

We show here that the i-AAA protease Yme1 has a role in folding of proteins in the intermembrane space of mitochondria and identify a number of endogenous proteins that aggregate in its absence. Thus the function of Yme1 in mitochondrial proteostasis extends beyond its role in proteolytic removal of misfolded and nonassembled inner membrane proteins.
Abstract
The vast majority of mitochondrial proteins are synthesized in the cytosol and transported into the organelle in a largely, if not completely, unfolded state. The proper function of mitochondria thus depends on folding of several hundreds of proteins in the various subcompartments of the organelle. Whereas folding of proteins in the mitochondrial matrix is supported by members of several chaperone families, very little is known about folding of proteins in the intermembrane space (IMS). We targeted dihydrofolate reductase (DHFR) as a model substrate to the IMS of yeast mitochondria and analyzed its folding. DHFR can fold in this compartment, and its aggregation upon heat shock can be prevented in an ATP-dependent manner. Yme1, an AAA (ATPases associated with diverse cellular activities) protease of the IMS, prevented aggregation of DHFR. Analysis of protein aggregates in mitochondria lacking Yme1 revealed the presence of a number of proteins involved in the establishment of mitochondrial ultrastructure, lipid metabolism, protein import, and respiratory growth. These findings explain the pleiotropic effects of deletion of YME1 and suggest an important role for Yme1 as a folding assistant, in addition to its proteolytic function, in the protein homeostasis of mitochondria
INTRODUCTION
The information needed for an unfolded polypeptide chain to reach its correct, biologically active form is encoded in its primary sequence. However, folding of the majority of newly synthesized proteins in the crowded environment of the cell depends on an elaborate system of cellular folding helpers, the molecular chaperones (Bukau et al., 2006; Mayer, 2010; Hartl et al., 2011). Molecular chaperones not only assist de novo folding, but also step in when cells are under stress to prevent unfolding and aggregation. In addition, chaperones can also mediate disaggregation and refolding of their substrates. Proteins that fail to fold correctly are degraded by a number of cellular proteases, including those of the ubiquitin-proteasome and the autophagy systems (Chen et al., 2011). Failure to maintain proteostasis is associated with a number of severe human disorders, most notably neurodegenerative diseases (Balch et al., 2008). Such intricate protein quality control systems have been observed in all kingdoms of life and are present in almost every subcompartment of the cell (Ron and Walter, 2007; Jonikas et al., 2009; Voos, 2009; Baker and Haynes, 2011; Baker et al., 2011; Bender et al., 2011; Smith et al., 2011).
Mitochondria are essential cell organelles delineated by two membranes: the outer and the inner mitochondrial membranes. The two membranes define two aqueous subcompartments: the intermembrane space (IMS) and the innermost matrix. Mitochondria occupy many hub positions in cellular metabolism, ranging from ATP production and biogenesis of Fe-S clusters to regulation of cell death (Nunnari and Suomalainen, 2012). The vast majority of their proteins are encoded in the nuclear genome, synthesized on cytosolic ribosomes with specific mitochondrial targeting signals, and then transported into the organelle with the help of one or more mitochondrial protein translocases (Neupert and Herrmann, 2007; Chacinska et al., 2009; Endo and Yamano, 2009). Mitochondrial protein translocases are complex molecular machines that recognize various mitochondrial targeting signals and then translocate and sort proteins into the mitochondrial subcompartment in which they fulfill their functions. The protein conducting channels of the mitochondrial protein translocases only allow transport of completely or largely unfolded proteins. Thus the function of mitochondria depends on the folding of many hundreds of proteins upon their release from the translocation channels. In this respect, folding upon translocation resembles folding of cytosolic proteins upon their exit from the ribosome tunnel.
Proteins residing in the mitochondrial matrix are synthesized with cleavable N-terminal matrix targeting signals (MTS), also called presequences. They are transported across both mitochondrial membranes in a completely unfolded state by the concerted action of the translocase of the outer mitochondrial membrane (TOM) and translocase of the inner mitochondrial membrane (TIM23) complexes in the outer and inner membranes, respectively. In the matrix, members of several families of molecular chaperones help newly imported proteins to fold through ATP- and cochaperone-regulated cycles of binding and release. Mitochondrial heat shock protein 70 (mtHsp70; known as Ssc1 in yeast) functions as part of the ATP-dependent import motor of the TIM23 complex to drive unidirectional transport into the matrix and subsequently assists in the folding process of these substrate proteins (Kang et al., 1990; Horst et al., 1997; Mapa et al., 2010). In the matrix, the ATPase activity of mtHsp70, and thereby its folding ability, is regulated by two cochaperones, the J-protein Mdj1 and the nucleotide exchange factor Mge1 (Rowley et al., 1994; Westermann et al., 1995). The Hsp60-Hsp10 system mediates ATP-dependent folding of single protein molecules enclosed in a cage (Ostermann et al., 1989; Tang et al., 2006). Furthermore, Hsp78, a member of the Clp/Hsp100 family, cooperates with mtHsp70 in the refolding of denatured proteins under stress conditions (Schmitt et al., 1995). Hsp78 can also dissolve small aggregates and promote degradation by the Pim1/LON protease of proteins that fail to refold (Voos, 2009). In mitochondria of higher eukaryotes, members of the Hsp90 and small heat shock chaperone families have also been identified (Felts et al., 2000; Morrow et al., 2010; Altieri et al., 2012).
Proteins residing in the IMS are transported across the outer membrane in a largely, if not completely, unfolded state. However, no member of the known chaperone families has been identified in this compartment, and little is known about folding of proteins in the IMS in general. Whereas only a single import pathway directing proteins to the matrix has been identified, several import pathways exist by which proteins reach the IMS. One group of proteins residing in the IMS are transported with bipartite targeting signals. The N-terminal MTSs target these proteins toward the matrix via the TOM-TIM23 pathway; an additional stop-transfer signal leads to arrest of translocation at the level of the inner membrane, and the proteins are laterally released by the TIM23 complex into the inner membrane. Such proteins can either remain anchored in the inner membrane or are cleaved by specific processing peptidases at the level of the inner membrane and released as soluble proteins into the IMS (Glick et al., 1992; Herrmann and Hell, 2005; Mokranjac and Neupert, 2008; Chacinska et al., 2009). Members of a second group of IMS proteins contain conserved cysteine motifs. They cross the outer membrane through the TOM complex and are captured in the IMS by the Mia40-Erv1 disulfide relay system. Mia40 (Tim40), in its oxidized form, specifically recognizes such proteins and introduces disulfide bonds into them. Erv1 then regenerates the oxidized, active form of Mia40. This recently identified import pathway not only drives vectorial movement of proteins across the outer membrane, but introduction of disulfide bonds seems to be sufficient for at least some of the substrates of this pathway to adopt their fold (Hell, 2008; Stojanovski et al., 2008; Koehler and Tienson, 2009; Sideris and Tokatlidis, 2010; Herrmann and Riemer, 2012). A similar folding-trap mechanism has also been proposed for import and folding of a third group of IMS proteins. A typical example of this group is cytochrome c, which is translocated in its apo form and, upon addition of the heme group catalyzed by cytochrome c heme lyase, folds and is thereby retained in the IMS (Dumont et al., 1991). Similarly, Ccs1 introduces a disulfide bond and a copper ion into Sod1, trapping the enzyme in the IMS (Field et al., 2003). How the rest of proteins that reside in the IMS fold remains unknown. Notably, the isolated AAA (ATPases associated with diverse cellular activities) domain of the i-AAA protease Yme1, a component of the mitochondrial protein quality control (reviewed in Baker et al., 2011), which exposes its catalytic domain into the IMS, has been suggested to have a chaperone-like activity in vitro (Leonhard et al., 1999). This suggestion was recently supported by the observation that the folding and/or assembly of subunit II of cytochrome c oxidase is dependent on Yme1 (Fiumera et al., 2009).
In the present study, we set out to investigate folding of proteins in the IMS. To this end, we expressed mouse dihydrofolate reductase (DHFR) in the IMS of yeast mitochondria and analyzed its folding. DHFR is a frequently used model substrate for investigating import and folding in mitochondria (Gaume et al., 1998; Vestweber and Schatz, 1988; Junker et al., 2005; Yamano et al., 2008). We observed that DHFR can fold in the IMS. Under heat-stress conditions, aggregation of DHFR in the IMS was prevented in an ATP-dependent manner. Interestingly, we found that folding of DHFR in the IMS depended on the presence of Yme1. Protein aggregates isolated from mitochondria lacking Yme1 were found to contain several mitochondrial proteins with a role in the establishment of mitochondrial ultrastructure, lipid metabolism, respiratory growth, and protein import. These data explain the pleiotropic effects of deletion of YME1 and suggest an important role of Yme1 as a folding assistant, in addition to its proteolytic function, in the protein homeostasis of mitochondria.
