

Abstract
Integrin αIIbβ3 has emerged as an important therapeutic target for thrombotic vascular diseases owing to its pivotal role in mediating platelet aggregation through interaction with adhesive ligands. In the search for effective anti-thrombotic agents that can be administered orally without inducing the high-affinity ligand binding state, we recently discovered via high-throughput screening of 33,264 compounds a novel, αIIbβ3-selective inhibitor (RUC-1) of adenosine-5′-diphosphate (ADP) -induced platelet aggregation that exhibits a different chemical scaffold and mode of binding with respect to classical Arg-Gly-Asp (RGD)-mimicking αIIbβ3 antagonists. Most importantly, RUC-1 and its higher-affinity derivative, RUC-2, do not induce major conformational changes in the protein β3 subunit or prime the receptor to bind ligand. To identify additional αIIbβ3-selective chemotypes that inhibit platelet aggregation through similar mechanisms, we screened in silico over 2.5 million commercially available, ‘lead-like’ small molecules based on complementarity to the predicted binding mode of RUC-2 into the RUC-1-αIIbβ3 crystal structure. This first reported structure-based virtual screening application to the αIIbβ3 integrin led to the identification of 2 αIIbβ3-selective antagonists out of 4 tested, which compares favorably with the 0.003 % “hit rate” of our previous high-throughput chemical screening study. The newly identified compounds, like RUC-1 and RUC-2, showed specificity for αIIbβ3 compared to αVβ3 and did not prime the receptor to bind ligand. They thus may hold promise as αIIbβ3 antagonist therapeutic scaffolds.
Keywords: Integrins, Virtual screening, Anti-thrombotic agents, Platelet aggregation, Small molecules
Introduction
Heart attacks and strokes caused by platelet-mediated thrombosis are leading causes of death or serious, long-term disabilities worldwide [1]. Although the incidence of these conditions in developed countries is declining, it is increasing in developing ones [2, 3]. Integrin αIIbβ3 is the most abundant adhesion/aggregation receptor on the surface of platelets and mediates platelet aggregation by binding the adhesive plasma proteins fibrinogen and von Willebrand factor [4]. These interactions are responsible for the arrest of bleeding at sites of vascular injury. However, in diseased states, they may result in thrombus formation, and consequent heart attacks and strokes, providing a rationale for therapeutic blockade of integrin αIIbβ3.
Recent αIIbβ3 crystal structures have provided detailed information about the primary binding site of the endogenous fibrinogen ligand recognized by its receptor during platelet aggregation [5]. In these structures, the fibrinogen C-terminal γ-chain peptide sequence GAKQAGDV binds to the αIIbβ3 binding pocket lying between the αIIb and β3 subunits, in part, by interacting with the αIIb subunit through a basic moiety and with the β3 subunit through a negatively charged aspartate that coordinates the Mg2+ ion in the metal ion-dependent adhesion site (MIDAS). Similar interactions account, in part, for the ability of small peptides containing the Arg-Gly-Asp (RGD) cell adhesion sequence to inhibit fibrinogen binding to αIIbβ3 [6].
Several potent αIIbβ3 antagonists have been developed as anti-platelet agents, of which three intravenously administered drugs are currently licensed for human use [7]. These are: (1) the Fab fragment of a chimeric, humanized version of the monoclonal mouse antibody 7E3 (abciximab), (2) a conformationally-constrained cyclic heptapeptide containing an αIIbβ3-selective Lys-Gly-Asp (KGD) motif (eptifibatide), and (3) a non-peptide, small-molecule RGD-mimicking compound (tirofiban). Recent crystal structures of αIIbβ3 bound to eptifibatide, tirofiban, and another RGD-mimetic, L-739758, confirmed binding of these agents to the αIIb subunit through their basic moieties and to the β3 MIDAS Mg2+ ion through their carboxyl groups, suggesting that their mechanisms of action are similar [5].
A series of orally-active αIIbβ3 antagonists were developed patterned after the RGD sequence, but none of them received regulatory approval because they failed to demonstrate efficacy as antithrombotic agents and some of them were paradoxically associated with a 30–35 % increased risk of death [8, 9]. In addition, they produced thrombocytopenia in a small percentage of patients [10]. It has been hypothesized that the increased risk of thrombosis with these compounds is due to their inducing the αIIbβ3 receptor to adopt a high-affinity ligand state such that, when their plasma levels declined, the receptor was able to spontaneously bind endogenous ligands, resulting in platelet aggregation [8, 9]. Similarly, it has been postulated that the conformation(s) induced by these agents expose regions of the receptor to which some patients have preformed antibodies, resulting in antibody coating their platelets, leading to thrombocytopenia [10].
We recently discovered a novel inhibitor of αIIbβ3-mediated platelet aggregation (RUC-1; IC50 = 13 ± 5 µM) via high-throughput screening of a chemical library of 33,264 small molecules [11]. RUC-1 is specific for αIIbβ3 relative to the closely related integrin αVβ3 and, unlike the RGD-based antagonists, it does not induce a major conformational change in the β3 subunit as demonstrated by the binding of monoclonal antibodies selective for ligand-induced binding sites, gel filtration, and dynamic light scattering [11, 12]. Moreover, unlike eptifibatide and an RGDS peptide, preincubation of αIIbβ3 with RUC-1 did not “prime” the receptor to bind its ligand fibrinogen [11, 12]. Structural studies demonstrated that RUC-1 binds exclusively to the αIIb subunit through direct interactions with residues D224 and Y190 as well as water-mediated interactions with residue D232 [12]. A more potent derivative of RUC-1, RUC-2 (IC50 = 0.096 ± 0.011 µM) [13, 14], was obtained via structure-based chemical optimization. The recently solved RUC2-αIIbβ3 crystal structure [13, 14] confirmed that RUC-2 binds to the αIIb subunit much like RUC-1, but in addition it exhibits a novel mode of binding to the β3 subunit involving a charge-charge interaction with the side chain of the β3 E220 residue usually involved in the coordination of the MIDAS Mg2+ ion, thus displacing the latter from its site. RUC-2 is also specific for αIIbβ3 compared to αVβ3 and does not prime the receptor to bind fibrinogen [14]. Notably, RUC-1 and RUC-2 exhibit unique binding modes to αIIbβ3 which differ from that of the RGD-like molecules, with potential advantages in that they induce less conformational changes in the αIIbβ3 β3 subunit [12].
