

Abstract
Estimating the colloidal stability of polymeric nanoparticles (NPs) in biological environments is critical for designing optimal preparations and to clarify the fate of these devices after administration. To characterize and quantify the physical stability of nanodevices suitable for biomedical applications, spherical NPs composed of poly-lactic acid (PLA) and poly-methyl-methacrylate (PMMA), in the range 100–200 nm, were prepared. Their stability in salt solutions, biological fluids, serum and tissue homogenates was analyzed by dynamic light scattering (DLS). The PMMA NPs remained stable in all fluids, while PLA NPs aggregated in gastric juice and spleen homogenate. The proposed stability test is therefore useful to see in advance whether NPs might aggregate when administered in vivo. To assess colloidal stability ex vivo as well, spectrophotofluorimetric analysis was employed, giving comparable results to DLS.
Keywords: Nanoparticles, Poly-lactic acid, Poly-methyl-methacrylate, Nanomedicine, Gastrointestinal fluids, Serum, Homogenates
Introduction
Parenteral and oral administrations are the most common delivery routes used for pharmaceuticals. Parenteral delivery allows direct administration of the drug to the whole cardiovascular system, while oral administration ensures better compliance as it is less invasive. However, the different pH and enzyme-rich gastric fluids encountered by the drug in the gastrointestinal tract, as well as the limited drug absorption due to the gastrointestinal barrier, strongly limit this administration route. Whichever route is chosen, the development of molecules for the treatment of diseases with unfavorable prognosis is often limited by drug bioavailability, rapid clearance, difficulty crossing biological barriers and—last but not least—by severe side effects. To overcome these hurdles, the use of biocompatible drug-loaded nanoparticles (NPs) has been proposed (Alexis et al. 2008).
Polymeric NPs are an innovative generation of nanovectors with great potential as drug carriers and tracking devices for targeted delivery to specific sites (Riehemann et al. 2009). They improve the gastric stability of therapeutics (Mohs et al. 2009) suggesting that they could be employed for oral drug delivery (Kuentz 2008). These nanoformulations may also favor the transport of drugs across the mucosal barrier, promoting their entry into the circulation (Win and Feng 2005).
Introducing such devices for a more effective drug delivery involves new problems, however, such as NP stability, drug release mechanisms, and NP interactions with the mononuclear phagocytes in the bloodstream, directly after injection or after crossing the gastrointestinal barrier. While the interaction of NPs with serum proteins has been investigated (Aggarwal et al. 2009), with particular attention to opsonization, and many theories have been proposed concerning the release mechanisms (Arifin et al. 2006), the stability of NPs in biological fluids has not been fully characterized. It is essential to understand whether and how the different body environments influence NP stability, to design optimal nanoformulations, and to identify their fate after administration.
In general, nanoformulation stability is classified in terms of chemical stability (i.e., the chemical degradation of the polymeric NPs), long-term “pharmaceutical stability” (i.e., the changes in the quality of a drug in time under the influence of environmental factors) (Muthu and Feng 2009), and physical or colloidal stability. While the long-term “pharmaceutical stability” (Muthu and Feng 2009) and chemical stability have been assessed (Lemoine et al. 1996; Avgoustakis et al. 2002), and different degradation mechanisms have been identified depending on NPs dimensions and polymer reactivity (von Burkersroda et al. 2002), the colloidal stability still remains poorly investigated (Win and Feng 2005; Ruenraroengsak et al. 2010; Madlova et al. 2009).
The aim of this study is to shed light on the physical stability of NPs for biomedical applications, stressing the need for in vitro/ex vivo testing of their stability, before in vivo studies are performed. Measuring the NPs size distribution in time by dynamic laser light scattering (DLS), the formation of aggregates in different biological fluids was monitored. As ex vivo stability is also needed, spectrophotofluorimetric (SPF) analysis was used and its ability to predict NP stability was qualitatively checked by comparison with DLS data.
Poly-lactic acid (PLA) and poly-methyl-methacrylate (PMMA) were selected as prototypes for NP preparations, since both polymers are biocompatible and approved by the US Food and Drug administration (Oh 2011; Squire et al. 2008). Their use permits a comparison of a hydrolytically degradable material, PLA, and a non-degradable one, PMMA (Devalapally et al. 2007; Kumari et al. 2010; Cui et al. 2009; Prokop and Davidson 2008). The stability of these polymer NPs was investigated in the stock solutions usually adopted for their storage and in biological fluids mimicking the environments they meet after administration. In particular, saliva, gastric juice, intestinal fluid, lysosomal fluid, serum and tissue homogenates were examined.