RESULTS
Expression and localization of model substrates in mitochondria
To gain insight into folding of proteins in the mitochondrial IMS, we expressed mouse DHFR in yeast and analyzed its folding in this compartment. DHFR was targeted to the IMS (IMS-DHFR) using the bipartite targeting signal of yeast cytochrome b2 (Figure 1A). For comparison, a hybrid protein was expressed in which the stop-transfer signal of cytochrome b2 was deleted, thus targeting DHFR to the matrix (matrix-DHFR). In addition, we expressed a mutant of DHFR that does not fold (DHFRmut; Vestweber and Schatz, 1988) both in the IMS and in the matrix. With IMS-DHFR, we observed two forms, the intermediate (i) form, in which only the MTS was proteolytically removed by mitochondrial processing peptidase, and the mature (m) form, in which the stop-transfer signal was additionally removed by the inner membrane peptidase (IMP; Geissler et al., 2000 and references therein; Figure 1B). Matrix-DHFR is proteolytically processed only once, to the m-form. Expression of the DHFR constructs did not compromise the integrity of mitochondria, as the expression levels of various mitochondrial proteins, Tim50, cytochrome b2, Tom22, and Ssc1, present in the different mitochondrial subcompartments were indistinguishable among the four types of mitochondria. To verify that the DHFR constructs were located in the correct mitochondrial subcompartments, we subjected isolated mitochondria to digitonin fractionation (Figure 1, C–F). Incubation of intact mitochondria with proteinase K (PK) led to degradation of surface-exposed Tom70, but not to degradation of any of the DHFR constructs. Mitochondria were then treated, in the presence of PK, with increasing concentrations of digitonin to successively open the outer and inner membranes. Both the wild-type and mutant versions of IMS-DHFR were degraded when the outer membrane was open, as evidenced by degradation of IMS-exposed inner membrane protein Tim50 (Figure 1, C and E). In contrast, matrix-DHFRWT and matrix-DHFRmut behaved like the matrix protein Hep1 (Figure 1, D and F). Taken together, these results show that all four model substrates were translocated into mitochondria and sorted to the expected subcompartments of the organelle.
FIGURE 1:

Generation and localization of DHFR constructs in mitochondria. (A) Schematic representation of model substrates. Wild-type and nonfolding mutant versions of mouse DHFR (C7S, S42C, N49C) were fused C-terminally to amino acid residues 1–107 of yeast cytochrome b2, targeting it to the IMS (IMS-DHFR). Constructs with deleted stop-transfer signal are targeted to the matrix (matrix-DHFR). p, bipartite targeting signal of cytochrome b2. Cleavage by the mitochondrial processing peptidase (MPP) leads to the intermediate-sized i-form; cleavage by the IMP leads to the mature m-form. (B) Mitochondria were isolated and analyzed by SDS–PAGE and immunodecoration using antibodies against the indicated proteins. DHFR constructs: i-form and m-form. (C–F) Mitochondria were incubated with increasing amounts of digitonin (0.005–0.1%) in the presence of PK for 25 min on ice. Samples were analyzed by SDS–PAGE and immunodecoration using antibodies against DHFR and the indicated mitochondrial marker proteins (Tom70, outer membrane; Tim50, IMS; Hep1, matrix). (G) Isolated mitochondria were solubilized with Triton X-100 and incubated with PK for 20 min at 0°C in the presence and absence of methotrexate (Mtx). Samples were analyzed by SDS–PAGE and immunodecoration with antibodies against DHFR. sf, stable fragment upon protease digestion.
We then asked whether the model substrates attained their mature fold in mitochondria. We lysed mitochondria with Triton X-100 and subjected them to PK treatment (Figure 1G). IMS-DHFRWT, as well as matrix-DHFRWT, yielded a protease-resistant fragment of circa 25 kDa, corresponding to folded DHFR (Gaume et al., 1998). In contrast, both DHFRmut constructs were completely degraded by PK. Thus DHFR can fold both in the IMS and in the matrix. Binding of methotrexate, a folate analogue, stabilizes the DHFR fold even further. Indeed, in the presence of methotrexate, with both IMS-DHFRWT and matrix-DHFRWT, more of the stable fragments were generated. As was expected, neither of the DHFRmut constructs was stabilized by methotrexate.
We conclude that DHFR can be stably expressed in the IMS, as well as in the matrix, and that it can fold properly in both mitochondrial subcompartments.
Requirements for folding of DHFR in IMS and matrix
We analyzed the effects of nucleotides and heat shock on aggregation of DHFR in mitochondria. The ATP levels of mitochondria isolated from cells expressing the various DHFR constructs were manipulated before they were subjected to a heat shock at 42°C for 3 min. A separate set of samples was kept at 25°C as a control. Subsequently, mitochondria were lysed with Triton X-100, and pellet and supernatant fractions representing aggregated and soluble proteins, respectively, were separated by centrifugation. At 25°C, IMS-DHFRWT was exclusively found in the soluble fraction, irrespective of the presence of ATP. In contrast, upon heat shock, IMS-DHFRWT distributed between soluble and aggregated fractions (Figure 2A, left panel). Interestingly, the i- and m-forms behaved differently. The i-form, still anchored to the inner membrane, was found predominantly in the aggregate fraction, irrespective of the presence or absence of ATP. The m-form, soluble in the IMS, was predominantly found in the soluble fraction in the presence of ATP but mostly aggregated in its absence. Thus, upon heat shock, the aggregation of DHFR in the IMS could be prevented in an ATP-dependent process. On the other hand, in the matrix, DHFR was found mostly in the soluble fraction, and only minor ATP-dependent, aggregation was observed upon heat shock (Figure 2A, right panel).
FIGURE 2:
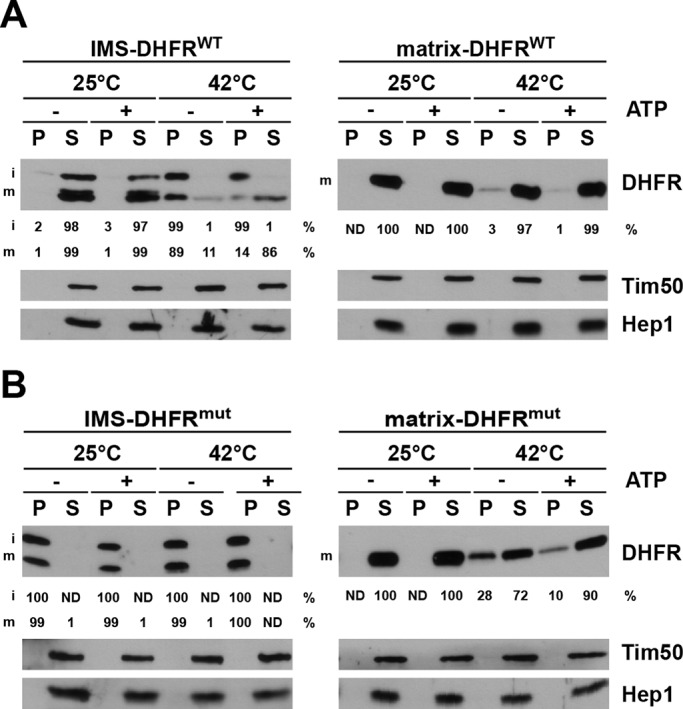
Aggregation of wild-type and mutant DHFR constructs in IMS and matrix. (A and B) Isolated mitochondria were incubated under conditions to increase or decrease the mitochondrial ATP levels and then exposed to 25°C or 42°C for 3 min. Mitochondria were then solubilized with Triton X-100–containing buffer and soluble (S) and aggregate (P, pellet) fractions separated by centrifugation and analyzed by SDS–PAGE and immunodecoration using the indicated antibodies. The DHFR signals were quantified in supernatant and pellet fractions and expressed as percentages of total. ND, not detectable.
A different picture was seen when folding of DHFRmut was analyzed (Figure 2B). IMS-DHFRmut aggregated even without heat treatment, and was present under all conditions entirely in the pellet fraction (Figure 2B, left panel). On the other hand, matrix-DHFRmut was found completely in the soluble fractions at 25°C (Figure 2B, right panel). On heat shock, part of it was found in the aggregate fraction. This aggregate was more prominent in the absence of ATP than in its presence, in agreement with the ATP-dependent action of molecular chaperones in the matrix.
Taken together, these experiments demonstrate that DHFR can fold in the IMS and that prevention of its aggregation upon heat shock is an ATP-dependent process. Furthermore, the folding capacity of the IMS is apparently lower than that of the mitochondrial matrix, at least when folding of DHFR is considered.
Folding helpers of DHFR in the IMS
To identify candidates with chaperone-like activity in the IMS that help DHFR to fold, we sought to identify binding partners of IMS-DHFR. We generated yeast strains expressing C-terminally His6-tagged variants of IMS- and matrix-DHFR, incubated solubilized mitochondria with NiNTA-agarose beads, and subsequently analyzed proteins bound to the beads by mass spectrometry (MS; Figure 3A). Mitochondria isolated from a yeast strain transformed with the empty plasmid served as a control. We found Yme1, Mgr1, and Mgr3 in elution fractions with IMS-DHFR, but not with matrix-DHFR nor with mitochondria not expressing any DHFR construct. Yme1, an AAA protease, is a component of the mitochondrial protein quality control system exposing its catalytic domain into the IMS (Baker et al., 2011). Mgr1 and Mgr3 have recently been implicated in the mitochondrial protein quality control system as well, and it has been speculated that they serve as adaptors to target substrates to Yme1 (Dunn et al., 2006, 2008).
FIGURE 3:

Identification of potential folding helpers of IMS-DHFR. (A) Workflow of NiNTA pulldown of His-tagged DHFR constructs and subsequent quantitative analysis by label-free MS. (B) Isolated mitochondria were solubilized with digitonin-containing buffer in the absence of nucleotides or in the presence of ATP or ADP. Samples were incubated with NiNTA-Agarose beads and specifically bound proteins were eluted with Laemmli buffer containing 500 mM imidazole. Total (10%) and bound (100%) fractions were analyzed by SDS–PAGE and immunodecoration with the indicated antibodies.
We went on to confirm the MS data by Western blotting (Figure 3B). A pulldown with NiNTA-agarose was performed, and fractions corresponding to total material and eluate were analyzed by SDS–PAGE and immunodecoration. Yme1 was indeed specifically copurified with His-tagged IMS-DHFR. Interestingly, we observed copurification of Yme1 with IMS-DHFR only if mitochondria were solubilized in the absence of added nucleotides or in the presence of ADP, but not if solubilization was carried out in the presence of ATP (Figure 3B). This behavior is typical for chaperone-substrate interactions in which ADP stabilizes interactions, whereas in the presence of ATP, interactions are transient and therefore usually not detectable. Interestingly, immunodecoration with antibodies against Hsp60 and Hsp70 demonstrated weak but specific and ATP-dependent copurification of these matrix chaperones with matrix-DHFR. In conclusion, IMS-DHFR binds specifically and in an ATP-dependent manner to Yme1, suggesting a role for the AAA protease in the quality control of this protein in vivo.