Here, we describe the results of a structure-based in silico screening study aimed at identifying inhibitors of αIIbβ3-mediated platelet aggregation by emulating the unique mode of binding of RUC-2 to αIIbβ3, with the goal of identifying alternative, possibly improved, therapeutic scaffolds. To the best of our knowledge, this is the first structure-based virtual screening study involving integrin αIIbβ3 reported in the literature. Specifically, we screened a large, putatively unbiased library of over 2.5 million commercially available, ‘lead-like’ small-molecules based on complementarity to the predicted mode of binding of RUC-2 into the crystal structure of the RUC-1-αIIbβ3 complex [12]. The study led to the identification of small-molecule chemotypes specific for αIIbβ3 that inhibit ADP-induced platelet aggregation, but do not induce a high-affinity ligand binding conformation in the receptor. When the RUC-2/αIIbβ3 complex crystal structure [14] became available after these studies began, the virtual screening was repeated using this structure, but no additional hits were discovered.
Methods
Ligand set
Compounds of the ‘lead-like’ subset of the ZINC database [15] were used for the molecular docking study. At the time this screen was performed (March 22th, 2011) this subset contained over 2.5 million commercially available small-molecules derived from a large, potentially unbiased library of commercially available compounds selected using the following criteria: calculated log P between 2.5 and 3.5, molecular weight between 250 and 350, and number of rotatable bonds between 5 and 7. For each molecule, the following information was accessible through the ZINC database: (1) up to 1,000 conformations pre-generated using the program OMEGA [16]; (2) van der Waals parameters derived from the all-atom AMBER force-field [17]; and (3) partial atomic charges and transfer free-energies calculated using AMSOL [18, 19].
Molecular docking screen
Docking calculations were performed with DOCK3.5.54 [20–22] using the crystal structure of the closed headpiece of integrin αIIbβ3 in complex with RUC-1 (PDB ID: 3NIF [12]) or RUC-2 (PDB ID: 3T3M [13, 14]). All non-protein atoms, except for the adjacent to MIDAS (ADMIDAS) and the synergistic metal binding site (SyMBS) metal ions, were removed from the PDB, and the most probable protonation state at pH 7.4 was assigned to the ionizable protein residues. Forty-five matching spheres, labeled for chemical matching based on the local protein environment, were used to indicate putative RUC-2 atom positions within the αIIbβ3 binding pocket. Ligand sampling was obtained using a bin size of 0.2 Å, a bin size overlap of 0.1 Å, and a distance tolerance of 1.2 Å for both the binding site matching spheres and the docked molecule (default parameters). The time of calculation was ~1.6 h using 312 2.9 GHz cores for each virtual screening experiment. An energy-based score corresponding to the sum of the receptor-ligand electrostatic and van der Waals interaction energies, corrected for ligand desolvation, was used to evaluate the docking of each molecule of the ligand set into the αIIbβ3 binding pocket. Specifically, the electrostatic energies in the binding site were calculated using the program Delphi [23] and partial charges from the united atom AMBER force-field [24] for all protein atoms and ions. A van der Waals grid based on the same force-field was generated with the program CHEMGRID. The ligand desolvation energy was estimated based on transfer free-energy between solvents of dieletrics 78 and 2, taking into account the extent to which the ligand is buried in the receptor binding site [25, 26]. The best energy-scored conformations of the ligand set received 100 steps of rigid-body energy minimization. A small fraction (0.6 %) of the 500 top-scoring docking compounds was discarded from the virtual screening against the predicted binding mode of RUC-2 into the RUC-1-αIIbβ3 crystal structure because these molecules appeared to have missing or disconnected atoms. No broken molecules were found among the 500 top-scoring docking compounds derived from the virtual screening against the RUC-2-αIIbβ3 crystal structure.
Compound sourcing, characterization, and solubilization
Five compounds, MSSM-1–5, were selected from the 500 top-scoring docking compounds based on the criteria specified in the “Results and discussion” session. Compound MSSM-1 was purchased from Enzo Life Science (http://www.enzolifesciences.com/), MSSM-5 was purchased from Toronto Research Chemical (http://www.trc-canada.com/), and compounds MSSM-2–4 were obtained from the National Cancer Institute of the National Institutes of Health. Compounds MSSM-1 and MSSM-3 were soluble in DMSO and MSSM-2 and MSSM-5 were soluble in water, but compound MSSM-4 was not soluble in DMSO, water, or Tris/HCl-saline buffer and so was not studied further. Compound MSSM-1 and MSSM-5 were 98 and 97 % pure according to their manufacturers, as established by HPLC and C18-reverse phase thin layer chromatography, respectively. The HPLC purification utilized a 0.1 % aqueous-TFA/acetonitrile solvent system, running a 30 min linear gradient of 0–20 % acetonitrile. The MSSM-2 and MSSM-3 were purified by HPLC on a C18 reverse phase column (Gemini) using a trifluoroacetic acid/acetonitrile gradient. Their final purities were 97 and 94 %, respectively, as judged by analytical LC/MS (Waters Acquity UPLC coupled to a Thermo LTQ mass spectrometer). Mass spectroscopic analysis of the active compounds MSSM-1 and MSSM-2 yielded the expected masses (Figure S1) and 1H NMR analysis of compounds MSSM-1,2,3, and 5 yielded the expected spectra (Figure S2).
Similarity calculations
Similarities between the 4 tested ligands and the 2,691 annotated αIIbβ3 integrin ligands in the ChEMBL database (http://www.ebi.ac.uk/chembl) were quantified by calculating their Tanimoto similarity coefficients to the nearest neighbors, using extended connectivity fingerprint maximum distances 4 (ECFP4) generated with the open source Java library for chemical fingerprints called jCompoundMapper [27]. The Tanimoto similarity coefficients were calculated using an in-house script in R language (see Supporting Information). For the generation of the fingerprints, all molecules were converted from a PDB file format to their corresponding SDFile using the program Corina [28]. To calculate Tanimoto similarity coefficients for RUC-1 and RUC-2, these compounds were removed together with their 18 congeners from the list of 2,691 annotated αIIbβ3 ligands contained in the ChEMBL database. The same methodology was used to run similarity calculations between the RUC-1/RUC-2 compounds and the lead-like subset of the ZINC database updated on February 6th, 2012 (4,554,059 entries).
