

Abstract
Objectives
This study aimed to investigate the clinicopathological changes induced by corticosteroid therapy in immunoglobulin (Ig)G4-related tubulointerstitial nephritis (TIN).
Methods
We studied six IgG4-related TIN patients receiving renal biopsies before and after corticosteroid therapy. Their clinical data and histological findings were evaluated before and after therapy.
Results
Elevated serum creatinine levels rapidly improved after corticosteroid therapy except for two patients, in whom it persisted. Abnormal radiological findings improved in all patients, although focal cortical atrophy persisted in three. Histologically, TIN-like dense lymphoplasmacytic infiltration, interstitial fibrosis, IgG4-positive plasma cell, CD4+CD25+ T cell, and Foxp3+ cell infiltration were characteristic before therapy. After therapy, the area with cell infiltration decreased and regional fibrosis became evident in the renal interstitium. The number of IgG4-positive plasma cells and Foxp3+ cells significantly diminished even in the early stage of therapy, whereas low to moderate numbers of CD4+ and CD8+ T cells still infiltrated where inflammation persisted in the later stage.
Conclusions
Our study shows that persistent renal insufficiency associated with macroscopic atrophy and microscopic fibrosis is not so rare in IgG4-related TIN. Pathologically, the behavior of regulatory T cells during the clinical course is quite similar to that of IgG4-positive plasma cells, and the behavior pattern of those cells is distinctive.
Keywords: IgG4-related disease, Tubulointerstitial nephritis, IgG4-positive plasma cell, Regulatory T cell, Corticosteroid therapy
Introduction
Immunoglobulin (Ig)G4-related disease (IgG4-RD) is a recently recognized systemic inflammatory disease with multiorgan involvement [1–5], including the kidney. Since 2004, accumulated case reports and case series have defined the radiographic and histopathological characteristic findings of IgG4-related kidney disease [6–20]. Two large studies [21, 22] demonstrated clinicopathological features of IgG4-related tubulointerstitial nephritis (TIN) that included high levels of serum IgG4, IgG, and IgE; hypocomplementemia; and TIN with copious IgG4-positive plasma cell infiltration with fibrosis. However, the clinical course and histological changes occurring after corticosteroid therapy have not yet been well characterized. In addition, although IgG4-RD is thought to be steroid responsive, not all cases achieve recovery of normal renal function [23], with, for example, one case report describing a patient who required maintenance hemodialysis despite corticosteroid therapy [24]. Therefore, in order to establish the optimal treatment plan to prevent progressive kidney damage, renal re-biopsy seems to be necessary. We undertook this study to evaluate the influence of corticosteroid therapy on the clinical and histopathological findings in IgG4-related TIN.
Materials and methods
Patients and materials
Between 1 September 2005 and 31 August 2010, we identified 11 IgG4-RD patients with kidney involvement, including renal parenchymal and pelvic lesions. We diagnosed IgG4-RD according to the provisional diagnostic criteria for IgG4-RD of Masaki et al. [25]. Renal biopsies were performed in six patients in whom IgG4-related TIN was highly suspected because of renal dysfunction, elevation of renal tubular markers, or abnormal imaging finding, and we diagnosed them with IgG4-related TIN based on diagnostic criteria proposed by Kawano et al. [26] and Raissian et al. [22]. Two patients (patients 1 and 5 in Table 1) had been included in our earlier studies [27, 28], two (patient 1 and 6) in a report by Saeki et al. [21], and one (patient 1) in a report by Yamaguchi et al. [29].
Table 1.
Clinicopathological characteristics of six patients with immunoglobulin (Ig)G4-related tubulointerstitial nephritis
| No. | Age/sex | Cr at pre-Tx (N 0.6–1) | IgG (N 870–1,600) | IgG4 (N < 105) | IgE (N < 250) | CH50 (N 32–47) | C3 (N 65–135) | C4 (N 13–35) | ANA | RF | U-β2MG (N < 250) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 59/M | 1.15 | 2,850 | 1,470 | 456 | 34 | 81 | 16 | (−) | (−) | 335 |
| 2 | 79/M | 0.54 | 4,756 | 409 | 457 | 8 | 41 | 3 | (+) | (−) | 496 |
| 3 | 77/W | 0.59 | 2,256 | 984 | 292 | 60 | 110 | 27 | (+) | (−) | 78 |
| 4 | 68/M | 1.90 | 3,830 | 736 | 242 | 3 | 33 | 1 | (+) | (+) | 7,375 |
| 5 | 75/W | 2.55 | 3,695 | 486 | 1,226 | 2 | 18 | 2 | (+) | (−) | 35,490 |
| 6 | 68/M | 7.26 | 4,661 | 1,120 | 335 | 5 | 10 | 7 | (+) | (+) | 3,240 |
| No. | Age/sex | U-NAG (N < 10) | Eosinophilia | Allergy | U-Pr | CT findings | Initial renal biopsy findings | Initial Tx of post-biopsy | Cr at re-biopsy (N 0.6–1) | Period between initial biopsy and re-biopsy (months) | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 59/M | 1.6 | (+) | (−) | (−) | LDLs | fTIN | PSL 30 | 1.06 | 14 | |
| 2 | 79/M | 19 | (−) | (−) | (−) | (−) | fTIN + EndPGN | PSL 20 | 0.51 | 7 | |
| 3 | 77/W | 0.4 | (−) | Rhinitis | (−) | LDLs + P | fTIN | PSL 20 | 0.63 | 4 | |
| 4 | 68/M | 8.8 | (−) | BA | (−) | LDLs | fTIN + IgAGN | PSL 30 | 1.20 | 4 | |
| 5 | 75/W | 5.9 | (−) | (−) | (+) | S | dTIN + HSPN | PSL 30 | 1.05 | 4 | |
| 6 | 68/M | 16.9 | (−) | (−) | (+) | S | dTIN + EndPGN | PSL 30 | 1.45 | 1 |
Conversion factor for Cr: mg/dl to μmol/l, ×88.4
ANA antinuclear antibody, BA bronchial asthma, CH50 serum CH50 at initial renal biopsy (U/ml), Cr serum creatinine (mg/dl), CT computed tomography, C3 serum C3 at initial renal biopsy (mg/dl), C4 serum C4 at initial renal biopsy (mg/dl), dTIN diffuse tubulointerstitial nephritis, EndPGN endocapillary proliferative glomerulonephritis fTIN focal tubulointerstitial nephritis, HSPN Henoch–Schönlein purpura nephritis, IgAGN IgA nephropathy, IgG serum immunoglobulin G at initial renal biopsy (mg/dl), IgG4 serum immunoglobulin G4 at initial renal biopsy (mg/dl), IgE serum immunoglobulin E at initial renal biopsy (IU/ml), LDLs multiple low-density lesions of the renal parenchyma, P diffuse thickening of the renal pelvic wall, post-biopsy post-renal biopsy, PSL prednisolone mg/day, re-biopsy renal re-biopsy, RF rheumatoid factor, S diffuse bilateral renal swelling, Tx treatment, U-β2MG urine β2-microglobulin at initial renal biopsy (μg/l), U-NAG urine N-acetyl-β-d-glucosaminidase at initial renal biopsy (U/l), U-Pr proteinuria
All six patients underwent re-biopsy while receiving corticosteroid therapy, as re-evaluation of the extent of cell infiltration and fibrosis was necessary to assess the future dosage regimen of corticosteroid. One patient (patient 1 in Table 1) underwent re-biopsy 14 months after the start of therapy, one (patient 2) 7 months later, three (patients 3, 4, and 5) 4 months later, and one (patient 6) only 1 month later. These biopsy specimens were obtained randomly, not specifically from the mass lesions. We examined these 12 specimens histologically and immunohistochemically. With regard to the extent of the renal interstitial lesion, diffuse TIN was defined as being present when ≥80% of the renal interstitium in renal biopsy specimens was affected and focal TIN when <80% was affected. We also retrospectively evaluated the clinical and radiographic findings of these six patients. Serial laboratory data during the clinical course were analyzed, and the computed tomography (CT) findings of renal lesions were investigated before and after corticosteroid therapy in all patients. This study received institutional ethics board approval, and informed consent for all data and samples was obtained from each patient. The research was in compliance with the Declaration of Helsinki.
