

Rb1 plays multiple roles in regulating key aspects of the cell cycle to ensure genomic integrity. Horizontal cells missing Rb1 have an elevated DNA content, sustain DNA damage, and persist as giant cells for several months.
Abstract
The retinoblastoma tumor susceptibility gene, Rb1, is a key regulator of the cell cycle, and mutations in this gene have been found in many human cancers. Prior studies showed that retina-specific knockout of Rb1 in the mouse results in the formation of abnormally large horizontal cells, but the development, fate, and genomic status of these cells remain unknown. In this study, we conditionally inactivate Rb1 in early retinal progenitors and show that the loss of Rb1 leads to the rapid degeneration of most retinal cells except horizontal cells, which persist as giant cells with aberrant centrosome content, DNA damage, and polyploidy/aneuploidy. We observed inappropriate cell cycle entry of Rb1-deficient horizontal cells during the first postnatal weeks, which dropped off abruptly by P30. Despite extensive DNA damage in Rb1-deficient horizontal cells, these cells can still enter mitosis. Adult Rb1-deficient horizontal cells display elevated DNA content (5N–34N) that varied continuously, suggesting the presence of aneuploidy. We also found evidence of supernumerary and disoriented centrosomes in a rare population of mitotic cells in the mutant retinas. Overall our data demonstrate that horizontal cells are a remarkably robust cell type and can survive for months despite extensive DNA damage and elevated genome content.
INTRODUCTION
Retinoblastoma is the most common ocular malignancy in children (Corson and Gallie, 2007). The causative gene, RB1, and its associated pathway have been implicated in the development of the majority of human neoplasms (Weinberg, 1995). Unlike human patients, mice carrying Rb1 mutations do not form tumors (Clarke et al., 1992; Jacks et al., 1992; Lee et al., 1992; Maandag et al., 1994). Instead, retinoblastoma formation in mice requires inactivation of Rb1 and another Rb family member, either p107 or p130 (Robanus-Maandag et al., 1998; Chen et al., 2004; MacPherson et al., 2004; Zhang et al., 2004). It has been proposed that intrinsic genetic compensation of Rb family members in mice, but not in humans, may account for the species-specific differences in retinoblastoma susceptibility (Donovan et al., 2006). Despite intensive study, the cell of origin of retinoblastoma remains elusive. A variety of retinoblastoma mouse models have suggested that one of two retinal cell types—horizontal or amacrine cells—may represent the cell of origin (Robanus-Maandag et al., 1998; Chen et al., 2004; Dannenberg et al., 2004; Dyer and Bremner, 2005; Laurie et al., 2006; Ajioka et al., 2007; Johnson et al., 2007). In contrast, studies of human retinoblastoma have implicated cone photoreceptor precursors as the potential cell of origin (Bogenmann et al., 1988; Hurwitz et al., 1990; Munier et al., 1994; Glubrecht et al., 2009; Santagata et al., 2009; Xu et al., 2009; Ganguly and Shields, 2010). This uncertainty underscores the need for further study of the Rb1 retinal phenotype.
Loss of Rb1 in the mouse results in the formation of enlarged cells in a number of tissues, including retina, cerebellum, skeletal muscle, and liver (Williams et al., 1994; Zacksenhaus et al., 1996; Marino et al., 2003; Mayhew et al., 2005). This phenomenon has been most carefully studied in the mouse liver, where the enlarged hepatocytes also showed increased ploidy secondary to a lack of coordination between DNA replication licensing and entry into mitosis (Bourgo et al., 2011). In the present study, we generated a retina-specific knockout of Rb1 to examine the development, fate, and genomic status of horizontal cells in the absence of Rb1. We found that the loss of Rb1 leads to deregulation of many cell cycle control mechanisms during postnatal development and gives rise to abnormally large horizontal cells in the adult retina that have an elevated DNA content and show persistent DNA damage.
RESULTS
Postnatal development of abnormally large horizontal cells in Rb1-deficient retina
Previous studies reported the presence of abnormally large horizontal cells in Rb1-deficient mouse retinas (MacPherson et al., 2004). To characterize these cells in more detail, we crossed the Rb1lox/lox strain to Six3-Cre transgenic mice to generate Six3-Cre+;Rb1lox/lox (Rb conditional knockout, RbCKO) mice (Furuta et al., 2000; Vooijs et al., 2002). In this RbCKO mouse model, the overall thickness of the retina was severely reduced in adult mice, with marked cell loss in all three nuclear layers (Figure 1B). In addition, we observed very large cells in the sclerad portion of the inner nuclear layer (INL) that were immunoreactive for the horizontal cell marker calbindin (Figure 1, B and D). These findings are consistent with the previously reported large horizontal cell phenotype seen in Pax6-Cre+;Rb1lox/lox retinas (MacPherson et al., 2004).These large cells were not found in control retina (Figure 1, A and C). A smaller number of large cells were also seen in the ganglion cell layer (unpublished data).