Platelet function assays
RUC-1, RUC-2, 7E3, tirofiban, and eptifibatide were obtained as previously described [12, 14]. The following assays were all carried out as previously described [11, 12, 14]: platelet adhesion to fibrinogen; adhesion of HEK293 cells expressing αVβ3 to vitronectin; and platelet aggregation to ADP (5 µM) using citrated platelet-rich plasma. The vehicles used to solubilize the compounds (saline or DMSO at 0.3 % final concentration) did not affect the central values of these assays.
Priming assay
To assess the ability of compounds identified to induce the high affinity, ligand binding state of the αIIbβ3 receptor we employed a modified version of the assay developed by Du et al. [29]. Washed platelets in HEPES-modified Tyrode’s buffer were incubated with the compounds for 20 min at room temperature (RT), fixed with 1 % paraformaldehyde for 40 min at RT, incubated with 5 mM glycine for 5 min at RT, washed X4, resuspended in buffer containing 2 mM Ca2+ and 1 mM Mg2+, incubated with Alexa 488-conjugated fibrinogen (200 µg/ml; Invitrogen) (with or without 10 µM eptifibatide) for 30 min at 37 °C, washed, diluted ten-fold, and analyzed by flow cytometry. The net fluorescence was calculated by determining the percentage of platelets with fluorescence values greater than 25 arbitrary units and subtracting the percentage in the untreated samples. In the 3 separate experiments, the mean ± SD values in the untreated samples were 4 ± 3 %.
Results and discussion
The mode of binding of αIIbβ3 co-crystallized small-molecule antagonists
Figure 1 shows the chemical structures of select αIIbβ3 non-peptide, small-molecule antagonists that have been studied in complex with αIIbβ3 by X-ray crystallography. They are the RGD-mimicking antagonists (S)-2-(butylsulfonamino)-3-(4-[4-(piperidin-4-yl)butoxy]phenyl) propanoic acid (tirofiban) and 3-{[5-(2-Piperidin-4-yl-ethyl)-thieno [2,3-b]thiophene-2-carbonyl]-amino}-2-(pyridine-3-sulfonylamino)-propionic acid (L-739758), as well as the newly discovered 2-ethyl-5-(piperazin-1-yl)-7H-[1,3,4]thiadiazolo[3,2-a] pyrimidin-7-one (RUC-1) and 2-amino-N-(3-(5-oxo-7-(piperazin-1-yl)-5H-[1,3,4]thiadiazolo[3,2-a]pyrimidin-2-yl)phenyl) acetamide (RUC-2). Figure 2 illustrates the differences and similarities between the modes of binding to αIIbβ3 of tirofiban (PDB ID: 2VDM [5]), L-739758 (PDB ID: 2VC2 [5]), RUC-1 (PDB ID: 3NIF [12]), and RUC-2 (PDB ID: 3T3M [13, 14]). Specifically, both tirofiban and L-739758 exhibit a binding mode similar to RGD peptides (Fig. 2A, B, respectively), with the ligand lying between the αIIb and β3 subunits, and mostly stabilized through strong, polar interactions with the D224 residue of the αIIb subunit, the β3 MIDAS Mg2+ ion, and the R214 residue of the β3 subunit. Although RUC-1 (Fig. 2C) and RUC-2 (Fig. 2D) also form an interaction between their basic moieties and the D224 residue of the αIIb subunit (in addition to a π-π stacking interaction with αIIb Y190, and water-mediated interactions with αIIb D232), they do not participate in the coordination of the MIDAS Mg2+ ion within the β3 subunit. While RUC-1 binds exclusively to the αIIb subunit, the crystal structure of the RUC-2-αIIbβ3 complex [13, 14] confirms similar interactions with the αIIb subunit, but reveals a charge-charge interaction of the β3 E220 residue with RUC-2 instead of the MIDAS Mg2+ ion, which was missing from the site.
Fig. 1.

Chemical structures of known antagonists co-crystallized with αIIbβ3 integrin
Fig. 2.

Crystallographic binding modes of antagonists tirofiban, L-739758, RUC-1, and RUC-2 (A, B, C and D, respectively). The αIIb and β3 subunits are shown as light blue and white cartoons, respectively. Side chains of protein residues interacting with the ligands are shown as sticks. ADMIDAS and SyMBS ions are shown as white spheres while the MIDAS ion is shown as a grey sphere. Ligands are shown as orange sticks, while black dot lines indicate hydrogen bonds
Virtual screen and compound selection
A workflow of the structure-based virtual screening approach is shown in Fig. 3. We initially screened over 2.5 million commercially available, “lead-like” compounds from the ZINC database [15] based on complementarity with the predicted binding mode of the newly identified RUC-2 compound into the RUC-1-αIIbβ3 crystal structure [12]. Subsequently, when the crystal structure of the RUC-2-αIIbβ3 complex (PDB ID: 3T3M [14]) became available, we performed an additional screen using this structure. The protein was kept rigid while each “lead-like” compound was docked into the binding pocket in an average of 425 orientations relative to the receptor, and an average of 2,500 conformations for each orientation. A score was assigned to each molecule and configuration within the binding pocket based on van der Waals and electrostatic complementarity with the receptor, corrected for ligand desolvation. In the initial screen, the 500 top-scoring docking hits (Table S1 of Supporting Information; 0.02 % of the docked library) were visually inspected. Ligands for experimental testing were selected based on chemotype diversity, chemico-physical properties, and other features that the molecular docking screen does not take into account. Molecules were singled out based on the following criteria: (1) The presence of hydrogen bond interactions between the ligand and both the αIIb D224 and the β3 E220 residues, or as an alternative, the αIIb D232 and β3 E220 residues; (2) chemotype diversity; and (3) purchasability. In the initial screen using the RUC-1 crystal structure, thirteen small molecules were extracted from the set of 500 best-scored compounds (see Table S1) based on the first criterion. These molecules corresponded to the DOCK scoring ranks 6 (MSSM-1 in Table 1), 45 (MSSM-2 in Table 1), 122, 141 (MSSM-3 in Table 1), 163, 238, 299, 336, 360, 385, 393 (MSSM-4 in Table 1), 400 (MSSM-5 in Table 1), and 433. Two of these (corresponding to DOCK scoring ranks 163 and 360) were eliminated because of chemical similarity to MSSM-3 and MSSM-4, respectively. Of the remaining 11 molecules, those corresponding to DOCK scoring ranks 122, 238, 299, 336, 385, and 433 were either no longer commercially available (compounds 122 and 336) or unacceptably expensive (compounds 238, 299, 385, and 433). We purchased the 5 remaining compounds (MSSM-1–5), but one (MSSM-4) could not be tested in functional assays because it was insoluble in water, saline, and DMSO. Thus, we ultimately tested the function of 4 compounds (MSSM-1–3 and 5).