Single immunostaining
Bouin’s fluid- or formalin-fixed and paraffin-embedded renal specimens of six patients with IgG4-related TIN were used for the immunostaining of IgG4, CD138, CD4, CD8, and Foxp3. The immunostaining was performed using a monoclonal antibody against human IgG4 (Zymed Laboratory, San Francisco, CA, USA), CD138 (AbD Serotec, Oxford, UK), CD4 (Nichirei, Tokyo, Japan), CD8 (Nichirei), and Foxp3 (AbD Serotec). The deparaffinized sections were microwaved in citrate buffer (pH 6.0) for 15 min. Cells positive for IgG4, CD138, CD4, CD8, or Foxp3 were counted in five different high-power fields (HPF: 10× eyepiece and 40× lens) with intense cell infiltration.
Dual fluorescent immunostaining of CD4 and CD25
All Bouin’s fluid-fixed and paraffin-embedded renal specimens were used for dual fluorescent immunostaining of CD4 and CD25. The deparaffinized sections were microwaved in citrate buffer (pH 6.0) for 20 min and incubated with normal donkey serum for protein blocking for 30 min. The specimens were incubated with a mouse monoclonal antibody to CD25 (Leica Microsystems, Wetzlar, Germany) and a rabbit monoclonal antibody to CD4 (Spring Bioscience, CA, USA) overnight at 4°C. Then, the specimens were incubated for 1 h at room temperature with Alexa Fluor 488-labeled donkey anti-mouse IgG antibodies and Alexa Fluor 594-labeled donkey anti-rabbit IgG antibodies (Molecular Probes, Carlsbad, CA, USA) and observed under a laser microscope and digitally merged. No positive staining was observed when the primary antibodies were replaced with normal donkey serum in the negative control of the staining procedures.
Statistical analysis
Statistical analysis was performed using the Wilcoxon signed rank test for continuous non-normally distributed data. Significant differences were defined as P < 0.05.
Results
Patient profiles
We analyzed six patients [four men and two women; average age 71.0 (range 59–79) years] with IgG4-related TIN. None had been treated with corticosteroid or any other immunosuppressants before the diagnosis. All patients had other organ involvement associated with IgG4-RD: four (66.7%) dacryoadenitis and/or sialoadenitis, six (100%) multiple lymphadenopathy, and three (50.0%) lung lesions. Pancreatic lesion was detected in only one patient (16.7%). Prostate, liver, and joint lesion was detected in one patient each. All patients were treated with prednisolone at an initial dose of 20–30 mg/day after the initial renal biopsy (Table 1).
Laboratory findings and changes associated with corticosteroid therapy
At presentation, all patients showed elevated levels of serum IgG4 (average 867.5 mg/dl, range 409–1,470 mg/dl, normal range <105 mg/dl) and IgG (average 3,675 mg/dl, range 2,256–4,756 mg/dl, normal range 870–1,700 mg/dl). Five patients (83.3%) showed elevated serum IgE levels (average 501.3 IU/ml, range 242–1,226 IU/ml, normal range <250 IU/ml), and only one patient had eosinophilia (eosinophils >5%). Four patients (66.7%) had hypocomplementemia. Antinuclear antibodies were positive in five patients (83.3%) and rheumatoid factor in two (33.3%). Serum creatinine concentration was elevated in four patients (66.7%). Urine β2-microglobulin (β2-MG) excretion was elevated in five patients (83.3%) and urine N-acetyl-β-d-glucosaminidase (NAG) level in two (33.3%), whereas two patients (33.3%) had proteinuria (Table 1). In three patients who showed severe renal dysfunction (serum creatinine >1.5 mg/dl) before corticosteroid therapy (patients 4, 5, and 6 in Table 1), serum creatinine rapidly declined within 1 month after the start of therapy. However, some degree of renal dysfunction persisted in two of them (Fig. 1a). Four patients had hypocomplementemia before corticosteroid therapy (patients 2, 4, 5, and 6 in Table 1), and corticosteroid therapy promptly improved serum complement levels within 2 months in three of them; recovery of serum complement level was very slow in one other (Fig. 1b). On the other hand, urine β2-MG and NAG concentrations fluctuated despite the corticosteroid therapy (data not shown).
Fig. 1.