Role of Yme1 in folding of DHFR in the IMS
To analyze the role of Yme1 in folding of IMS-DHFR, we expressed DHFR constructs in cells lacking Yme1. Interestingly, we observed higher levels of IMS-DHFR in mitochondria lacking Yme1 than in wild-type (Figure 4A). In contrast, absence of Yme1 had no effect on the levels of matrix-DHFR, further supporting a specific role of Yme1 in the biogenesis of IMS-DHFR. The higher expression levels of IMS-DHFR in the absence of Yme1 are likely due to reduced degradation of IMS-DHFR, in agreement with the known proteolytic role of Yme1 in protein quality control in the IMS (Leonhard et al., 2000). The levels of other mitochondrial proteins analyzed were not affected by deletion of YME1 (Figure 4A).
FIGURE 4:

Effect of Yme1 on aggregation of DHFR fusion proteins. (A) Isolated mitochondria (5 and 15 μg) were analyzed by SDS–PAGE and immunodecoration with indicated antibodies. (B and C) Mitochondria were solubilized with Triton X-100–containing buffer, and soluble (S) and aggregate (P, pellet) fractions were separated by centrifugation. Samples were analyzed by SDS–PAGE followed by immunodecoration with the indicated antibodies.
Next we analyzed the folding state of the DHFR constructs in mitochondria lacking Yme1 (Figure 4B). Isolated mitochondria were solubilized, and soluble and aggregated fractions were separated by centrifugation. In the absence of Yme1, IMS-DHFR was found in the aggregate fraction, even under nonstress conditions (Figure 4B). This demonstrates that Yme1 is not simply involved in degradation of DHFR but also argues for its role in folding of this model substrate. Interestingly, the two forms of IMS-DHFR behaved differently. The i-form was recovered predominantly in the aggregate fraction, whereas the m-form was distributed equally between soluble and aggregate fractions. Thus both the membrane-anchored and soluble forms of DHFR require Yme1 for folding, but it seems that the membrane-anchored form is more dependent on Yme1. In contrast, both forms were completely soluble in the presence of Yme1, as also shown above. Importantly, deletion of YME1 had no effect on folding of DHFR in the matrix. Matrix-DHFR remained soluble in both types of mitochondria (Figure 4C).
Taken together, these data demonstrate a specific role of Yme1 in the folding of DHFR in the IMS, independent of its function in degradation, strongly suggesting a chaperone-like activity of the i-AAA protease in vivo.
Aggregation of proteins in mitochondria lacking Yme1
To identify endogenous substrates of Yme1, we analyzed the protein aggregation in mitochondria lacking Yme1, using stable isotope labeling with amino acids in cell culture (SILAC) and MS. To this end, wild-type cells and cells lacking Yme1 were grown in medium with 13C and 15N (“heavy”) and 12C and 14N (“light”) lysine. Mitochondria were isolated and solubilized, and soluble and aggregate fractions were separated by centrifugation. The aggregate fractions from both types of mitochondria were mixed and separated by SDS–PAGE; this was followed by Lys-C digestion and analysis by MS. Table 1 contains mitochondrial proteins that, in at least two out of four experiments, showed 1.6-fold or higher aggregation propensity in mitochondria lacking Yme1. Thus these proteins represent the most likely candidates for endogenous substrates of chaperone activity of Yme1. Importantly, the list predominantly contains proteins known to reside in the IMS, demonstrating a specific role of Yme1 in folding in this compartment. Interestingly, among the proteins identified were Ups2, a recently identified soluble IMS protein involved in lipid metabolism (Osman et al., 2009a; Tamura et al., 2009, 2012); Nde1, the NADH:ubiquinone dehydrogenase anchored in the inner membrane (Augustin et al., 2005); and prohibitins, protein and lipid scaffolds in the inner membrane (Osman et al., 2009b). These three proteins were previously shown to be proteolytic substrates of Yme1 (Augustin et al., 2005; Kambacheld et al., 2005; Dunn et al., 2006, 2008; Potting et al. 2010). Our data suggest that Yme1 may also be involved in their folding. Another protein enriched in the aggregates of mitochondria lacking Yme1 was Cox2, a previously shown proteolytic, but also a possible folding, substrate of Yme1 (Nakai et al., 1995; Pearce and Sherman, 1995; Weber et al., 1996; Fiumera et al., 2009).
TABLE 1:
Proteins that aggregate in Δyme1 mitochondria.
| Open reading frame | Protein | Submitochondrial locationa | Transmembrane domainsa | Cofactor | Functionb | |
|---|---|---|---|---|---|---|
| 1 | Q0250 | COX2 | IM-IMS | 2 | Copper | Subunit II of cytochrome c oxidase, mitochondrially encoded |
| 2 | YAL039C | CYC3 | IMS | — | Heme, iron | Cytochrome c heme lyase (holocytochrome c synthase), attaches heme to apo-cytochrome c in the IMS |
| 3 | YBL095W | — | Not known | 1 | — | Unknown |
| 4 | YBR262C | AIM5 | IM-IMS | 1 | — | Subunit of mitochondrial IM organizing system (MitOS/MICOS/MINOS), role in maintenance of cristae junctions and IM architecture |
| 5 | YBR282W | MRPL27 | Matrix | — | — | Mitochondrial ribosomal protein of the large subunit |
| 6 | YCL044C | MGR1 | IM-IMS | 2 | — | Subunit of mitochondrial i-AAA protease, which degrades misfolded mitochondrial proteins, binds to substrates to facilitate proteolysis, and is required for growth of rho0 cells |
| 7 | YCR071C | IMG2 | Matrix | — | — | Mitochondrial ribosomal protein of the large subunit |
| 8 | YDL174C | DLD1 | IM-IMS | 1 | FAD, zinc | d-lactate dehydrogenase, oxidizes d-lactate to pyruvate |
| 9 | YDR316W | OMS1 | IM-IMS | 1 | — | With conserved methyltransferase motif, multicopy suppressor of respiratory defects caused by OXA1 mutations |
| 10 | YFL036W | RPO41 | Matrix | — | — | RNA polymerase; enhancing DNA bending and melting to facilitate preinitiation open-complex formation |
| 11 | YFR011C | AIM13 | IMS | — | — | Subunit of mitochondrial IM organizing system (MitOS/MICOS/MINOS), role in maintenance of cristae junctions and IM architecture |
| 12 | YGL057C | GEP7 | IM | 1 | — | Unknown function; null mutant exhibits respiratory growth defect and synthetic interactions with prohibitin (Phb1) and Gem1 |
| 13 | YGL068W | MNP1 | Matrix | — | — | Protein associated with mitochondrial nucleoid, required for normal respiratory growth |
| 14 | YGR029W | ERV1 | IMS | — | FAD | Flavin-linked sulfhydryl oxidase, oxidizes Mia40p as part of the disulfide relay system |
| 15 | YGR076C | MRPL25 | Matrix | — | — | Mitochondrial ribosomal protein of the large subunit |
| 16 | YGR132C | PHB1 | IM-IMS | 1 | — | Inner mitochondrial membrane chaperone that stabilizes newly synthesized proteins |
| 17 | YGR174C | CBP4 | IM-IMS | 1 | — | Required for assembly of cytochrome bc1 complex; interacts with the Cbp3-Cbp6 complex and newly synthesized cytochrome b to promote assembly of cytochrome b into cytochrome bc1 complex |
| 18 | YGR286C | BIO2 | Matrix | — | Iron, sulfur | Biotin synthase, catalyzes the conversion of dethiobiotin to biotin |
| 19 | YHL021C | AIM17 | Not known | — | Iron | Unknown; null mutant displays reduced frequency of mitochondrial genome loss |
| 20 | YHR005C-A | TIM10 | IMS | — | Zinc | Essential IMS protein, forms a complex with Tim9 that delivers hydrophobic proteins to TIM22 complex for insertion into the IM |
| 21 | YHR024C | MAS2 | Matrix | — | Zinc | Large subunit of mitochondrial processing protease, essential processing enzyme, cleaves the N-terminal targeting sequences from mitochondrially imported proteins |
| 22 | YIL155C | GUT2 | IM-IMS | 1 | FAD | Mitochondrial glycerol-3-phosphate dehydrogenase |
| 23 | YJL066C | MPM1 | Not known | — | — | Unknown function, no hydrophobic stretches |
| 24 | YJR045C | SSC1 | Matrix | — | ATP | Hsp70 family ATPase, constituent of the import motor component of TIM23 complex, involved in protein translocation and folding |
| 25 | YJR048W | CYC1 | IMS | — | Heme, iron | Electron carrier of mitochondrial intermembrane space, transfers electrons from ubiquinone-cytochrome c oxidoreductase to cytochrome c oxidase during cellular respiration |
| 26 | YJR100C | AIM25 | Not known | — | — | Unknown function, null mutant viable/displays elevated rate of mitochondrial genome loss |
| 27 | YKL138C | MRPL31 | Matrix | — | — | Mitochondrial ribosomal protein of the large subunit |
| 28 | YKL150W | MCR1 | OM/IMS | 1/- | FAD, NAD | Mitochondrial NADH-cytochrome b5 reductase, involved in ergosterol biosynthesis |
| 29 | YKR016W | FCJ1 | IM-IMS | 1 | — | Orthologue of mammalian mitofilin, essential role in maintenance of cristae junctions and IM architecture, component of mitochondrial IM organizing system (MitOS/MICOS/MINOS) |
| 30 | YLL027W | ISA1 | Matrix | — | — | Required for maturation of mitochondrial (4Fe-4S) proteins |
| 31 | YLR168C | UPS2 | IMS | — | — | Role in regulation of phospholipid metabolism by inhibiting conversion of phosphatidylethanolamine to phosphatidylcholine |
| 32 | YLR203C | MSS51 | Matrix | — | — | Translational activator for the mitochondrial COX1 mRNA; influences COX1 mRNA translation and Cox1 assembly into cytochrome c oxidase |
| 33 | YML025C | YML6 | Matrix | — | — | Mitochondrial ribosomal protein of the large subunit |
| 34 | YMR115W | MGR3 | IM-IMS | 1 | — | Subunit of mitochondrial i-AAA protease, which degrades misfolded mitochondrial proteins, binds to substrates to facilitate proteolysis, and is required for growth of rho0 cells |
| 35 | YMR145C | NDE1 | IM-IMS | 1 | FAD, NAD | Mitochondrial external NADH dehydrogenase, catalyzes oxidation of cytosolic NADH, providing it to the respiratory chain |
| 36 | YMR203W | TOM40 | OM-IMS | β-barrel | — | Component of the TOM complex, responsible for recognition and initial import steps for all mitochondrially directed proteins |
| 37 | YNL100W | AIM37 | IM-IMS | 2 | — | Subunit of mitochondrial IM organizing system (MitOS/MICOS/MINOS), role in maintenance of cristae junctions and IM architecture |
| 38 | YNR018W | RCF2 | IM-IMS | 2 | — | Cytochrome c oxidase subunit; role in assembly of respiratory supercomplexes; required for late-stage assembly of the Cox12 and Cox13 and for cytochrome c oxidase activity |
| 39 | YNR020C | ATP23 | IMS | — | Zinc | Metalloprotease of the IM, required for processing of Atp6; role in assembly of the F0 sector of the F1F0 ATP synthase complex |
| 40 | YOR020C | HSP10 | Matrix | — | — | Matrix cochaperonin that inhibits the ATPase activity of Hsp60; involved in protein folding and sorting in mitochondria; similarity to Escherichia coli groES |
| 41 | YOR211C | MGM1 | IM-IMS/IMS | 1/- | GTP | GTPase; complex with Ugo1 and Fzo1; required for mitochondrial morphology and genome maintenance; long and short form; homologue of human OPA1 involved in autosomal dominant optic atrophy |
Wild-type and Δyme1 cells were grown in medium containing light or heavy lysine, mitochondria were isolated, and soluble and aggregate fractions were separated by centrifugation. Aggregate fractions were analyzed by MS. Four biological replicates were analyzed. The table contains alphabetically sorted mitochondrial proteins that showed at least 1.6-fold higher aggregation propensity in mitochondria lacking Yme1 in at least two of the experiments. Only proteins with at least two different quantified peptides were considered.