Fig. 3.

A workflow of the structure-based virtual screening approach applied to a model of the RUC-2/αIIbβ3 complex
Table 1.
Chemical structures of the 4 tested compounds alongside their DOCK scoring rank from the virtual screening experiment carried out using the predicted mode of binding of RUC-2 to the RUC-1-αIIbβ3 crystal structure, their percentage inhibition of αIIbβ3-mediated platelet adhesion to fibrinogen, and when available, their IC50 value for inhibition of platelet aggregation
| Cpd (Rank) |
Structure | % Inhibition of adhesion at 100 µM |
IC50 (µM) | ECFP4- based Tc |
Most similar known αIIbβ3 ligand |
|---|---|---|---|---|---|
| RUC-1 | 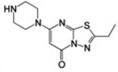 |
92±8 | 11.5 ± 1 | 0.16 | 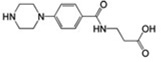 |
| RUC-2 | 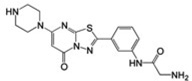 |
102±5 | 0.15 ± 0.01 | 0.2 | 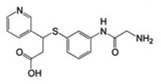 |
| MSSM-1 (6) | 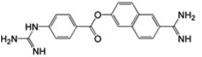 |
61±6 | 12.5 ± 1.19 | 0.21 | 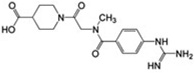 |
| MSSM-2 (45) | 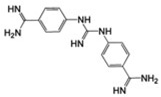 |
36±9 | 47.7 ± 4.7 | 0.32 | 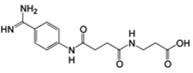 |
| MSSM-3 (141) | 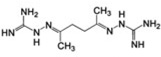 |
10±3 | – | 0.15 | 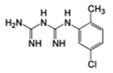 |
| MSSM-4 (393) | 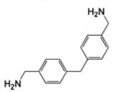 |
ND | – | 0.15 | 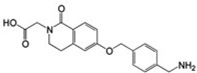 |
| MSSM-5 (400) | 0±0 | – | 0.15 |
Also listed is the extended connectivity fingerprint maximum distance 4 (ECFP4)-based Tanimoto similarity coefficients (Tc) to the most similar known αIIbβ3 integrin ligand in the ChEMBL database
In the subsequent screen using the RUC-2 crystal structure [14], approximately half of the resulting 500 top-scoring docking hits were identical to those derived from the initial virtual screen, although not necessarily with a corresponding scoring rank. These included 12 (highlighted in blue color in Table S2) of the 13 molecules we had selected based on specific hydrogen bonding interactions with both αIIb D224 (or D232) and β3 E220. Of the remaining half top-scoring docking hits, 10 additional molecules were selected based on the aforementioned interactions (highlighted in pink color in Table S2). These molecules corresponded to the DOCK scoring ranks 156, 161, 179, 202, 210, 320, 350, 364, 424 and 450. Among them, those corresponding to DOCK scoring ranks 161, 179, 210, 320, 350, 424 and 450 were no longer commercially available, while 156, 202 and 364 were unacceptably expensive, leaving us with no additional molecules to test from the virtual screening experiment using the RUC-2-αIIbβ3 crystal structure.
Inhibitory activity, binding specificity, and “priming” effect
As shown in Fig. 4 and Table 1, two compounds (MSSM-1 and MSSM-2) were active in the platelet adhesion assay. In 3 separate experiments, conducted at a concentration of 100 µM, MSSM-1 produced 61 ± 6 % (mean ± SD) and MSSM-2 produced 36 ± 9 % inhibition of platelet adhesion to fibrinogen, respectively, whereas MSSM-3 and MSSM-5 produced 10 ± 3 % and no inhibition, respectively. RUC-1 and RUC-2 were more potent inhibitors of platelet adhesion at 100 µM, and at 10 µM, RUC-2 still produced 99 ± 9 % inhibition, whereas RUC-1, MSSM-1 and MSSM-2 produced much less or no inhibition. For further comparison, tirofiban at 10 µM produced 100 ± 6 % inhibition.
Fig. 4.

Effect of RUC-1, RUC-2, MSSM-1 and MSSM-2 on adhesion of platelets to immobilized fibrinogen at different concentrations. Washed platelets in buffer containing 1 mM Ca2+ and 1 mM Mg2+ were allowed to adhere to microtiter wells precoated with purified fibrinogen (50 µg/ml) in the absence or presence of the indicated compounds. After 1 h at RT the wells were washed and the adherent platelets detected by lysing them with 0.1 % Triton X-100 and assaying the released acid phosphatase activity
Since MSSM-1 and MSSM-2 showed the most activity in the adhesion assay, they were further tested for their ability to inhibit the initial wave of ADP-induced platelet aggregation of citrated platelet-rich plasma (Fig. 5, Table 1). Their IC50 values were 12.5 ± 1.19 µM and 47.7 ± 4.7 µM (n = 3), respectively. By comparison, the IC50 values (n = 3) for RUC-1 and RUC-2 were 11.5 ± 1 µM and 0.15 ± 0.01 µM, respectively, when tested against the same platelet-rich plasma samples. It must be noted, however, that unlike RUC-2, RUC-1 and MSSM-1–2 derive directly from screenings and have not been further optimized. The IC50 values for mAb 7E3 determined in two of the above experiments were both 0.10 ± 0.01 µM.
Fig. 5.