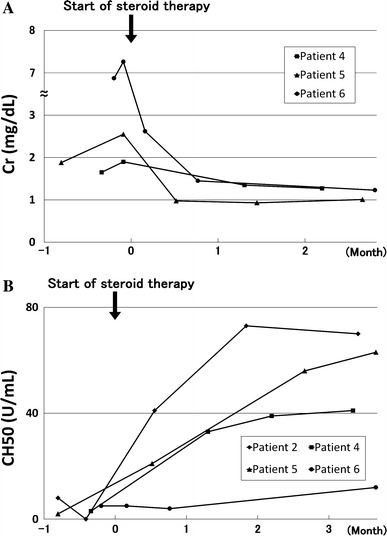
Patient’s clinical course. Serum creatinine concentrations rapidly declined within 1 month after corticosteroid therapy in patients 4, 5, and 6, who showed severe renal dysfunction before therapy (a). Hypocomplementemia was improved by corticosteroid therapy within 2 months in patients 2, 4, and 5, but response to therapy was insufficient in patient 6 (b)
Computed tomography findings and changes associated with corticosteroid therapy
Computed tomography revealed some radiologic findings of the renal parenchymal lesions in five patients. Among them, multiple low-density lesions on enhanced CT (Fig. 2a, c, e) were the most frequent findings and were observed in three patients (patients 1, 3, and 4 in Table 1), one of whom also had diffuse thickening of the renal pelvic wall (Fig. 2c). On the other hand, the renal parenchymal lesion observed in two patients (patients 5 and 6 in Table 1) who underwent only plain CT because of severe renal dysfunction before corticosteroid therapy was diffuse bilateral renal swelling (Fig. 2g, i). No radiological abnormal findings were apparent in only one patient (patient 2 in Table 1).
Fig. 2.
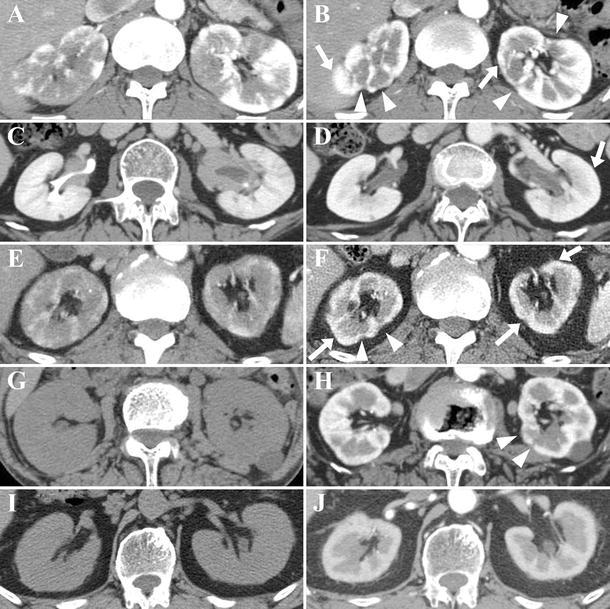
Contrast-enhanced (a–f, h, j) or plain (g, i) computed tomography (CT) findings of immunoglobulin (Ig)G4-related renal lesions and changes induced by corticosteroid therapy. Multiple low-density lesions on enhanced CT (a patient 1, c patient 3, e patient 4), diffuse bilateral renal swelling on plain CT (g patient 5, i patient 6), and diffuse thickening of the renal pelvic wall (c) were observed before therapy. Contrast enhancement of the renal cortex recovered after therapy in all patients with multiple low-density lesions (b, d, farrows show recovering lesions), though some atrophic areas of decreased enhancement remained (b, farrowheads show atrophic lesions). Diffuse bilateral renal swelling was improved with small areas of decreased enhancement 4 months after therapy in patient 5 (harrowheads show lesions with decreased enhancement) and with relatively uniform contrast enhancement 1 month after therapy in patient 6 (j). Diffuse thickening of the renal pelvic wall became less marked 4 months after therapy in patient 3 (d)
On the whole, corticosteroid therapy quickly ameliorated renal lesions. Three patients with multiple low-density lesions on enhanced CT showed recovery of contrast enhancement of the renal cortex after therapy. However, scar-like focal cortical atrophy persisted in two of them (Fig. 2b, f). Two patients with diffuse bilateral renal swelling showed apparent improvement of the renal swelling after therapy (Fig. 2h, j). A few small scar-like focal cortical atrophy persisted in one of them (Fig. 2h), whereas relatively uniform contrast enhancement was observed in the other (Fig. 2j). Diffuse thickening of the renal pelvic wall became thinner after therapy in patient 3 (Fig. 2d).
Histological and immunohistochemical findings and changes associated with corticosteroid therapy
Histological findings of six renal specimens before corticosteroid therapy were as follows: Dense lymphoplasmacytic infiltration and sparse distribution of atrophic renal tubules in the renal interstitium were present (Fig. 3a–c). The findings of renal tubulitis were mild (Fig. 3a, e, f). The interstitial lesions were often localized, and the border of the lesion was fairly clear (Fig. 3b). On the other hand, the interstitial lesion was diffuse in two patients (patients 5 and 6 in Table 1) with severe renal dysfunction whose plain CT images before therapy showed diffuse bilateral renal swelling (Fig. 3c). In one case (patient 6), inflammation extended beyond the renal capsule (Fig. 3d). Fibrosis with occasional infiltrating cells was observed (Fig. 3e, f). Lymph follicles were not evident in any patient. Glomerular lesions were observed in four: one showed Henoch–Schönlein purpura nephritis [28], another IgA nephropathy, and the other two focal endocapillary proliferation. Immunohistochemically, in addition to IgG4-positive plasma cells (Fig. 5a), Foxp3+ cells (Fig. 5b) and CD4+CD25+ T cells (Fig. 5c) were detected in the interstitial lesions of IgG4-related TIN. CD4+ T cells (Fig. 6a) and CD8+ T cells (Fig. 6b) were also found there.
Fig. 3.
