

Abstract
In modeling blood–brain barrier (BBB) passage, in silico models have yielded ~80% prediction accuracy, and are currently used in early drug discovery. Being derived from molecular structural information only, these models do not take into account the biological factors responsible for the in vivo outcome. Passive permeability and P-glycoprotein (Pgp, ABCB1) efflux have been successfully recognized to impact xenobiotic extrusion from the brain, as Pgp is known to play a role in limiting the BBB penetration of oral drugs in humans. However, these two properties alone fail to explain the BBB penetration for a significant number of marketed central nervous system (CNS) agents. The Biopharmaceutics Drug Disposition Classification System (BDDCS) has proved useful in predicting drug disposition in the human body, particularly in the liver and intestine. Here we discuss the value of using BDDCS to improve BBB predictions of oral drugs. BDDCS class membership was integrated with in vitro Pgp efflux and in silico permeability data to create a simple 3-step classification tree that accurately predicted CNS disposition for more than 90% of 153 drugs in our data set. About 98% of BDDCS class 1 drugs were found to markedly distribute throughout the brain; this includes a number of BDDCS class 1 drugs shown to be Pgp substrates. This new perspective provides a further interpretation of how Pgp influences the sedative effects of H1-histamine receptor antagonists.
Keywords: Brain disposition, Data mining, Drug discovery, Rules of thumb
1. Introduction
Delivery of new drug candidates to the CNS is a challenging problem in drug development. It is important to design drugs that target the CNS when it is the site of action. It is also important to prevent peripherally acting drugs from accessing the CNS where they could exert undesirable and potentially harmful side effects. Being one of the oldest issues in drug discovery and QSAR (quantitative structure-activity relationship), significant efforts are made in academia and industry to develop screens for optimization and prioritization, which include in vitro assays and computational models to evaluate CNS penetration.
QSAR studies have been instrumental in early phases of drug discovery, serving as fast and inexpensive tools to prioritize new molecular entities (NMEs) as candidate CNS agents. BBB disposition has been linked to molecular properties such as drug lipophilicity (LogP and LogD7.4) [1,2], molecular weight [1–3], and the tendency to form hydrogen bonds quantified as polar surface area (PSA) [4] or as a function of nitrogen and oxygen counts [5]. These approaches led to simple rules of thumb, or to more elaborate in silico models for predicting CNS disposition yielding ~80% accuracies for early phase screening [5–7]. However, we believe that compound optimization based simply on molecular properties restricts the portion of chemical space explored. Therefore, anchoring brain penetration directly to biological factors may result in a more flexible selection process for drug discovery, which is desirable when designing compound libraries or optimizing interesting candidates.
After characterizing the expression of Pgp in the mouse brain, Cordon-Cardo et al. first suggested that Pgp might play a role in vivo in limiting brain penetration of xenobiotics [8]. Pgp is an efflux transporter, also known as ABCB1, which uses energy derived from ATP hydrolysis to transport its substrates against a concentration gradient [9]. The importance of Pgp in CNS penetration has been established using knockout mice [10]. Mahar Doan and coworkers experimentally tested the efflux ratio (ER) of 48 CNS and 45 non-CNS drugs, reporting that CNS agents are less likely to be Pgp substrates and more likely to be highly permeable drugs [11]. By analyzing 119 marketed CNS drugs and 108 proprietary CNS drug candidates, Wager and colleagues [12] confirmed the latter observations. Going from candidates to marketed CNS drugs, these investigators observed a marked increase in the number of highly permeable compounds (from 51% to 75%) and Pgp non-substrates (from 55% to 75%). Moreover, only about 40% of CNS drug candidates were both highly permeable and Pgp non-substrates, as opposed to 70% of successfully CNS marketed drugs. This indicates that both high permeability and lack of Pgp efflux are desirable for CNS drug candidates to become marketed CNS drugs. Surprisingly, 20% of marketed CNS drugs analyzed exhibited both unfavorable permeability and Pgp efflux. Until now, no explanations have been proposed for these observations.
The Biopharmaceutics Drug Disposition Classification System (BDDCS) has been successful in predicting in vivo absorption, disposition, and drug–drug interactions of marketed drugs [13–15]. BDDCS is a four class system based on extent of metabolism and solubility measures that has been used to explain the role of transporters in pharmacokinetics and their interplay with metabolizing enzymes in the liver and intestine (Fig. 1).
Fig. 1.
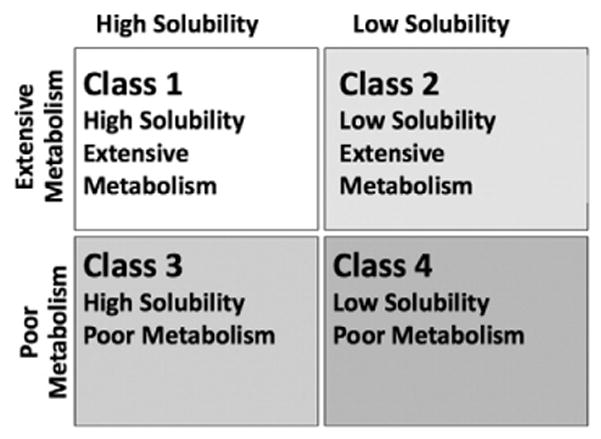
The Biopharmaceutics Drug Disposition Classification System (BDDCS).
Based on observations in the intestine and liver, BDDCS class 1 drugs appear to be the most biopharmaceutically desirable class due to their high intestinal absorption and lack of clinically relevant uptake and efflux transporter effects [13,15]. Nine of the 14 drugs with unfavorable in vitro permeability and Pgp profile reported by Wager and coworkers have BDDCS classification data available in the data set of oral drugs that we recently published [16]; of these, 7 (78%) are BDDCS class 1 drugs. This suggests that, analogous to what has been observed in the intestine and liver, class 1 drugs may not be significantly influenced by transporters in the brain.
Here, we investigate the use of BDDCS in predicting the brain disposition of 153 orally administered drugs for which Pgp efflux data are available. Using BDDCS, in vitro Pgp efflux ratio values, and in silico calculated permeability (VolSurf+ descriptor CACO2), we define three rules of thumb to predict brain penetration, which for the present data set are accurate over 90% of the time. The model outperforms in silico strategies for predicted BBB permeability [5,17,18], which for the 153 drugs were accurate around 80% of the time. Simple molecular properties such as those used in the Norinder-Haeberlein [5] and Lipinski BBB [18] rules of thumb for brain disposition were analyzed with respect to BDDCS classification, Pgp impact, and in silico predicted permeability. The use of this framework in predicting the sedative effect of antihistaminic drugs is also discussed.
2. Criteria and rationale for data collection
2.1. Efflux ratio
The Pgp impact on drug permeability, evaluated in terms of efflux ratio (ER), is defined as follows:
where Papp(a→b) is permeability from the apical to the basolateral side (absorption) and Papp(b→a) is permeability from the basolateral to the apical side (secretion). For Pgp substrates, the absorption should be decreased and the secretion increased in cell lines over-expressing Pgp, as opposed to wild type cell lines, yielding ER values greater than 1. In 2001, Polli and colleagues [19] published a collection of over 60 molecules tested for Pgp transport and inhibition, suggesting that an efflux ratio value ≥2 should be adopted to identify Pgp substrates. However, in confirmatory studies using a Pgp inhibitor for non-substrates, the authors noted that a few drugs having ER<2 (loratadine, diltiazem, and neostigmine) were Pgp substrates. For all of the oral drugs included in our previously analyzed BDDCS data set [16], we carried out an extensive literature search for ER values in MDCK-MDR1 cell lines. We were able to find at least one ER value for 160 oral drugs from 16 different articles [11,12,19–32], resulting in a database of 318 ER values. About 77% of the ERs were derived from MDCK-MDR1 cell lines originating from the Netherlands Cancer Institute (Borst cell lines), while the remaining 23% were derived from MDCK-MDR1 cell lines originating from the National Institute of Health (NIH cell lines). We noticed that in our data set that ER values from NIH cell lines tended to be higher than those from Borst cell lines, in agreement with the reported difference in Pgp expression levels for the two cell lines [33]. In the Results section of this paper, we address the criteria for defining Pgp substrates and non-substrates in these cell lines. The 318 efflux ratios for the 160 drugs are tabulated in Supplemental Table S1.
2.2. BDDCS classification
BDDCS class assignments were based on extent of metabolism and FDA solubility. Highly metabolized drugs (equal or above 70% in humans) were assigned to classes 1 and 2, while highly soluble drugs were assigned to classes 1 and 3 (Fig. 1). Solubility class is defined based on the dose number (Do):
where HDS is the highest dose strength approved for commercial use, and LWS is the lowest water solubility at 37 °C in the pH range from 1 to 7.5. When Do is equal or below 1, the drug is considered highly soluble. We have recently provided a detailed exhaustive description of the criteria [34] and discussed the selection of the drugs included in the oral drugs data set [16].
2.3. BBB penetration data
We conducted an exhaustive search for blood–brain barrier (BBB) penetration data for the 160 oral drugs where BDDCS class and Pgp ER data were available to classify the drugs as either brain-penetrating (BBB+) or having little if any ability to cross the BBB (BBB−). Marketed CNS agents were directly assigned to the BBB+ class without further investigation. When the unbound drug brain/plasma ratios in humans were available, these values were used for defining the BBB class; drugs with values greater than or equal to 0.1 were assigned as BBB+, otherwise, they were assigned as BBB−.
When the unbound brain/plasma ratio of a drug in humans was unavailable, then either the unbound CSF/plasma ratio in humans or brain penetration data from other species was used. We recognize that interspecies differences and using CSF/plasma ratios in place of brain/plasma ratios may affect the BBB assignment outcome [35,36]. Therefore, we created a prioritization scheme of how to define the BBB class when more than one of these values was available. From an analysis of rat unbound brain/plasma and CSF/plasma ratio data for 39 drugs [36], we noted that, even when up to a five-fold difference was found between these data, the BBB assignment was in agreement for 35 out of 39 drugs. By comparing human and rat brain penetration data, we found that the human CSF/plasma ratio was a better surrogate for the human unbound brain/plasma ratio than any available data from other species. For example, verapamil is a BBB+ drug (human brain/plasma ratio=0.55) [37]; its human CSF/plasma ratio of 1.13 [36] consistently classifies it as a BBB+ drug, while its rat brain/plasma ratio of 0.053 [36] misclassifies it as a BBB− drug. Hence, when human brain/plasma ratio data were unavailable, we used the human CSF/plasma ratio information for assigning the BBB class over any other species data (including brain/plasma ratios). Due to the maximum five-fold numerical difference observed between CSF/plasma and brain/plasma ratios for the same drugs, we expect no significant incongruence in the BBB assignment for drugs with CSF/plasma ratios greater than or equal to 0.5 (or at least 33% distributed in the CSF). When numerical data were unavailable in humans and other species, qualitative phrases like “distribution in brain negligible” were used to assign the drug as BBB−. Qualitative phrases like “extensively distributed in brain tissues” or evidence of CNS side-effects were used to assign the drug as BBB+.
Diltiazem was excluded from our analysis since it was found to be controversial; it is reported to exhibit a “decreasing concentration gradient in the following organs: liver, kidney, lung, spleen, heart and brain” [38], yet Naito et al. [39] reported that the CSF/plasma ratio for diltiazem was from 0.05 to 0.2. The prodrug famciclovir was also excluded since the BDDCS classification was relative to the dosed compound, not to the active species (penciclovir) that penetrates the BBB. Five other drugs had to be excluded because there was no BBB penetration data available that matched any of these criteria. The original BBB penetration data for the remaining 153 oral drugs can be found in References [11,12, 21,36–111]. The BBB assignments for the data set can be found in Supplemental Tables S1 and S2.
2.4. In silico calculations
Even though our preference was to relate in vitro properties with human data, an in silico parameter for passive permeability had to be adopted due to the unavailability of consistent in vitro data for all the 153 drugs. The molecular descriptor CACO2 generated using VolSurf+ [112], a computational procedure specifically designed to produce descriptors related to pharmacokinetic properties [17], was used as a surrogate for in vitro passive permeability data. Our previous work has suggested that the VolSurf+ CACO2 descriptor derived from the Caco-2 cell permeation studies was a suitable surrogate for passive permeability [16,34]. The CACO2 model is a qualitative model trained using a thousand compound in-house data set [113,114].
To compare how our framework improved the brain penetration prediction of current state-of-the-art in silico rules of thumb and models, we included several other descriptors. In analogy with CACO2, the VolSurf+ descriptor LgBB is trained on a five hundred compound in-house data set, and it is based on the methodology presented by Crivori et al. [113,115]. We further calculated the parameters necessary to predict brain penetration following the Norinder-Haeberlein [5] and Lipinski BBB [18] rules of thumb. These included calculated LogP (CLogP), molecular weight (MW), hydrogen bond acceptor counts (HBA), hydrogen bond donor count (HBD), and number of violation to Lipinski Rule of 5 (Ro5) that were taken from our previous work [34], and counts of oxygen and nitrogen that were extrapolated from the SMILES structure of the drugs as reported in WOMBAT-PK [116,117]. Predicted brain penetration values according to Norinder-Haeberlein [5] and Lipinski BBB [18] rules of thumbs were also reported. Norinder and Haeberlein provided two rules for brain penetration: (i) when the sum of nitrogens and oxygens in the molecule (N+O) is five or less, the drug is considered BBB+ (brain/plasma ratio>0.1), and (ii) when CLogP minus N+O is positive, brain/plasma ratio is greater than unity (brain/plasma ratio >1). We only included the first rule in our comparison since the second rule exceeds our criteria for BBB assignment. Lipinski BBB, as reported by Pajouhesh and Lenz [18], proposed that a drug is unlikely to be BBB+ if:
Molecular weight>400
LogP>5
Hydrogen bond acceptor count>7
Hydrogen bond donor count>3
To evaluate any possible influence of pKa on brain penetration, we performed and report the lowest in silico pKa calculations for the acid and basic centers of the 153 drugs using the software MoKa [118,119]. All the in silico data used are available in Supplemental Tables S2, S5, and S6.
