1 | 28 ± | 360 ± 10 |
| 5:2:3/5:5 | 86 ± 0 | −13 ± 0 | 1.0 ± 0.4 | 140 ± 50 |
| 5:2:3/7:3 | 85 ± 0 | −16 ± 1 | 1.5 ± 0.2 | 200 ± 30 |
| 5:2:3/8:3 | 92 ± 1 | −14 ± 1 | 1.7 ± 0.4 | 220 ± 50 |
Structural compositions of each oligomer (allyl:phenyl:octyl/thiol:phenyl).
Taken as the max(tan δ).
Reported at T = 35 °C.
The structure-property relationship between siloxane oligomer composition and observed network behavior persists when looking at the mechanical test data presented in Table 3. For a given ene-functional oligomer, the elasticity, represented by percentage elongation at break (%El) increases markedly with decreasing crosslink density. The rise in elongation with decreasing thiol-functional monomers is a two-fold phenomenon. First, the lower crosslink density allows for greater flexibility of the network since fewer tethering points exist to restrict movement or serve as failure sites through the rupture of covalent bonds. Secondly, in the thiol-functionalized oligomers, a decrease in crosslink sites is accompanied by an increase in secondary interactions, as the concentration of benzene-functional repeats increases relative to thiol-functional repeats. Therefore, with increasing diphenyl repeats in either the thiol-functionalized oligomer or allyl-functionalized oligomer, higher elongation values are observed. The relationship between rising secondary interactions from increased diphenyl repeats in the allyl-functionalized oligomer is clearly shown in Figure 5a, while the same trend from increased diphenyl repeats in the thiolated oligomer is apparent in Figure 5b.
TABLE 3.
Comprehensive results (avg±st dev) of the MTS evaluation of photopolymerized samples conducted in this study. The systems were formulated with 0.5 wt% IR184 and irradiated by UV processing (9 rpm, 100% light intensity). Sample thickness was 1.0 mm. Mechanical testing was performed in sets of 5 per formulation, but only a single specimen was tested by FTIR to confirm complete conversion of functional groups.
| Ene/Thiol (x:y:z/x:y)a | %C=C Conversion | TEB (kJ/m3) | E (MPa) | σU (kPa) | El |
|---|---|---|---|---|---|
| 5:4:1/5:5 | 86 | 205 ± 50 | 0.4 ± 0.0 | 350 ± 50 | 84 ± 7 |
| 5:4:1/7:3 | 90 | 150 ± 10 | 1.3 ± 0.1 | 600 ± 30 | 48 ± 4 |
| 5:4:1/8:3 | 93 | 150 ± 20 | 1.8 ± 0.1 | 670 ± 60 | 41 ± 4 |
| 5:3:2/5:5 | 86 | 85 ± 15 | 0.4 ± 0.0 | 240 ± 40 | 63 ± 5 |
| 5:3:2/7:3 | 90 | 110 ± 20 | 1.5 ± 0.1 | 530 ± 40 | 40 ± 5 |
| 5:3:2/8:3 | 94 | 96 ± 10 | 1.9 ± | 590 ± 30 | 31 ± 2 |
| 5:2:3/5:5 | 87 | 55 ± 9 | 0.3 ± 0.0 | 170 ± 20 | 60 ± 4 |
| 5:2:3/7:3 | 93 | 68 ± 8 | 0.8 ± 0.0 | 320 ± 20 | 42 ± 3 |
| 5:2:3/8:3 | 95 | 50 ± 10 | 1.1 ± 0.1 | 310 ± 50 | 29 ± 3 |
Structural compositions of each oligomer (allyl:phenyl:octyl/thiol:phenyl).
FIGURE 5.

Stress versus strain plots for networks formed under two scenarios. a) A single thiol oligomer (1:1 SiSH DP) polymerized with the three ene oligomers, and b) a single ene oligomer (5:3:2 SiNHC=C DP DO) polymerized with the three thiol oligomers. All samples were formulated with 0.5 wt% IR184 and photopolymerized via single pass UV processing (9 rpm, 100% light intensity). For clarity, not all data points are shown.
In contrast to elasticity, ultimate strength shows a consistent rise with increasing crosslink density. A similar argument can be used, however, to account for this trend. With increasing crosslink density comes a higher concentration of covalent bonds that must absorb energy, rupture, and ultimately allow network failure. Figure 5b best illustrates this phenomenon. As the number of thiol-functional repeats increases, so too does ultimate strength. The difference in strength between networks formulated with the 1:1 SiSH DP oligomer and the 3:1 SiSH DP oligomer is drastic. However, little disparity is noted between the 3:1 SiSH DP and the 2:1 SiSH DP network behavior given the slight alteration in their average degrees of thiol functionalization (8 thiols per chain vs. 7 thiols per chain, respectively). Since secondary contribute significantly to the overall viscoelastic behavior of the polymer, increasing the concentration of functionalities that can hydrogen bond or pi-pi stack generally increases ultimate strength. While an increase is certainly observed, the impact of secondary interactions on ultimate strength is less significant than that of covalently formed crosslinks. As seen in Figure 5a, increasing the concentration of phenyl-functional monomers in the SiNHC=C DP DO oligomer has only a limited effect.