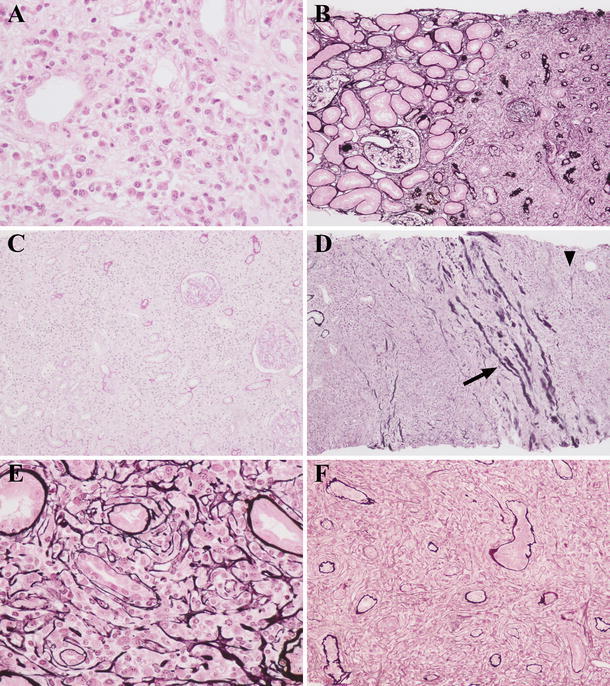
Light microscopy findings of the renal interstitium before corticosteroid therapy. Severe lymphoplasmacytic infiltration with tubular atrophy was observed (a patient 6, b patient 4, c patient 6). Interstitial lesions were often focal, and the borderline between lesion and nonlesion was fairly clear (b). In two patients with severe renal dysfunction, interstitial lesion was diffuse (c). Inflammatory lesion beyond the renal capsule was detected (d patient 6, arrow shows the renal capsule, and arrowhead shows inflammation beyond the renal capsule). A characteristic fibrosis that appeared to surround infiltrating cells was observed (e patient 4, f patient 5) [a Hematoxylin and eosin (H&E) staining ×400; b, d periodic acid-methenamine-silver (PAM)-H&E staining ×100; c periodic acid-Schiff (PAS) staining ×100; e, f PAM-H&E staining ×400]
Fig. 5.
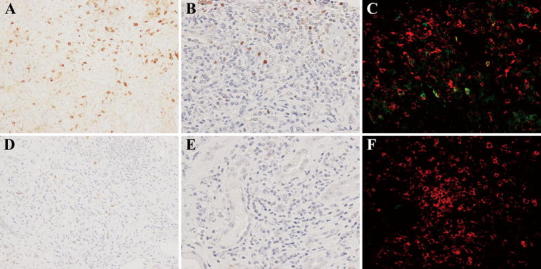
Changes in immunoglobulin (Ig)G4-positive plasma cell, Foxp3+ cell, and CD4+CD25+ cell infiltration before (a–c) and 1 month after (d–f) corticosteroid therapy in patient 6. Compared with the pretreatment specimens (a), the number of IgG4-positive plasma cells in the lesions was markedly diminished in the posttreatment specimens (d). In the same way, compared with the pretreatment specimens (b), the number of Foxp3+ cells obviously decreased in the posttreatment specimens (e). Some CD4+CD25+ cells in the lesions were observed before treatment (c), whereas almost none were detected after therapy (f) [a, d IgG4 ×200, b, e Foxp3 ×400, c, f CD4 (red) and CD25 (green) ×400]
Fig. 6.
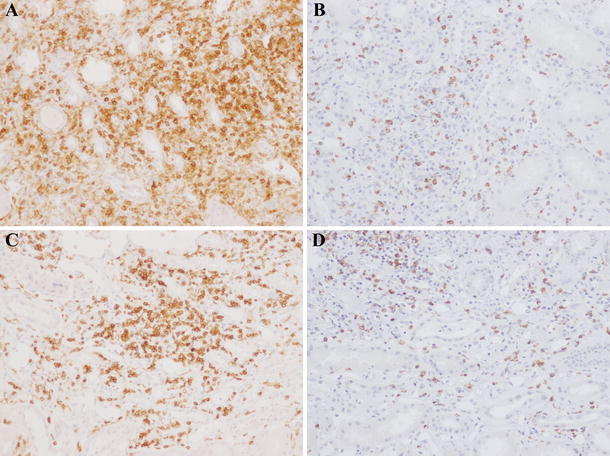
Changes in CD4+ and CD8+ T cell infiltration before (a, b) and 4 months after (c, d) corticosteroid therapy in patient 5. The extent of CD4+ T cell (a, c) and CD8+ T cell (b, d) infiltration in the lesions remained largely constant, even after therapy (a, c CD4 ×100, b, d CD8 ×100)
The re-biopsy specimen obtained 1 month after corticosteroid therapy from patient 6 showed that areas with intense cell infiltration remained (Fig. 4a), but infiltration of IgG4-positive plasma cells (Fig. 5d), Foxp3+ cells (Fig. 5e), and CD4+CD25+ T cells (Fig. 5f) considerably decreased, and there was little obvious fibrosis of the renal interstitium (Fig. 4a). The re-biopsy specimen 14 months after therapy from patient 1 demonstrated an obvious decrease of cell infiltration except for small localized infiltrations, whereas patchy marked fibrosis remained (Fig. 4b). In the re-biopsy specimens 4 months after therapy from patient 5 who showed severe renal dysfunction before therapy, we observed mixed lesions where cell infiltration was dense or fibrosis was predominant or neither of these features was noted (Fig. 4c, d, e, respectively). In the re-biopsy specimens 4 months after therapy from patient 3 who showed normal renal function before therapy, we observed the same three components as in patient 5, but fibrosis was not marked. In one patient with diffuse renal interstitial cell infiltration and sparse distribution of atrophic tubules (patient 6), tubular atrophy and sparseness of tubular distribution seemed to partially improve after therapy (Figs. 3c, 4f). In the immunostaining specimens, cell count of various infiltrating cells in the lesions with intense cell infiltration before and after corticosteroid therapy revealed that the characteristic infiltrating cells, such as IgG4-positive plasma cells and Foxp3+ cells, showed significant decreases after corticosteroid therapy (Figs. 5d, e, 7a–c). Other CD4+ T cells and CD8+ T cells were relatively persistent, even in the later stage of therapy (Figs. 6c, d, 7d, e). The average number of IgG4-positive plasma cells in five different HPF decreased from 156.4/HPF (range 102–210) to 2.0/HPF (range 0–3) in patient 1; 41.8/HPF (range 36–60) to 7.2/HPF (range 1–15) in patient 2; 50.2/HPF (range 25–77) to 12.2/HPF (range 5–36) in patient 3; 27.6/HPF (range 9–46) to 5.0/HPF (range 3–11) in patient 4; 62.2/HPF (range 45–80) to 4.8/HPF (range 3–7) in patient 5; and 73.2/HPF (range 50–108) to 20.8/HPF (range 19–22) in patient 6 (Fig. 7a). In the same way, the average number of Foxp3+ cells decreased from 35.4/HPF (range 26–52) to 8.4/HPF (range 1–16) in patient 1; 25.4/HPF (range 18–45) to 3.8/HPF (range 2–6) in patient 2; 18.0/HPF (range 10–23) to 1.2/HPF (range 0–3) in patient 3; 41.8/HPF (range 36–46) to 4.8/HPF (range 4–6) in patient 4; 33.2/HPF (range 30–35) to 3.0/HPF (range 0–5) in patient 5; and 23.0/HPF (range 19–30) to 3.2/HPF (range 1–5) in patient 6 (Fig. 7c).