FIGURE 1:
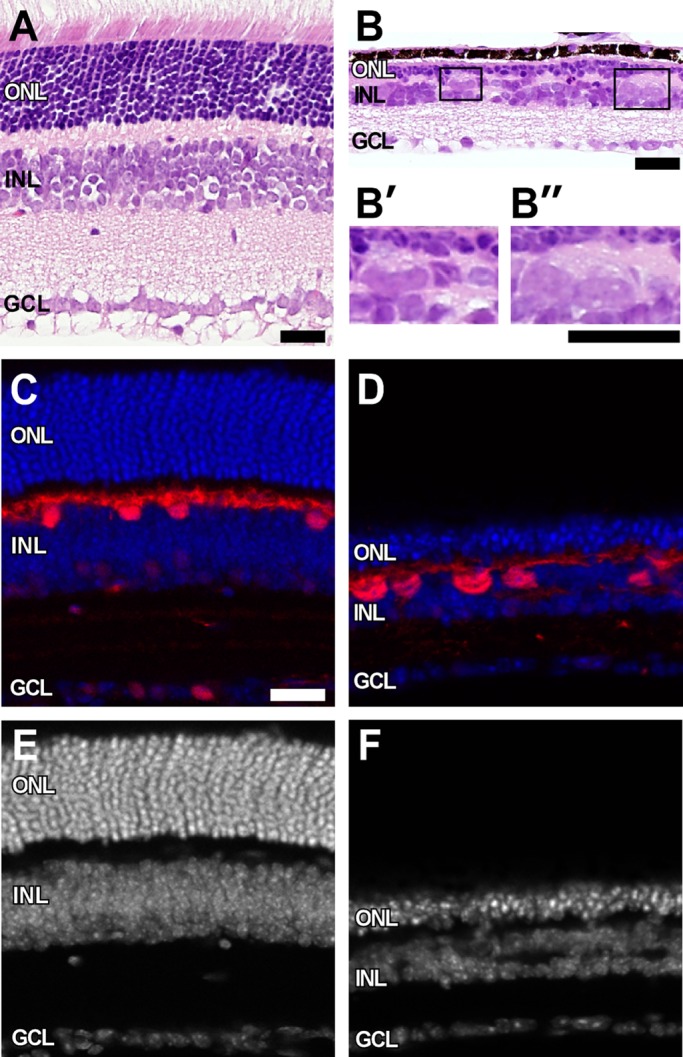
Giant horizontal cells in Rb1-deficient retina. H&E-stained sections show marked thinning of all three nuclear layers in RbCKO retina at P26 (B) relative to controls (A). (B′, B′′) Boxed areas in B show grossly enlarged cells at the sclerad edge of the INL. Calbindin immunohistochemistry (red) confirms the identity of these enlarged cells in the mutant retina (D) compared with the control (Six3-Cre+;Rb1Iox/+; C). Sections were counterstained with DAPI (blue, white) to visualize nuclei in control (C, E) and RbCKO (D, F) retinas. GCL, ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer. Scale bars, 25 μm.
Next we quantified the size of the abnormally large horizontal cells by measuring the cross-sectional area of the nuclei from STYOX green-stained cells in control and RbCKO retina at P26 (Figure 2, A–D). Cells with a cross-sectional area in the 15- to 165-μm2 range showed similar frequencies in both genotypes (Figure 2, C–E). However, a second population of nuclei was observed in the INL of RbCKO retina that had a much larger cross-sectional area (180–585 μm2; Figure 2E, inset, red bars). These findings confirm that the horizontal cells in the adult RbCKO retina can be up to several times the size of control horizontal cells.