Materials and methods
Materials
2-Ethylhexanoic acid tin(II) salt (Sn(Oct)2), polystyrene standards from 500 to 2,000,000 Da and 3,6-dimethyl-1,4-dioxane-2,5-dione (d,l-lactide, LT, further recrystallized in extra dried Toulene puriss) were purchased from Sigma Aldrich (Buchs, Switzerland). Toluene was obtained from Riedel-de Haen (Basel, Switzerland). Methyl methacrylate, tetrahydrofuran (THF), cyclohexane, potassium peroxydisulfate, Tween® 80, dextrose alpha-d-(+)-glucose anhydrous, sodium hydrogen carbonate, 1-dodecanol, HPLC grade acetonitrile and orthophosphoric acid, sodium dl-lactate and pepsin from porcine gastric mucosa were purchased from Fluka-Chemie AG (Germany). Dichloromethane and Rhodamine B base were purchased from Acros Organics (Basel, Switzerland). Cholesteryl Bodipy® was obtained from molecular probes (Milan, Italy).
Synthesis of NPs
PLA was synthesized by ring opening polymerization of LT according to Yu et al. (2009). The reaction was carried out at 130 °C for 3 h in order to limit side reactions such as trans-esterification and cyclization. Sn(Oct)2 was used as catalyst and dl-lactic acid as co-catalyst. In a first step, 25 g of LT were melted at 100 °C using an oil bath in a stirred, round-bottomed, one-neck 100 mL reactor flask. Then, 70 and 30 mg of catalyst and co-catalyst, respectively, were added and the temperature was raised up to reaction condition. After reaction, the polymer was dissolved in dichloromethane and precipitated in cyclohexane in order to remove residual monomer and impurities such as cyclic compounds. After purification, the polymer was dried in vacuum at 250 mbar at 25 °C and characterized by size exclusion chromatography (SEC).
NPs of PLA loaded with Cholesteryl Bodipy® were produced by flash nanoprecipitation (Liu et al. 2008). In brief, a THF solution of 1 % (w/v) PLA and 0.001 % (w/v) Cholesteryl Bodipy® was mixed in a 0.1 % (w/v) Tween® 80 distilled water solution, using a 4-inlet vortex mixer in which the streams were tangentially mixed (Liu et al. 2008). Altering streams of distilled water and THF, at flow rates of 100 and 10 mL/min, respectively, were employed. THF was removed by evaporation at 125 mbar at room temperature for 6 h. The residual THF was quantified by gas chromatographic analysis, as described in “Gas chromatography” section. The Tween® 80 concentration in the PLA NPS suspension was then adjusted to 0.25 % (w/v) in order to reach the same concentrations of Tween® 80 and PMMA. NPs were characterized by TEM and DLS.
PMMA NPs loaded with Rhodamine B base were prepared by emulsion polymerization. In brief, a solution containing 2 % (w/v) of methyl methacrylate, 0.02 % (w/v) of Rhodamine B base, and 5 % of Tween® 80 was kept under magnetic stirring at 80 °C. Then, 0.05 % (w/v) of potassium peroxydisulfate was added and the reaction was carried out for 4 h. The PMMA NPS were then diluted 1:20 (v/v) in water to obtain a suspension with the same particle and Tween® 80 concentration as the PLA ones. PMMA molecular weight was characterized by SEC (after centrifugation of the NPs at 16,000×g for 10 min) and particle size and morphology were investigated by DLS and SEM.
Size exclusion chromatography (SEC)
The molecular weight distribution of PMMA and PLA was characterized by SEC (Agilent, 1100 series) equipped with ultraviolet and differential refractive index detectors. A pre-column and two PLgel 5 μm MIXED-C columns (Polymer Laboratories, length 300 mm and diameter 7.5 mm, measuring range 2,000–2,000,000 Da) were employed. Chloroform was used as eluent at a flow rate of 1 mL/min and the temperature was set at 30 °C. The polymer was dissolved in chloroform employing a concentration of 1 mg/mL. Universal calibration was applied, based on poly(styrene) standards, adopting the Mark–Houwink constants for P(dl)LA. (Yu et al. 2009).