3. Elaboration, analysis, and application of rules for brain disposition prediction
3.1. Pgp and brain disposition
Of the 160 oral drugs for which Pgp ER data were available, 63 had more than one reported ER value in Borst cell lines. Adopting the ER threshold of 2 as suggested by Polli and coworkers [19], 87% of these 63 drugs were classified coherently as either Pgp substrates or non-substrates. Only 4 drugs had more than one reported ER in NIH cell lines. Of these 4 drugs, 3 of them were inconsistently classified using the threshold of 2. For 53 drugs where values were available in both Borst and NIH cell lines, the agreement in assignment was only 67%. This suggests that another threshold should be adopted for the NIH cell lines. For the 53 drugs where at least one reported ER value was available from both the Borst and NIH cell lines, ERs in NIH cell lines were on average 4.26-fold higher than those in the Borst cell lines. Therefore, when assigning the Pgp class, we adopted a threshold of 2 for Borst cell lines, and 8.5 for NIH cell lines. When more than one ER value was available for the same drug in the same cell line, the threshold values were then applied to the averages. Using these criteria, the two systems agreed about 81% of the time. For the 10 drugs where there was no agreement (atomoxetine, chlorpromazine, citalopram, clozapine, metoclopramide, olanzapine, ranitidine, risperidone, sertraline, and tiagabine), assignment was based on the Borst cell line data. Even though we adopted an ER value of 8.5 as the threshold for defining Pgp impact on brain penetration for data from the NIH cell lines, the appropriateness of this threshold should be further investigated.
The assumption that Pgp efflux is the only factor limiting brain penetration, was found to be correct 78% of the time for this data set (Fig. 2): 63% of the data set were Pgp non-substrates that are BBB+, while 15% were Pgp substrates that are BBB− (Fig. 2A). In particular, 80% of BBB+ molecules were Pgp non-substrates (Fig. 2C), and 72% of BBB− were Pgp substrates (Fig. 2D). However, it must be noted that around 51% of Pgp substrates were BBB+ (Fig. 2E), thus being a Pgp substrate does not imply being BBB−, while 92% of Pgp non-substrates were BBB+ (Fig. 2F).
Fig. 2.
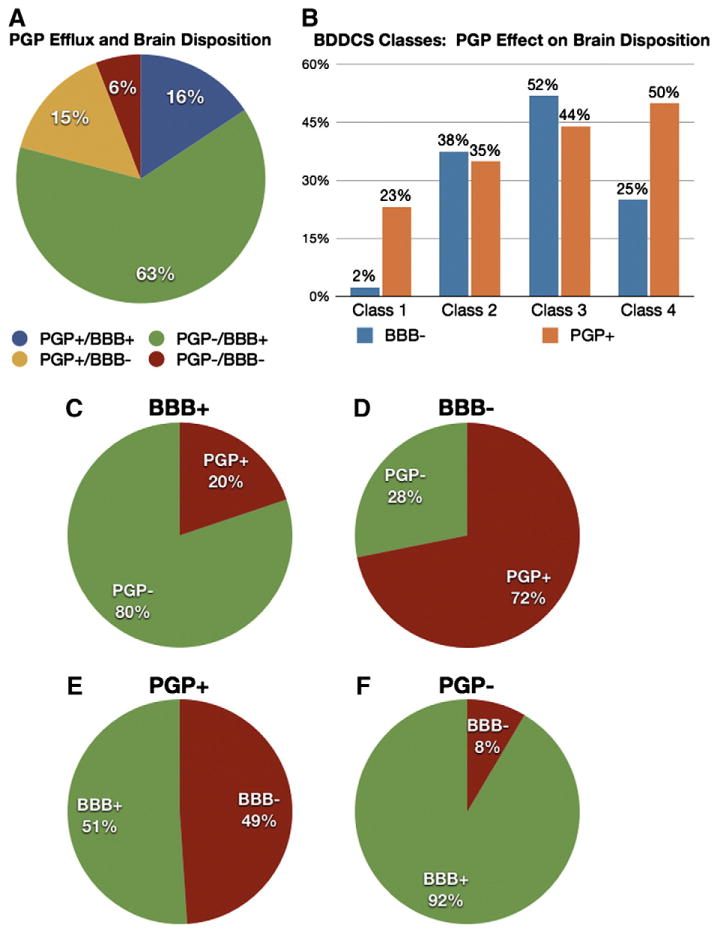
Pie and bar charts showing the relative percentage of the drugs in the data set classified based on their ability to cross the blood brain barrier, Pgp profile, and BDDCS class.
Interestingly, the absence of Pgp effect on brain penetration was mainly associated with BDDCS class 1 drugs (Fig. 2B), where 98% of the time class 1 drugs were BBB+, even if 23% of them were Pgp substrates, therefore 88% of Pgp substrates in class 1 were BBB+. In contrast, 75% of the Pgp substrates in BDDCS classes 2, 3, and 4, were BBB−.
3.2. In silico permeability and brain disposition
Due to a lack of in vitro permeability data, we evaluated the relationship between in silico permeability and brain disposition by using the calculated octanol/water partition coefficient CLogP [120] and the VolSurf+ descriptor CACO2. LogP has been highly used in medicinal chemistry as a surrogate for permeability. We have previously demonstrated that the VolSurf+ descriptor CACO2 can be used to distinguish highly versus poorly metabolized/permeable compounds [16]. Therefore, we evaluated the suitability of this descriptor as a filter for poorly permeable compounds. In Fig. 3, we use box plots [121] and receiver-operating characteristic (ROC) curves [122] to show the distribution of BBB+ and BBB− classes with respect to CLogP and calculated CACO2 permeability. In agreement with Wager and coworkers [12], we found no significant difference in CLogP distribution for the two sets (Fig. 3A and C). In contrast, a marked difference between BBB+ and BBB− with respect to CACO2 permeability was recognized (Fig. 3A and B). We observed that if a value of CACO2 descriptor equal to −0.3 is adopted, 40.6% of BBB− molecules are filtered with a loss of only 3.3% of BBB+ (Fig. 3A).
Fig. 3.
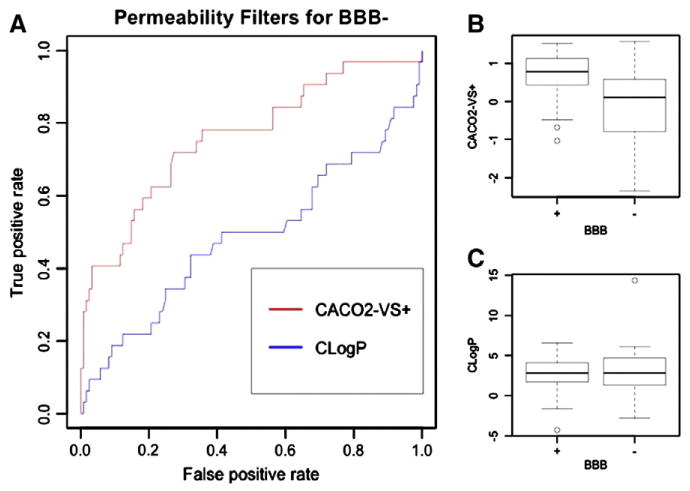
A) Receiver-operating characteristic (ROC) curves for CACO2 permeability and CLogP used as classifiers to discriminate BBB+ and BBB− classes, where BBB− is the targeted class (class to be predicted). Each point in the ROC curve represents a descriptor value (either CACO2 or CLogP) and the fraction of true positives (correctly classified BBB− drugs) and false positives (incorrectly classified BBB− drugs) associated, if that descriptor value is used as the cut-off to discriminate the two classes. B) CLogP distributions of BBB+ and BBB− drugs. The box is delimited by the 1st and the 3rd quartiles, the line inside the box is the median, the whiskers correspond to 5th and 95th percentiles, and the dots are outliers. C) VolSurf+ CACO2 descriptor distributions of BBB+ and BBB− drugs. The box is delimited by the 1st and the 3rd quartiles, the line inside the box is the median, the whiskers correspond to 5th and 95th percentiles, and the dots are outliers.
Others have linked brain penetration to pKa due to its effect on passive permeability; that is, neutral species are expected to be favored in crossing biological membranes, as opposed to charged species [123]. In particular, this effect appears to be marked for acid centers having low pKa (under 5.5), since these drugs (e.g., Class 2 NSAIDs) are mostly negatively charged at physiological pH. We estimated the lowest acid pKa for drugs in our data set using MoKa (Supplemental Table S2). Six (cefuroxime, cetirizine, fexofenadine, methotrexate, pravastatin, and sulfasalazine) of the 32 (18.7%) BBB− drugs had a calculated acid pKa under 5.5, while this was true for only 5 (gabapentin, indomethacin, rosuvastatin, tiagabine, and warfarin) out of 121 BBB+ drugs (4.1%). However, of the 6 BBB− drugs with low acid pKa, all but cetirizine were predicted to be low permeability (VolSurf+ CACO2 descriptor below −0.3) while this was true for only 2 (gabapentin and rosuvastatin) BBB+ drugs with low acid pKa. Hence, even if based on these data an influence of pKa on brain penetration can be recognized, it appears to be taken into account by the VolSurf+ CACO2 descriptor.
3.3. Generation of BDDCS-based BBB rules
The data collected were used to elaborate a classification tree for predicting CNS disposition (Fig. 4). The classification tree was based on 153 oral drugs and was created using the data mining software Orange Canvas [124]. In order to have a straightforward framework, three different two-level variables were chosen: BDDCS class (either Class 1 or Classes 2, 3, and 4), Pgp Impact (“+” for substrates, “−” for non-substrates), and calculated permeability (“+” if the VolSurf+ CACO2 descriptor was above −0.3, otherwise “−”). The rationale for the choice of the threshold for the CACO2 descriptor is presented above. The following options were used for building the classification tree: attribute selection based on gain ratio, no binarization, stop splitting nodes with a majority class of 90%, post-pruning (recursively merge leaves with the same majority class, m=3).
Fig. 4.
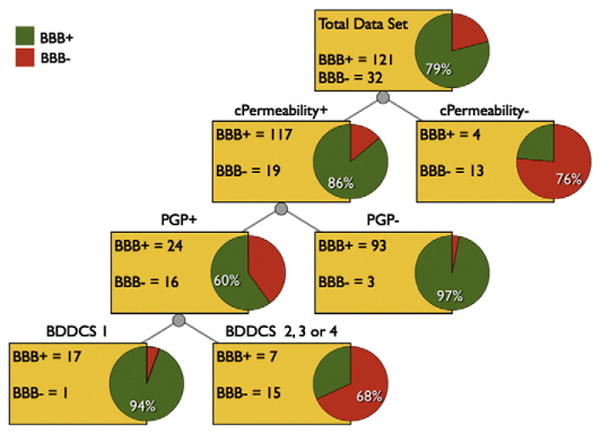
Pipeline of rules for predicting brain disposition. Rules are presented as a classification tree, and are based on the drug BDDCS class, Pgp profile, and the VolSurf+ descriptor CACO2 value (cPermeability is+if it is above −0.3, “−” if it is below). Pie chart red represents BBB− fraction of drug population evaluated; pie chart green represents BBB+ fraction.
Being based on a sequence of three two-level variables, it can be also simply considered as a pipeline of rules of thumb. By classifying drugs first based on calculated permeability, then Pgp efflux, and finally BDDCS Classification, the model is correct for 138 out of 153 drugs, gaining an accuracy of 90.2% for this data set (Figs. 4 and 5). We address 15 incorrectly classified drugs in following Outliers Analysis section. As previously discussed, the use of a filter for low permeability allows correct classification of a significant portion of BBB− drugs with only few BBB+ drugs being misclassified. Drugs that do not seem to be limited by passive permeability predominantly (93.7%) cross the BBB if they are Pgp non-substrates. Evaluating only permeable Pgp substrate drugs (the last level in Fig. 4), BDDCS Class 1 drugs were generally capable of penetrating the BBB (94.4%), while the remaining classes of drugs were predominantly BBB− (68.1%).
Fig. 5.
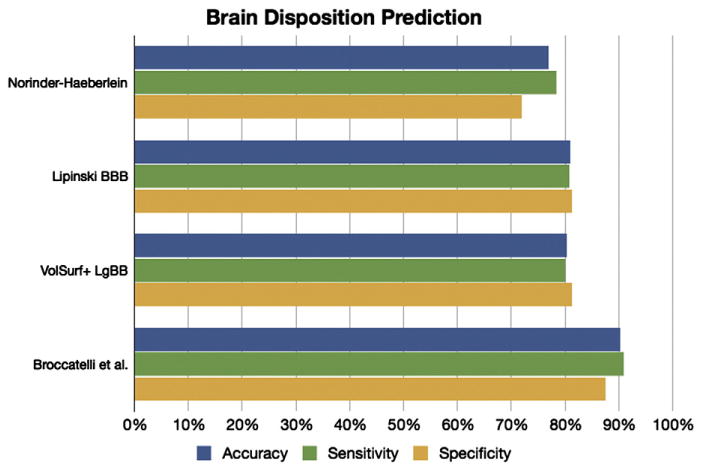
Accuracy of blood–brain barrier crossing prediction for the different methods. The sensitivity is the accuracy in predicting BBB+ drugs, while specificity is the accuracy in predicting BBB− drugs.