Inhibition of ADP-induced platelet aggregation. Compounds were incubated at the indicated concentrations with citrated platelet-rich plasma for 15 min at 37 °C and then ADP (5 µM) was added and aggregation monitored by the change in light transmission. The initial slopes of platelet aggregation were measured and the IC50s determined by comparison with the untreated sample. Data shown are mean ± SD (n = 3), except for mAb 7E3, which is shown as the means of 2 separate experiments
The specificity of MSSM-1 and MSSM-2 for αIIbβ3 was assessed by analyzing their ability to inhibit the interaction of the closely related integrin receptor, αVβ3, with its ligand, vitronectin (Fig. 6). The αVβ3-specific mAb LM609 inhibited adhesion of HEK293 cells expressing αVβ3 to vitronectin by 101 ± 2 % (n = 4) at 80 µg/ml and the anti-αVβ3 + αIIbβ3 mAb 7E3 inhibited adhesion by 97 ± 6 % (n = 4), whereas mAb 10E5 inhibited adhesion by 3 ± 6 % (n = 4; Fig. 6). In sharp contrast, MSSM-1 at 300 µM produced only 8 ± 9 % (n = 4) inhibition and MSSM-2 at the same concentration produced only 11 ± 14 % inhibition (n = 4). For comparison, RUC-1 at 100 µM produced 8 ± 11 % (n = 4) inhibition and RUC-2 at 10 µM produced 7 ± 8 % inhibition (n = 4). Thus, like both RUC-1 and RUC-2, MSSM-1 and MSSM-2 showed specificity for αIIbβ3 compared with αVβ3.
Fig. 6.

Effect of EDTA, mAbs 7E3, 10E5, LM609, RUC-1, RUC-2, MSSM-1, and MSSM-2 on adhesion of HEK293 cells expressing αVβ3 to immobilized vitronectin. HEK293 cells expressing αVβ3 were added in buffer containing 1 mM Mg2+ to microtiter wells precoated with vitronectin (5 µg/ml). After 1 h at RT, the wells were washed and the adherent cells detected by lysing the cells with Triton X-100 and measuring released acid phosphatase activity
We also tested the “priming” effect of MSSM-1 and MSSM-2, that is, their ability to induce αIIbβ3 to adopt a high-affinity ligand binding conformation as judged by their inducing platelet αIIbβ3 to bind fibrinogen (Fig. 7). Incubation of washed platelets with tirofiban (0.5 µM), an RGDS peptide (100 µM), or eptifibatide (1 µM) followed by fixation in paraformaldehyde and washing, increased fluorescent fibrinogen binding to platelets as judged by the percentage of platelets with fluorescence values above those in the absence of the agents. The increases were 31.8 ± 14.1, 34.6 ± 15 %, and 31 ± 18.1 respectively (n = 5 for all). The specificity of the binding was established by the ability of eptifibatide or EDTA to block the binding. In sharp contrast neither MSSM-1 nor MSSM-2 at 300 µM produced an increase in the percentage of platelets binding fibrinogen (0 ± 0.4 and 0 ± 0 %, respectively; n = 5). For comparison, RUC-1 (100 µM) produced a 14.4 ± 6.7 % (n = 5) increase and RUC-2 (1 µM) produced a 3 ± 2.7 % (n = 5) increase. Based on these data we concluded that MSSM-1 and MSSM-2, like RUC-1 and RUC-2, demonstrate minimal priming activity when compared to tirofiban, the peptide RGDS, and eptifibatide.
Fig. 7.

Priming effects of compounds. Tirofiban (0.5 µM), an RGDS peptide (100 µM), eptifibatide (1 µM), RUC-1 (100 µM), RUC-2 (1 µM), MSSM-1 (300 µM), or MSSM-2 (300 µM) were added to washed platelets and then the platelets were fixed with 1 % paraforrmaldeyde. After quenching the paraformaldehyde with glycine and washing, fluorescent fibrinogen (200 µg/ml) was added for 30 min at 37 °C and then, after washing again, bound fluorescent fibrinogen was detected by flow cytometry. The percentage of platelets with fluorescence values above 25 arbitrary units (AU) was recorded. The data shown is the percentage of platelets with values above 25 AU in the presence of each compound, minus the percentage in the untreated platelet sample. The latter averaged 4 ± 3 %
In summary, based on the platelet adhesion and aggregation data, half of the compounds we tested demonstrated biologically meaningful inhibition of the receptor. This value can be compared to the inhibitory activity of 14 % of the compounds selected from a structure-based virtual screening application to the highly homologous αVβ3 integrin, with IC50 values ranging from 30 to 200 µM [30]. Although docking scoring functions are often unable to rank order ligands by affinity and/or potency, we note that the identified inhibitors MSSM-1 and MSSM-2 exhibited the highest rank (6th and 45th, respectively) among the docked ~2.5 million compounds based on complementarity to the predicted binding mode of RUC-2 in the RUC-1-αIIbβ3 crystal structure.
Predicted binding modes
As shown in Fig. 8, MSSM-1–5 were predicted to interact with key interaction sites of RUC-2 (Fig. 8A), including αIIb D224 (or alternatively D232), αIIb Y190, and β3 E220. Compounds MSSM-1 and MSSM-2 (Fig. 8B, C, respectively) formed a hydrogen bond with the αIIb D224 residue, whereas MSSM-3, MSSM-4 and MSSM-5 (Fig. 8D, E, F, respectively) interacted with the αIIb D232 residue. The importance of an interaction with D224 instead of D232 is suggested by the greater ability of MSSM-1 and MSSM-2 to inhibit platelet adhesion to fibrinogen.
Fig. 8.