Fig. 4.
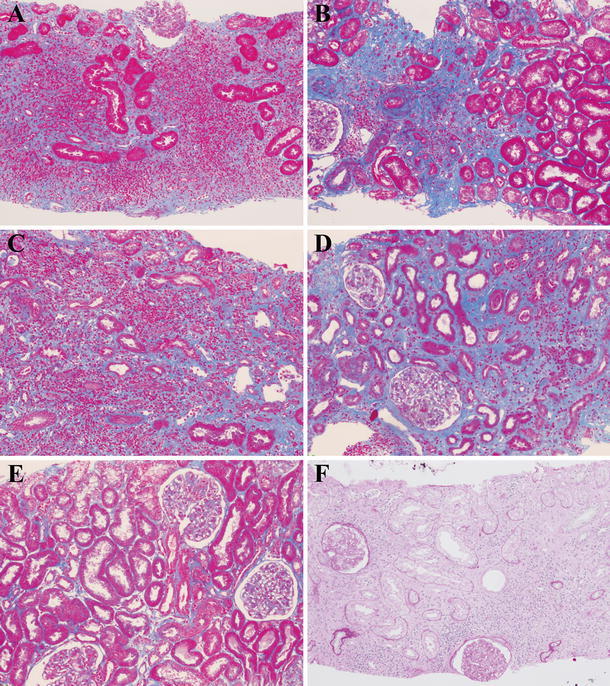
Changes in light microscopy findings of the renal interstitium after corticosteroid therapy. Azan staining showed that interstitial fibrosis was not evident 1 month after corticosteroid therapy in patient 6 (a), whereas localized severe fibrosis was observed 14 months after therapy in patient 1 (b). There were mixed lesions where cell infiltration was dense (c) or fibrosis was predominant (d) or neither of them was noted (e) 4 months after therapy in patient 5. Interstitium in the posttreatment specimen of patient 6 seemed to show recovery of some renal tubules 1 month after corticosteroid therapy (f) [a–e Azan staining ×100, f periodic acid-Schiff (PAS) staining ×100]
Fig. 7.
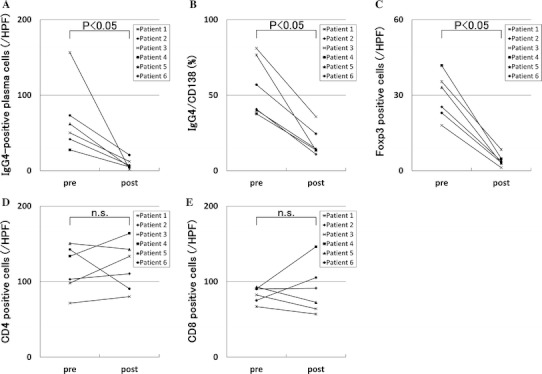
Changes in numbers of infiltrating cells before and after corticosteroid therapy. In six IgG4-related TIN cases, the average of IgG4-positive plasma cell numbers and the ratio of IgG4-positive cells to CD138-positive cells in the lesions significantly decreased after compared with before corticosteroid therapy (a, b, respectively). Similarly, average of Foxp3+ cell numbers significantly decreased (c). However, average of CD4+ and CD8+ T cell numbers did not change (d, e, respectively)
Discussion
This study examined the clinicopathological features in patients with IgG4-related TIN before and after corticosteroid therapy. The characteristic laboratory findings before therapy were elevated serum IgE levels or eosinophilia, high incidence of hypocomplementemia, and hyper-β2 microglobulinuria. Renal insufficiency was also frequent, and almost all patients with IgG4-related TIN had some radiographic abnormalities. Therapy with 20–30 mg/day of prednisolone rapidly improved renal function in three patients with severe renal dysfunction, whereas some renal insufficiency persisted in two. Radiographic abnormalities persisted in half of the patients. Histologically, infiltration of CD4+CD25+ T cells and Foxp3+ cells in addition to IgG4-positive plasma cells was characteristic in IgG4-related TIN. Notably, IgG4-positive plasma cells, Foxp3+ cells, and CD4+CD25+ cells had already decreased 1 month after corticosteroid therapy in one case (patient 6), whereas small to moderate numbers of CD4+ T cells and CD8+ T cells infiltrated where inflammation persisted in all patients. In the later stage after treatment, patchy or regional fibrosis remained to some extent.
In this study, radiographic findings of renal lesions were almost the same as those noted in past reports [21, 30]. Multiple low-density lesions on enhanced CT, diffuse bilateral renal swelling, and thickening of the renal pelvic wall were the major features. Past reports mentioned that corticosteroid therapy was effective in improving the appearance of renal lesions on imaging study, although cortical scar or renal capsular dimpling persisted in a small number of cases [30–33]. In our study, although some renal parenchymal lesions showed recovery of contract enhancement, others progressed to scar-like atrophy in three of six cases. In contrast to past studies [30–33], both imaging study and histological analysis were performed before and after corticosteroid therapy in all patients in this study. Our data might suggest that these atrophic lesions in imaging study correspond to the histological fibrotic lesions. In addition, recovery of contrast enhancement might relate to the partially normalized renal interstitium observed in the posttreatment specimens of patients 5 and 6, who showed diffuse TIN and renal tubular atrophy before therapy. In this way, IgG4-related TIN could leave macroscopic atrophy and microscopic fibrosis, which might explain why renal function did not totally recover after corticosteroid therapy in our patients with severe renal insufficiency. The possibility of these sequelae must be considered when determining the corticosteroid dose and optimal timing of the initiation of treatment.