FIGURE 2:

Morphometric analysis of INL cells. (A, B) SYTOX green counterstained sections were used to hand trace the nuclear outlines of cells in the INL. (B′) Boxed area in B shows large and irregularly shaped nuclei in the INL of RbCKO retina. (C, D) Representative digitized images from traced SYTOX Green–labeled nuclei were used to calculate the cross-sectional nuclear area of INL cells (unshaded) for control (Six3-Cre+;Rb1lox/+; C) and RbCKO (D) retinas at P26. (E) Frequency histogram showing the distribution of cross-sectional nuclear area for cells in the INL of control (black) and RbCKO retinas (red). Cells from RbCKO show a distribution that extends toward much larger areas (inset). Scale bars, 20 μm.
To determine the fate of the large horizontal cells in RbCKO retinas, we examined histological sections at multiple postnatal time points. We found that the grossly enlarged horizontal cells persist in the mutant retina for several months (Supplemental Figure S1). We also observed that the shapes of the horizontal cell bodies and nuclei in the mutant retina were irregular and dysmorphic at these later time points (Supplemental Figure S1). In addition, the RbCKO retina continued to degenerate, with a loss of cells in all three nuclear layers. SYTOX green nuclear morphometric analysis of the INL at 4.5 mo of age revealed similar trends in the frequency and distribution of nuclear area as those observed at 1 mo of age (P26), although the second population of nuclei in RbCKO retina had a narrower distribution (Supplemental Figure S2). In summary, these data are consistent with persistence of the abnormal RbCKO horizontal cells for several months after birth.
To quantify the time course of horizontal cell enlargement, we measured the area of calbindin-positive horizontal cells from dissociated control and RbCKO retinas at multiple postnatal time points (Figure 3A). Calbindin labels horizontal cells very strongly but also weakly marks a subset of amacrine and retinal ganglion cells (Haverkamp and Wassle, 2000). Therefore only very strongly labeled cells that displayed the characteristic horizontal cell nuclear morphology were imaged and measured. We found that the mean area (± SD) in control horizontal cells increased gradually from P7 (62.5 ± 12.7 μm2) to P30 (84.1 ± 28.3 μm2; Figure 3B, black symbols), consistent with cellular maturation that occurs over this time period and in accordance with prior results (Ajioka et al., 2007). Whereas the size of Rb1-deficient horizontal cells was not significantly different from that of controls at P7, we observed a progressive increase in the cross-sectional area of horizontal cells from P7 (72.7 ± 21.3 μm2) to P30 (203.6 ± 115.8 μm2; Figure 3B, red symbols). To evaluate the percentage of horizontal cells, we dissociated RbCKO and control retinas and immunostained them for calbindin and counterstained them with 4′,6-diamidino-2-phenylindole (DAPI) at P7 and P10. We counted an average of 5000 DAPI-stained cells in three independent experiments for each age and genotype and scored the number of calbindin-positive horizontal cells in each. We found that the percentage of horizontal cells was not significantly different between the two genotypes at either age examined (Supplemental Figure S3). These results demonstrate that the percentage of horizontal cells remains constant, and changes in horizontal cell size begin during the second postnatal week and progress at least through P30 in RbCKO retinas.
FIGURE 3:

Knockout of E2f1 suppresses the enlarged horizontal cell phenotype observed in RbCKO retinas during the postnatal period. (A) Representative images of dissociated retinal cells from control (Six3-Cre+;Rb1lox/+, Rb1lox/+, Rb1lox/lox; left) and RbCKO (right) immunostained with calbindin (red) at various postnatal time points. DAPI staining is in blue. (B) Quantification of horizontal cell area in control (black), RbCKO (red) and Rb1;E2f1 double knockout (DKO, green; ***p < 0.001, one-way ANOVA followed by Tukey's post hoc test). Shown is the mean (dashed blue line) ± SD of three independent experiments for each age and genotype. Not significant, n.s. Scale bar, 10 μm.
Cellular degeneration in the Pax6-Cre+;Rb1lox/lox retina can be rescued by simultaneous knockout of E2f1, suggesting that the degeneration is mediated, in part, by the unopposed activity of E2f1 (Chen et al., 2007). To determine whether changes in horizontal cell area are also E2f1 dependent, we crossed RbCKO onto an E2f1−/− background and measured the area of horizontal cells in the double knockout at P15 and P30. We found no enlargement of horizontal cells at either time point, indicating that the abnormal increase in horizontal cell size in the RbCKO retina is E2f1 dependent (Figure 3B, green symbols).