aIM: inner membrane. OM: outer membrane. Submitochondrial localization and transmembrane domains according to the Uniprot database (www.uniprot.org).
bFunction according to the Saccharomyces Genome Database (www.yeastgenome.org).
We used Western blotting to verify some of the identified substrates of Yme1. We first compared the steady-state levels of a number of identified proteins in wild-type mitochondria and mitochondria lacking Yme1. Deletion of YME1 led to an approximately twofold increase of the steady-state levels of Erv1, Mcs10/Mos1/Mio10, and Mcs27/Aim37, suggesting that these proteins are also proteolytic substrates of Yme1 (Figure 5A). The steady-state levels of other proteins identified in the aggregates from mitochondria lacking Yme1 were essentially the same in both types of mitochondria. These include Mcs19/Aim13, Fcj1, Phb2, Gut2, and Dld1. Also, steady-state levels of a number of other mitochondrial proteins residing in various mitochondrial subcompartments were unaffected by deletion of YME1. For analysis of aggregation of these proteins, wild-type mitochondria and mitochondria lacking Yme1 were solubilized, and soluble and aggregate fractions were separated by centrifugation. Erv1 was indeed found in the aggregate fraction from mitochondria lacking Yme1 but not in wild-type mitochondria (Figure 5B). Folding of Erv1 in vitro has recently been shown to require introduction of disulfide bonds by Mia40, followed by insertion of FAD (Kallergi et al., 2012). Data presented here show that Yme1 has a role in this process that probably extends beyond proteolytic removal of incorrectly folded Erv1. In addition, Phb2, Gut2, Mcs19, and Dld1 were also specifically found in aggregates from mitochondria lacking Yme1. Incubation of isolated mitochondria for 3 min at 42°C induced a stronger aggregation of the majority of the identified substrates (Figure 5B). Notably, the aggregation of other substrates, such as Mcs27 and Fcj1, became obvious in mitochondria lacking Yme1.
FIGURE 5:

Effect of Yme1 on aggregation of mitochondrial proteins. (A) Mitochondria (5 and 15 μg) were analyzed by SDS–PAGE and immunodecoration with the indicated antibodies. (B) Mitochondria were preincubated for 3 min at 25 or 42°C and solubilized with Triton X-100–containing buffer, and soluble (S) and aggregate (P, pellet) fractions were separated by centrifugation. Samples were analyzed by SDS–PAGE followed by immunodecoration with the indicated antibodies. The DHFR signals were quantified in supernatant and pellet fractions and expressed as percentages of total. ND, not detectable. (C and D) Submitochondrial localization of Mpm1. Mitochondria harboring a myc-tagged version of Mpm1 were subjected to (C) digitonin fractionation, as described in Figure 1, and (D) carbonate extraction (CE). Samples were analyzed by SDS–PAGE and immunodecoration with the indicated antibodies. (E) Steady-state levels of Mpm1 in wild-type and ∆yme1 strain. Mitochondria (5, 15, and 30 μg) were analyzed as in (A). (F) Aggregation of Mpm1 in ∆yme1 cells. Isolated mitochondria were analyzed as in (B).
Mpm1 showed the highest aggregation propensity of all proteins in the absence of Yme1. Interestingly, this conserved but ill-characterized mitochondrial protein was found to copurify with the very recently identified mitochondrial contact site (MICOS)/mitochondrial inner-membrane organizing system (MINOS)/mitochondrial organizing structure (MitOS) complex, which is important for the ultrastructure of mitochondria (Hoppins et al., 2011), suggesting an interesting function for this protein. We therefore analyzed the submitochondrial localization of Mpm1 and the potential role of Yme1 in its folding. Mitochondria were isolated from a yeast strain harboring a chromosomally myc-tagged version of Mpm1 and were subjected to digitonin fractionation in the presence of PK, as described above. Mpm1 was protected against protease digestion in intact mitochondria but became accessible as soon as the outer membrane was opened (Figure 5C). Furthermore, Mpm1 was found in the soluble fraction upon carbonate extraction (Figure 5D), in agreement with the lack of any predictable transmembrane segments in the sequence. These results demonstrate that Mpm1 localizes to the IMS. The localization of Mpm1 is thus in agreement with Mpm1 being a substrate of Yme1.
We next analyzed the endogenous levels of Mpm1 in wild-type and in mitochondria lacking Yme1. Deletion of YME1 had no effect on the steady-state levels of Mpm1, suggesting that Mpm1 is not merely a proteolytic substrate of Yme1, if at all (Figure 5E). Indeed, when aggregation of Mpm1 was analyzed in wild-type mitochondria and in mitochondria lacking Yme1, a clear difference was observed (Figure 5F). Whereas in wild-type mitochondria, Mpm1 was found entirely in the soluble fraction, a considerable portion of Mpm1 aggregated in mitochondria lacking Yme1. These results further confirm the SILAC analysis and identify Mpm1 as an endogenous substrate of the chaperone-like activity of Yme1.
An analysis of the endogenous substrates of Yme1, listed in Table 1, does not point to any particular characteristic but rather suggests a high degree of flexibility of Yme1 with respect to substrates handled. A number of proteins identified in the aggregates from mitochondria lacking Yme1 are anchored in the inner membrane with one or multiple transmembrane segments, in agreement with the previous reports that Yme1 is involved in degradation of nonassembled or misfolded proteins of the mitochondrial inner membrane that expose domains into the IMS (Baker et al., 2011). However several of the identified proteins are soluble in the IMS. Furthermore, an in silico analysis suggests that substrates of all known different IMS import pathways are present among the identified proteins. Also, some of the identified proteins have known small molecule cofactors, whereas others do not. The repertoire of Yme1 substrates is thus obviously far more diverse than previously anticipated.
Taken together, the results presented here demonstrate that Yme1 has an important role in the folding of proteins in the IMS, in addition to its previously identified role in proteolytic degradation of proteins in this compartment.
DISCUSSION
The IMS of mitochondria contains a considerable number of proteins with important functions in mitochondrial respiration and transport of proteins, lipids, ions, and metabolites, as well as in apoptosis (Herrmann and Riemer, 2010). These proteins are synthesized in the cytosol, transported across the outer membrane in an unfolded state, and need to fold in the IMS. However, no member of the known chaperone families has been identified in the IMS, raising the question of how proteins fold in this compartment. To analyze folding of proteins in the IMS in general, we targeted DHFR, a protein frequently used to analyze import into and folding in mitochondria, to this compartment. Our analysis led to the identification of Yme1, the i-AAA protease that is anchored in the inner membrane and exposes its functional domains into the IMS, as an important factor for folding of proteins in the IMS. Yme1 contains a typical AAA domain, with Walker A and Walker B boxes and the second region of homology, followed by the proteolytic domain (reviewed in Gerdes et al., 2012). Yme1 has primarily been analyzed in light of its ATP-dependent proteolytic activity (Weber et al., 1996; Leonhard et al., 2000; Kominsky et al., 2002; Dunn et al., 2006; Graef et al., 2007; Potting et al., 2010; Elliott et al., 2012). However, an in vitro experiment suggested a chaperone-like activity of its isolated AAA domain (Leonhard et al., 1999). This finding was recently supported by a study suggesting that Yme1 promotes folding and/or assembly of Cox2 in vivo (Fiumera et al., 2009).