Gas chromatography
Gas chromatography was employed to verify the complete removal of THF from the PLA latex. A Hewlett-Packard gas chromatograph HP6890 apparatus was used, equipped with a (crosslinked 5 % PH ME Siloxane) 30 m × 0.3 mm × 0.25 μm (USA) HP column and TCD detector. Helium was used as carrier gas at a flow rate of 10 mL/min. The samples were injected as prepared after nanoprecipitation. The injection and detector temperature were 250 °C and the column temperature was raised from 60 to 250 °C. THF water mixtures with a known amount of THF were injected for calibration.
Dynamic laser light scattering (DLS)
NPs size was measured by DLS at 37 °C performed with a Zeta Nano ZN (Malvern Instruments, UK). Each cuvette was filled with NP suspensions and the different solutions (stock solutions, gastrointestinal fluids, serum and tissue homogenates) in a 1:1 (v/v) ratio. In other words, the DLS analysis was performed with a concentration of 5 × 10−4 (v/v). Multiple scattering was ruled out by performing analysis in more dilute conditions (up to 5 × 10−6 v/v) which gave the same result. The data are the average of three runs of the same sample where the standard deviation (SD) was always below 5 %.
Transmission electron microscopy (TEM)
To evaluate the PLA NP size and morphology, one drop of the NP suspension was diluted in 10 mL water and 40 μL of this solution were deposited on a copper grid (400 mesh) previously covered with a 8 nm thick carbon layer. After 30 s, the droplet was removed and images of the slices were taken with a FEI Morgagni 268 transmission electron microscope (FEI Company, USA).
High performance liquid chromatography (HPLC)
PLA degradation was evaluated by means of an HPLC apparatus (1200 series, Agilent) equipped with a C18 column (3.9 mm × 150 mm, particle size 3.5 μm, Eclipse XDB, Agilent) and an ultraviolet detector (210 nm). Water and acetonitrile, acidified with 0.1 % (v/v) phosphoric acid, were used as mobile phases (flow rate 1 mL/min) to quantify the lactic acid (Codari et al. 2010) After precipitation of the NPs via centrifugation at 13,000×g, the supernatant was recovered and injected as such in the HPLC.
SPF analysis
SPF analysis was performed to investigate the colloidal stability over time. PLA and PMMA NPs were diluted 1:1 (v/v) with the different stock solutions, gastrointestinal fluids, serum and tissue homogenates and incubated at 37 °C for different times. The fluorescence intensities (FI) of the suspensions were determined every 10 min for 60 h, using a spectrophotofluorimeter (EnSpire™ Multilabel Plate Reader, Perkin Elmer, USA) at 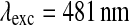 and
and 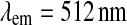 for cholesteryl Bodipy® and at
for cholesteryl Bodipy® and at 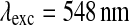 and
and 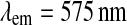 for Rhodamine B base. All the experiments were run in triplicate in Corning® 96-well plates. Data are expressed as scaled values with respect to their initial value and as averages of the three runs. The SD among the samples was always lower than 5 %, so it was omitted from the graphs.
for Rhodamine B base. All the experiments were run in triplicate in Corning® 96-well plates. Data are expressed as scaled values with respect to their initial value and as averages of the three runs. The SD among the samples was always lower than 5 %, so it was omitted from the graphs.
Stock solutions and biological fluids
The NPs were incubated in stock solutions, gastrointestinal fluids, and tissue homogenates. The following stock solutions were used: distilled water, 5 mM phosphate buffered saline (PBS) and aqueous solutions of NaCl (0.9 % w/v), and dextrose (5 % w/v). Simulated gastrointestinal fluids were prepared according to US Pharmacopeia XXIV, 2006 and their composition and pH are summarized in Table 1. Saliva contained 0.1 mM mucin, and gastric juice 0.9 mM pepsin; intestinal fluid was prepared without pancreatin.
Table 1.