The main histological findings before corticosteroid therapy were consistent with previously published histological features of IgG4-related TIN [21, 22, 34]. In addition, extension of inflammation beyond the renal capsule reported by Yamaguchi et al. [29] was also observed and seemed to correspond to the extension of lesions beyond the pancreatic capsule in autoimmune pancreatitis (AIP). Zen et al. [35] reported infiltration of CD4+CD25+ T cells and Foxp3+ cells in the lesions of IgG4-related pancreatitis and cholangitis and that those lesions had significantly increased levels of Th2 and regulatory T-cell cytokines. Nakashima et al. [36] also reported increased Th2 and regulatory cytokines in the lesions of IgG4-related TIN. In this study, we similarly confirmed the presence of CD4+CD25+ T and Foxp3+ cells in the renal interstitium where lymphocytes and IgG4-positive plasma cells infiltrated. Accordingly, the presence of CD4+CD25+ T or Foxp3+ cell infiltration might be another distinctive finding of IgG4-related TIN. As Houghton and Troxell [37] reported that an abundant infiltration of IgG4-positive plasma cell is not so specific for IgG4-related TIN, it will be necessary to survey the presence or absence of these regulatory cells in TIN diseases other than IgG4-related TIN.
We focused on histological and immunohistochemical changes of IgG4-related TIN during the clinical course of corticosteroid therapy. The area with infiltrating cells in the renal interstitium decreased with the passage of time after therapy was initiated. Conversely, more obvious regional fibrosis was observed in the re-biopsy specimens, although there was the possibility of sampling bias due to randomly performed biopsies. Reflecting these histological findings, the radiological lesions to some degree showed recovery following therapy, whereas in some parts, there was progression to scar formation. These findings suggest the need to search for ways to prevent fibrosis in addition to control of inflammation. Whether early initiation of corticosteroid therapy can prevent fibrosis remains to be verified, as there was no untreated control group in this study and so this point could not be concluded definitively. However, comparing posttreatment findings of patients 3 and 5, both of whom underwent re-evaluation 4 months after the start of therapy, macroscopic atrophy and microscopic fibrosis were more marked in patient 5 with severe renal dysfunction before therapy than in patient 3 with normal renal function then. This difference implies that early initiation of corticosteroid therapy prevents fibrosis to some extent. The possibility should also be considered that corticosteroid therapy alone is insufficient to prevent residual fibrosis despite early initiation. Other treatment options should be examined, as rituximab therapy, for example, has been reported to be effective in achieving clinical improvement in IgG4-RD [38, 39]. The characteristic cells, including CD4+CD25+ T cells, Foxp3+ cells, and IgG4-positive plasma cells, seemed to disappear quickly after corticosteroid initiation and did not reappear as long as corticosteroid was administered, whereas other CD4+ T and CD8+ T cells persisted for a long time in lesions where cell infiltration was still observed. This finding suggests that we cannot precisely evaluate IgG4-positive plasma cell infiltration in diagnosing IgG4-related TIN if corticosteroid therapy has already been initiated. The pathogenic significance of this finding remains unclear. More accumulation of clinical and histological data of IgG4-related TIN, including recurrences during corticosteroid tapering or after cessation of corticosteroid therapy and basic research based on those data, are essential to elucidate the roles of these cells in the pathogenesis of IgG4-related TIN.
Biomarkers that could be used as a goal for treating IgG4-related TIN have not yet been established. It was reported that in AIP, serum IgG4, IgG, and circulating immune complex decreased after corticosteroid therapy and increased at relapse, whereas serum C3 and C4 levels showed reciprocal changes [40]. Tabata et al. [41] suggested that the measurement of serial serum IgG4 levels was useful to determine disease activity of IgG4-RD. Nevertheless, whether normalization of serum IgG4 levels could be a treatment goal is unclear, as it is not always observed despite apparent clinical remission [41]. Cutoff values of these markers as a goal should be further examined. Nishi et al. [42] reported that elevated urine NAG and/or α1-microglobulin concentrations were useful markers for detecting renal abnormalities in their AIP patients with or without clinically detected renal involvement. However, neither urine β2-MG nor NAG entirely responded to corticosteroid therapy and fluctuated despite the continuation of treatment in our study. Thus, reliable markers showing a goal for treatment remain to be identified. Further studies are required to seek such markers, including novel candidates.
The small number of cases is a limitation of our study that relates to the rarity of this disease, and the inconsistent follow-up times of radiological and histological data in each patient is another limitation. However, with increasing worldwide awareness of IgG4-related TIN as a distinct disease entity [21, 22], we can expect larger studies that include many more cases and anticipate that, with more patients enrolled in each stage of corticosteroid therapy, especially in the very early and later stages, the dynamics of various infiltrating cells during the clinical course will be better clarified.
In conclusion, our investigations suggest that, clinically, persistent renal insufficiency associated with macroscopic atrophy and microscopic fibrosis is not so rare in IgG4-related TIN. Pathologically, the behavior of regulatory T cells during the clinical course of corticosteroid therapy is quite similar to that of IgG4-positive plasma cells, and the distinctive behavior pattern of those cells may provide a clue to the mechanisms underlying this disease. Further studies are required to elucidate the total picture of this disease, including its clinical and pathogenic aspects.
Acknowledgments
This work was supported by Health and Labour Sciences Research Grants for the Study of Intractable Diseases from the Ministry of Health, Labor and Welfare, Japan. We thank John Gelblum for his critical reading of the manuscript.
Conflict of interest
None.