In Fig. 5, we show the accuracy in predicting brain penetration for our data set using our classification tree (Table 1) as well as the VolSurf+ descriptor LgBB, and Lipinski BBB and Norinder-Haeberlein rules of thumb. Notably, in silico methods solely based on the molecular structure have accuracy of ~80%, with Lipinski BBB rules and LgBB being reasonably accurate and well-balanced. These methods appear to be well-suited for early phase drug discovery compound prioritization. The rule-based system derived from our classification tree outperforms these other models. However, our approach cannot be directly compared with the other models, as it requires several in vitro measures. Therefore, we recommend adopting it as guidelines during advanced stage drug discovery.
Table 1.
List of 153 oral drugs and data used for brain penetration prediction.
| Drug | Pgp Substrate | BDDCS Class | CACO2_VSa | cPermeabilityb | Predicted BBB | Actual BBB |
|---|---|---|---|---|---|---|
| Acebutolol | + | 1 | −0.05 | + | + | + |
| Alprazolam | − | 1 | 1.16 | + | + | + |
| Alprenolol | − | 1 | 0.75 | + | + | + |
| Amantadine | − | 3 | 0.59 | + | + | + |
| Amisulpride | + | 4 | −0.01 | + | − | + |
| Amitriptyline | − | 1 | 1.40 | + | + | + |
| Amoxapine | − | 1 | 0.98 | + | + | + |
| Amprenavir | + | 2 | 0.25 | + | − | − |
| Antipyrine | − | 1 | 1.22 | + | + | + |
| Aprepitant | + | 2 | 0.04 | + | − | + |
| Astemizole | + | 2 | 1.06 | + | − | − |
| Atenolol | − | 3 | −0.08 | + | + | + |
| Atomoxetine | + | 1 | 1.13 | + | + | + |
| Biperiden | − | 1 | 1.22 | + | + | + |
| Bromazepam | − | 1 | 0.90 | + | + | + |
| Bromocriptine | + | 1 | 0.29 | + | + | + |
| Bupropion | − | 1 | 1.03 | + | + | + |
| Buspirone | − | 2 | 0.43 | + | + | + |
| Caffeine | − | 1 | 0.70 | + | + | + |
| Carbamazepine | − | 2 | 1.29 | + | + | + |
| Cefuroxime | − | 3 | −1.14 | − | − | − |
| Cetirizine | + | 3 | 0.04 | + | − | − |
| Chloroquine | + | 3 | 0.94 | + | − | + |
| Chlorothiazide | − | 4 | −0.48 | − | − | − |
| Chlorpheniramine | − | 1 | 1.18 | + | + | + |
| Chlorpromazine | + | 1 | 1.31 | + | + | + |
| Cimetidine | + | 3 | 0.45 | + | − | − |
| Citalopram | − | 2 | 0.90 | + | + | + |
| Clarithromycin | + | 3 | −1.11 | − | − | − |
| Clemastine | − | 1 | 1.40 | + | + | + |
| Clomipramine | + | 1 | 1.41 | + | + | + |
| Clonazepam | − | 1 | 0.78 | + | + | + |
| Clonidine | − | 3 | 1.05 | + | + | + |
| Clozapine | + | 2 | 1.52 | + | − | + |
| Colchicine | + | 1 | 0.59 | + | + | − |
| Cyclobenzaprine | − | 1 | 1.45 | + | + | + |
| Cyclosporin A | + | 2 | −0.77 | − | − | − |
| Desipramine | − | 1 | 1.12 | + | + | + |
| Desloratadine | + | 2 | 1.16 | + | − | − |
| Dexamethasone | + | 1 | 0.07 | + | + | + |
| Diazepam | − | 1 | 1.28 | + | + | + |
| Digoxin | + | 3 | −1.64 | − | − | − |
| Diphenhydramine | − | 1 | 1.36 | + | + | + |
| Dipyridamole | + | 2 | −0.56 | − | − | − |
| Domperidone | + | 2 | 0.36 | + | − | − |
| Donepezil | − | 2 | 0.97 | + | + | + |
| Doxepin | − | 1 | 1.25 | + | + | + |
| Duloxetine | − | 1 | 1.13 | + | + | + |
| Eletriptan | + | 1 | 0.62 | + | + | + |
| Enoxacin | − | 4 | −0.68 | − | − | + |
| Ergotamine | + | 1 | −0.19 | + | + | + |
| Erythromycin | + | 3 | −0.91 | − | − | − |
| Escitalopram | − | 1 | 0.93 | + | + | + |
| Ethosuximide | − | 1 | 0.62 | + | + | + |
| Etoposide | + | 3 | −0.92 | − | − | − |
| Fexofenadine | − | 3 | −0.34 | − | − | − |
| Fluoxetine | − | 1 | 0.76 | + | + | + |
| Fluphenazine | − | 2 | 0.60 | + | + | + |
| Fluvoxamine | − | 1 | −0.07 | + | + | + |
| Gabapentin | − | 3 | −0.42 | − | − | + |
| Galantamine | − | 1 | 0.37 | + | + | + |
| Guanfacine | − | 3 | 0.79 | + | + | + |
| Haloperidol | − | 2 | 0.66 | + | + | + |
| Hydrocodone | − | 1 | 0.62 | + | + | + |
| Hydroxyzine | − | 1 | 1.00 | + | + | + |
| Imipramine | − | 1 | 1.36 | + | + | + |
| Indinavir | + | 2 | 0.00 | + | − | − |
| Indomethacin | − | 2 | 0.15 | + | + | + |
| Itraconazole | − | 2 | 0.98 | + | + | − |
| Ketoconazole | − | 2 | 0.97 | + | + | − |
| Labetalol | + | 1 | −0.39 | − | − | − |
| Lamotrigine | − | 2 | 0.57 | + | + | + |
| Loperamide | + | 3 | 0.85 | + | − | − |
| Loratadine | − | 2 | 1.57 | + | + | − |
| Lorazepam | − | 1 | 0.64 | + | + | + |
| Maprotiline | − | 1 | 1.24 | + | + | + |
| Meprobamate | − | 1 | 0.24 | + | + | + |
| Methotrexate | − | 3 | −2.35 | − | − | − |
| Methylphenidate | − | 1 | 0.95 | + | + | + |
| Methylprednisolone | + | 1 | −0.01 | + | + | + |
| Metoclopramide | − | 3 | 0.40 | + | + | + |
| Metoprolol | − | 1 | 0.40 | + | + | + |
| Mexilitene | − | 1 | 0.79 | + | + | + |
| Mirtazapine | − | 1 | 1.21 | + | + | + |
| Morphine | − | 1 | 0.03 | + | + | + |
| Naloxone | − | 1 | −0.07 | + | + | + |
| Naltrexone | − | 1 | −0.06 | + | + | + |
| Nelfinavir | + | 2 | 0.44 | + | − | − |
| Neostigmine | + | 3 | 0.58 | + | − | − |
| Nicardipine | − | 1 | 0.81 | + | + | + |
| Nifedipine | − | 2 | 0.89 | + | + | + |
| Nimodipine | − | 2 | 0.69 | + | + | + |
| Nitrazepam | − | 2 | 0.65 | + | + | + |
| Nitrendipine | − | 2 | 0.80 | + | + | + |
| Nordazepam | − | 1 | 1.14 | + | + | + |
| Nortriptyline | − | 1 | 1.19 | + | + | + |
| Olanzapine | − | 2 | 1.34 | + | + | + |
| Oxcarbazepine | − | 2 | 0.86 | + | + | + |
| Oxprenolol | − | 1 | 0.61 | + | + | + |
| Oxycodone | − | 1 | 0.24 | + | + | + |
| Paliperidone | + | 4 | −0.17 | + | − | + |
| Paroxetine | + | 1 | 0.58 | + | + | + |
| Phenytoin | − | 2 | 0.52 | + | + | + |
| Pravastatin acid | − | 3 | −0.85 | − | − | − |
| Prazosin | + | 1 | 0.47 | + | + | + |
| Primidone | − | 2 | 0.81 | + | + | + |
| Promazine | − | 1 | 1.25 | + | + | + |
| Promethazine | − | 1 | 1.29 | + | + | + |
| Propranolol | − | 1 | 0.74 | + | + | + |
| Pyridostigmine | − | 3 | 0.53 | + | + | + |
| Quetiapine | − | 1 | 0.85 | + | + | + |
| Quinidine | + | 1 | 0.53 | + | + | + |
| Ramelteon | − | 1 | 1.24 | + | + | + |
| Ranitidine | + | 3 | 0.45 | + | − | − |
| Reboxetine | − | 1 | 0.98 | + | + | + |
| Reserpine | + | 1 | 0.59 | + | + | + |
| Riluzole | − | 1 | 0.77 | + | + | + |
| Risperidone | − | 1 | 0.45 | + | + | + |
| Ritonavir | + | 2 | 0.12 | + | − | − |
| Rivastigmine | − | 1 | 1.05 | + | + | + |
| Rizatriptan | − | 1 | 0.51 | + | + | + |
| Ropinirole | − | 1 | 0.73 | + | + | + |
| Rosuvastatin | − | 3 | −1.04 | − | − | + |
| Saquinavir | + | 2 | 0.09 | + | − | − |
| Scopolamine | − | 1 | 0.23 | + | + | + |
| Selegiline | − | 1 | 1.44 | + | + | + |
| Sertraline | + | 1 | 1.25 | + | + | + |
| Simvastatin acid | − | 2 | 1.06 | + | + | + |
| Sulfasalazine | − | 2 | −0.81 | − | − | − |
| Sulpiride | − | 3 | −0.24 | + | + | + |
| Sumatriptan | − | 1 | 0.44 | + | + | + |
| Tacrine | − | 1 | 0.90 | + | + | + |
| Talinolol | + | 3 | 0.22 | + | − | − |
| Temazepam | − | 1 | 0.79 | + | + | + |
| Terfenadine | + | 2 | 0.86 | + | − | − |
| Thioridazine | + | 1 | 1.29 | + | + | + |
| Tiagabine | − | 2 | 0.34 | + | + | + |
| Topiramate | − | 3 | 0.06 | + | + | + |
| Trazodone | − | 2 | 1.21 | + | + | + |
| Trifluoperazine | − | 1 | 1.07 | + | + | + |
| Trimethoprim | + | 3 | 0.46 | + | − | + |
| Trimipramine | − | 2 | 1.42 | + | + | + |
| Tropisetron | − | 1 | 0.99 | + | + | + |
| Varenicline | − | 3 | 0.52 | + | + | + |
| Venlafaxine | − | 1 | 0.91 | + | + | + |
| Verapamil | + | 1 | 0.78 | + | + | + |
| Warfarin | − | 2 | 0.36 | + | + | + |
| Zaleplon | − | 2 | 1.03 | + | + | + |
| Zidovudine | − | 1 | −0.49 | − | − | + |
| Ziprasidone | + | 2 | 0.90 | + | − | + |
| Zolmitriptan | + | 1 | 0.36 | + | + | + |
| Zolpidem | − | 1 | 1.39 | + | + | + |
| Zonisamide | − | 1 | 0.43 | + | + | + |
VolSurf+ descriptor CACO2 for passive permeability.
cPermeability is positive when VolSurf+ CACO2 descriptor is above −0.3.
3.4. Outliers analysis
Fifteen drugs (10% of the data set) were incorrectly predicted. Eleven of these: amisulpride, aprepitant, chloroquine, clozapine, enoxacin, gabapentin, paliperidone, rosuvastatin, trimethoprim, ziprasidone, and zidovudine were false negatives (incorrectly predicted as BBB−). Four drugs (enoxacin, gabapentin, rosuvastatin, and zidovudine) had false negative predictions due to poor passive permeability calculations. The remaining seven drugs were false negatives since they were not BDDCS class 1. Saturable uptake processes have been implicated in the transport of amisulpride [125] and gabapentin [126,127] into the brain. Four other false negatives have been reported to be substrates for uptake transporters expressed in the brain [128]: rosuvastatin (OAT3, OATP1A2, OATP1B1, and OATP2B1) [129–131], zidovudine (OAT1, OAT2, OAT3, and OAT4) [132], trimethoprim (OCT2) [133], and chloroquine (OCT2) [133]. Paliperidone is a false negative since its BBB assignment is BBB+ class based on its category as a neurodrug [12], even though its brain/plasma ratio in rats is 0.04 [134], numerically making it BBB− based on our stated criteria. Clozapine and ziprasidone were incorrectly predicted as BBB− because they are non-class 1 Pgp substrates as noted above. However, they are borderline Pgp substrates: ER was under 2.5 in the Borst cell line and under 2 in the NIH cell line and is only predicted as BBB− since we chose to make the assignment only based on Borst cell line data when differences in prediction was observed for Borst versus NIH cell lines.
The remaining 4 of the 15 incorrectly predicted drugs (colchicine, itraconazole, ketoconazole, and loratadine) are false positives. Itraconazole, ketoconazole, and loratadine were assigned as Pgp non-substrates in our study based on their ER values. However, loratadine’s average ER values are borderline with respect to the thresholds adopted in this work (1.9 compared to 2 in Borst cell lines and 7.4 compared to 8.5 in NIH cell lines). Moreover, Polli and colleagues [19] confirmed the influence of Pgp efflux in loratadine’s permeability via the potent Pgp inhibitor elacridar. They also recognized that the MDCK-MDR1 system used tends to be insensitive to highly permeable drugs, such as ketoconazole and itraconazole. Ketoconazole has been confirmed to be a Pgp substrate in the Caco-2 cell line at low, but not at high, concentrations [135], suggesting that Pgp transport may be saturable for this drug.