Elevated DNA content in Rb1-deficient horizontal cells
The nuclear enlargement observed in hepatocytes missing Rb1 is accompanied by changes in ploidy or DNA content (Mayhew et al., 2005). Typically, flow cytometry approaches can be utilized to measure ploidy in individual groups of cells. Initially, we used flow cytometry to measure DNA content in dissociated retinal cells from adult (P21) RbCKO and control mice. Using this approach, we observed that there was a shift in the cell population that contained a higher DNA content; however, the peak was quite broad (unpublished data). Moreover, the lack of specific cell surface markers for horizontal cells to measure DNA content by flow cytometry prompted us to use an alternative method. We therefore deployed a cytofluorometric approach (see Materials and Methods) to evaluate changes in DNA content in RbCKO horizontal cells. To validate our approach of measuring DNA content in retinal cells using DAPI, we performed a series of controls (Supplemental Figure S4). First, we determined whether there were differences in DNA content in adult control horizontal cells by normalizing the integrated density measurement (IDM) values of the control calbindin-positive cells to the mean of the IDM values in the control calbindin-negative (reference) population at P35. We found no significant difference in the DNA content of control horizontal cells compared with the reference population (Supplemental Figure S4A). Next we determined whether there were differences in DNA content in adult RbCKO calbindin-negative cells by normalizing the mean IDM values of this reference cell population to the mean of the IDM values in the control calbindin-negative population at P35. This analysis revealed that calbindin-negative cells in the adult RbCKO retina are also diploid (Supplemental Figure S4B). Finally, we tested the hypothesis that mitotic wild-type retinal progenitors would have a 4N DNA content compared with nonmitotic cells. P0 wild-type retinas were dissociated and stained with the mitotic marker phospho–histone H3 Ser-10 (PH3) and DAPI. PH3+ IDM values were normalized to PH3− measurement values. Indeed, mitotic retinal progenitor cells have double the amount of DNA found in nonmitotic cells (p < 0.0001; Supplemental Figure S4C). These data demonstrate that DNA content can be reliably measured using a cytofluorometric approach.
Next we analyzed adult (P35) RbCKO horizontal cells to test whether changes in DNA content were present. We found that the mean DNA content in calbindin-negative cells in RbCKO retina (2.2N ± 0.88) was unchanged compared with calbindin-positive cells in control retina (2N ± 0.74; p = 0.57), in agreement with morphometric analyses (red triangle vs. gray circle in Figure 4). In striking contrast, the mean DNA content in calbindin-positive cells in RbCKO retina (12N ± 6.8) was markedly increased compared with control calbindin-positive cells (p < 0.0001; gray triangle vs. gray circle in Figure 4). Of interest, the DNA content of Rb1-deficient horizontal cells did not cluster around multiples of 2N (i.e., 4N, 8N, 16N) as previously reported in Rb1-deficient hepatocytes (Mayhew et al., 2005) but varied widely between 5N and 34N. Taken together, these results indicate that the DNA content in adult Rb1-deficient horizontal cells is elevated and varies over a wide range, suggesting the presence of aneuploid and/or polyploid cells that have partially replicated their genome.
FIGURE 4:
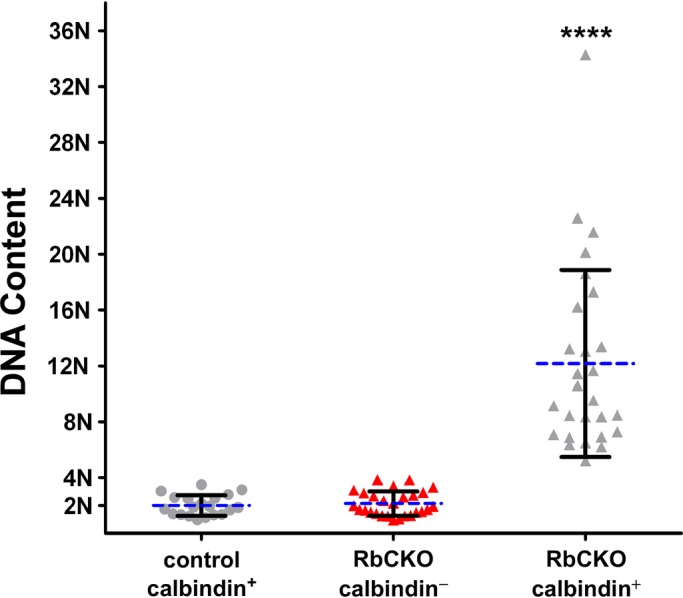
Elevated ploidy in adult Rb1-deficient horizontal cells. DNA content was measured using cytofluorometric analysis (see Materials and Methods) and normalized to calbindin-positive horizontal cells in control (Six3-Cre+;Rb1lox/+, Rb1lox/+) retina (gray circles) at P35. DNA content is significantly increased in RbCKO calbindin-positive horizontal cells (gray triangles, ****p < 0.0001, one-way ANOVA followed by Tukey's post hoc test) but remains near diploid in RbCKO calbindin-negative cells (red triangles). Shown is the mean (dashed blue line) ± SD of three independent experiments for each age and genotype.