Composition of the synthetic biological fluids used to simulate the physicochemical environment of the gastrointestinal tract
| Composition | Saliva | Gastric juice | Intestinal fluid | Lysosomal fluid |
|---|---|---|---|---|
| Calcium chloride dihydrate | 40.0 | – | – | 0.2 |
| Mucin | 0.1 | – | – | – |
| Potassium chloride | 50.0 | – | – | – |
| Sodium chloride | 70.0 | 300.0 | – | 549.3 |
| Sodium phosphate dibasic | 40.0 | – | – | 5.0 |
| Urea | 170.0 | – | – | – |
| Pepsin | – | 0.9 | – | – |
| Hydrochloric acid | – | 840.0 | – | – |
| Sodium hydroxide | – | – | – | 1500.4 |
| Magnesium chloride hexahydrate | – | – | – | 0.1 |
| Sodium sulfate | – | – | – | 2.7 |
| Citric acid | – | – | – | 1082.7 |
| Glycine | – | – | – | 7.9 |
| Sodium citrate tribasic dihydrate | – | – | – | 2.6 |
| Tartaric acid disodium salt dihydrate | – | – | – | 3.9 |
| Sodium lactate | – | – | – | 7.6 |
| Sodium pyruvate | – | – | – | 7.8 |
| Sodium hydrogen carbonate | – | – | 800.1 | – |
| pH | 5.6 | 1.2 | 8.1 | 3.8 |
Concentrations are reported in mM
Male CD1 mice, 18–20 g body weight (Charles River Italy, Calco, Como, Italy) were used in order to prepare murine serum and tissue homogenates. Procedures involving animals and their care were conducted in conformity with national and international laws and policies (EEC Council Directive 86609, OJ L358, 1, 12 December 1987; Italian Legislative Decree 116/92, Gazzetta Ufficiale della Repubblica Italiana n. 10, 18 February 1992; Guide for the Care and Use of Laboratory Animals, US National Research Council, 1996). Animals were housed at constant temperature (22 ± 2 °C) and relative humidity (60 ± 5 %) and were fed ad libitum with a standard diet. They were then anesthetized and blood was collected from the retroorbital plexus. They were then killed by cervical dislocation and the brain, spleen, and liver was excised. Tissues were homogenized in 5 mM PBS, pH 7.4, in a 1:4 (w/v) ratio and centrifuged at 9,000×g for 10 min at 4 °C (Rizzardini et al. 1998). The supernatant was collected and stored at −20 °C. Before use, homogenates were diluted 1:10 (v/v) with distilled water, centrifuged at 1,100×g for 10 min at 25 °C, and the supernatant recovered in order to obtain a transparent solution.
Results and discussion
PLA and PMMA NPs were characterized by DLS in terms of size, polydispersity index and zeta potential. PLA NPs produced by nanoprecipitation showed a larger size and polydispersity than the PMMA ones (Table 2). TEM and SEM analysis showed that PLA and PMMA NPs were spherical and good agreement was found with the DLS results (Fig. 1). Both NP suspensions had a negative zeta potential (Table 2) due to the residual charges of the KPS initiator for PMMA NPs and the dissociation of the carboxylic end groups of the lactic acid for PLA NPs. The larger zeta potential value of the PMMA NPs was confirmed by the higher critical coagulation concentration of the PMMA NPs, which are thus expected to exhibit higher stability. The molecular weight, evaluated by SEC, was 100 and 50 kDa for PMMA and PLA, respectively.
Table 2.
Physico-chemical features of fluorescent-loaded polymeric NPs in distilled water
| Polymer | z-average (nm) | Polydispersity index | Zeta potential (mV) | Critical coagulation concentration (M) |
|---|---|---|---|---|
| PLA | 196.0 ± 0.6 | 0.173 ± 0.003 | −18.5 ± 0.9 | 0.31 |
| PMMA | 95.7 ± 2.1 | 0.040 ± 0.01 | −38.4 ± 2.5 | 0.43 |
Fig. 1.

TEM of PLA NPs (a) and PMMA NPs (b)
The NPs were incubated at 37 °C in different stock solutions, gastrointestinal fluids, serum and tissue homogenates in order to study their colloidal stability. The NPs dimension was monitored by DLS over time to evaluate the occurrence of cluster formation, through aggregation phenomena, which causes a shift of the particle size distribution (PSD) toward higher values. In stock solutions, size distribution did not vary for PLA and PMMA preparations, which thus proved to be stable (Fig. 2a, b) in media in which they are typically stored.
Fig. 2.