Five out of 122 extensively metabolized (class 1 plus class 2) drugs in our study had calculated CACO2 descriptor below −0.3 defining them as poorly permeable drugs. Four of these 5 extensively metabolized drugs (dipyridamole, labetalol, sulfasalazine, and zidovudine) were found to be metabolized mainly by non-CYP enzymes [42]. Thus, it could be possible that the overlap between extensive metabolism and high permeability could be less accurate when the major metabolizing enzyme is not a CYP.
3.5. Molecular properties and brain disposition
The biological factors that influence CNS penetration were analyzed with respect to the parameters on which the Lipinski BBB and Norinder-Haeberlein criteria were defined. For classes BBB+, BBB−, Pgp+, Pgp−, BDDCS class 1, BDDCS classes 2-3-4, cPermeability+ and cPermeability−, the average and the standard deviation of several drug properties were calculated and are reported in Fig. 6. Molecular properties considered were CLogP, molecular weight, and the oxygen, nitrogen, hydrogen bond acceptor, and hydrogen bond donor counts. It has been reported that a high polar surface area and/or high molecular weight increase the likelihood of an NME being BBB− [136]. When we analyzed compounds that have a high molecular weight and/or high counts of nitrogen and oxygen (surrogate for polar surface area), we found that these compounds are also likely to be Pgp substrates, having low permeabilities, and/or being non-BDDCS class 1 drugs. The results of our work suggest that these latter properties are the driving force for these drugs being BBB−. Hence, we believe that these molecular properties are predicting the NMEs’ permeabilities, ability to interact with Pgp, and/or its BDDCS classification (metabolism and solubility), and as a consequence their BBB outcome. No significant difference in terms of average CLogP was observed for Pgp substrates and non-substrates, BDDCS classes 1 versus classes 2, 3 and 4, and BBB+ versus BBB−drugs.
Fig. 6.
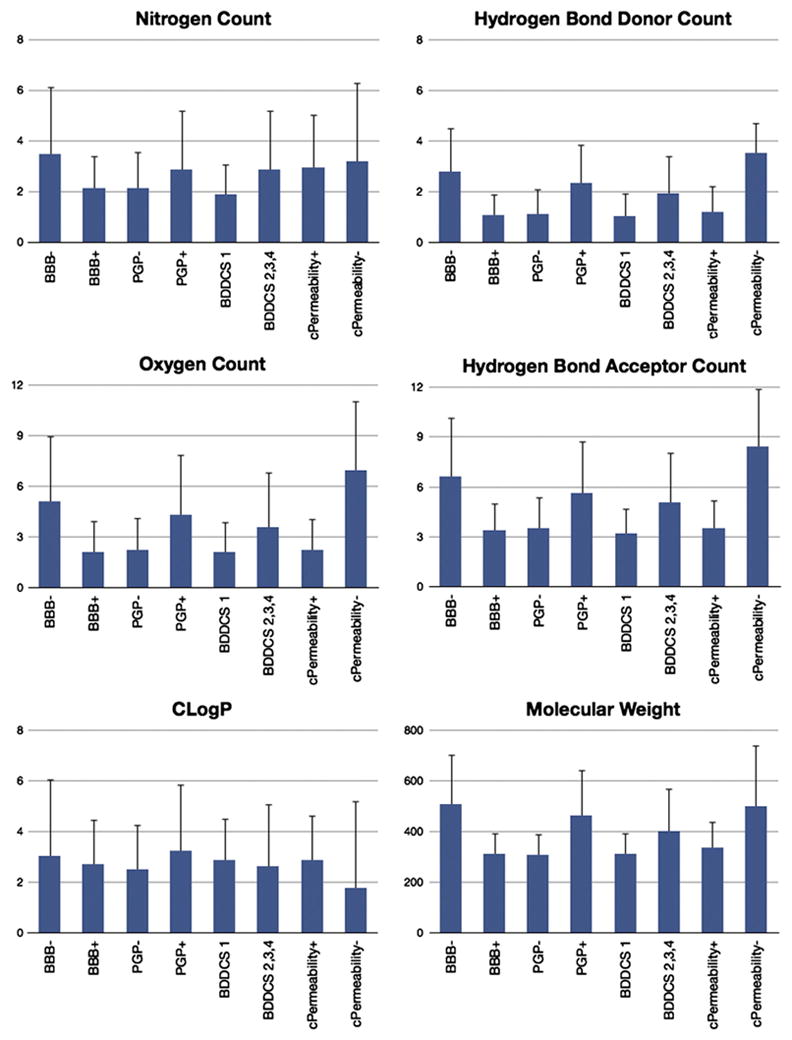
Average and standard deviation for the following drug properties: nitrogen, hydrogen bond donor, oxygen and hydrogen bond acceptor counts, molecular weight, CLogP. cPermeability is “+” if VolSurf+ CACO2 descriptor is over −0.3, otherwise it is “−”.
Surprisingly, based on this data set, there seems to be a relationship between the Lipinski rules of 5 (Ro5) violations [137] and Pgp efflux: drugs having no violation to Ro5s tend to be Pgp non-substrates 79% of the time (based on 121 drugs), drugs having one violation are Pgp substrates 61% (based on 23 drugs), and drugs having two or more violations are Pgp substrates 89% (based on 9 drugs). Since our data set is mostly composed of marketed CNS drugs, it is possible that these observations may be skewed due to a sampling issue. To test this, we analyzed the relationship between Ro5 and Pgp for 34 drugs excluded from our data set for noncompliant criteria but with available efflux data. Twenty-five of the 34 drugs had no Ro5 violations, with only 68% of them being Pgp non-substrates. This decrease (from 79% to 68%) may indicate an artifact associated with Pgp efflux optimization during the drug discovery process for CNS drugs. On the other hand, 3 out of 5 (60%) drugs with one Lipinski Ro5 violation and all the 4 drugs having 2 or more Lipinski violations were Pgp substrates. Therefore, a drug having more than one Ro5 violation is highly likely to be a Pgp substrate, while the contrary is not necessarily true.
3.6. Case Study: predicting the brain disposition of antihistamines with BDDCS
The sedative effect of first generation antihistamines [138] has been associated with brain penetration, where drugs are believed not to be Pgp substrates [139]. We previously evaluated 64 H1-histamine receptor antagonists and the severity of sedation as a side effect associated to these drugs [140]. We noted that Pgp transport is the major difference between sedating and non-sedating antihistamines, and that it can be used to predict the drug’s sedative effect in combination with the adjusted dose for that drug. Using our classification tree, 22 H1 antagonists exerting sedative effect were accurately predicted to be BBB+, and all the 6 non-sedating H1 antagonists were predicted as BBB− (Fig. 7 and Supplemental Table S7). Amitriptyline and risperidone have conflicting reports on being Pgp substrates (Supplemental Table S1). However, according to our classification tree, they are correctly predicted to be BBB+ and to exert CNS sedative effects as they are BDDCS class 1 drugs.
Fig. 7.
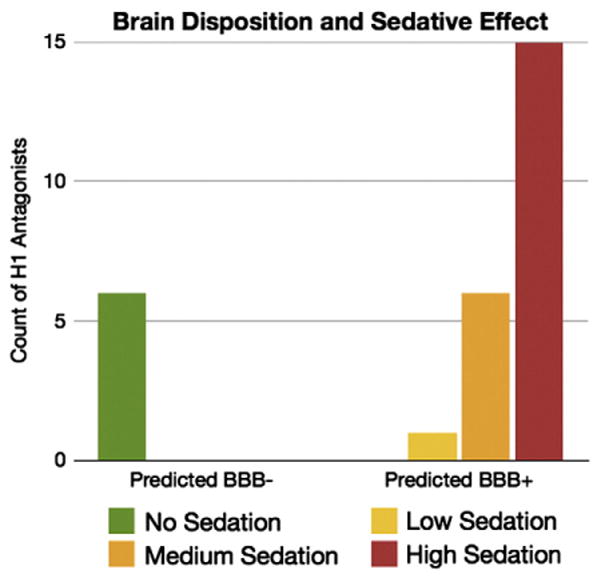
Predicted blood–brain barrier penetration for 28 drugs with affinity for H1 receptor and associated severity of sedative effect. BBB− drugs do not exert CNS side-effects.
4. Integration of findings and recommendations
We hereby introduce a novel perspective for brain disposition evaluation for orally administered drugs. In the past, Pgp efflux and passive permeability had been identified as crucial factors for CNS penetration. Pgp is a known gatekeeper capable of limiting exposure of CNS targets to those drugs that are Pgp substrates. This is contrary to what is observed in the leakier gut membrane, where Pgp only limits the permeability of poorly permeable/poorly metabolized compounds (BDDCS classes 3 and 4). In knockout mice studies, it has been shown that Pgp-mediated efflux reduces the brain penetration of class 1 substrate drugs, like verapamil [141] and bromocriptine [142]. However, even when Pgp efflux affects BDDCS class 1 drugs, the extent of the drug penetrating the brain is usually at least 10% of the plasma concentration, which characterizes it as a BBB+ drug. Consequently, 98% of BDDCS class 1 drugs in our data set are BBB+. In contrast, Pgp substrates within BDDCS classes 2, 3 and 4 were incapable of crossing the BBB around 75% of the time (and 84% if the ambiguous data on loratadine, ketoconazole, and itraconazole were not considered). For about 40% of BBB− drugs, we determined that the major limiting factor was passive permeability. Even though adequate in vitro permeability data was unavailable for our data set, we found that the in silico calculated permeability (Vol-Surf+ descriptor CACO2) is a suitable surrogate. Three simple criteria– calculated permeability, Pgp efflux, and BDDCS assignment – were employed in a classification tree that correctly classified over 90% of our 153 drug data set, with the prediction accuracy being over 87% for both BBB+ and BBB− drugs.
From our data mining, important considerations about the in vitro systems used and the role of uptake transporters in the brain have emerged. We noted that not only the cell lines used but also the source from where it is retrieved can strongly influence the experimental outcome. We observed a marked difference between the efflux ratios of certain drugs in two commonly used in vitro Pgp systems, the Borst and NIH MDCK-MDR1 cell lines. We have observed that ER values tend to be significantly higher for Pgp substrates in the NIH cell lines in agreement with the higher protein expression levels reported [33]. For this latter point, we recommend that different thresholds for defining Pgp impact may need to be defined but the numerical basis (particularly for the NIH cell line) should be further investigated. As pointed out previously by Polli and colleagues [19], highly permeable Pgp substrates tend to not be recognized in the MDCK-MDR1 system. Being highly permeable, these compounds will be either BDDCS class 1 or 2 drugs. Since BDDCS class 1 drugs appeared not to be affected by Pgp in the brain, we suggest that only BDDCS class 2 drugs should be further tested at lower concentrations in vitro or even in an in vivo Pgp knockout model to confirm the Pgp effect on its disposition.
There may be some non-BDDCS class 1 Pgp substrate drugs that may be able or can be optimized to cross the BBB. Data suggests that this may be attained if the drug has a good affinity for uptake transporters in the brain. Further works needs to be done to characterize the important uptake transporters in the brain and good in vitro systems for evaluating these uptake transporters should be implemented. These could avoid prematurely excluding interesting non-BDDCS class 1 Pgp substrate candidate drugs.
Increasing attention has recently been placed on investigating the role of BCRP (ABCG2) on limiting the brain penetration of drugs. It may be possible that BCRP efflux should be incorporated into our classification tree for improving large scale brain penetration prediction. Our results suggest that BCRP is not a major determinant for a significant fraction of drugs in this data set. Ignoring the importance of BCRP efflux should result in a high number of false positive predictions. We only observed 5 false positives, where 4 of them are probably Pgp substrates not recognized by the in vitro assay as discussed above. It may be that BCRP limits the brain penetration of some of the drugs in our data set by acting in tandem with Pgp as suggested by Polli et al. for lapatinib [143].
A link between BDDCS class 1 drugs and CNS side effects emerged when we applied our classification tree to a set of 28 drugs with affinity for the H1 antihistamine receptor. All the predicted BBB+ drugs were known to have sedative effects, while all the BBB− drugs were non-sedating. Furthermore, BDDCS class 1 amitriptyline and risperidone, which have been reported to be Pgp substrates, were predicted to be BBB+ and they are, in fact, sedating. For this reason, while the design of BDDCS class 1 drugs is desirable when the target is the brain, BDDCS class 1 drugs should be avoided or carefully monitored when the intended target is peripheral. In this latter case, selecting those BDDCS class 2 NMEs that are P-gp substrates is likely to result in reduced CNS-related side effects.
The theoretical basis for BDDCS is that the extent of metabolism predicts high versus low permeability rate. A high permeability rate could be observed due to paracellular pathway, transcellular pathway, and/or an uptake transporter. It has already been pointed out that extensive metabolism may not occur if the drug is predominantly absorbed via a paracellular pathway or uptake transport [16,144]. Therefore, extensive metabolism is likely to be a good predictor of passive permeability when transcellular transit is the main pathway for absorption [16,145]. We did observe that 5 extensively metabolized drugs in our data set (~4%) had low calculated permeabilities according to our defined criteria for brain penetration. Four of these 5 drugs are known to be mainly processed by non-CYP enzymes, contrary to what is expected for the majority of marketed drugs [146]. Even though a certain degree of inaccuracy for in silico calculations is expected, we are struck by these findings where we believe it is not a coincidence; that is, passive transcellular permeability may be predicted by CYP metabolism, but not necessarily by non-CYP metabolism. We are currently further investigating this hypothesis.