References
- 1.Hamano H, Kawa S, Horiuchi A, Unno H, Furuya N, Akamatsu T, et al. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J Med. 2001;344:732–738. doi: 10.1056/NEJM200103083441005. [DOI] [PubMed] [Google Scholar]
- 2.Hamano H, Kawa S, Ochi Y, Unno H, Shiba N, Wajiki M, et al. Hydronephrosis associated with retroperitoneal fibrosis and sclerosing pancreatitis. Lancet. 2002;359:1403–1404. doi: 10.1016/S0140-6736(02)08359-9. [DOI] [PubMed] [Google Scholar]
- 3.Yamamoto M, Harada S, Ohara M, Suzuki C, Naishiro Y, Yamamoto H, et al. Clinical and pathological differences between Mikulicz’s disease and Sjögren’s syndrome. Rheumatology (Oxford) 2005;44:227–234. doi: 10.1093/rheumatology/keh447. [DOI] [PubMed] [Google Scholar]
- 4.Takahira M, Kawano M, Zen Y, Minato H, Yamada K, Sugiyama K. IgG4-related chronic sclerosing dacryoadenitis. Arch Ophthalmol. 2007;125:1575–1578. doi: 10.1001/archopht.125.11.1575. [DOI] [PubMed] [Google Scholar]
- 5.Cheuk W, Yuen HK, Chan JK. Chronic sclerosing dacryoadenitis: part of the spectrum of IgG4-related sclerosing disease? Am J Surg Pathol. 2007;31:643–645. doi: 10.1097/01.pas.0000213445.08902.11. [DOI] [PubMed] [Google Scholar]
- 6.Uchiyama-Tanaka Y, Mori Y, Kimura T, Sonomura K, Umemura S, Kishimoto N, et al. Acute tubulointerstitial nephritis associated with autoimmune-related pancreatitis. Am J Kidney Dis. 2004;43:e18–e25. doi: 10.1053/j.ajkd.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 7.Takeda S, Haratake J, Kasai T, Takaeda C, Takazakura E. IgG4-associated idiopathic tubulointerstitial nephritis complicating autoimmune pancreatitis. Nephrol Dial Transplant. 2004;19:474–476. doi: 10.1093/ndt/gfg477. [DOI] [PubMed] [Google Scholar]
- 8.Nakamura H, Wada H, Origuchi T, Kawakami A, Taura N, Aramaki T, et al. A case of IgG4-related autoimmune disease with multiple organ involvement. Scand J Rheumatol. 2006;35:69–71. doi: 10.1080/03009740500499484. [DOI] [PubMed] [Google Scholar]
- 9.Watson SJ, Jenkins DA, Bellamy CO. Nephropathy in IgG4-related systemic disease. Am J Surg Pathol. 2006;30:1472–1477. doi: 10.1097/01.pas.0000213308.43929.97. [DOI] [PubMed] [Google Scholar]
- 10.Saeki T, Saito A, Yamazaki H, Emura I, Imai N, Ueno M, et al. Tubulointerstitial nephritis associated with IgG4-related systemic disease. Clin Exp Nephrol. 2007;11:168–173. doi: 10.1007/s10157-007-0464-9. [DOI] [PubMed] [Google Scholar]
- 11.Saeki T, Nishi S, Ito T, Yamazaki H, Miyamura S, Emura I, et al. Renal lesions in IgG4-related systemic disease. Intern Med. 2007;46:1365–1371. doi: 10.2169/internalmedicine.46.0183. [DOI] [PubMed] [Google Scholar]
- 12.Katano K, Hayatsu Y, Matsuda T, Miyazaki R, Yamada K, Kawano M, et al. Endocapillary proliferative glomerulonephritis with crescent formation and concurrent tubulointerstitial nephritis complicating retroperitoneal fibrosis with a high serum level of IgG4. Clin Nephrol. 2007;68:308–314. doi: 10.5414/cnp68308. [DOI] [PubMed] [Google Scholar]
- 13.Murashima M, Tomaszewski J, Glickman JD. Chronic tubulointerstitial nephritis presenting as multiple renal nodules and pancreatic insufficiency. Am J Kidney Dis. 2007;49:e7–e10. doi: 10.1053/j.ajkd.2006.10.025. [DOI] [PubMed] [Google Scholar]
- 14.Yoneda K, Murata K, Katayama K, Ishikawa E, Fuke H, Yamamoto N, et al. Tubulointerstitial nephritis associated with IgG4-related autoimmune disease. Am J Kidney Dis. 2007;50:455–462. doi: 10.1053/j.ajkd.2007.05.018. [DOI] [PubMed] [Google Scholar]
- 15.Cornell LD, Chicano SL, Deshpande V, Collins AB, Selig MK, Lauwers GY, et al. Pseudotumors due to IgG4 immune-complex tubulointerstitial nephritis associated with autoimmune pancreatocentric disease. Am J Surg Pathol. 2007;31:1586–1597. doi: 10.1097/PAS.0b013e318059b87c. [DOI] [PubMed] [Google Scholar]
- 16.Aoki A, Sato K, Itabashi M, Takei T, Yoshida T, Arai J, et al. A case of Mikulicz’s disease complicated with severe interstitial nephritis associated with IgG4. Clin Exp Nephrol. 2009;13:367–372. doi: 10.1007/s10157-008-0127-5. [DOI] [PubMed] [Google Scholar]
- 17.Morimoto J, Hasegawa Y, Fukushima H, Uesugi N, Hisano S, Saito T, et al. Membranoproliferative glomerulonephritis-like glomerular disease and concurrent tubulointerstitial nephritis complicating IgG4-related autoimmune pancreatitis. Intern Med. 2009;48:157–162. doi: 10.2169/internalmedicine.48.1339. [DOI] [PubMed] [Google Scholar]
- 18.Saeki T, Imai N, Ito T, Yamazaki H, Nishi S. Membranous nephropathy associated with IgG4-related systemic disease and without autoimmune pancreatitis. Clin Nephrol. 2009;71:173–178. doi: 10.5414/cnp71173. [DOI] [PubMed] [Google Scholar]
- 19.Dhobale S, Bedetti C, Killian P, Ilyas M, Liput J, Jasnosz K, et al. IgG4 related sclerosing disease with multiple organ involvements and response to corticosteroid treatment. J Clin Rheumatol. 2009;15:354–357. doi: 10.1097/RHU.0b013e3181b5d631. [DOI] [PubMed] [Google Scholar]
- 20.Kuroda N, Nakamura S, Miyazaki K, Inoue K, Ohara M, Mizuno K, et al. Chronic sclerosing pyelitis with an increased number of IgG4-positive plasma cells. Med Mol Morphol. 2009;42:236–238. doi: 10.1007/s00795-008-0425-8. [DOI] [PubMed] [Google Scholar]
- 21.Saeki T, Nishi S, Imai N, Ito T, Yamazaki H, Kawano M, et al. Clinicopathological characteristics of patients with IgG4-related tubulointerstitial nephritis. Kidney Int. 2010;78:1016–1023. doi: 10.1038/ki.2010.271. [DOI] [PubMed] [Google Scholar]