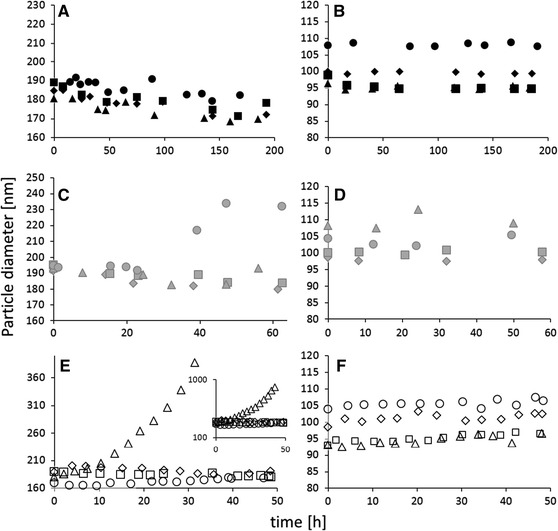
Effect of biological fluids on particle size—DLS measurements. PLA (a, c, e) and PMMA (b, d, f). Stock solutions: distilled water (filled diamond), PBS (filled square), isotonic (filled triangle), dextrose (filled circle); gastrointestinal fluids: synthetic intestinal fluid (shaded diamond), artificial lysosomal fluid (shaded square), saliva (shaded triangle), gastric juice (shaded circle); homogenates: serum (open diamond), brain (open square), spleen (open triangle), liver (open circle)
The stability of NPs in synthetic gastrointestinal fluids was evaluated for 60 h to simulate maximum persistence in the gastrointestinal tract after oral administration. To create the different chemical environments of the gastrointestinal tract (i.e., mouth, stomach, and intestine), fluids usually employed to evaluate the stability of drug formulations were prepared as described in the United States Pharmacopeia (US Pharmacopeia XXIV, 2006) (Table 1). No significant change in PSD was observed for PMMA and PLA preparations incubated at 37 °C with saliva (Fig. 2c, d). In synthetic gastric juice, significant aggregation of PLA NPs began after 24 h incubation (Fig. 2c), leading to an increase of about 20 % of the mean of the PSD, while PMMA NPs remained stable probably thanks to their higher zeta potential (Fig. 2d). Intestinal stability was investigated by incubating NPs in artificial intestinal fluid, prepared without pancreatin, and in lysosomal fluid. The PSDs of PLA and PMMA NPs were not affected proving their stability (Fig. 2c, d).
NP stability in serum and tissue homogenates was considered for 48 h in order to mimic a typical persistence in the body. Homogenates represent the best possibility to get an idea of the effect of the organs or body fluids on the particles.
If NPs aggregate in serum homogenate for instance, this would mean that they could aggregate also upon injection in blood (as serum is a blood component). Not only clots could be formed but also the drug delivery properties of NPs would be affected. On one hand, drug release from single NPs is different than the drug release from a NPs cluster, as the drug has to diffuse throughout different “layers” of particles. On the other hand, the NPs residence time in the body would be affected: if NPs are larger than a certain threshold, they are easily uptaken by white cells (He et al. 2010). As shown in Fig. 2e, PLA NPs remained stable in serum, brain, and liver homogenates indicating that the interaction with serum proteins or the presence of hepatic detoxifying enzymes did not alter their physical stability. PLA NPs, however, significantly aggregated in the spleen homogenate after approximately 15–16 h incubation, forming large clusters of approximately 800 nm after 48 h. PMMA NPs remained stable in all the homogenates for the whole analysis (Fig. 2f) due to their higher zeta potential.
More than on the reasons leading to aggregation, connected to the complex environment regulated by enzymes, salts, and pH, focus should be set on the fact that NPs underwent aggregation in some cases. This clearly shows that the proposed systematic in vitro/ex vivo stability test is essential when dealing with new drug delivery nanoformulations: for any new NP preparation designed for in vivo studies, the colloidal stability should be assessed before in vivo experiments are realized, in order to avoid dangerous aftermath due to aggregates or clots formation. To perform different ex vivo studies as the one of this study, in which the samples were quite diluted, DLS may not be adopted because of the samples’ opacity and the presence of various other particulate substances, similar in size to the NPs. As in in vivo studies, spectrophotofluorimetry is commonly employed to evaluate the NP biodistribution, a correlation between NP fluorescence and colloidal stability was investigated.
For this purpose, fluorescent probes were loaded into the NPs. For PLA, cholesteryl Bodipy® was chosen for its high hydrophobicity, an important issue when dealing with nanoprecipitation (Fessi et al. 1989; Mora-Huertas et al. 2010). The same dye could not be employed for the PMMA formulation, as it is not resistant to radical exposure, for this reason Rhodamine B base was selected.