Even though there is an overlap between BCS and BDDCS class assignment for many drugs, it should not be assumed that BCS will be as good a predictor for brain penetration as BDDCS. According to our framework, when the drug is a Pgp substrate, it is excluded from the brain if it is a non-BDDCS class 1 drug. As mentioned above, BCS class 1 drugs can be BDDCS class 3 drugs if their absorptive mechanism is primarily via uptake transport (e.g. digoxin [147] and neostigmine [148]) or the paracellular pathway (e.g. sotalol [149]) in the intestine. Being that digoxin, neostigmine, and sotalol are Pgp substrates [19,23,150], they are correctly classified as being BBB−[42,62,151] using BDDCS while incorrect predictions arise using BCS. Hence, we recommend using caution when considering the use of BCS for predicting brain disposition.
Using in silico models such as Lipinski’s CNS rules and the VolSurf+ descriptor LgBB, one can achieve good prediction accuracies of about 80% that are suitable for compound prioritization and elimination in the early phases of drug discovery. In advanced phases, properties such as Pgp profile, in vitro permeability, and solubility should be assessed. At this stage of drug discovery, our decision scheme can better serve as a guideline for optimizing interesting candidates in a flexible way, as opposed to pure in silico-based structural considerations that constrict the portion of the chemical space being explored. Computational models for Pgp substrate/non-substrate could be incorporated, together with an in silico BDDCS model [16] and the VolSurf+ CACO2 model, to implement the computational equivalent of this decision scheme. With enough data and adequate sampling, such Pgp substrate/non-substrate models are quite possible, given that such models already exist for Pgp inhibitor/non-inhibitor data [152].
Simple molecular descriptors have been used for decades to predict CNS disposition. Even though our approach outperforms these models, it is interesting that different perspectives can achieve an overlapping amount of success in their predictions. This led us to investigate if the molecular descriptors were in fact linked to the biological factors influencing brain penetration. We observed that, on average, the molecular weight and the number of heteroatoms (either expressed as oxygens, nitrogens, hydrogen bond acceptors, and hydrogen bond donors) not only increased the chances of being BBB−, but also of being a Pgp substrate, having a low permeability, and being a non-BDDCS class 1 drug. Even if these rules of thumb are generally successful in their predictions and apply to more than half of the known drugs, they have large standard deviations and cannot be considered infallible. Drug discovery should not be strictly guided by such simple descriptors, but also by less intuitive properties. We suggest that for highly metabolized drugs, BBB penetration in humans can be optimized by increasing their solubility. Even if linking these two properties is not intuitive, Alelyunas et al. [153] found that, starting from the experimental solubility measures for 98 marketed CNS drugs, over 85% of them are highly soluble. Other recent work [154] suggests that solubility optimization can be achieved by manipulating the molecular symmetry of the drugs. Adopting such strategies, rather than drastically changing the molecular weight or number of heteroatoms, could result in optimal disposition properties while having a smaller impact on the bioactivity of interesting CNS candidates.
We also found that the probability of being a Pgp substrate increases with the number of violations to the Lipinski rules of five (Ro5) [137]. Approximately 79% of our drugs with no Ro5 violations are Pgp non-substrates, while about 89% of those with 2 or more violations are Pgp substrates. When an additional 34 drugs with known efflux ratios were analyzed, the correspondence between the lack of Pgp efflux and no Ro5 violations weakened, while Ro5 violations, particularly 2 or more, were confirmed to be a good predictor of Pgp efflux. Ro5 was developed to highlight good bioavailability via passive permeability. Therefore, a number of two or more Ro5 violations are likely to identify drugs that are not only Pgp substrates but also in BDDCS classes 3 and 4 (thus corresponding to drugs with suboptimal permeability). Shugarts and Benet have shown that Pgp can limit the intestinal fraction absorbed for class 3 and 4 drugs [15]. This observation explains what has been recently reported by Varma and colleagues: the Lipinski Ro5 models bioavailability because of its effect on the fraction absorbed in the intestine, but not the fraction metabolized in the gut and liver [155]. Shugarts and Benet have observed that most of the BDDCS class 3 and 4 drugs on the market can be absorbed in the intestine because they are substrates for uptake transporters in the gut [15]. Hence, we suggest that high-value NMEs usually considered unsuitable because they are not included in the physicochemical space for optimal oral bioavailability (e.g., two or more Ro5 violations) could be further investigated by understanding and optimizing their affinity for uptake transporters in the gut. Such an approach may lead to a larger area of chemical space being available for drug discovery.
5. Future prospects of BDDCS in drug discovery
Within the last decade, our laboratory has investigated the relationship between drug transport and drug disposition in the human body, particularly with respect to transporter-enzyme interplay. The transformation of information into knowledge has been possible in part due to a simple four class system: the BDDCS. We recognized that classes 1, 2, and 3 have different profiles with respect to brain disposition, as well as protein binding [16], intestinal absorption [15], and potential drug–drug interactions [15]. An in silico approach to predicting BDDCS class drugs solely based on their molecular structure has been proposed [16]. Using such knowledge, it is possible to guide the drug discovery process from the early phases in order to avoid facing toxicity and drug–drug interaction problems that usually emerge in the last stages, when failures becomes very expensive. In Fig. 8, we provide BDDCS-based strategies for achieving these goals.
Fig. 8.
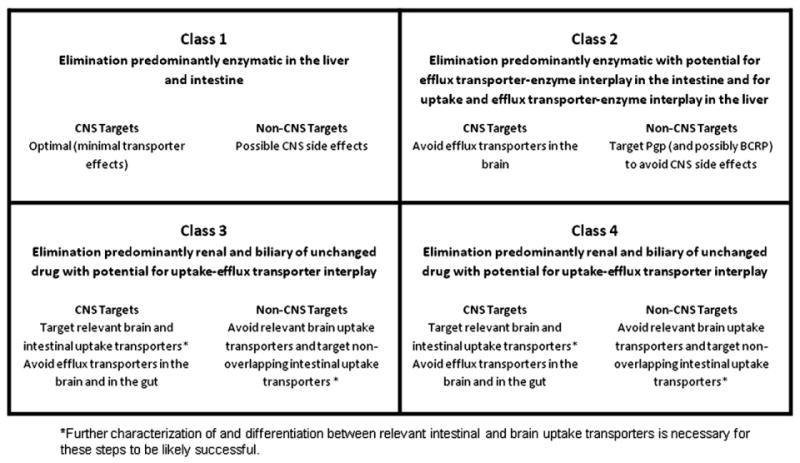
BDDCS in drug discovery.
We suggest that when looking for CNS drugs, NMEs predicted as class 1 drugs should be prioritized, since they will likely have no problem with intestinal absorption, protein binding, and brain penetration. In contrast, when the candidate drug is for peripheral use, class 1 may need to be avoided, in order to exclude CNS side effects. In these cases, Pgp may even need to be switched from an antitarget to a target, as to prevent brain entry, a mechanism observed for antihistamines [140]. Class 2 Pgp substrate drugs could be ideal peripheral candidates, since they easily penetrate the gut membrane by passive permeability. For these drugs, in vitro permeability testing may be unnecessary. However, it is important to identify the fraction of the NME dose that is metabolized in the intestine and in the liver, to forecast potential toxicity and drug–drug interaction (transporter–enzyme interplay). Nevertheless, since BDDCS class 2 drugs are highly lipophilic, these NMEs should be tested for hERG, CYP, and Pgp inhibition, which have been linked to the lipophilicity of the drug [152,156,157]. Not likely to have issues related to metabolism, BDDCS class 3 NMEs should be optimized to have good binding affinity to intestinal uptake transporters in order to be well absorbed. Use of this biological knowledge could represent a gateway for exploring new portions of the chemical space that were previously unexplored by medicinal chemists.
Supplementary Material
Acknowledgments
This work was supported in part by NIH grants GM-61390, GM-75900 and GM-90457 (LZB), by NIH grants GM-095952, MH-084690, and CA-118100, and by the Villum Foundation CDSB (TIO), by an AFPE Pre-Doctoral Fellowship and a NIH Kirschstein-NRSA Training Grant T32-GM007175 (CAL), and by Molecular Discovery, Ltd. (FB).
Abbreviations
- BDDCS
Biopharmaceutics drug disposition classification system
- BBB
Blood–brain barrier
- Pgp
P-glycoprotein
- CNS
Central nervous system
Appendix A
Supplementary data to this article can be found online at doi:10. 1016/j.addr.2011.12.008.
References
- 1.Hansch C, Steward AR, Iwasa J. The correlation of localization rates of benzeneboronic acids in brain and tumor tissue with substituent constants. Mol Pharmacol. 1965;1:87–92. [PubMed] [Google Scholar]
- 2.Hansch C, Bjorkroth JP, Leo A. Hydrophobicity and central nervous system agents: on the principle of minimal hydrophobicity in drug design. J Pharm Sci. 1987;76:663–687. doi: 10.1002/jps.2600760902. [DOI] [PubMed] [Google Scholar]
- 3.Levin VA. Relationship of octanol/water partition coefficient and molecular weight to rat brain capillary permeability. J Med Chem. 1980;23:682–684. doi: 10.1021/jm00180a022. [DOI] [PubMed] [Google Scholar]
- 4.Ghose AK, Herbertz T, Hudkins RL, Dorsey BD, Mallamo JP. Knowledge-based, central nervous system (CNS) lead selection and lead optimization for CNS drug discovery. ACS Chem Neurosci. 2012;3:50–68. doi: 10.1021/cn200100h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Norinder U, Haeberlein M. Computational approaches to the prediction of the blood–brain distribution. Adv Drug Deliv Rev. 2002;54:291–313. doi: 10.1016/s0169-409x(02)00005-4. [DOI] [PubMed] [Google Scholar]
- 6.Clark DE. In silico prediction of blood–brain barrier permeation. Drug Discov Today. 2003;8:927–933. doi: 10.1016/s1359-6446(03)02827-7. [DOI] [PubMed] [Google Scholar]
- 7.Ecker GF, Noe CR. In silico prediction models for blood–brain barrier permeation. Curr Med Chem. 2004;11:1617–1628. doi: 10.2174/0929867043365071. [DOI] [PubMed] [Google Scholar]
- 8.Cordon-Cardo C, O’Brien JP, Casals D, Rittman-Grauer L, Biedler JL, Melamed MR, Bertino JR. Multidrug-resistance gene (P-glycoprotein) is expressed by endothelial cells at blood–brain barrier sites. Proc Natl Acad Sci. 1989;86:695–698. doi: 10.1073/pnas.86.2.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cornwell MM, Tsuruo T, Gottesman MM, Pastan I. ATP-binding properties of P glycoprotein from multidrug-resistant KB cells. FASEB J. 1987;1:51–54. doi: 10.1096/fasebj.1.1.2886389. [DOI] [PubMed] [Google Scholar]
- 10.Schinkel AH, Wagenaar E, Mol CA, van Deemter L. P-glycoprotein in the blood–brain barrier of mice influences the brain penetration and pharmacological activity of many drugs. J Clin Invest. 1996;97:2517–2524. doi: 10.1172/JCI118699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mahar Doan KM, Humphreys JE, Webster LO, Wring SA, Shampine LJ, Serabjit-Singh CJ, Adkison KK, Polli JW. Passive permeability and P-glycoprotein-mediated efflux differentiate central nervous system (CNS) and non-CNS marketed drugs. J Pharmacol Exp Ther. 2002;303:1029–1037. doi: 10.1124/jpet.102.039255. [DOI] [PubMed] [Google Scholar]
- 12.Wager TT, Ramalakshmi YC, Hou X, Troutman MD, Verhoest PR, Villalobos A, Will Y. Defining desirable central nervous system drug space through the alignment of molecular properties, in vitro ADME, and safety attributes. ACS Chem Neurosci. 2010;1:420–434. doi: 10.1021/cn100007x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu CY, Benet LZ. Predicting drug disposition via application of BCS: transport/absorption/elimination interplay and development of a biopharmaceutics drug disposition classification system. Pharm Res. 2005;22:11–23. doi: 10.1007/s11095-004-9004-4. [DOI] [PubMed] [Google Scholar]
- 14.Custodio JM, Wu CY, Benet LZ. Predicting drug disposition, absorption/elimination/ transporter interplay and the role of food on drug absorption. Adv Drug Deliv Rev. 2008;60:717–733. doi: 10.1016/j.addr.2007.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shugarts S, Benet LZ. The role of transporters in the pharmacokinetics of orally administered drugs. Pharm Res. 2009;26:2039–2054. doi: 10.1007/s11095-009-9924-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Broccatelli F, Cruciani G, Benet LZ, Oprea TI. BDDCS class prediction for new molecular entities. Mol Pharmaceutics. 2012;9:570–580. doi: 10.1021/mp2004302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cruciani G, Crivori P, Carrupt P-A, Testa B. Molecular fields in quantitative structure-permeation relationships: the VolSurf approach. J Mol Struct (THEO-CHEM) 2000;503:7–30. [Google Scholar]