- 22.Raissian Y, Nasr SH, Larsen CP, Colvin RB, Smyrk TC, Takahashi N, et al. Diagnosis of IgG4-related tubulointerstitial nephritis. J Am Soc Nephrol. 2011;22:1343–1352. doi: 10.1681/ASN.2011010062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tsubata Y, Akiyama F, Oya T, Ajiro J, Saeki T, Nishi S, et al. IgG4-related chronic tubulointerstitial nephritis without autoimmune pancreatitis and the time course of renal function. Intern Med. 2010;49:1593–1598. doi: 10.2169/internalmedicine.49.3787. [DOI] [PubMed] [Google Scholar]
- 24.Saida Y, Homma N, Hama H, Ueno M, Imai N, Nishi S, et al. A case of IgG4-related tubulointerstitial nephritis showing the progression of renal dysfunction after a cure for autoimmune pancreatitis. Nippon Jinzo Gakkai Shi. 2010;52:73–9 (in Japanese, abstract in English). [PubMed]
- 25.Masaki Y, Dong L, Kurose N, Kitagawa K, Morikawa Y, Yamamoto M, et al. Proposal for a new clinical entity, IgG4-positive multiorgan lymphoproliferative syndrome: analysis of 64 cases of IgG4-related disorders. Ann Rheum Dis. 2009;68:1310–1315. doi: 10.1136/ard.2008.089169. [DOI] [PubMed] [Google Scholar]
- 26.Kawano M, Saeki T, Nakashima H, Nishi S, Yamaguchi Y, Hisano S, et al. Proposal for diagnostic criteria for IgG4-related kidney disease. Clin Exp Nephrol. 2011;15:615–626. doi: 10.1007/s10157-011-0521-2. [DOI] [PubMed] [Google Scholar]
- 27.Kim F, Yamada K, Inoue D, Nakajima K, Mizushima I, Kakuchi Y, et al. IgG4-related tubulointerstitial nephritis and hepatic inflammatory pseudotumor without hypocomplementemia. Intern Med. 2011;50:1239–1244. doi: 10.2169/internalmedicine.50.5102. [DOI] [PubMed] [Google Scholar]
- 28.Ito K, Yamada K, Mizushima I, Aizu M, Fujii H, Mizutomi K, et al. Henoch–Schönlein purpura nephritis in a patient with IgG4-related disease: a possible association. Clin Nephrol. 2012 (in press). [DOI] [PubMed]
- 29.Yamaguchi Y, Kanetsuna Y, Honda K, Yamanaka N, Kawano M, Nagata M. Characteristic tubulointerstitial nephritis in IgG4-related disease. Hum Pathol. 2011 [Epub ahead of print]. [DOI] [PubMed]
- 30.Takahashi N, Kawashima A, Fletcher JG, Chari ST. Renal involvement in patients with autoimmune pancreatitis: CT and MR imaging findings. Radiology. 2007;242:791–801. doi: 10.1148/radiol.2423060003. [DOI] [PubMed] [Google Scholar]
- 31.Khalili K, Doyle DJ, Chawla TP, Hanbidge AE. Renal cortical lesions in patients with autoimmune pancreatitis: a clue to differentiation from pancreatic malignancy. Eur J Radiol. 2008;67:329–335. doi: 10.1016/j.ejrad.2007.07.020. [DOI] [PubMed] [Google Scholar]
- 32.Sohn JH, Byun JH, Yoon SE, Choi EK, Park SH, Kim MH, et al. Abdominal extrapancreatic lesions associated with autoimmune pancreatitis: radiological findings and changes after therapy. Eur J Radiol. 2008;67:497–507. doi: 10.1016/j.ejrad.2007.08.018. [DOI] [PubMed] [Google Scholar]
- 33.Triantopoulou C, Malachias G, Maniatis P, Anastopoulos J, Siafas I, Papailiou J. Renal lesions associated with autoimmune pancreatitis: CT findings. Acta Radiol. 2010;51:702–707. doi: 10.3109/02841851003738846. [DOI] [PubMed] [Google Scholar]
- 34.Nishi S, Imai N, Yoshida K, Ito Y, Saeki T. Clinicopathological findings of immunoglobulin G4-related kidney disease. Clin Exp Nephrol. 2011 [Epub ahead of print]. [DOI] [PubMed]
- 35.Zen Y, Fujii T, Harada K, Kawano M, Yamada K, Takahira M, et al. Th2 and regulatory immune reactions are increased in immunoglobin (sic) G4-related sclerosing pancreatitis and cholangitis. Hepatology. 2007;45:1538–1546. doi: 10.1002/hep.21697. [DOI] [PubMed] [Google Scholar]
- 36.Nakashima H, Miyake K, Moriyama M, Tanaka A, Watanabe M, Abe Y, et al. An amplification of IL-10 and TGF-β in patients with IgG4-related tubulointerstitial nephritis. Clin Nephrol. 2010;73:385–391. doi: 10.5414/cnp73385. [DOI] [PubMed] [Google Scholar]
- 37.Houghton DC, Troxell ML. An abundance of IgG4+ plasma cells is not specific for IgG4-related tubulointerstitial nephritis. Mod Pathol. 2011;24:1480–1487. doi: 10.1038/modpathol.2011.101. [DOI] [PubMed] [Google Scholar]
- 38.Khosroshahi A, Bloch DB, Deshpande V, Stone JH. Rituximab therapy leads to rapid decline of serum IgG4 levels and prompt clinical improvement in IgG4-related systemic disease. Arthritis Rheum. 2010;62:1755–1762. doi: 10.1002/art.27435. [DOI] [PubMed] [Google Scholar]
- 39.Zaidan M, Cervera-Pierot P, Seigneux S, Dahan K, Fabiani B, Callard P, et al. Evidence of follicular T-cell implication in a case of IgG4-related systemic disease with interstitial nephritis. Nephrol Dial Transplant. 2011;26:2047–2050. doi: 10.1093/ndt/gfr097. [DOI] [PubMed] [Google Scholar]
- 40.Muraki T, Hamano H, Ochi Y, Komatsu K, Komiyama Y, Arakura N, et al. Autoimmune pancreatitis and complement activation system. Pancreas. 2006;32:16–21. doi: 10.1097/01.mpa.0000188308.75043.e4. [DOI] [PubMed] [Google Scholar]
- 41.Tabata T, Kamisawa T, Takuma K, Egawa N, Setoguchi K, Tsuruta K, et al. Serial changes of elevated serum IgG4 levels in IgG4-related systemic disease. Intern Med. 2011;50:69–75. doi: 10.2169/internalmedicine.50.4321. [DOI] [PubMed] [Google Scholar]
- 42.Nishi H, Shibagaki Y, Hirano K, Akahane K, Kido R, Nangaku N, et al. Laboratory and imaging features of kidney involvement in autoimmune pancreatitis: incidence, correlation, and steroid therapy response. Clin Nephrol. 2010;73:253–259. doi: 10.5414/cnp73253. [DOI] [PubMed] [Google Scholar]