It must be kept in mind that polymer degradation and dye release can significantly bias the SPF analysis, while this was not an issue with DLS. Considering though that PMMA is non-degradable, only PLA degradation driven by hydrolysis under biological conditions could influence the analysis. Thus, the chemical stability of PLA NPs was considered analyzing the released amount of lactic acid by reversed phase chromatography. (Codari et al. 2010) In the first 7 days, no PLA degradation was observed for any of the salt solutions. Only after 14 days an amount of lactic acid ranging from 2 to 4 % was found, reflecting a slight degradation of the polymer (Table 3). PLA NPs incubated with gastrointestinal fluids, serum or tissue homogenates for 48–60 h did not degrade since no traces of lactic acid were detectable. Thus, these results indicate that degradation was not an issue. Moreover, since diffusion decreases with increasing molecular weight (Masaro and Zhu 1999), dye diffusion from NPs was negligible on account of the high molecular weight of the probes used. As a further argument, an order of magnitude calculation allows to estimate the time needed for such a diffusion to occur. Considering the particle size d = 100 nm and the diffusion coefficient of the dye assumed comparable to a hydrophobic drug and equal to 10–20 m2/s (Polakovic et al. 1999), one can calculate an approximate characteristic time of diffusion equal to 106 s, which is by far higher than the time of analysis considered (105 s).
Table 3.
Lactic acid release from PLA NPs after 14 days of incubation with different salt solutions
| Solution | Lactic acid (ppm) | PLA converted to lactic acid (%) |
|---|---|---|
| Water | 8 ± 2 | 1.6 ± 0.4 |
| 0.9 % Sodium chloride | 12 ± 3 | 2.4 ± 0.6 |
| 5 % Dextrose | 12 ± 2 | 2.4 ± 0.4 |
| 5 mM Phosphate buffered solution, pH 7.4 | 19 ± 4 | 3.8 ± 0.8 |
As shown in Fig. 3, the FI signals remained stable over the whole analysis for all fluids except for the PLA NPs incubated with gastric juice and spleen homogenate (Fig. 3c, e), where the FI signal decreased with time. FI changes occurred only in biological samples where DLS already showed aggregation. Thus, it can be concluded that dye diffusion was indeed negligible, since otherwise the FI signal would have increased with time, and that the signal drop was due to quenching consequent to particle aggregation. Hence, there is a correlation between the information on suspension stability provided by DLS and the FI analysis.
Fig. 3.

Effect of biological fluids on NP stability—FI measurements. Stock solutions: distilled water (filled diamond), PBS (filled square), isotonic (filled triangle), dextrose (filled circle); gastrointestinal fluids: Synthetic intestinal fluid (shaded diamond), artificial lysosomal fluid (shaded square), saliva (shaded triangle), gastric juice (shaded circle); homogenates: serum (open diamond), brain (open square), spleen (open triangle), liver (open circle)
Figure 3c, e shows that the FI signal drops 30 % after approximately 22 and 12 h, respectively. These are roughly the times of the start of aggregation established by DLS (Fig. 2c, e). Far from providing a quantitative correlation, these findings do confirm a qualitative accordance between the techniques and thus the possibility of using spectrophotofluorimetry to verify NP stability in ex vivo samples.
Conclusions
To design effective polymeric NPs for drug delivery, their stability has to be assessed thoroughly not only in stock solutions but also in fluids mimicking biological ones and in organ homogenates. Colloidal stability and polymer degradation of NPs in synthetic gastrointestinal fluids, serum and tissue homogenates was evaluated for 48–60 h, to reproduce their typical time of persistence in the body, and up to 200 h in stock solutions. While PMMA NPs remained stable in all the fluids, PLA NPs aggregated in gastric juice and in spleen homogenate.
These data also indicate that DLS is a powerful tool to evaluate NP colloidal stability in vitro/ex vivo experiments and that SPF analysis can be used to assess the stability as well. Qualitative accordance was found between DLS and spectrophotofluorimetry proving that this latter technique can indeed be used when switching from in vitro to ex vivo analysis to overcome opacity of the biological samples. These stability evaluation procedures should be therefore adopted for any new NP formulation before attempting in vivo studies.
Acknowledgments
This study was partly funded by the Telethon Foundation, Project n. GGP 10120, by the European Community’s Seventh Framework Program (FP7/2007-2013) under grant agreement n. 212043 and by AIRC—Associazione Italiana per la Ricerca sul Cancro, Special Program Molecular Clinical Oncology 5 per mille, Project n. 10016. All these funders have not roles in the study design and in the decision to submit the paper for publication. The authors acknowledge the support by the Electron Microscopy of ETH Zurich (EMEZ). Furthermore they thank Marco Furlan for taking the TEM pictures.