- 18.Pajouhesh H, Lenz GR. Medicinal chemical properties of successful central nervous system drugs. NeuroRx. 2005;2:541–553. doi: 10.1602/neurorx.2.4.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Polli JW, Wring SA, Humphreys JE, Huang L, Morgan JB, Webster LO, Serabjit-Singh CS. Rational use of in vitro P-glycoprotein assays in drug discovery. J Pharmacol Exp Ther. 2001;299:620–628. [PubMed] [Google Scholar]
- 20.Troutman MD, Thakker DR. Novel experimental parameters to quantify the modulation of absorptive and secretory transport of compounds by P-glycoprotein in cell culture models of intestinal epithelium. Pharm Res. 2003;20:1210–1224. doi: 10.1023/a:1025001131513. [DOI] [PubMed] [Google Scholar]
- 21.Chen C, Hanson E, Watson JW, Lee JS. P-glycoprotein limits the brain penetration of nonsedating but not sedating H1-antagonists. Drug Metab Dispos. 2003;31:312–318. doi: 10.1124/dmd.31.3.312. [DOI] [PubMed] [Google Scholar]
- 22.Chen C, Mireles RJ, Campbell SD, Lin J, Mills JB, Xu JJ, Smolarek TA. Differential interaction of 3-hydroxy-3-methylglutaryl-CoA reductase inhibitors with ABCB1, ABCC2, and OATP1B1. Drug Metab Dispos. 2005;33:537–546. doi: 10.1124/dmd.104.002477. [DOI] [PubMed] [Google Scholar]
- 23.Wang Q, Rager JD, Weinstein K, Kardos PS, Dobson GL, Li J, Hidalgo IJ. Evaluation of the MDR-MDCK cell line as a permeability screen for the blood–brain barrier. Int J Pharm. 2005;288:349–359. doi: 10.1016/j.ijpharm.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 24.Kim WY, Benet LZ. P-glycoprotein (P-gp/MDR1)-mediated efflux of sex-steroid hormones and modulation of P-gp expression in vitro. Pharm Res. 2004;21:1284–1293. doi: 10.1023/b:pham.0000033017.52484.81. [DOI] [PubMed] [Google Scholar]
- 25.Eriksson UG, Dorani H, Karlsson J, Fritsch H, Hoffmann KJ, Olsson L, Sarich TC, Wall U, Schutzer KM. Influence of erythromycin on the pharmacokinetics of ximelagatran may involve inhibition of P-glycoprotein-mediated excretion. Drug Metab Dispos. 2006;34:775–782. doi: 10.1124/dmd.105.008607. [DOI] [PubMed] [Google Scholar]
- 26.Huang L, Wang Y, Grimm S. ATP-dependent transport of rosuvastatin in membrane vesicles expressing breast cancer resistance protein. Drug Metab Dispos. 2006;34:738–742. doi: 10.1124/dmd.105.007534. [DOI] [PubMed] [Google Scholar]
- 27.Baltes S, Gastens AM, Fedrowitz M, Potschka H, Kaever V, Loscher W. Differences in the transport of the antiepileptic drugs phenytoin, levetiracetam and carbamazepine by human and mouse P-glycoprotein. Neuropharmacology. 2007;52:333–346. doi: 10.1016/j.neuropharm.2006.07.038. [DOI] [PubMed] [Google Scholar]
- 28.Carrara S, Reali V, Misiano P, Dondio G, Bigogno C. Evaluation of in vitro brain penetration: optimized PAMPA and MDCKII-MDR1 assay comparison. Int J Pharm. 2007;345:125–133. doi: 10.1016/j.ijpharm.2007.05.057. [DOI] [PubMed] [Google Scholar]
- 29.Obradovic T, Dobson GG, Shingaki T, Kungu T, Hidalgo IJ. Assessment of the first and second generation antihistamines brain penetration and role of P-glycoprotein. Pharm Res. 2007;24:318–327. doi: 10.1007/s11095-006-9149-4. [DOI] [PubMed] [Google Scholar]
- 30.Summerfield SG, Read K, Begley DJ, Obradovic T, Hidalgo IJ, Coggon S, Lewis AV, Porter RA, Jeffrey P. Central nervous system drug disposition: the relationship between in situ brain permeability and brain free fraction. J Pharmacol Exp Ther. 2007;322:205–213. doi: 10.1124/jpet.107.121525. [DOI] [PubMed] [Google Scholar]
- 31.de Souza J, Benet LZ, Huang Y, Storpirtis S. Comparison of bidirectional lamivudine and zidovudine transport using MDCK, MDCK-MDR1, and Caco-2 cell monolayers. J Pharm Sci. 2009;98:4413–4419. doi: 10.1002/jps.21744. [DOI] [PubMed] [Google Scholar]
- 32.Feng B, Mills JB, Davidson RE, Mireles RJ, Janiszewski JS, Troutman MD, de Morais SM. In vitro P-glycoprotein assays to predict the in vivo interactions of P-glycoprotein with drugs in the central nervous system. Drug Metab Dispos. 2008;36:268–275. doi: 10.1124/dmd.107.017434. [DOI] [PubMed] [Google Scholar]
- 33.Park MS, Okochi H, Benet LZ. Is ciprofloxacin a substrate of P-glycoprotein? Arch Drug Inf. 2011;4:1–9. doi: 10.1111/j.1753-5174.2010.00032.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Benet LZ, Broccatelli F, Oprea TI. BDDCS applied to over 900 drugs. AAPS J. 2011;13:519–547. doi: 10.1208/s12248-011-9290-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kodaira H, Kusuhara H, Ushiki J, Fuse E, Sugiyama Y. Kinetic analysis of the cooperation of P-glycoprotein (P-gp/Abcb1) and breast cancer resistance protein (Bcrp/Abcg2) in limiting the brain and testis penetration of erlotinib, flavopiridol, and mitoxantrone. J Pharmacol Exp Ther. 2010;333:788–796. doi: 10.1124/jpet.109.162321. [DOI] [PubMed] [Google Scholar]
- 36.Friden M, Winiwarter S, Jerndal G, Bengtsson O, Wan H, Bredberg U, Hammarlund-Udenaes M, Antonsson M. Structure-brain exposure relationships in rat and human using a novel data set of unbound drug concentrations in brain interstitial and cerebrospinal fluids. J Med Chem. 2009;52:6233–6243. doi: 10.1021/jm901036q. [DOI] [PubMed] [Google Scholar]
- 37.Sasongko L, Link JM, Muzi M, Mankoff DA, Yang X, Collier AC, Shoner SC, Unadkat JD. Imaging P-glycoprotein transport activity at the human blood–brain barrier with positron emission tomography. Clin Pharmacol Ther. 2005;77:503–514. doi: 10.1016/j.clpt.2005.01.022. [DOI] [PubMed] [Google Scholar]
- 38.McAuley BJ, Schroeder JS. The use of diltiazem hydrochloride in cardiovascular disorders. Pharmacotherapy. 1982;2:121–133. doi: 10.1002/j.1875-9114.1982.tb04518.x. [DOI] [PubMed] [Google Scholar]
- 39.Naito K, Nagao T, Otsuka M, Harigaya S, Nakajima H. Penetration into and elimination from the cerebrospinal fluid of diltiazem, a calcium antagonist, in anesthetized rabbits. Arzneimittelforschung. 1986;36:25–28. [PubMed] [Google Scholar]
- 40.Zaman R, Jack DB, Kendall MJ. The penetration of acebutolol and its major metabolite, diacetolol, into human cerebrospinal fluid and saliva. Br J Clin Pharmacol. 1981;12:427–429. doi: 10.1111/j.1365-2125.1981.tb01239.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brenner M, Haass A, Jacobi P, Schimrigk K. Amantadine sulphate in treating Parkinson’s disease: clinical effects, psychometric tests and serum concentrations. J Neurol. 1989;236:153–156. doi: 10.1007/BF00314331. [DOI] [PubMed] [Google Scholar]
- 42.Lacy CF, Armstrong LL, Goldman MP, Lance LL. Lexi-Comp’s Drug Information Handbook 2011–2012: A Comprehensive Resource for All Clinicians and Healthcare Professionals. 20. Lexi-Comp, Inc; Hudson, Ohio: 2011. [Google Scholar]
- 43.Edwards JE, Brouwer KR, McNamara PJ. GF120918, a P-glycoprotein modulator, increases the concentration of unbound amprenavir in the central nervous system in rats. Antimicrob Agents Chemother. 2002;46:2284–2286. doi: 10.1128/AAC.46.7.2284-2286.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cheng Z, Zhang J, Liu H, Li Y, Zhao Y, Yang E. Central nervous system penetration for small molecule therapeutic agents does not increase in multiple sclerosis- and Alzheimer’s disease-related animal models despite reported blood–brain barrier disruption. Drug Metab Dispos. 2010;38:1355–1361. doi: 10.1124/dmd.110.033324. [DOI] [PubMed] [Google Scholar]
- 45.Kurz H. The permeation of drugs across the so-called blood–brain-barrier at low temperature. Experientia. 1964;20:96–97. doi: 10.1007/BF02151263. [DOI] [PubMed] [Google Scholar]
- 46.Simons FE, Fraser TG, Reggin JD, Simons KJ. Comparison of the central nervous system effects produced by six H1-receptor antagonists. Clin Exp Allergy. 1996;26:1092–1097. [PubMed] [Google Scholar]
- 47.Cruickshank JM, Neil-Dwyer G, Cameron MM, McAinsh J. Beta blockers and the central nervous system (CNS). Abstract. 6th Sci. Meet. Int. Soc. Hypertension; Goteborg, Sweden. 1979. [Google Scholar]
- 48.Preskorn SH, Othmer SC. Evaluation of bupropion hydrochloride: the first of a new class of atypical antidepressants. Pharmacotherapy. 1984;4:20–34. doi: 10.1002/j.1875-9114.1984.tb03306.x. [DOI] [PubMed] [Google Scholar]
- 49.Nehlig A, Daval JL, Debry G. Caffeine and the central nervous system: mechanisms of action, biochemical, metabolic and psychostimulant effects. Brain Res Rev. 1992;17:139–170. doi: 10.1016/0165-0173(92)90012-b. [DOI] [PubMed] [Google Scholar]
- 50.Kossmann T, Hans V, Stocker R, Imhof HG, Joos B, Trentz O, Morganti-Kossmann MC. Penetration of cefuroxime into the cerebrospinal fluid of patients with traumatic brain injury. J Antimicrob Chemother. 1996;37:161–167. doi: 10.1093/jac/37.1.161. [DOI] [PubMed] [Google Scholar]
- 51.Cetirizine Hydrochloride. Pfizer Inc; New York, NY: 1999. Product Information: Zyrtec®. [Google Scholar]
- 52.Adjene JO, Adenowo TK. Histological studies of the effects of chronic administration of chloroquine on the inferior colliculus of the adult Wistar rat. J Med Biomed Res. 2005;4:83–87. [Google Scholar]
- 53.Chlorothiazide Sodium Intravenous. Merck & Co., Inc; West Point, PA: 1999. Product Information: Diuril®. [Google Scholar]
- 54.Gilman AG, Goodman LS, Rall TW, Murad F. Goodman and Gilman’s The Pharmacological Basis of Therapeutics. 7. Macmillan Publishing Co; New York, NY: 1985. [Google Scholar]
- 55.Lin JH. Pharmacokinetic and pharmacodynamic properties of histamine H2-receptor antagonists. Relationship between intrinsic potency and effective plasma concentrations. Clin Pharmacokinet. 1991;20:218–236. doi: 10.2165/00003088-199120030-00004. [DOI] [PubMed] [Google Scholar]
- 56.Anonymous. Anon: Phase III Profiles, I. Biomega Corporation; Skokie, IL: 1990. Clarithromycin; pp. 1–4. [Google Scholar]
- 57.Turner RB, Sperber SJ, Sorrentino JV, O’Connor RR, Rogers J, Batouli AR, Gwaltney JM., Jr Effectiveness of clemastine fumarate for treatment of rhinorrhea and sneezing associated with the common cold. Clin Infect Dis. 1997;25:824–830. doi: 10.1086/515546. [DOI] [PubMed] [Google Scholar]
- 58.Desrayaud S, Guntz P, Scherrmann JM, Lemaire M. Effect of the P-glycoprotein inhibitor, SDZ PSC 833, on the blood and brain pharmacokinetics of colchicine. Life Sci. 1997;61:153–163. doi: 10.1016/s0024-3205(97)00370-6. [DOI] [PubMed] [Google Scholar]
- 59.Lensmeyer GL, Wiebe DA, Carlson IH, Subramanian R. Concentrations of cyclosporin A and its metabolites in human tissues postmortem. J Anal Toxicol. 1991;15:110–115. doi: 10.1093/jat/15.3.110. [DOI] [PubMed] [Google Scholar]
- 60.Balis FM, Lester CM, Chrousos GP, Heideman RL, Poplack DG. Differences in cerebrospinal fluid penetration of corticosteroids: possible relationship to the prevention of meningeal leukemia. J Clin Oncol. 1987;5:202–207. doi: 10.1200/JCO.1987.5.2.202. [DOI] [PubMed] [Google Scholar]
- 61.Schwartz MA, Koechlin BA, Postma E, Palmer S, Krol G. Metabolism of diazepam in rat, dog, and man. J Pharmacol Exp Ther. 1965;149:423–435. [PubMed] [Google Scholar]
- 62.Mayer U, Wagenaar E, Dorobek B, Beijnen JH, Borst P, Schinkel AH. Full blockade of intestinal P-glycoprotein and extensive inhibition of blood–brain barrier P-glycoprotein by oral treatment of mice with PSC833. J Clin Invest. 1997;100:2430–2436. doi: 10.1172/JCI119784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Au-Yeung SC, Rurak DW, Gruber N, Riggs KW. A pharmacokinetic study of diphenhydramine transport across the blood–brain barrier in adult sheep: potential involvement of a carrier-mediated mechanism. Drug Metab Dispos. 2006;34:955–960. doi: 10.1124/dmd.105.007898. [DOI] [PubMed] [Google Scholar]
- 64.Stein MB, Black B, Brown TM, Uhde TW. Lack of efficacy of the adenosine re-uptake inhibitor dipyridamole in the treatment of anxiety disorders. Biol Psychiatry. 1993;33:647–650. doi: 10.1016/0006-3223(93)90105-m. [DOI] [PubMed] [Google Scholar]
- 65.Domperidone Maleate. Janssen-Ortho; Toronto, Ontario, Canada: 1999. Product Information: Motilium®. [Google Scholar]
- 66.Scheld WM. Quinolone therapy for infections of the central nervous system. Rev Infect Dis. 1989;11(Suppl 5):S1194–S1202. doi: 10.1093/clinids/11.supplement_5.s1194. [DOI] [PubMed] [Google Scholar]
- 67.Perrin VL. Clinical pharmacokinetics of ergotamine in migraine and cluster headache. Clin Pharmacokinet. 1985;10:334–352. doi: 10.2165/00003088-198510040-00004. [DOI] [PubMed] [Google Scholar]
- 68.Bergan T, Hellum KB, Schreiner A, Digranes A, Josefsson K. Passage of erythromycin into human suction skin blisters. Curr Ther Res. 1982;32:597–603. [Google Scholar]