Predicted binding modes of the 5 ligands identified by the docking screens (B, C, D, E and F) compared to the RUC-2 binding pose (A). The αIIb subunit is shown as a light blue cartoon with the side chains of residues as sticks. The β3 subunit is shown in white cartoon with the side chain of E220 as sticks. ADMIDAS and SyMBS ions are shown as white spheres. The five ligands are shown as orange sticks and the co-crystallized antagonist RUC-2 is shown using cyan sticks (A) or cyan lines (B, C, D, E and F). Black dot lines indicate hydrogen bonds
Novelty
MSSM-1 and MSSM-2 are significantly different from annotated αIIbβ3 ligands in the ChEMBL database (http://www.ebi.ac.uk/chembl), as quantified by comparing their topological fingerprints and their calculated Tanimoto similarity coefficient (Tc) [31] values (Table 1). The latter identifies nearest neighbors based on extended connectivity fingerprint maximum distance 4 (ECFP4), with a value of 0 indicating that the compounds are maximally dissimilar and 1 indicating that they are maximally similar [32]. MSSM-1 and MSSM-2 demonstrated ECFP4-based Tc values of 0.21 and 0.32, respectively to all of the annotated αIIbβ3 small-molecule ligands in the ChEMBL database, which is below 0.40, the value above which molecules are considered reasonably similar [33]. These data suggested the novelty of both agents. However, MSSM-1 itself is annotated in the ChEMBL database as a potent serine protease (C1r) inhibitor (cited under the names of 6-[amino(imino)methyl]-2-naphthyl 4-{[amino(imino)methyl] amino}benzoate, futhan, or nafamostat) [34, 35], and has been used clinically as nafamostat mesylate in Asia to treat pancreatitis [36]. Moreover, a further literature search unexpectedly revealed that MSSM-1, under the nafamostat designation, has actually been reported to inhibit platelet aggregation induced by ADP [37] [with an IC50 very similar to the one we obtained (9.3 ± 2.8 µM vs our 12.5 ± 1.2 µM)] and fibrinogen binding to ADP-stimulated human platelets [38]. In addition, the chemical space of this compound has been exploited to obtain more potent, classical, RGD-like αIIbβ3 antagonists. Specifically, compounds 4-(6-amidino-2-naphthylaminocarbonyl)phenoxyacetic acid and 4-(6-amidino-2-naphthalenecarboxamido)phenoxyacetic acid, which had the guanidine group of nafamostat replaced by a carboxylic acid inhibited ADP-induced aggregation of human platelet-rich plasma with IC50 values of 0.05 and 0.07 µM, respectively [39]. The higher affinity of these compounds can be explained based on current structural knowledge as most likely reflecting the ability of their carboxylic acid moieties to directly coordinate the MIDAS Mg2+ ion, and therefore to induce the unwanted high-affinity ligand binding conformation in the receptor.
MSSM-2 is known as 4-({(E)-amino[(4-carbamimidoylphenyl) amino] methylidene} amino) benzenecarboximidamide, and has been reported to have an antileukemic effect in mouse models according to information that is accessible through the Developmental Therapeutics Program of the National Cancer Institute. The most similar annotated αIIbβ3 small-molecule ligand to this compound in the ChEMBL database, 3-[3-(4-Carbamimidoyl-phenylcarbamoyl)-propionylamino]-propionic acid [40], is a substituted β amino acid, RGD-like derivative (Table 1). Based on its structure, and unlike MSSM-2, this compound is expected to interact with the MIDAS Mg2+ ion within the β3 subunit through its carboxylic group, and therefore to produce unwanted conformational changes in the receptor.
In order to assess whether comparable results could have been achieved using a much simpler approach, similarity calculations were run between the RUC-1/RUC-2 compounds and the currently available lead-like subset of the ZINC database (updated on February 6th, 2012 with 4,554,059 entries). Neither MSSM-1 nor MSSM-2 would have been identified by this approach considering that their Tc values compared to RUC-2 are 0.096 and 0.078, respectively. Even the compounds most similar to RUC-1 and RUC-2 (C23695127 and C63922479) had Tc values of 0.37 and 0.25, respectively [33]. It must be noted, however, that the compound with a Tc of 0.37 with respect to RUC-1 is a RUC-1 derivative.
Conclusions
Although three αIIbβ3 antagonists (abciximab, eptifibatide and tirofiban) are currently used in clinical practice as effective anti-platelet agents, they can only be administered intravenously and the two small molecules prime the receptor to assume the high affinity ligand binding conformation. Our structure-based virtual screening and compound selection criteria allowed us to efficiently identify small-molecule chemotypes with a very favorable hit-rate of 50 %. To the best of our knowledge, this is the first report of a structure-based virtual screen to identify an inhibitor of integrin αIIbβ3. Most importantly, it uses novel structural information and a unique hypothesis of a novel mechanism of inhibitory action that does not require coordination of the MIDAS Mg2+ ion within the β3 subunit. Thus, the current study shows the potential of structure-based virtual screening against new αIIbβ3 integrin crystal structures to explore novel ligand binding mechanisms, and possibly identify anti-platelet agents with better benefit-to-risk profiles. We were pleased to note that the chemical scaffold of one of the two active compounds (MSSM-1) has already been utilized to develop inhibitors of αIIbβ3-mediated platelet aggregation, though it required producing compounds that interact with the MIDAS Mg2+ ion [38, 39] and thus may have some of the undesirable features of other compounds in this group [8, 9]. To the best of our knowledge, the other compound (MSSM-2) has never been identified as inhibiting platelet aggregation or the binding of adhesive proteins to αIIbβ3 or platelets. Following our mechanistic hypothesis that the inhibitory action of these compounds does not involve engaging the MIDAS Mg2+ ion, these identified ligands are promising new starting points for structure-based ligand optimization, which is an ongoing effort of our laboratories.
Supplementary Material
Acknowledgments
The authors wish to thank Drs. Mihaly Mezei and Davide Provasi from Mount Sinai School of Medicine for technical assistance with similarity calculations, Joseph Fernandez and Nagarajan Chandramouli from the Rockefeller University Proteomics Resource Center for mass spectroscopy analysis and compound purification, Yufeng Wei from the Rockefeller University Spectroscopy Resource Center for NMR analysis, and Mayte Suarez-Farinas for statistical analysis. This work was supported by NHLBI grant HL019278, a Clinical and Translational Science Award 2UL1RR024143 to Rockefeller University, and funds from Stony Brook University. Computations were run on resources available through the Scientific Computing Facility of Mount Sinai School of Medicine.
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s10822-012-9594-6) contains supplementary material, which is available to authorized users.
Contributor Information
Ana Negri, Department of Structural and Chemical Biology, Mount Sinai School of Medicine, 1425 Madison Avenue, Box 1677, New York, NY 10029-6574, USA.
Jihong Li, Laboratory of Blood and Vascular Biology, Rockefeller University, New York, NY, USA.
Sarasija Naini, Laboratory of Blood and Vascular Biology, Rockefeller University, New York, NY, USA.
Barry S. Coller, Laboratory of Blood and Vascular Biology, Rockefeller University, New York, NY, USA
Marta Filizola, Email: marta.filizola@mssm.edu, Department of Structural and Chemical Biology, Mount Sinai School of Medicine, 1425 Madison Avenue, Box 1677, New York, NY 10029-6574, USA.