We observed that wild-type Yme1 interacted with IMS-DHFR in an ATP-dependent manner in vivo, even though the previous in vitro experiments suggested an ATP-independent interaction of Yme1 with substrates (Leonhard et al., 1999). We believe that the two studies cannot be directly compared. The previous study used an in vitro approach with in vitro synthesized DHFR constructs, which were imported into isolated mitochondria. Under those conditions, it was only possible to detect an interaction between mutant versions of Yme1 with the mutant version of DHFR. No interaction between wild-type proteins, which were used in the same experiment, was seen, suggesting that in vitro and in vivo conditions were not identical.
We identified a number of endogenous mitochondrial proteins in whose folding and/or degradation Yme1 plays a role. Among the proteins that aggregate in the absence of Yme1 were proteins that are involved in essentially all of the above described functions of the IMS, explaining the pleiotropic effects of deletion of YME1. We found Ups2, a protein with a role in phospholipid metabolism (Osman et al., 2009a; Tamura et al., 2009, 2012) and a proteolytic substrate of Yme1 (Potting et al., 2010). Our data suggest that Yme1 is involved in the folding of Ups2 as well. Thus, impaired biogenesis of Ups2 may be one of the reasons for the altered phospholipid composition of mitochondria lacking Yme1 (Nebauer et al., 2007). Furthermore, Dld1, Gut2, and Cox2 are required for growth of yeast cells on respiratory media (Steinmetz et al., 2002); their aggregation in the absence of Yme1 can explain the inability of Δyme1 cells to grow on nonfermentable carbon sources at higher temperatures (Thorsness et al., 1993). Cells lacking Yme1 display abnormal mitochondrial morphology, in both yeast (Campbell and Thorsness, 1998) and mammalian cells (Stiburek et al., 2012). This may be explained by aggregation and/or altered endogenous levels of various components of the recently identified MICOS/MINOS/MitOS complex (Harner et al., 2011; Hoppins et al., 2011; von der Malsburg et al., 2011). Interestingly, Mpm1, the IMS protein with the highest propensity to aggregate in the absence of Yme1, was implicated in the same complex (Hoppins et al., 2011). Similarly, impaired biogenesis of prohibitins, protein and lipid scaffolds in the inner membrane (Osman et al., 2009b), and Erv1 and Tim10, essential components of mitochondrial protein translocation systems, could well explain the phenotypes of YME1 deletion. We conclude that Yme1 occupies one of the hub positions in an intricate network that controls the physiology of mitochondria.
The presence of Yme1 had no influence on the endogenous levels of the majority of proteins that aggregated in its absence. This underlines the notion that Yme1 has a dual function, as a chaperone and as a protease. In other systems controlling folding and degradation, these processes are separated, such that degradation takes place only upon unsuccessful folding (Voos, 2009; Sauer and Baker, 2011). Indeed, whether AAA proteases in general can function either as degradation or refolding machines remains to be determined (Sauer and Baker, 2011). How Yme1 balances these two activities is unclear. Its AAA domain may recognize unfolded domains and, in a first step, act in the folding mode, attempting to fold the unfolded protein, possibly in multiple cycles. Yme1 might, however, keep substrates in a folding-competent conformation or even assist in disaggregation of aggregates, thereby enabling new rounds of folding. If the folding reaction is unsuccessful, the protein will be degraded. Notably, the m-AAA protease can also uncouple its ATPase and proteolytic activities. ATP-dependent membrane dislocation of the precursor of cytochrome c peroxidase, Ccp1, by the m-AAA protease occurs independently of its proteolytic activity (Tatsuta et al., 2007). Furthermore, complete degradation by m-AAA protease can be prevented by stable folding of its substrates, allowing the enzyme to function in processing, rather than in degradation, mode (Bonn et al., 2011). Thus the mitochondrial AAA proteases can apparently switch between different functional modes, likely depending on the nature of a substrate being handled.
Are there additional chaperones in the IMS of mitochondria? No obvious ATP-dependent candidates have been found so far. On the other hand, not all chaperones are necessarily ATP-dependent. Examples include trigger factor in bacteria and all small heat shock proteins (Ferbitz et al., 2004; Haslbeck et al., 2005). Although these chaperones are apparently not present in the mitochondrial IMS, it is not possible to rule out the presence of other ATP-independent chaperones. Another possibility is that at least some of the substrates start to fold using the inner face of the translocation channel of the TOM complex as a folding platform. Indeed, evidence has been provided that the TOM complex can bind nonfolded proteins and prevent their aggregation (Esaki et al., 2003). It should be noted, though, that the estimated dimensions of the translocation channel of the TOM complex would clearly not allow complete folding of any protein, since at most one or two helices could be accommodated in the channel (Ahting et al., 1999). Folding could then continue on the components of translocation systems in the IMS. Indeed, the small TIM complexes were proposed to act as chaperones to prevent aggregation of precursors of hydrophobic membrane proteins passing through the IMS and to mediate their transport to the translocases in the inner and outer membranes (Neupert and Herrmann, 2007; Chacinska et al., 2009; Endo and Yamano, 2009). Recent evidence also suggests that the precursor form of Tim23 can partially fold on small TIM complexes, which could assist its proper delivery and integration into the inner membrane (Davis et al., 2007). Furthermore, recent NMR and x-ray structures showed α-helical folding of substrates bound to Mia40 (Kawano et al., 2009; Banci et al., 2010). The IMS domains of Tim23 and/or Tim50 could possibly also serve as a folding platform for proteins laterally sorted by the TIM23 complex. Indeed, the IMS domain of Tim23 seems to be intrinsically disordered (Gevorkyan-Airapetov et al., 2009; de la Cruz et al., 2010), and it has been suggested that such domains have chaperone-like functions (Dyson and Wright, 2005). Yme1 could be one of the proteins in the IMS that already engages the incoming polypeptide chains during translocation and possibly starts helping them to fold. Interestingly, import of human polynucleotide phosphorylase into yeast mitochondria was reported to depend on Yme1 (Rainey et al., 2006). Still, how general the involvement of Yme1 in import of proteins into IMS is remains to be determined. In any case, folding of proteins in the IMS does not seem to resemble folding in other cellular compartments, and several different, probably interconnected, folding pathways may exist. A detailed analysis of the folding pathways of individual proteins of the IMS will likely reveal even more surprises.
MATERIALS AND METHODS
Plasmids, yeast strains, and cell growth
The construct IMS-DHFR, which codes for the first 107 amino acids of yeast cytochrome b2, fused to the coding sequence of full-length mouse DHFR in pYES2 vector (Invitrogen, Carlsbad, CA), was described before (Popov-Celeketic et al., 2011). The construct matrix-DHFR lacks the 19–amino acid stretch comprising the sorting signal of yeast cytochrome b2. The DHFRmut constructs contain three point mutations (C7S/S42C/N49C; Vestweber and Schatz, 1988) in the DHFR moieties, and were, like C-terminal His6 tags, introduced by standard molecular biology techniques. pYES2 plasmid is a 2 μ vector that allows expression of proteins in yeast cells from the regulatable GAL promoter. All constructs were transformed into haploid wild-type yeast strain YPH499. The Δyme1 strain was generated by replacing the corresponding gene in YPH499 with the KAN cassette via homologous recombination. C-terminal myc-tagging of Mpm1 was likewise performed by homologous recombination into the chromosome, using pYM5 vector.
Yeast cells were grown at 30°C. DHFR constructs were expressed in yeast cells on selective lactate medium containing 0.1% glucose upon addition of 0.5% galactose, and mitochondria were isolated 2 h later. For SILAC assays, cells were grown on selective lactate medium containing either normal or heavy lysine isotope 13C615N2 (Sigma-Aldrich, St. Louis, MO). Otherwise, cells were grown in complete lactate medium.
Aggregation assay
For assessing the aggregation of proteins in vivo, isolated mitochondria were solubilized with 0.5% Triton X-100, and soluble and aggregate fractions were separated by centrifugation at 18,000 × g for 10 min and subsequently analyzed by SDS–PAGE and immunodecoration or by MS. Where indicated, isolated mitochondria were preincubated for 10 min at 25°C, either in the presence of 1.5 mM ATP and 1.5 mM NADH to increase the mitochondrial ATP levels or with 0.1U/ml apyrase and 8 μM oligomycin to deplete ATP. Mitochondria were then incubated for 3 min at 25°C or 42°C prior to solubilization.