- 69.Piredda S, Monaco F. Ethosuximide in tears, saliva, and cerebrospinal fluid. Ther Drug Monit. 1981;3:321–323. doi: 10.1097/00007691-198104000-00001. [DOI] [PubMed] [Google Scholar]
- 70.Hande KR, Wedlund PJ, Noone RM, Wilkinson GR, Greco FA, Wolff SN. Pharmacokinetics of high-dose etoposide (VP-16-213) administered to cancer patients. Cancer Res. 1984;44:379–382. [PubMed] [Google Scholar]
- 71.Zhao R, Kalvass JC, Yanni SB, Bridges AS, Pollack GM. Fexofenadine brain exposure and the influence of blood–brain barrier P-glycoprotein after fexofenadine and terfenadine administration. Drug Metab Dispos. 2009;37:529–535. doi: 10.1124/dmd.107.019893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Curry SH, Whelpton R, de Schepper PJ, Vranckx S, Schiff AA. Kinetics of fluphenazine after fluphenazine dihydrochloride, enanthate and decanoate administration to man. Br J Clin Pharmacol. 1979;7:325–331. doi: 10.1111/j.1365-2125.1979.tb00941.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chen C. Physicochemical, pharmacological and pharmacokinetic properties of the zwitterionic antihistamines cetirizine and levocetirizine. Curr Med Chem. 2008;15:2173–2191. doi: 10.2174/092986708785747625. [DOI] [PubMed] [Google Scholar]
- 74.Letendre SL, Capparelli EV, Ellis RJ, McCutchan JA. Indinavir population pharmacokinetics in plasma and cerebrospinal fluid. The HIV Neurobehavioral Research Center Group. Antimicrob Agents Chemother. 2000;44:2173–2175. doi: 10.1128/aac.44.8.2173-2175.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bannwarth B, Netter P, Lapicque F, Pere P, Thomas P, Gaucher A. Plasma and cerebrospinal fluid concentrations of indomethacin in humans. Relationship to analgesic activity. Eur J Clin Pharmacol. 1990;38:343–346. doi: 10.1007/BF00315572. [DOI] [PubMed] [Google Scholar]
- 76.Tucker RM, Denning DW, Dupont B, Stevens DA. Itraconazole therapy for chronic coccidioidal meningitis. Ann Intern Med. 1990;112:108–112. doi: 10.7326/0003-4819-112-2-108. [DOI] [PubMed] [Google Scholar]
- 77.Ketoconazole. Janssen Pharmaceutica, Inc; Titusville, NJ: 1998. Product Information: Nizoral® Tablets. [Google Scholar]
- 78.Product Information: Labetalol HCl Oral Tablets. Eon Labs, Inc; Laurelton, NY: 2004. [Google Scholar]
- 79.Elkiweri IA, Zhang YL, Christians U, Ng KY, Tissot van Patot MC, Henthorn TK. Competitive substrates for P-glycoprotein and organic anion protein transporters differentially reduce blood organ transport of fentanyl and loperamide: pharmaco-kinetics and pharmacodynamics in Sprague–Dawley rats. Anesth Analg. 2009;108:149–159. doi: 10.1213/ane.0b013e31818e0bd1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Loratadine/Pseudoephedrine. Schering Corporation; Kenilworth, NJ: 1999. Product Information: Claritin-D® 12 HOUR. [Google Scholar]
- 81.Methotrexate. Xanodyne Pharmacal, Inc; Florence, KY: 2003. Product Information: Methotrexate LPF®. [Google Scholar]
- 82.Chen TC, Mackic JB, McComb JG, Giannotta SL, Weiss MH, Zlokovic BV. Cellular uptake and transport of methylprednisolone at the blood–brain barrier. Neurosurgery. 1996;38:348–354. doi: 10.1097/00006123-199602000-00023. [DOI] [PubMed] [Google Scholar]
- 83.Liu X, Van Natta K, Yeo H, Vilenski O, Weller PE, Worboys PD, Monshouwer M. Unbound drug concentration in brain homogenate and cerebral spinal fluid at steady state as a surrogate for unbound concentration in brain interstitial fluid. Drug Metab Dispos. 2009;37:787–793. doi: 10.1124/dmd.108.024125. [DOI] [PubMed] [Google Scholar]
- 84.Metoprolol. Novartis Pharmaceutical; East Hanover, NJ: 1999. Product Information: Lopressor®. [Google Scholar]
- 85.Ohara S, Hayashi R, Momoi H, Miki J, Yanagisawa N. Mexiletine in the treatment of spasmodic torticollis. Mov Disord. 1998;13:934–940. doi: 10.1002/mds.870130612. [DOI] [PubMed] [Google Scholar]
- 86.Aweeka F, Jayewardene A, Staprans S, Bellibas SE, Kearney B, Lizak P, Novakovic-Agopian T, Price RW. Failure to detect nelfinavir in the cerebrospinal fluid of HIV-1-infected patients with and without AIDS dementia complex. J Acquir Immune Defic Syndr Hum Retrovirol. 1999;20:39–43. doi: 10.1097/00042560-199901010-00006. [DOI] [PubMed] [Google Scholar]
- 87.Takakura S, Sogabe K, Satoh H, Mori J, Fujiwara T, Totsuka Z, Tokuma Y, Kohsaka M. Nilvadipine as a neuroprotective calcium entry blocker in a rat model of global cerebral ischemia. A comparative study with nicardipine hydrochloride. Neurosci Lett. 1992;141:199–202. doi: 10.1016/0304-3940(92)90894-d. [DOI] [PubMed] [Google Scholar]
- 88.Uchida S, Yamada S, Nagai K, Deguchi Y, Kimura R. Brain pharmacokinetics and in vivo receptor binding of 1,4-dihydropyridine calcium channel antagonists. Life Sci. 1997;61:2083–2090. doi: 10.1016/s0024-3205(97)00881-3. [DOI] [PubMed] [Google Scholar]
- 89.Manjunath K, Venkateswarlu V. Pharmacokinetics, tissue distribution and bio-availability of nitrendipine solid lipid nanoparticles after intravenous and intra-duodenal administration. J Drug Target. 2006;14:632–645. doi: 10.1080/10611860600888850. [DOI] [PubMed] [Google Scholar]
- 90.McDevitt DG. Comparison of pharmacokinetic properties of beta-adrenoceptor blocking drugs. Eur Heart J. 1987;8(Suppl M):9–14. doi: 10.1093/eurheartj/8.suppl_m.9. [DOI] [PubMed] [Google Scholar]
- 91.Battino D, Estienne M, Avanzini G. Clinical pharmacokinetics of antiepileptic drugs in paediatric patients. Part II. Phenytoin, carbamazepine, sulthiame, lamotrigine, vigabatrin, oxcarbazepine and felbamate. Clin Pharmacokinet. 1995;29:341–369. doi: 10.2165/00003088-199529050-00004. [DOI] [PubMed] [Google Scholar]
- 92.Maher VM, Thompson GR. HMG CoA reductase inhibitors as lipid-lowering agents: five years experience with lovastatin and an appraisal of simvastatin and pravastatin. Q J Med. 1990;74:165–175. [PubMed] [Google Scholar]
- 93.Zhou L, Schmidt K, Nelson FR, Zelesky V, Troutman MD, Feng B. The effect of breast cancer resistance protein and P-glycoprotein on the brain penetration of flavopiridol, imatinib mesylate (Gleevec), prazosin, and 2-methoxy-3-(4-(2-(5-methyl-2-phenyloxazol-4-yl)ethoxy)phenyl)propanoic acid (PF-407288) in mice. Drug Metab Dispos. 2009;37:946–955. doi: 10.1124/dmd.108.024489. [DOI] [PubMed] [Google Scholar]
- 94.Taylor EA, Jefferson D, Carroll JD, Turner P. Cerebrospinal fluid concentrations of propranolol, pindolol and atenolol in man: evidence for central actions of beta-adrenoceptor antagonists. Br J Clin Pharmacol. 1981;12:549–559. doi: 10.1111/j.1365-2125.1981.tb01264.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ochs HR, Greenblatt DJ, Lloyd BL, Woo E, Sonntag M, Smith TW. Entry of quinidine into cerebrospinal fluid. Am Heart J. 1980;100:341–346. doi: 10.1016/0002-8703(80)90148-9. [DOI] [PubMed] [Google Scholar]
- 96.Kagevi I, Wahlby L. CSF concentrations of ranitidine. Lancet. 1985;1:164–165. doi: 10.1016/s0140-6736(85)91932-4. [DOI] [PubMed] [Google Scholar]
- 97.Product Information: Reserpine Oral Tablets. Eon Labs, Inc; Laurelton, NY: 2004. [Google Scholar]
- 98.Kravcik S, Gallicano K, Roth V, Cassol S, Hawley-Foss N, Badley A, Cameron DW. Cerebrospinal fluid HIV RNA and drug levels with combination ritonavir and saquinavir. J Acquir Immune Defic Syndr. 1999;21:371–375. [PubMed] [Google Scholar]
- 99.Wang C, Quan LH, Guo Y, Liu CY, Liao YH. Uptake and biodistribution of rizatriptan to blood and brain following different routes of administration in rats. Int J Pharm. 2007;337:155–160. doi: 10.1016/j.ijpharm.2006.12.039. [DOI] [PubMed] [Google Scholar]
- 100.Ose A, Kusuhara H, Endo C, Tohyama K, Miyajima M, Kitamura S, Sugiyama Y. Functional characterization of mouse organic anion transporting peptide 1a4 in the uptake and efflux of drugs across the blood–brain barrier. Drug Metab Dispos. 2010;38:168–176. doi: 10.1124/dmd.109.029454. [DOI] [PubMed] [Google Scholar]
- 101.Chen C, Lin J, Smolarek T, Tremaine L. P-glycoprotein has differential effects on the disposition of statin acid and lactone forms in mdr1a/b knockout and wild-type mice. Drug Metab Dispos. 2007;35:1725–1729. doi: 10.1124/dmd.107.015677. [DOI] [PubMed] [Google Scholar]
- 102.Serra G, Forgione A, D’Aquila PS, Collu M, Fratta W, Gessa GL. Possible mechanism of antidepressant effect of L-sulpiride. Clin Neuropharmacol. 1990;13(Suppl 1):S76–S83. doi: 10.1097/00002826-199001001-00009. [DOI] [PubMed] [Google Scholar]
- 103.Shingaki T, Hidalgo IJ, Furubayashi T, Sakane T, Katsumi H, Yamamoto A, Yamashita S. Nasal delivery of P-gp substrates to the brain through the nose–brain pathway. Drug Metab Pharmacokinet. 2011;26:248–255. doi: 10.2133/dmpk.DMPK-10-RG-108. [DOI] [PubMed] [Google Scholar]
- 104.Badcock NR, Osborne GA, Nyman TL, Sansom LN, Russell WJ, Frewin DB. Plasma and cerebrospinal fluid concentration of temazepam following oral drug administration. Eur J Clin Pharmacol. 1990;38:153–155. doi: 10.1007/BF00265975. [DOI] [PubMed] [Google Scholar]
- 105.Terfenadine. Marion Merrell Dow; Kansas City, MO: 1996. Product Information: Seldane®. [Google Scholar]
- 106.McHenry MC, Fieker DH. Newer antibacterial and antimycotic drugs. Critical appraisal. Med Clin North Am. 1978;62:873–897. doi: 10.1016/s0025-7125(16)31745-x. [DOI] [PubMed] [Google Scholar]
- 107.Reeves DS, Wilkinson PJ. The pharmacokinetics of trimethoprim and tri-methoprim/sulphonamide combinations, including penetration into body tissues. Infection. 1979;7(Suppl 4):S330–S341. doi: 10.1007/BF01639009. [DOI] [PubMed] [Google Scholar]
- 108.Benya TJ, Wagner JG. Rapid equilibration of warfarin between rat tissue and plasma. J Pharmacokinet Biopharm. 1975;3:237–255. doi: 10.1007/BF01066920. [DOI] [PubMed] [Google Scholar]
- 109.Garattini S, Bertele V. Efficacy, safety and cost of new drugs acting on the central nervous system. Eur J Clin Pharmacol. 2003;59:79–84. doi: 10.1007/s00228-003-0569-3. [DOI] [PubMed] [Google Scholar]
- 110.Crowe S, Cooper DA, Chambers DE. Managing HIV. Part 4: Primary therapy. 4.1 Antiretroviral therapies for HIV. Med J Aust. 1996;164:290–295. [PubMed] [Google Scholar]
- 111.Ferrari MD, Roon KI, Lipton RB, Goadsby PJ. Oral triptans (serotonin 5-HT (1B/1D) agonists) in acute migraine treatment: a meta-analysis of 53 trials. Lancet. 2001;358:1668–1675. doi: 10.1016/S0140-6736(01)06711-3. [DOI] [PubMed] [Google Scholar]
- 112.VolSurf+, version 1.0.4. Molecular Discovery Ltd; London, U.K: available from. www.moldiscovery.com/soft_vsplus.php. [Google Scholar]
- 113.VolSurf+ Manual. Molecular Discovery Ltd; London, U.K: available from. www.molddiscovery.com/docs/vsplus/ [Google Scholar]
- 114.Cruciani G, Meniconi M, Carosati E, Zamora I, Mannhold R. VOLSURF: a tool for drug ADME-properties prediction. In: van de Waterbeemd H, Lennernäs H, Artursson P, editors. Drug Bioavailability: Estimation of Solubility, Permeability, Absorption and Bioavailability. WILEY-VCH; 2003. pp. 406–419. [Google Scholar]
- 115.Crivori P, Cruciani G, Carrupt PA, Testa B. Predicting blood–brain barrier permeation from three-dimensional molecular structure. J Med Chem. 2000;43:2204–2216. doi: 10.1021/jm990968+. [DOI] [PubMed] [Google Scholar]
- 116.WOMBAT-PK. Sunset Molecular Discovery LLC; New Mexico, U.S.A: 2010. available from. www.sunsetmolecular.com. [Google Scholar]
- 117.Olah M, Rad R, Ostopovici L, Borá A, Hadaruga N, Hadaruga D, Moldovan R, Fulias A, Mracec M, Oprea TI. WOMBAT and WOMBAT-PK: bioactivity databases for lead and drug discovery. In: Schreiber SL, Kapoor TM, Wess G, editors. Chemical Biology: From Small Molecules to Systems Biology and Drug Design. WILEY-VCH; 2007. pp. 780–786. [Google Scholar]
- 118.MoKa, version 1.1.0. Molecular Discovery Ltd; London, U.K: available from. www.moldiscovery.com/soft_moka.php. [Google Scholar]
- 119.Milletti F, Storchi L, Sforna G, Cruciani G. New and original pKa prediction method using grid molecular interaction fields. J Chem Inf Model. 2007;47:2172–2181. doi: 10.1021/ci700018y. [DOI] [PubMed] [Google Scholar]
- 120.Leo AJ. Calculating log Poct from structures. Chem Rev. 1993;93:1281–1306. [Google Scholar]
- 121.Williamson DF, Parker RA, Kendrick JS. The box plot: a simple visual method to interpret data. Ann Intern Med. 1989;110:916–921. doi: 10.7326/0003-4819-110-11-916. [DOI] [PubMed] [Google Scholar]
- 122.Zweig MH, Campbell G. Receiver-operating characteristic (ROC) plots: a fundamental evaluation tool in clinical medicine. Clin Chem. 1993;39:561–577. [PubMed] [Google Scholar]
- 123.Manallack DT. The pK(a) distribution of drugs: application to drug discovery. Perspect Med Chem. 2008;1:25–38. [PMC free article] [PubMed] [Google Scholar]
- 124.Orange Canvas, version 2.0b. www.www.orange.biolab.si.