Conflict of interest
All authors disclose any actual or potential conflict of interest including any financial, personal or other relationships with other people or organizations within 3 years of beginning the work submitted that could inappropriately influence (bias) their work.
Glossary
- NPs
Nanoparticles
- DLS
Dynamic light scattering
- PLA
Poly-lactic acid
- PMMA
Poly-methyl-methacrylate
- Sn(Oct)2
2-Ethylhexanoic acid tin(II) salt
- LT
S,S-3,6-Dimethyl-1,4-dioxane-2,5-dione
- SEC
Size exclusion chromatography
- THF
Tetrahydrofuran
- TEM
Transmission electron microscopy
- SEM
Scanning electron microscopy
- FI
Fluorescence intensity
- HPLC
High pressure liquid chromatography
- PSD
Particle size distribution
References
- Aggarwal P, Hall JB, McLeland CB, Dobrovolskaia MA, McNeil SE. Nanoparticle interaction with plasma proteins as it relates to particle biodistribution, biocompatibility and therapeutic efficacy. Adv Drug Deliv Rev. 2009;61(6):428–437. doi: 10.1016/j.addr.2009.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexis F, Pridgen E, Molnar LK, Farokhzad OC. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol Pharm. 2008;5(4):505–515. doi: 10.1021/mp800051m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arifin DY, Lee LY, Wang CH. Mathematical modeling and simulation of drug release from microspheres: Implications to drug delivery systems. Adv Drug Deliv Rev. 2006;58(12–13):1274–1325. doi: 10.1016/j.addr.2006.09.007. [DOI] [PubMed] [Google Scholar]
- Avgoustakis K, Beletsi A, Panagi Z, Klepetsanis P, Karydas AG, Ithakissios DS. PLGA-mPEG nanoparticles of cisplatin: in vitro nanoparticle degradation, in vitro drug release and in vivo drug residence in blood properties. J Control Release. 2002;79(1–3):123–135. doi: 10.1016/S0168-3659(01)00530-2. [DOI] [PubMed] [Google Scholar]
- Codari F, Moscatelli D, Storti G, Morbidelli M. Characterization of low-molecular-weight PLA using HPLC. Macromol Mater Eng. 2010;295(1):58–66. doi: 10.1002/mame.200900172. [DOI] [Google Scholar]
- Cui F, Qian F, Zhao Z, Yin L, Tang C, Yin C. Preparation, characterization, and oral delivery of insulin loaded carboxylated chitosan grafted poly(methyl methacrylate) nanoparticles. Biomacromolecules. 2009;10(5):1253–1258. doi: 10.1021/bm900035u. [DOI] [PubMed] [Google Scholar]
- Devalapally H, Chakilam A, Amiji MM. Role of nanotechnology in pharmaceutical product development. J Pharm Sci. 2007;96(10):2547–2565. doi: 10.1002/jps.20875. [DOI] [PubMed] [Google Scholar]
- Fessi H, Puisieux F, Devissaguet JP, Ammoury N, Benita S. Nanocapsule formation by interfacial polymer deposition following solvent displacement. Int J Pharm. 1989;55(1):R1–R4. doi: 10.1016/0378-5173(89)90281-0. [DOI] [Google Scholar]
- He CB, Hu YP, Yin LC, Tang C, Yin CH. Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials. 2010;31(13):3657–3666. doi: 10.1016/j.biomaterials.2010.01.065. [DOI] [PubMed] [Google Scholar]
- Kuentz M. Drug absorption modeling as a tool to define the strategy in clinical formulation development. AAPS J. 2008;10(3):473–479. doi: 10.1208/s12248-008-9054-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari A, Yadav SK, Yadav SC. Biodegradable polymeric nanoparticles based drug delivery systems. Colloids Surf B. 2010;75(1):1–18. doi: 10.1016/j.colsurfb.2009.09.001. [DOI] [PubMed] [Google Scholar]
- Lemoine D, Francois C, Kedzierewicz F, Preat V, Hoffman M, Maincent P. Stability study of nanoparticles of poly(epsilon-caprolactone), poly(d,l-lactide) and poly(d,l-lactide-co-glycolide) Biomaterials. 1996;17(22):2191–2197. doi: 10.1016/0142-9612(96)00049-X. [DOI] [PubMed] [Google Scholar]