NiNTA-agarose pulldown
Isolated mitochondria were incubated for 10 min at 25°C in the presence of 1 mM ADP or ATP or were depleted of nucleotides, as described above. Subsequently, mitochondria were solubilized for 20 min at 4°C with 1% (wt/vol) digitonin in 20 mM Tris (pH 8.0), 80 mM KCl, 10% glycerol, 20 mM imidazole, and 1 mM phenylmethylsulfonyl fluoride. After a clarifying spin, solubilized material was incubated with NiNTA-agarose beads (Qiagen, Valencia, CA) on an overhead roller for 60 min at 4°C. After three washing steps, proteins specifically bound to the beads were eluted with Laemmli buffer containing 500 mM imidazole. Samples were analyzed by SDS–PAGE, which was followed by immunodecoration or label-free quantitative MS. For the latter purpose, 12 mg of mitochondria were used per sample. The gel was stained with Coomassie, and bands were manually excised and digested with trypsin, as described before (Wilm et al., 1996; Shevchenko et al., 2000), with minor modifications. For the MS analysis, tryptic peptides were injected in an Ultimate 3000 HPLC system (LC Packings, Des Moines, IA), as described elsewhere (Forne et al., 2012). The effluent from the HPLC was directly electrosprayed into a LTQ-Orbitrap mass spectrometer (Thermo Fisher Scientific, Lafayette, CO). The MS instrument was operated in data-dependent mode. Survey full-scan MS spectra (from m/z 300–2000) were acquired in the Orbitrap with resolution R = 60,000 at m/z 400 (after accumulation to a “target value” of 500,000 in the linear ion trap). The six most intense peptide ions with charge states between 2 and 4 were sequentially isolated to a target value of 10,000, fragmented by collision-induced dissociation (CID), and recorded in the linear ion trap. For all measurements with the Orbitrap detector, three lock-mass ions were used for internal calibration (Olsen et al., 2005). Typical MS conditions were: spray voltage = 1.5 kV; no sheath and auxiliary gas flow; heated capillary temperature = 200ºC; normalized CID energy = 35%; activation q = 0.25; activation time = 30 ms. Proteins were identified using Mascot 2.3.02 (database: Swissprot 57.10; taxonomy: Saccharomyces cerevisiae; MS tolerance: 10 ppm; MS/MS tolerance: 0.5 Da; peptide false discovery rate [FDR]: 0.1; protein FDR: 0.01; minimum peptide length: 5; variable modifications: oxidation [M]; fixed modifications: carbamidomethyl [C]), and protein fold-change was quantified using spectral counting (Scaffold 3.0.9.1). Proteins used for quantification were filtered as follows: peptide thresholds: 95.0%; protein thresholds: 99.0%; 2 peptides minimum. Proteins that showed at least fivefold enrichment in pulldown from mitochondria containing His-tagged IMS-DHFR over the two other samples were considered to be positive hits.
Identification of aggregating proteins by SILAC and MS
Wild-type YPH499 and Δyme1 strain were grown in medium containing normal and heavy lysine (Lys8:13C615N2-L-lysine • HCl, 13C6H1415N2O2). Mitochondria were isolated, and an aggregation assay was performed with ATP-depleted mitochondria, as described above. The aggregate fractions of wild-type and Δyme1 mitochondria grown on heavy and light lysine, respectively, were pooled prior to separation by SDS–PAGE. In the other set of samples, aggregates from wild-type mitochondria isolated from cells grown on light lysine were mixed with aggregates from mitochondria isolated from Δyme1 cells grown on heavy lysine. The SDS–PAGE separation, in-gel digestion, and MS analysis were performed as described above, with the exception of the protease (Lys-C instead of trypsin).
Maxquant 1.0.13.13 in combination with Mascot 2.3.02 was used for protein identification and quantification. Conditions for identification using Mascot were as described above. Conditions for Maxquant were: peptides for protein quantification, unique and razor; Min. peptides, 1; Min. ratio count, 2; Multiplicity, 2; Heavy labels, Lys8. A complete list of identified proteins is given in the Supplemental Material. Mitochondrial proteins that showed in at least two out of four independent experiments a 1.6-fold or higher aggregation ratio in mitochondria lacking Yme1 compared with wild-type, as judged from the SILAC ratios of at least two different peptides, are included in Table 1.
Miscellaneous
A serum against Yme1 was generated in rabbits by injecting a peptide corresponding to the last 20 residues of Yme1, coupled to KLH (keyhole limpet hemocyanin). The specific antibodies were affinity-purified before use. Western blots were scanned using ImageScanner (GE Healthcare, Waukesha, WI) and quantified with the accompanying ImageMaster 1D Elite software. Previously published procedures were used for isolation of mitochondria (Daum et al., 1982), protease treatment of DHFR (Gaume et al., 1998), and digitonin fractionation (Mokranjac et al., 2003).
Supplementary Material
Acknowledgments
We gratefully acknowledge Christiane Kotthoff, Marica Malesic, Zdenka Stanic, and Petra Robisch for their expert technical assistance; Thomas Langer, Takashi Tatsuta, and Max Harner for generously providing plasmids and antibodies; and Deutsche Forschungsgemeinschaft (SFB 594) and the German-Israeli Foundation for financial support. B.S. is a recipient of a BayEFG fellowship.
Abbreviations used:
- AAA
ATPases associated with diverse cellular activities
- CID
collision-induced dissociation
- DHFR
dihydrofolate reductase
- FDR
false discovery rate
- i-
intermediate
- IMP
inner membrane peptidase
- IMS
intermembrane space
- m-
mature
- MS
mass spectrometry
- MTS
matrix targeting signal
- OM
outer membrane
- PK
proteinase K
- SILAC
stable isotope labeling with amino acids in cell culture
- TIM
translocase of the inner mitochondrial membrane
- TOM
translocase of the outer mitochondrial membrane
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-05-0420) on September 19, 2012.
REFERENCES
- Ahting U, Thun C, Hegerl R, Typke D, Nargang FE, Neupert W, Nussberger S. The TOM core complex: the general protein import pore of the outer membrane of mitochondria. J Cell Biol. 1999;147:959–968. doi: 10.1083/jcb.147.5.959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altieri DC, Stein GS, Lian JB, Languino LR. TRAP-1, the mitochondrial Hsp90. Biochim Biophys Acta. 2012;1823:767–773. doi: 10.1016/j.bbamcr.2011.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustin S, Nolden M, Muller S, Hardt O, Arnold I, Langer T. Characterization of peptides released from mitochondria: evidence for constant proteolysis and peptide efflux. J Biol Chem. 2005;280:2691–2699. doi: 10.1074/jbc.M410609200. [DOI] [PubMed] [Google Scholar]
- Baker BM, Haynes CM. Mitochondrial protein quality control during biogenesis and aging. Trends Biochem Sci. 2011;36:254–261. doi: 10.1016/j.tibs.2011.01.004. [DOI] [PubMed] [Google Scholar]
- Baker MJ, Tatsuta T, Langer T. Quality control of mitochondrial proteostasis. Cold Spring Harb Perspect Biol. 2011;3:a007559. doi: 10.1101/cshperspect.a007559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balch WE, Morimoto RI, Dillin A, Kelly JW. Adapting proteostasis for disease intervention. Science. 2008;319:916–919. doi: 10.1126/science.1141448. [DOI] [PubMed] [Google Scholar]
- Banci L, et al. Molecular chaperone function of Mia40 triggers consecutive induced folding steps of the substrate in mitochondrial protein import. Proc Natl Acad Sci USA. 2010;107:20190–20195. doi: 10.1073/pnas.1010095107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender T, Lewrenz I, Franken S, Baitzel C, Voos W. Mitochondrial enzymes are protected from stress-induced aggregation by mitochondrial chaperones and the Pim1/LON protease. Mol Biol Cell. 2011;22:541–554. doi: 10.1091/mbc.E10-08-0718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonn F, Tatsuta T, Petrungaro C, Riemer J, Langer T. Presequence-dependent folding ensures MrpL32 processing by the m-AAA protease in mitochondria. EMBO J. 2011;30:2545–2556. doi: 10.1038/emboj.2011.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukau B, Weissman J, Horwich A. Molecular chaperones and protein quality control. Cell. 2006;125:443–451. doi: 10.1016/j.cell.2006.04.014. [DOI] [PubMed] [Google Scholar]
- Campbell CL, Thorsness PE. Escape of mitochondrial DNA to the nucleus in yme1 yeast is mediated by vacuolar-dependent turnover of abnormal mitochondrial compartments. J Cell Sci. 1998;111:2455–2464. doi: 10.1242/jcs.111.16.2455. [DOI] [PubMed] [Google Scholar]
- Chacinska A, Koehler CM, Milenkovic D, Lithgow T, Pfanner N. Importing mitochondrial proteins: machineries and mechanisms. Cell. 2009;138:628–644. doi: 10.1016/j.cell.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Retzlaff M, Roos T, Frydman J. Cellular strategies of protein quality control. Cold Spring Harb Perspect Biol. 2011;3:a004374. doi: 10.1101/cshperspect.a004374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daum G, Bohni PC, Schatz G. Import of proteins into mitochondria. Cytochrome b2 and cytochrome c peroxidase are located in the intermembrane space of yeast mitochondria. J Biol Chem. 1982;257:13028–13033. [PubMed] [Google Scholar]