- 125.Hartter S, Huwel S, Lohmann T, Abou El Ela A, Langguth P, Hiemke C, Galla HJ. How does the benzamide antipsychotic amisulpride get into the brain? An in vitro approach comparing amisulpride with clozapine. Neuropsychopharmacology. 2003;28:1916–1922. doi: 10.1038/sj.npp.1300244. [DOI] [PubMed] [Google Scholar]
- 126.Luer MS, Hamani C, Dujovny M, Gidal B, Cwik M, Deyo K, Fischer JH. Saturable transport of gabapentin at the blood–brain barrier. Neurol Res. 1999;21:559–562. doi: 10.1080/01616412.1999.11740975. [DOI] [PubMed] [Google Scholar]
- 127.Thurlow RJ, Hill DR, Woodruff GN. Comparison of the uptake of [3H]-gabapentin with the uptake of L-[3H]-leucine into rat brain synaptosomes. Br J Pharmacol. 1996;118:449–456. doi: 10.1111/j.1476-5381.1996.tb15424.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Nishimura M, Naito S. Tissue-specific mRNA expression profiles of human ATP-binding cassette and solute carrier transporter superfamilies. Drug Metab Pharmacokinet. 2005;20:452–477. doi: 10.2133/dmpk.20.452. [DOI] [PubMed] [Google Scholar]
- 129.Ho RH, Tirona RG, Leake BF, Glaeser H, Lee W, Lemke CJ, Wang Y, Kim RB. Drug and bile acid transporters in rosuvastatin hepatic uptake: function, expression, and pharmacogenetics. Gastroenterology. 2006;130:1793–1806. doi: 10.1053/j.gastro.2006.02.034. [DOI] [PubMed] [Google Scholar]
- 130.Windass AS, Lowes S, Wang Y, Brown CD. The contribution of organic anion transporters OAT1 and OAT3 to the renal uptake of rosuvastatin. J Pharmacol Exp Ther. 2007;322:1221–1227. doi: 10.1124/jpet.107.125831. [DOI] [PubMed] [Google Scholar]
- 131.Kitamura S, Maeda K, Wang Y, Sugiyama Y. Involvement of multiple transporters in the hepatobiliary transport of rosuvastatin. Drug Metab Dispos. 2008;36:2014–2023. doi: 10.1124/dmd.108.021410. [DOI] [PubMed] [Google Scholar]
- 132.Takeda M, Khamdang S, Narikawa S, Kimura H, Kobayashi Y, Yamamoto T, Cha SH, Sekine T, Endou H. Human organic anion transporters and human organic cation transporters mediate renal antiviral transport. J Pharmacol Exp her. 2002;300:918–924. doi: 10.1124/jpet.300.3.918. [DOI] [PubMed] [Google Scholar]
- 133.Zolk O, Solbach TF, Konig J, Fromm MF. Structural determinants of inhibitor interaction with the human organic cation transporter OCT2 (SLC22A2) Naunyn Schmiedebergs Arch Pharmacol. 2009;379:337–348. doi: 10.1007/s00210-008-0369-5. [DOI] [PubMed] [Google Scholar]
- 134.Aravagiri M, Yuwiler A, Marder SR. Distribution after repeated oral administration of different dose levels of risperidone and 9-hydroxy-risperidone in the brain and other tissues of rat. Psychopharmacology. 1998;139:356–363. doi: 10.1007/s002130050726. [DOI] [PubMed] [Google Scholar]
- 135.Elsby R, Surry DD, Smith VN, Gray AJ. Validation and application of Caco-2 assays for the in vitro evaluation of development candidate drugs as substrates or inhibitors of P-glycoprotein to support regulatory submissions. Xenobiotica. 2008;38:1140–1164. doi: 10.1080/00498250802050880. [DOI] [PubMed] [Google Scholar]
- 136.Clark DE. Rapid calculation of polar molecular surface area and its application to the prediction of transport phenomena. 2. Prediction of blood–brain barrier penetration. J Pharm Sci. 1999;88:815–821. doi: 10.1021/js980402t. [DOI] [PubMed] [Google Scholar]
- 137.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 138.Timmerman H. Histamine H1 blockers: from relative failures to blockbusters within series of analogues. In: Fischer J, Ganellin CR, editors. Analogue-based Drug Discovery. Wiley-VCH; 2006. [Google Scholar]
- 139.Chishty M, Reichel A, Siva J, Abbott NJ, Begley DJ. Affinity for the P-glycoprotein efflux pump at the blood–brain barrier may explain the lack of CNS side-effects of modern antihistamines. J Drug Target. 2001;9:223–228. doi: 10.3109/10611860108997930. [DOI] [PubMed] [Google Scholar]
- 140.Broccatelli F, Carosati E, Cruciani G, Oprea TI. Transporter-mediated efflux influences CNS side effects: ABCB1, from antitarget to target. Mol Inform. 2010;29:16–26. doi: 10.1002/minf.200900075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Hendrikse NH, Schinkel AH, de Vries EG, Fluks E, Van der Graaf WT, Willemsen AT, Vaalburg W, Franssen EJ. Complete in vivo reversal of P-glycoprotein pump function in the blood–brain barrier visualized with positron emission tomography. Br J Pharmacol. 1998;124:1413–1418. doi: 10.1038/sj.bjp.0701979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Vautier S, Lacomblez L, Chacun H, Picard V, Gimenez F, Farinotti R, Fernandez C. Interactions between the dopamine agonist, bromocriptine and the efflux protein, P-glycoprotein at the blood–brain barrier in the mouse. Eur J Pharm Sci. 2006;27:167–174. doi: 10.1016/j.ejps.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 143.Polli JW, Olson KL, Chism JP, John-Williams LS, Yeager RL, Woodard SM, Otto V, Castellino S, Demby VE. An unexpected synergist role of P-glycoprotein and breast cancer resistance protein on the central nervous system penetration of the tyrosine kinase inhibitor lapatinib (N-{3-chloro-4-[(3-fluorobenzyl)oxy] phenyl}-6-[5-({[2-(methylsulfonyl)ethyl]amino}methyl)-2-furyl]-4-quinazolinamine; GW572016) Drug Metab Dispos. 2009;37:439–442. doi: 10.1124/dmd.108.024646. [DOI] [PubMed] [Google Scholar]
- 144.Chen ML, Yu L. The use of drug metabolism for prediction of intestinal permeability. Mol Pharm. 2009;6:74–81. doi: 10.1021/mp8001864. [DOI] [PubMed] [Google Scholar]
- 145.Larregieu CA, Benet LZ. Evaluating the link between passive transcellular permeability and extent of drug absorption and metabolism in humans. Abstract. 25th AAPS Annual Meeting and Exposition; Washington, DC. 2011. [Google Scholar]
- 146.Bjornsson TD, Callaghan JT, Einolf HJ, Fischer V, Gan L, Grimm S, Kao J, King SP, Miwa G, Ni L, Kumar G, McLeod J, Obach RS, Roberts S, Roe A, Shah A, Snikeris F, Sullivan JT, Tweedie D, Vega JM, Walsh J, Wrighton SA. The conduct of in vitro and in vivo drug–drug interaction studies: a Pharmaceutical Research and Manufacturers of America (PhRMA) perspective. Drug Metab Dispos. 2003;31:815–832. doi: 10.1124/dmd.31.7.815. [DOI] [PubMed] [Google Scholar]
- 147.Yao HM, Chiou WL. The complexity of intestinal absorption and exsorption of digoxin in rats. Int J Pharm. 2006;322:79–86. doi: 10.1016/j.ijpharm.2006.05.030. [DOI] [PubMed] [Google Scholar]
- 148.Kim MK, Shim CK. The transport of organic cations in the small intestine: current knowledge and emerging concepts. Arch Pharm Res. 2006;29:605–616. doi: 10.1007/BF02969273. [DOI] [PubMed] [Google Scholar]
- 149.Alt A, Potthast H, Moessinger J, Sickmuller B, Oeser H. Biopharmaceutical characterization of sotalol-containing oral immediate release drug products. Eur J Pharm Biopharm. 2004;58:145–150. doi: 10.1016/j.ejpb.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 150.Liu W, Okochi H, Zhai S-D, Benet LZ. Sotalol’s permeability in cell, rat, and PAMPA studies. Abstract. 25th AAPS Annual Meeting and Exposition; Washington, DC. 2011. [Google Scholar]
- 151.Prichard BN, Tomlinson B. The additional properties of beta adrenoceptor blocking drugs. J Cardiovasc Pharmacol. 1986;8(Suppl 4):S1–S15. doi: 10.1097/00005344-198608004-00002. [DOI] [PubMed] [Google Scholar]
- 152.Broccatelli F, Carosati E, Neri A, Frosini M, Goracci L, Oprea TI, Cruciani G. A novel approach for predicting P-glycoprotein (ABCB1) inhibition using molecular interaction fields. J Med Chem. 2011;54:1740–1751. doi: 10.1021/jm101421d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Alelyunas YW, Empfield JR, McCarthy D, Spreen RC, Bui K, Pelosi-Kilby L, Shen C. Experimental solubility profiling of marketed CNS drugs, exploring solubility limit of CNS discovery candidate. Bioorg Med Chem Lett. 2010;20:7312–7316. doi: 10.1016/j.bmcl.2010.10.068. [DOI] [PubMed] [Google Scholar]
- 154.Ishikawa M, Hashimoto Y. Improvement in aqueous solubility in small molecule drug discovery programs by disruption of molecular planarity and symmetry. J Med Chem. 2011;54:1539–1554. doi: 10.1021/jm101356p. [DOI] [PubMed] [Google Scholar]
- 155.Varma MV, Obach RS, Rotter C, Miller HR, Chang G, Steyn SJ, El-Kattan A, Troutman MD. Physicochemical space for optimum oral bioavailability: contribution of human intestinal absorption and first-pass elimination. J Med Chem. 2010;53:1098–1108. doi: 10.1021/jm901371v. [DOI] [PubMed] [Google Scholar]
- 156.Lewis DF, Lake BG, Dickins M. Quantitative structure–activity relationships (QSARs) in CYP3A4 inhibitors: the importance of lipophilic character and hydrogen bonding. J Enzyme Inhib Med Chem. 2006;21:127–132. doi: 10.1080/14756360500532747. [DOI] [PubMed] [Google Scholar]
- 157.Vaz RJ, Klabunde T. Antitargets: prediction and prevention of drug side effects. In: Mannhold R, Kubinyi H, Folkers G, editors. Methods and Principles in Medicinal Chemistry. WILEY-VCH; 2008. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.