References
- 1.Lopez AD, Mathers CD, Ezzati M, Jamison DT, Murray CJL. Global burden of disease and risk factors, 2011/01/21 edn. Washington: World Bank; 2006. Measuring the global burden of disease and risk factors, 1990–2001. [PubMed] [Google Scholar]
- 2.Roger VL, Go AS, Lloyd-Jones DM, Adams RJ, Berry JD, Brown TM, Carnethon MR, Dai S, de Simone G, Ford ES, Fox CS, Fullerton HJ, Gillespie C, Greenlund KJ, Hailpern SM, Heit JA, Ho PM, Howard VJ, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Makuc DM, Marcus GM, Marelli A, Matchar DB, McDermott MM, Meigs JB, Moy CS, Mozaffarian D, Mussolino ME, Nichol G, Paynter NP, Rosamond WD, Sorlie PD, Stafford RS, Turan TN, Turner MB, Wong ND, Wylie-Rosett J. Heart disease and stroke statistics—2011 update: a report from the American Heart Association. Circulation. 2011;123(4):e18–e209. doi: 10.1161/CIR.0b013e3182009701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang G, Kong L, Zhao W, Wan X, Zhai Y, Chen LC, Koplan JP. Emergence of chronic non-communicable diseases in China. Lancet. 2008;372(9650):1697–1705. doi: 10.1016/S0140-6736(08)61366-5. [DOI] [PubMed] [Google Scholar]
- 4.Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110(6):673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 5.Springer TA, Zhu J, Xiao T. Structural basis for distinctive recognition of fibrinogen gammaC peptide by the platelet integrin alphaIIbbeta3. J Cell Biol. 2008;182(4):791–800. doi: 10.1083/jcb.200801146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Basani RB, D’Andrea G, Mitra N, Vilaire G, Richberg M, Kowalska MA, Bennett JS, Poncz M. RGD-containing peptides inhibit fibrinogen binding to platelet alpha(IIb)beta3 by inducing an allosteric change in the amino-terminal portion of alpha(IIb) J Biol Chem. 2001;276(17):13975–13981. doi: 10.1074/jbc.M011511200. [DOI] [PubMed] [Google Scholar]
- 7.Hagemeyer CE, Peter K. Targeting the platelet integrin GPIIb/IIIa. Curr Pharm Des. 2010;16(37):4119–4133. doi: 10.2174/138161210794519255. [DOI] [PubMed] [Google Scholar]
- 8.Cox D. Oral GPIIb/IIIa antagonists: what went wrong? Curr Pharm Des. 2004;10(14):1587–1596. doi: 10.2174/1381612043384673. [DOI] [PubMed] [Google Scholar]
- 9.Chew DP, Bhatt DL, Topol EJ. Oral glycoprotein IIb/IIIa inhibitors: why don’t they work? Am J Cardiovasc Drugs. 2001;1(6):421–428. doi: 10.2165/00129784-200101060-00002. [DOI] [PubMed] [Google Scholar]
- 10.Scirica BM, Cannon CP, Cooper R, Aster RH, Brassard J, McCabe CH, Charlesworth A, Skene AM, Braunwald E. Drug-induced thrombocytopenia and thrombosis: evidence from patients receiving an oral glycoprotein IIb/IIIa inhibitor in the Orbofiban in patients with unstable coronary syndromes-(OPUS-TIMI 16) trial. J Thromb Thrombolysis. 2006;22(2):95–102. doi: 10.1007/s11239-006-8669-4. [DOI] [PubMed] [Google Scholar]
- 11.Blue R, Murcia M, Karan C, Jirouskova M, Coller BS. Application of high-throughput screening to identify a novel alphaIIb-specific small-molecule inhibitor of alphaIIbbeta3-mediated platelet interaction with fibrinogen. Blood. 2008;111(3):1248–1256. doi: 10.1182/blood-2007-08-105544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu J, Negri A, Provasi D, Filizola M, Coller BS, Springer TA. Closed headpiece of integrin alphaIIbbeta3 and its complex with an alphaIIbbeta3-specific antagonist that does not induce opening. Blood. 2010;116(23):5050–5059. doi: 10.1182/blood-2010-04-281154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McCoy J, Shen M, Huang W, Coller B, Thomas CJ. Probe reports from the NIH molecular libraries program [Internet] Bethesda (MD): National Center for Biotechnology Information (US); 2010. Inhibitors of platelet integrin alphallbbeta3. 2010 Mar 27 [updated 2011 Mar 3] [PubMed] [Google Scholar]
- 14.Zhu J, Choi WS, McCoy JG, Negri A, Naini S, Li J, Shen M, Huang W, Bougie D, Rasmussen M, Aster R, Thomas CJ, Filizola M, Springer TA, Coller BS. Structure-guided design of a high-affinity platelet integrin alphaIIbbeta3 receptor antagonist that disrupts Mg(2)(+) binding to the MIDAS. Sci Trans Med. 2012;4(125):125ra132. doi: 10.1126/scitranslmed.3003576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Irwin JJ, Shoichet BK. ZINC-a free database of commercially available compounds for virtual screening. J Chem Inf Model. 2005;45:177–182. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boström J, Greenwood JR, Gottfries J. Assessing the performance of OMEGA with respect to retrieving bioactive conformations. J Mol Graph Model. 2003;21(5):449–462. doi: 10.1016/s1093-3263(02)00204-8. [DOI] [PubMed] [Google Scholar]
- 17.Weiner SJ, Kollman PA, Nguyen DT, Case DA. An all atom force field for simulations of proteins and nucleic acids. J Comp Chem. 1986;7(2):230–252. doi: 10.1002/jcc.540070216. [DOI] [PubMed] [Google Scholar]
- 18.Chambers CC, Hawkins GD, Cramer CJ, Truhlar DG. Model for aqueous solvation based on class IV atomic charges and first solvation shell effects. J Phys Chem. 1996;100:16385–16398. [Google Scholar]
- 19.Li JB, Zhu TH, Cramer CJ, Truhlar DG. New class IV charge model for extracting accurate partial charges from wave functions. J Phys Chem A. 1998;102:1820–1831. [Google Scholar]