- Davis AJ, Alder NN, Jensen RE, Johnson AE. The Tim9p/10p and Tim8p/13p complexes bind to specific sites on Tim23p during mitochondrial protein import. Mol Biol Cell. 2007;18:475–486. doi: 10.1091/mbc.E06-06-0546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Cruz L, Bajaj R, Becker S, Zweckstetter M. The intermembrane space domain of Tim23 is intrinsically disordered with a distinct binding region for presequences. Protein Sci. 2010;19:2045–2054. doi: 10.1002/pro.482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumont ME, Cardillo TS, Hayes MK, Sherman F. Role of cytochrome c heme lyase in mitochondrial import and accumulation of cytochrome c in Saccharomyces cerevisiae. Mol Cell Biol. 1991;11:5487–5496. doi: 10.1128/mcb.11.11.5487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn CD, Lee MS, Spencer FA, Jensen RE. A genomewide screen for petite-negative yeast strains yields a new subunit of the i-AAA protease complex. Mol Biol Cell. 2006;17:213–226. doi: 10.1091/mbc.E05-06-0585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn CD, Tamura Y, Sesaki H, Jensen RE. Mgr3p and Mgr1p are adaptors for the mitochondrial i-AAA protease complex. Mol Biol Cell. 2008;19:5387–5397. doi: 10.1091/mbc.E08-01-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- Elliott LE, Saracco SA, Fox TD. Multiple roles of the Cox20 chaperone in assembly of Saccharomyces cerevisiae cytochrome c oxidase. Genetics. 2012;190:559–567. doi: 10.1534/genetics.111.135665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo T, Yamano K. Multiple pathways for mitochondrial protein traffic. Biol Chem. 2009;390:723–730. doi: 10.1515/BC.2009.087. [DOI] [PubMed] [Google Scholar]
- Esaki M, Kanamori T, Nishikawa SI, Shin I, Schultz PG, Endo T. Tom40 protein import channel binds to non-native proteins and prevents their aggregation. Nat Struct Biol. 2003;10:988–94. doi: 10.1038/nsb1008. [DOI] [PubMed] [Google Scholar]
- Felts SJ, Owen BA, Nguyen P, Trepel J, Donner DB, Toft DO. The hsp90-related protein TRAP1 is a mitochondrial protein with distinct functional properties. J Biol Chem. 2000;275:3305–3312. doi: 10.1074/jbc.275.5.3305. [DOI] [PubMed] [Google Scholar]
- Ferbitz L, Maier T, Patzelt H, Bukau B, Deuerling E, Ban N. Trigger factor in complex with the ribosome forms a molecular cradle for nascent proteins. Nature. 2004;431:590–596. doi: 10.1038/nature02899. [DOI] [PubMed] [Google Scholar]
- Field LS, Furukawa Y, O'Halloran TV, Culotta VC. Factors controlling the uptake of yeast copper/zinc superoxide dismutase into mitochondria. J Biol Chem. 2003;278:28052–28059. doi: 10.1074/jbc.M304296200. [DOI] [PubMed] [Google Scholar]
- Fiumera HL, Dunham MJ, Saracco SA, Butler CA, Kelly JA, Fox TD. Translocation and assembly of mitochondrially coded Saccharomyces cerevisiae cytochrome c oxidase subunit Cox2 by Oxa1 and Yme1 in the absence of Cox18. Genetics. 2009;182:519–528. doi: 10.1534/genetics.109.101196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forne I, Ludwigsen J, Imhof A, Becker PB, Mueller-Planitz F. Probing the conformation of the ISWI ATPase domain with genetically encoded photoreactive crosslinkers and mass spectrometry. Mol Cell Proteomics. 2012;11:M111.012088. doi: 10.1074/mcp.M111.012088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaume B, Klaus C, Ungermann C, Guiard B, Neupert W, Brunner M. Unfolding of preproteins upon import into mitochondria. EMBO J. 1998;17:6497–6507. doi: 10.1093/emboj/17.22.6497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissler A, Krimmer T, Bomer U, Guiard B, Rassow J, Pfanner N. Membrane potential-driven protein import into mitochondria. The sorting sequence of cytochrome b2 modulates the ΔΨ-dependence of translocation of the matrix-targeting sequence. Mol Biol Cell. 2000;11:3977–3991. doi: 10.1091/mbc.11.11.3977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdes F, Tatsuta T, Langer T. Mitochondrial AAA proteases–towards a molecular understanding of membrane-bound proteolytic machines. Biochim Biophys Acta. 2012;1823:49–55. doi: 10.1016/j.bbamcr.2011.09.015. [DOI] [PubMed] [Google Scholar]
- Gevorkyan-Airapetov L, Zohary K, Popov-Celeketic D, Mapa K, Hell K, Neupert W, Azem A, Mokranjac D. Interaction of Tim23 with Tim50 is essential for protein translocation by the mitochondrial TIM23 complex. J Biol Chem. 2009;284:4865–4872. doi: 10.1074/jbc.M807041200. [DOI] [PubMed] [Google Scholar]
- Glick BS, Beasley EM, Schatz G. Protein sorting in mitochondria. Trends Biochem Sci. 1992;17:453–459. doi: 10.1016/0968-0004(92)90487-t. [DOI] [PubMed] [Google Scholar]
- Graef M, Seewald G, Langer T. Substrate recognition by AAA+ ATPases: distinct substrate binding modes in ATP-dependent protease Yme1 of the mitochondrial intermembrane space. Mol Cell Biol. 2007;27:2476–2485. doi: 10.1128/MCB.01721-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harner M, Korner C, Walther D, Mokranjac D, Kaesmacher J, Welsch U, Griffith J, Mann M, Reggiori F, Neupert W. The mitochondrial contact site complex, a determinant of mitochondrial architecture. EMBO J. 2011;30:4356–4370. doi: 10.1038/emboj.2011.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartl FU, Bracher A, Hayer-Hartl M. Molecular chaperones in protein folding and proteostasis. Nature. 2011;475:324–332. doi: 10.1038/nature10317. [DOI] [PubMed] [Google Scholar]
- Haslbeck M, Franzmann T, Weinfurtner D, Buchner J. Some like it hot: the structure and function of small heat-shock proteins. Nat Struct Mol Biol. 2005;12:842–846. doi: 10.1038/nsmb993. [DOI] [PubMed] [Google Scholar]
- Hell K. The Erv1-Mia40 disulfide relay system in the intermembrane space of mitochondria. Biochim Biophys Acta. 2008;1783:601–609. doi: 10.1016/j.bbamcr.2007.12.005. [DOI] [PubMed] [Google Scholar]
- Herrmann JM, Hell K. Chopped, trapped or tacked-protein translocation into the IMS of mitochondria. Trends Biochem Sci. 2005;30:205–211. doi: 10.1016/j.tibs.2005.02.005. [DOI] [PubMed] [Google Scholar]
- Herrmann JM, Riemer J. The intermembrane space of mitochondria. Antioxid Redox Signal. 2010;13:1341–1358. doi: 10.1089/ars.2009.3063. [DOI] [PubMed] [Google Scholar]
- Herrmann JM, Riemer J. Mitochondrial disulfide relay: redox-regulated protein import into the intermembrane space. J Biol Chem. 2012;287:4426–4433. doi: 10.1074/jbc.R111.270678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppins S, Collins SR, Cassidy-Stone A, Hummel E, Devay RM, Lackner LL, Westermann B, Schuldiner M, Weissman JS, Nunnari J. A mitochondrial-focused genetic interaction map reveals a scaffold-like complex required for inner membrane organization in mitochondria. J Cell Biol. 2011;195:323–340. doi: 10.1083/jcb.201107053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horst M, Oppliger W, Rospert S, Schonfeld HJ, Schatz G, Azem A. Sequential action of two hsp70 complexes during protein import into mitochondria. EMBO J. 1997;16:1842–1849. doi: 10.1093/emboj/16.8.1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonikas MC, et al. Comprehensive characterization of genes required for protein folding in the endoplasmic reticulum. Science. 2009;323:1693–1697. doi: 10.1126/science.1167983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junker JP, Hell K, Schlierf M, Neupert W, Rief M. Influence of substrate binding on the mechanical stability of mouse dihydrofolate reductase. Biophys J. 2005;89:L46–L48. doi: 10.1529/biophysj.105.072066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallergi E, et al. Targeting and maturation of Erv1/ALR in the mitochondrial intermembrane space. ACS Chem Biol. 2012;7:707–714. doi: 10.1021/cb200485b. [DOI] [PubMed] [Google Scholar]
- Kambacheld M, Augustin S, Tatsuta T, Müller S, Langer T. Role of the novel metallopeptidase MoP112 and saccharolysin for the complete degradation of proteins residing in different subcompartments of mitochondria. J Biol Chem. 2005;280:20132–20139. doi: 10.1074/jbc.M500398200. [DOI] [PubMed] [Google Scholar]
- Kang PJ, Ostermann J, Shilling J, Neupert W, Craig EA, Pfanner N. Requirement for hsp70 in the mitochondrial matrix for translocation and folding of precursor proteins. Nature. 1990;348:137–143. doi: 10.1038/348137a0. [DOI] [PubMed] [Google Scholar]
- Kawano S, Yamano K, Naoe M, Momose T, Terao K, Nishikawa S, Watanabe N, Endo T. Structural basis of yeast Tim40/Mia40 as an oxidative translocator in the mitochondrial intermembrane space. Proc Natl Acad Sci USA. 2009;106:14403–14407. doi: 10.1073/pnas.0901793106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehler CM, Tienson HL. Redox regulation of protein folding in the mitochondrial intermembrane space. Biochim Biophys Acta. 2009;1793:139–145. doi: 10.1016/j.bbamcr.2008.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kominsky DJ, Brownson MP, Updike DL, Thorsness PE. Genetic and biochemical basis for viability of yeast lacking mitochondrial genomes. Genetics. 2002;162:1595–1604. doi: 10.1093/genetics/162.4.1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonhard K, Guiard B, Pellecchia G, Tzagoloff A, Neupert W, Langer T. Membrane protein degradation by AAA proteases in mitochondria: extraction of substrates from either membrane surface. Mol Cell. 2000;5:629–638. doi: 10.1016/s1097-2765(00)80242-7. [DOI] [PubMed] [Google Scholar]
- Leonhard K, Stiegler A, Neupert W, Langer T. Chaperone-like activity of the AAA domain of the yeast Yme1 AAA protease. Nature. 1999;398:348–351. doi: 10.1038/18704. [DOI] [PubMed] [Google Scholar]
- Mapa K, Sikor M, Kudryavtsev V, Waegemann K, Kalinin S, Seidel CA, Neupert W, Lamb DC, Mokranjac D. The conformational dynamics of the mitochondrial Hsp70 chaperone. Mol Cell. 2010;38:89–100. doi: 10.1016/j.molcel.2010.03.010. [DOI] [PubMed] [Google Scholar]