Although clear correlations exist between siloxane structural compositions, elasticity, and ultimate strength, no such trends are obvious with respect to network toughness (TEB). Toughness is a measurement of the amount of energy a material can absorb on a volumetric basis prior to failure. Thus, increased toughness results when a balance is struck between the crosslink density and the degree of secondary interactions. In this study, that balance is best met with the 5:4:1 SiNHC=C DP DO oligomer being polymerized with the 1:1 SiSH DP oligomer. The low density of thiol repeats in the SiSH DP oligomer provides a low crosslink density that limits the brittleness and enhances the elongation at break. Furthermore, the high concentration of diphenyl repeats in both the SiNHC=C DP DO and SiSH DP oligomers give the network elevated ultimate strength and elongation with respect to other networks of equal crosslink density.
Conversely, the lowest observed toughness value was in the formulation with the exact opposite structural characteristics to the 5:4:1 SiNHC=C DP DO/1:1 SiSH DP network. The 5:2:3 SiNHC=C DP DO with the 3:1 SiSH DP contained the lowest concentration of diphenyl repeats for pi-pi stacking with the greatest number of thiol functionalities for establishing crosslinks. Although this network had the highest ultimate strength of networks made with the 5:2:3 SiNHC=C DP DO oligomer, it also had the lowest elongation. Additionally, the 5:2:3 SiNHC=C DP DO/3:1 SiSH DP network had the lowest strength of all the networks containing 3:1 SiSH DP. The combination of low strength with restricted elongation created a brittle material with low toughness.
In the final component of this study, the surface energies of the crosslinked thiol-ene networks were determined through static contact angle measurements. The results of the goniometer experiments are shown in Figure 6. The structural content of each oligomer again impacts the observed material properties; as the fraction of hydrophobic octyl-functional monomers increases, so does the contact angle. This trend holds regardless of the composition of the thiol oligomer. However, the overall concentration of hydrophobic or hydrophilic substituents within a thiol-ene formulation does not rise linearly with the degree of thiol-functionalization. Consequently, no regular trend exists to correlate the degree of thiol functionalization with contact angle for a given ene-functionalized oligomer.
FIGURE 6.

Contact angle measurements of the nine fully formulated networks (0.5 wt% IR184). Crosslinked networks were formed by irradiating samples via single pass UV processing (9 rpm, 100% light intensity).
CONCLUSIONS
The synthesis and characterization of thiol- and ene-functionalized siloxanes with systematically varied compositions as well as polymers formed from varying oligomer structures have been successfully carried out. The synthesis of polysiloxanes by condensation yields oligomers with low polydispersity and a near Poisson distribution. Furthermore, the material properties of polysiloxanes have been readily manipulated through the choice of pendant functionalities, as evidenced by thermomechanical, MTS, and surface energy measurements. Networks formed from oligomers with a high degree of reactive functionalities (i.e., thiols), tended to exhibit higher glass transition temperatures, rubbery moduli, ultimate strength, and Young’s moduli, but they also displayed lower elongation at break. The concentration of pendant groups capable of secondary interactions (i.e., pi-pi stacking or hydrogen bonding), tended to directly impact rubbery modulus, ultimate strength, and elongation at break, although not to the same extent as degree of thiol functionalization. Secondary interactions did, however, play a greater role in glass transition temperature than did crosslink density. Lastly, the concentration of octyl-functionalized repeats within the SiNHC=C DP DO oligomer significantly influenced the surface energy of crosslinked networks. Material hydrophobicity increased directly with the number of octyl-functional repeats at every crosslink density.
Acknowledgments
We acknowledge funding from the National Institute of Health, NIH grant DE018233 and from the Septodont Corporation.