- 20.Lorber DM, Shoichet BK. Flexible ligand docking using conformational ensembles. Protein Sci. 1998;7(4):938–950. doi: 10.1002/pro.5560070411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lorber DM, Shoichet BK. Hierarchical docking of databases of multiple ligand conformations. Curr Top Med Chem. 2005;5(8):739–749. doi: 10.2174/1568026054637683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuntz ID, Blaney JM, Oatley SJ, Langridge R, Ferrin TE. A geometric approach to macromolecule-ligand interactions. J Mol Biol. 1982;161(2):269–288. doi: 10.1016/0022-2836(82)90153-x. [DOI] [PubMed] [Google Scholar]
- 23.Nicholls A, Honig B. A rapid finite difference algorithm, utilizing successive over-relaxation to solve the Poisson–Boltzmann equation. J Comp Chem. 1991;12(4):435–445. [Google Scholar]
- 24.Weiner SJ, Kollman PA, Case DA, Singh UC, Ghio C, Alagona G, Profeta S, Weiner PA. A new force-field for molecular mechanical simulation of nucleic-acids and proteins. J Am Chem Soc. 1984;106:765–784. [Google Scholar]
- 25.Shoichet BK, Leach AR, Kuntz ID. Ligand solvation in molecular docking. Proteins Struct Funct Genet. 1999;34:4–16. doi: 10.1002/(sici)1097-0134(19990101)34:1<4::aid-prot2>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 26.Wei BQQ, Baase WA, Weaver LH, Matthews BW, Shoichet BK. A model binding site for testing scoring functions in molecular docking. J Mol Biol. 2002;322:339–355. doi: 10.1016/s0022-2836(02)00777-5. [DOI] [PubMed] [Google Scholar]
- 27.Hinselmann G, Rosenbaum L, Jahn A, Fechner N, Zell A. jCompoundMapper: an open source Java library and command-line tool for chemical fingerprints. J Cheminform. 2011;3(3):1–14. doi: 10.1186/1758-2946-3-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gasteiger J, Rudolph C, Sadowski J. Automatic generation of 3D Atomic coordinates for organic molecules. Tetrahedron Comp Method. 1990;3:537–547. [Google Scholar]
- 29.Du XP, Plow EF, Frelinger AL, III, O’Toole TE, Loftus JC, Ginsberg MH. Ligands “activate” integrin alpha IIb beta 3 (platelet GPIIb-IIIa) Cell. 1991;65(3):409–416. doi: 10.1016/0092-8674(91)90458-b. [DOI] [PubMed] [Google Scholar]
- 30.Zhou Y, Peng H, Ji Q, Qi J, Zhu Z, Yang C. Discovery of small molecule inhibitors of integrin alphavbeta3 through structure-based virtual screening. Bioorg Med Chem Lett. 2006;16(22):5878–5882. doi: 10.1016/j.bmcl.2006.08.061. [DOI] [PubMed] [Google Scholar]
- 31.Willett P, Barnard JM, Downs GM. Chemical similarity searching. J Chem Inf Comput Sci. 1998;38(6):983–996. [Google Scholar]
- 32.Rogers D, Brown RD, Hahn M. Using extended-connectivity fingerprints with Laplacian-modified Bayesian analysis in high-throughput screening follow-up. J Biomol Screen. 2005;10(7):682–686. doi: 10.1177/1087057105281365. [DOI] [PubMed] [Google Scholar]
- 33.Wawer M, Bajorath J. Similarity-potency trees: a method to search for SAR information in compound data sets and derive SAR rules. J Chem Inf Model. 2010;50(8):1395–1409. doi: 10.1021/ci100197b. [DOI] [PubMed] [Google Scholar]
- 34.Hays SJ, Caprathe BW, Gilmore JL, Amin N, Emmerling MR, Michael W, Nadimpalli R, Nath R, Raser KJ, Stafford D, Watson D, Wang K, Jaen JC. 2-amino-4H-3,1-benzoxazin-4-ones as inhibitors of C1r serine protease. J Med Chem. 1998;41(7):1060–1067. doi: 10.1021/jm970394d. [DOI] [PubMed] [Google Scholar]
- 35.Plummer JS, Cai C, Hays SJ, Gilmore JL, Emmerling MR, Michael W, Narasimhan LS, Watson MD, Wang K, Nath R, Evans LM, Jaen JC. Benzenesulfonamide derivatives of 2-substituted 4H-3,1-benzoxazin-4-ones and benzthiazin-4-ones as inhibitors of complement C1r protease. Bioorg Med Chem Lett. 1999;9(6):815–820. doi: 10.1016/s0960-894x(99)00095-5. [DOI] [PubMed] [Google Scholar]
- 36.Chang JH, Lee IS, Kim HK, Cho YK, Park JM, Kim SW, Choi MG, Chung IS. Nafamostat for prophylaxis against post-endoscopic retrograde cholangiopancreatography pancreatitis compared with gabexate. Gut Liver. 2009;3(3):205–210. doi: 10.5009/gnl.2009.3.3.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fuse I, Higuchi W, Toba K, Aizawa Y. Inhibitory mechanism of human platelet aggregation by nafamostat mesilate. Platelets. 1999;10(4):212–218. doi: 10.1080/09537109976040. [DOI] [PubMed] [Google Scholar]
- 38.Hodohara K, Fujiyama Y, Hosoda S, Yasunaga K. Inhibition of fibrinogen binding to ADP-stimulated human platelets by synthetic serine protease inhibitors. Blood Vessel. 1989;20:213–219. [Google Scholar]
- 39.Ono S, Inoue Y, Yoshida T, Ashimori A, Kosaka K, Imada T, Fukaya C, Nakamura N. Preparation and pharmacological evaluation of novel glycoprotein (Gp) IIb/IIIa antagonists. 1. The selection of naphthalene derivatives. Chem Pharm Bull (Tokyo) 1999;47(12):1685–1693. doi: 10.1248/cpb.47.1685. [DOI] [PubMed] [Google Scholar]
- 40.Bovy PR, Rico JG, Rogers TE, Tjoeng FS, Zablocki JA. Substituted β-amino acid derivatives useful as platelet aggregation inhibitors and intermediates thereof. 5239113. Patent number: 1993 Issuedate: 24 Aug 1993.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.