- Mayer MP. Gymnastics of molecular chaperones. Mol Cell. 2010;39:321–331. doi: 10.1016/j.molcel.2010.07.012. [DOI] [PubMed] [Google Scholar]
- Mokranjac D, Neupert W. Energetics of protein translocation into mitochondria. Biochim Biophys Acta. 2008;1777:758–762. doi: 10.1016/j.bbabio.2008.04.009. [DOI] [PubMed] [Google Scholar]
- Mokranjac D, Paschen SA, Kozany C, Prokisch H, Hoppins SC, Nargang FE, Neupert W, Hell K. Tim50, a novel component of the TIM23 preprotein translocase of mitochondria. EMBO J. 2003;22:816–825. doi: 10.1093/emboj/cdg090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow G, Kim HJ, Le Pecheur M, Kaul SC, Wadhwa R, Tanguay RM. Protection from aging by small chaperones: A trade-off with cancer? Ann NY Acad Sci. 2010;1197:67–75. doi: 10.1111/j.1749-6632.2009.05413.x. [DOI] [PubMed] [Google Scholar]
- Nakai T, Yasuhura T, Fujiki Y, Ohashi A. Multiple genes, including a member of the AAA family, are essential for degradation of unassembled subunit 2 of cytochrome c oxidase in yeast mitochondria. Mol Cell Biol. 1995;15:4441–4452. doi: 10.1128/mcb.15.8.4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nebauer R, Schuiki I, Kulterer B, Trajanoski Z, Daum G. The phosphatidylethanolamine level of yeast mitochondria is affected by the mitochondrial components Oxa1p and Yme1p. FEBS J. 2007;274:6180–6190. doi: 10.1111/j.1742-4658.2007.06138.x. [DOI] [PubMed] [Google Scholar]
- Neupert W, Herrmann JM. Translocation of proteins into mitochondria. Annu Rev Biochem. 2007;76:723–749. doi: 10.1146/annurev.biochem.76.052705.163409. [DOI] [PubMed] [Google Scholar]
- Nunnari J, Suomalainen A. Mitochondria: in sickness and in health. Cell. 2012;148:1145–1159. doi: 10.1016/j.cell.2012.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen JV, de Godoy LM, Li G, Macek B, Mortensen P, Pesch R, Makarov A, Lange O, Horning S, Mann M. Parts per million mass accuracy on an Orbitrap mass spectrometer via lock mass injection into a C-trap. Mol Cell Proteomics. 2005;4:2010–2021. doi: 10.1074/mcp.T500030-MCP200. [DOI] [PubMed] [Google Scholar]
- Osman C, Haag M, Potting C, Rodenfels J, Dip PV, Wieland FT, Brugger B, Westermann B, Langer T. The genetic interactome of prohibitins: coordinated control of cardiolipin and phosphatidylethanolamine by conserved regulators in mitochondria. J Cell Biol. 2009a;184:583–596. doi: 10.1083/jcb.200810189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osman C, Merkwirth C, Langer T. Prohibitins and the functional compartmentalization of mitochondrial membranes. J Cell Sci. 2009b;122:3823–3830. doi: 10.1242/jcs.037655. [DOI] [PubMed] [Google Scholar]
- Ostermann J, Horwich AL, Neupert W, Hartl FU. Protein folding in mitochondria requires complex formation with hsp60 and ATP hydrolysis. Nature. 1989;341:125–130. doi: 10.1038/341125a0. [DOI] [PubMed] [Google Scholar]
- Pearce DA, Sherman F. Degradation of cytochrome oxidase subunits in mutants of yeast lacking cytochrome c and suppression of the degradation by mutation of yme1. J Biol Chem. 1995;270:20879–20882. doi: 10.1074/jbc.270.36.20879. [DOI] [PubMed] [Google Scholar]
- Popov-Celeketic D, Waegemann K, Mapa K, Neupert W, Mokranjac D. Role of the import motor in insertion of transmembrane segments by the mitochondrial TIM23 complex. EMBO Rep. 2011;12:542–548. doi: 10.1038/embor.2011.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potting C, Wilmes C, Engmann T, Osman C, Langer T. Regulation of mitochondrial phospholipids by Ups1/PRELI-like proteins depends on proteolysis and Mdm35. EMBO J. 2010;29:2888–2898. doi: 10.1038/emboj.2010.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainey RN, Glavin JD, Chen HW, French SW, Teitell MA, Koehler CM. A new function in translocation for the mitochondrial i-AAA protease Yme1: import of polynucleotide phosphorylase into the intermembrane space. Mol Cell Biol. 2006;26:8488–8497. doi: 10.1128/MCB.01006-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- Rowley N, Prip-Buus C, Westermann B, Brown C, Schwarz E, Barrell B, Neupert W. Mdj1p, a novel chaperone of the DnaJ family, is involved in mitochondrial biogenesis and protein folding. Cell. 1994;77:249–259. doi: 10.1016/0092-8674(94)90317-4. [DOI] [PubMed] [Google Scholar]
- Sauer RT, Baker TA. AAA+ proteases: ATP-fueled machines of protein destruction. Annu Rev Biochem. 2011;80:587–612. doi: 10.1146/annurev-biochem-060408-172623. [DOI] [PubMed] [Google Scholar]
- Schmitt M, Neupert W, Langer T. Hsp78, a Clp homologue within mitochondria, can substitute for chaperone functions of mt-hsp70. EMBO J. 1995;14:3434–3444. doi: 10.1002/j.1460-2075.1995.tb07349.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevchenko A, Chernushevich I, Wilm M, Mann M. De novo peptide sequencing by nanoelectrospray tandem mass spectrometry using triple quadrupole and quadrupole/time-of-flight instruments. Methods Mol Biol. 2000;146:1–16. doi: 10.1385/1-59259-045-4:1. [DOI] [PubMed] [Google Scholar]
- Sideris DP, Tokatlidis K. Oxidative protein folding in the mitochondrial intermembrane space. Antioxid Redox Signal. 2010;13:1189–1204. doi: 10.1089/ars.2010.3157. [DOI] [PubMed] [Google Scholar]
- Smith MH, Ploegh HL, Weissman JS. Road to ruin: targeting proteins for degradation in the endoplasmic reticulum. Science. 2011;334:1086–1090. doi: 10.1126/science.1209235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinmetz LM, et al. Systematic screen for human disease genes in yeast. Nat Genet. 2002;31:400–404. doi: 10.1038/ng929. [DOI] [PubMed] [Google Scholar]
- Stiburek L, Cesnekova J, Kostkova O, Fornuskova D, Vinsova K, Wenchich L, Houstek J, Zeman J. YME1L controls the accumulation of respiratory chain subunits and is required for apoptotic resistance, cristae morphogenesis and cell proliferation. Mol Biol Cell. 2012;23:1010–1023. doi: 10.1091/mbc.E11-08-0674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojanovski D, Muller JM, Milenkovic D, Guiard B, Pfanner N, Chacinska A. The MIA system for protein import into the mitochondrial intermembrane space. Biochim Biophys Acta. 2008;1783:610–617. doi: 10.1016/j.bbamcr.2007.10.004. [DOI] [PubMed] [Google Scholar]
- Tamura Y, Endo T, Iijima M, Sesaki H. Ups1p and Ups2p antagonistically regulate cardiolipin metabolism in mitochondria. J Cell Biol. 2009;185:1029–1045. doi: 10.1083/jcb.200812018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura Y, Onguka O, Aiken Hobbs AE, Jensen RE, Iijima M, Claypool SM, Sesaki H. Role for two conserved intermembrane space proteins, Ups1p and Up2p, in intra-mitochondrial phospholipid trafficking. J Biol Chem. 2012;287:15205–15218. doi: 10.1074/jbc.M111.338665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang YC, Chang HC, Roeben A, Wischnewski D, Wischnewski N, Kerner MJ, Hartl FU, Hayer-Hartl M. Structural features of the GroEL-GroES nano-cage required for rapid folding of encapsulated protein. Cell. 2006;125:903–914. doi: 10.1016/j.cell.2006.04.027. [DOI] [PubMed] [Google Scholar]
- Tatsuta T, Augustin S, Nolden M, Friedrichs B, Langer T. m-AAA protease-driven membrane dislocation allows intramembrane cleavage by rhomboid in mitochondria. EMBO J. 2007;26:325–335. doi: 10.1038/sj.emboj.7601514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorsness PE, White KH, Fox TD. Inactivation of YME1, a member of the ftsH-SEC18-PAS1-CDC48 family of putative ATPase-encoding genes, causes increased escape of DNA from mitochondria in Saccharomyces cerevisiae. Mol Cell Biol. 1993;13:5418–5426. doi: 10.1128/mcb.13.9.5418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vestweber D, Schatz G. Point mutations destabilizing a precursor protein enhance its post-translational import into mitochondria. EMBO J. 1988;7:1147–1151. doi: 10.1002/j.1460-2075.1988.tb02924.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von der Malsburg K, et al. Dual role of mitofilin in mitochondrial membrane organization and protein biogenesis. Dev Cell. 2011;21:694–707. doi: 10.1016/j.devcel.2011.08.026. [DOI] [PubMed] [Google Scholar]
- Voos W. Mitochondrial protein homeostasis: the cooperative roles of chaperones and proteases. Res Microbiol. 2009;160:718–725. doi: 10.1016/j.resmic.2009.08.003. [DOI] [PubMed] [Google Scholar]
- Weber ER, Hanekamp T, Thorsness PE. Biochemical and functional analysis of the YME1 gene product, an ATP and zinc-dependent mitochondrial protease from S. cerevisiae. Mol Biol Cell. 1996;7:307–317. doi: 10.1091/mbc.7.2.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermann B, Prip-Buus C, Neupert W, Schwarz E. The role of the GrpE homologue, Mge1p, in mediating protein import and protein folding in mitochondria. EMBO J. 1995;14:3452–3460. doi: 10.1002/j.1460-2075.1995.tb07351.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilm M, Shevchenko A, Houthaeve T, Breit S, Schweigerer L, Fotsis T, Mann M. Femtomole sequencing of proteins from polyacrylamide gels by nano-electrospray mass spectrometry. Nature. 1996;379:466–469. doi: 10.1038/379466a0. [DOI] [PubMed] [Google Scholar]
- Yamano K, Kuroyanagi-Hasegawa M, Esaki M, Yokota M, Endo T. Step-size analyses of the mitochondrial Hsp70 import motor reveal the Brownian ratchet in operation. J Biol Chem. 2008;283:27325–27332. doi: 10.1074/jbc.M805249200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.