- Liu Y, Cheng CY, Prud’homme RK, Fox RO. Mixing in a multi-inlet vortex mixer (MIVM) for flash nano-precipitation. Chem Eng Sci. 2008;63(11):2829–2842. doi: 10.1016/j.ces.2007.10.020. [DOI] [Google Scholar]
- Madlova M, Jones SA, Zwerschke I, Ma Y, Hider RC, Forbes B. Poly(vinyl alcohol) nanoparticle stability in biological media and uptake in respiratory epithelial cell layers in vitro. Eur J Pharm Biopharm. 2009;72(2):437–443. doi: 10.1016/j.ejpb.2009.01.009. [DOI] [PubMed] [Google Scholar]
- Masaro L, Zhu XX. Physical models of diffusion for polymer solutions, gels and solids. Prog Polym Sci. 1999;24(5):731–775. doi: 10.1016/S0079-6700(99)00016-7. [DOI] [Google Scholar]
- Mohs AM, Duan HW, Kairdolf BA, Smith AM, Nie SM. Proton-resistant quantum dots: stability in gastrointestinal fluids and implications for oral delivery of nanoparticle agents. Nano Res. 2009;2(6):500–508. doi: 10.1007/s12274-009-9046-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mora-Huertas CE, Fessi H, Elaissari A. Polymer-based nanocapsules for drug delivery. Int J Pharm. 2010;385(1–2):113–142. doi: 10.1016/j.ijpharm.2009.10.018. [DOI] [PubMed] [Google Scholar]
- Muthu MS, Feng SS. Pharmaceutical stability aspects of nanomedicines. Nanomedicine (Lond) 2009;4(8):857–860. doi: 10.2217/nnm.09.75. [DOI] [PubMed] [Google Scholar]
- Oh JK. Polylactide (PLA)-based amphiphilic block copolymers: synthesis, self-assembly, and biomedical applications. Soft Matter. 2011;7(11):5096–5108. doi: 10.1039/c0sm01539c. [DOI] [Google Scholar]
- Polakovic M, Gorner T, Gref R, Dellacherie E. Lidocaine loaded biodegradable nanospheres II. Modelling of drug release. J Control Release. 1999;60(2–3):169–177. doi: 10.1016/S0168-3659(99)00012-7. [DOI] [PubMed] [Google Scholar]
- Prokop A, Davidson JM. Nanovehicular intracellular delivery systems. J Pharm Sci. 2008;97(9):3518–3590. doi: 10.1002/jps.21270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riehemann K, Schneider SW, Luger TA, Godin B, Ferrari M, Fuchs H. Nanomedicine—challenge and perspectives. Angew Chem Int Edn. 2009;48(5):872–897. doi: 10.1002/anie.200802585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzardini M, Zappone M, Villa P, Gnocchi P, Sironi M, Diomede L, Meazza C, Monshouwer M, Cantoni L. Kupffer cell depletion partially prevents hepatic heme oxygenase 1 messenger RNA accumulation in systemic inflammation in mice: role of interleukin 1beta. Hepatology. 1998;27(3):703–710. doi: 10.1002/hep.510270311. [DOI] [PubMed] [Google Scholar]
- Ruenraroengsak P, Cook JM, Florence AT. Nanosystem drug targeting: facing up to complex realities. J Control Release. 2010;141(3):265–276. doi: 10.1016/j.jconrel.2009.10.032. [DOI] [PubMed] [Google Scholar]
- Squire MW, Ludwig BJ, Thompson JR, Jagodzinski J, Hall D, Andes D. Premixed antibiotic bone cement—an in vitro comparison of antimicrobial efficacy. J Arthroplast. 2008;23(6):110–114. doi: 10.1016/j.arth.2008.03.014. [DOI] [PubMed] [Google Scholar]
- Burkersroda F, Schedl L, Gopferich A. Why degradable polymers undergo surface erosion or bulk erosion. Biomaterials. 2002;23(21):4221–4231. doi: 10.1016/S0142-9612(02)00170-9. [DOI] [PubMed] [Google Scholar]
- Win KY, Feng SS. Effects of particle size and surface coating on cellular uptake of polymeric nanoparticles for oral delivery of anticancer drugs. Biomaterials. 2005;26(15):2713–2722. doi: 10.1016/j.biomaterials.2004.07.050. [DOI] [PubMed] [Google Scholar]
- Yu YC, Storti G, Morbidelli M. Ring-opening polymerization of l,l-lactide: kinetic and modeling study. Macromolecules. 2009;42(21):8187–8197. doi: 10.1021/ma901359x. [DOI] [Google Scholar]