References
- 1.Jones RG, Ando W, Chojnowski J. In Silicon-Containing Polymers - The Science and Technology of Their Synthesis and Applications. Chapter 1. Springer - Verlag; Dordrecht, The Netherlands: 2000. pp. 3–35. [Google Scholar]
- 2.Zheng PW, McCarthy TJ. Langmuir. 2011;26:18585–18590. doi: 10.1021/la104065e. [DOI] [PubMed] [Google Scholar]
- 3.Krumpfer JW, McCarthy TJ. Langmuir. 2011;27:11514–11519. doi: 10.1021/la202583w. [DOI] [PubMed] [Google Scholar]
- 4.Sun F, Liao B, Zhang L, Du HG, Huang YD. J Appl Polym Sci. 2011;120:3604–3612. [Google Scholar]
- 5.Xu L, Dai CH, Chen L, Xie HD. J Polym Eng. 2011;31:369–374. [Google Scholar]
- 6.Swinburne ML, Willmot D, Patrick D. Eur J Orthodont. 2011;33:407–412. doi: 10.1093/ejo/cjq091. [DOI] [PubMed] [Google Scholar]
- 7.Mojsiewicz-Pienkowska K, Jamrogiewicz M, Zebrowska M, Sznitowska M, Centkowska K. J Pharm and Biomed Anal. 2011;56:131–138. doi: 10.1016/j.jpba.2011.04.011. [DOI] [PubMed] [Google Scholar]
- 8.Hosseinzadeh F, Galehassadi M, Mahkam M. J Appl Polym Sci. 2011;122:2368–2373. [Google Scholar]
- 9.Frisch EE. Sampe Journal. 1985;21:23–28. [Google Scholar]
- 10.Czerwiński W, Ostrowska-Gumkowska B, Kozakiewicz J, Kujawski W, Warszawski A. Desalination. 2004;163:207–214. [Google Scholar]
- 11.Zhang X, Lin G, Kumar SR, Mark JE. Polymer. 2009;50:5414–5421. [Google Scholar]
- 12.Scibiorek M, Gladkova NK, Chojnowski J. Polymer Bulletin. 2000;44:377–384. [Google Scholar]
- 13.Combe EC, BF, Douglas WH. Dental Biomaterials. Chapter 30. Kluwer Academic; Boston: 1999. pp. 281–303. [Google Scholar]
- 14.Hao XJ, Jeffery JL, Wilkie JS, Meijs GF, Clayton AB, Watling JD, Ho A, Fernandez V, Acosta C, Yamamoto H, Aly MGM, Parel JM, Hughes TC. Biomaterials. 2010;31:8153–8163. doi: 10.1016/j.biomaterials.2010.07.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Loth H, Foltin U. Journal of Controlled Release. 1998;54:273–282. doi: 10.1016/s0168-3659(97)00218-6. [DOI] [PubMed] [Google Scholar]
- 16.Kloosterboer JG. Adv Polym Sci. 1988;84:1–61. [Google Scholar]
- 17.Zwiers RJM, Dortant GCM. Appl Optics. 1985;24:4483–4488. doi: 10.1364/ao.24.004483. [DOI] [PubMed] [Google Scholar]
- 18.Cramer NB, Reddy SK, O’Brien AK, Bowman CN. Macromolecules. 2003;36:7964–7969. [Google Scholar]
- 19.Kloxin CJ, Scott TF, Bowman CN. Macromolecules. 2009;42:2551–2556. doi: 10.1021/ma802771b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reddy SK, Cramer NB, Bowman CN. Macromolecules. 2006;39:3673–3680. [Google Scholar]
- 21.Cramer NB, Bowman CN. J Polym Sci Part A: Polym Chem. 2001;39:3311–3319. [Google Scholar]
- 22.Hoyle CE, Lee TY, Roper T. J Polym Sci Part A: Polym Chem. 2004;42:5301–5338. [Google Scholar]
- 23.Hoyle CE, Bowman CN. Angew Chem-Int Edit. 2010;49:1540–1573. doi: 10.1002/anie.200903924. [DOI] [PubMed] [Google Scholar]
- 24.Iha RK, Wooley KL, Nystrom AM, Burke DJ, Kade MJ, Hawker CJ. Chem Rev. 2009;109:5620–5686. doi: 10.1021/cr900138t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Herczynska L, Lestel L, Boileau S, Chojnowski J, Polowinski S. Eur Polym J. 1999;35:1115–1122. [Google Scholar]
- 26.Schreck KM, Leung D, Bowman CN. Macromolecules. 2011;44:7520–7529. doi: 10.1021/ma201695x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.He JY, Nebioglu A, Zong ZG, Soucek MD, Wollyung KM, Wesdemiotis C. Macromol Chem Phys. 2005;206:732–743. [Google Scholar]
- 28.Flory PJ. Principles of Polymer Chemistry. Chapter 60. Cornell University Press; Ithaca, NY: 1953. pp. 432–492. [Google Scholar]
- 29.Hyde JF, Brown PL, Smith AL. J Am Chem Soc. 1960;82:5854–5858. [Google Scholar]