Rb1-deficient horizontal cells exhibit unscheduled genome replication
Mammalian cells use cell cycle control mechanisms to maintain a diploid state and prohibit proliferation of cells that sustain DNA damage or have changes in their DNA content (Davoli and de Lange, 2011). When these control mechanisms are breached, it is possible for cells to achieve an elevated DNA content and become polyploid and/or aneuploid. Rb1 plays a critical role in many of these control processes, and this has been well documented in a variety of model systems (Manning and Dyson, 2011) but has not been explored in mouse horizontal cells. To gain a better understanding of the role that Rb1 plays in these processes in horizontal cells, we examined cell cycle and DNA damage markers in RbCKO horizontal cells at different time points throughout the postnatal period.
First, in order to assess the extent and distribution of cycling cells in control and RbCKO retinas, we stained for Ki-67, a marker of cells that are in the cell cycle. We observed an absence of Ki-67 immunostaining from the central region of control retina at all of the ages tested (Figure 5, A, C, E, and G), although some Ki-67+ cells were detected in the periphery of control retinas at P7 (unpublished data). In contrast, we found robust Ki-67 labeling in the central region of RbCKO retinas in all three nuclear layers, which persisted through P30 (Figure 5, B, D, F, and H). In addition, we observed Ki-67+ cells in the area where horizontal cell bodies are located. To examine whether Rb1-deficient horizontal cells were in an active phase of the cell cycle, we double labeled sections with calbindin and Ki-67. Indeed, we found that RbCKO horizontal cells were immunopositive for Ki-67 during a 2-wk period postnatally (Figure 5, I–K). The expression of Ki-67 tapered off dramatically at P30 in RbCKO retina, and we observed only a few double-positive cells (Figure 5H′, white arrowhead). To quantify the frequency of horizontal cells that were cycling, we scored the number of calbindin;Ki-67 double-positive cells from dissociated RbCKO retina. We found that nearly 40% of horizontal cells are actively cycling from P7 (38.5 ± 5%) to P10 (42.4 ± 1.6%); however only 30.7 ± 3.2% are cycling at P15 (p < 0.05; Figure 5L). These findings demonstrate that Rb1-deficient horizontal cells are in an active phase of the cell cycle at a time before and during which differences in size are observed and at a time when DNA content was found to be elevated. These data suggest that Rb1 plays a role in preventing inappropriate cell cycle entry and aberrant ploidy development in differentiated mouse horizontal cells.
FIGURE 5:

Deregulated cell cycle control in RbCKO retina. (A–H) Immunostained sections from the central portion of the retina showing the pattern of Ki-67 immunoreactivity (red) in RbCKO that is absent from control (Six3-Cre+;Rb1lox/+, Rb1lox/+, Rb1lox/lox) retina at several postnatal time points. DAPI staining is in blue. (H′–K) Double-labeling for Ki-67 (red) and calbindin (green) reveals that horizontal cells remain in the cell cycle for several weeks in RbCKO retina. (L) Percentage of calbindin;Ki-67 double-positive cells from dissociated RbCKO retina between the first and second postnatal weeks (*p < 0.05, one-way ANOVA followed by Tukey's post hoc test). Shown is the mean ± SD of three independent experiments. Scale bars, A–H, 50 μm; H’–K, 10 μm.
To further confirm the Ki-67 findings, we examined S-phase entry by in vivo labeling with a thymidine analogue, 5-ethynyl-2′-deoxyuridine (EdU). EdU was injected intraperitoneally (IP) into RbCKO and control mice at P7 and P10. After 4 h, retinas were dissociated and stained for EdU (Figure 6, A–D), and the number of EdU+ cells was quantified at P7 and P10 (Figure 6E). EdU+ cells were sparse in control retina (0.14 ± 0.06%) at P7 but were readily observed in the RbCKO retina (10.9 ± 0.8%; p < 0.0001). At P10, EdU+ cells were exceedingly rare (1/17,000) in control retina but persisted in RbCKO retina (2.1 ± 0.2%).
FIGURE 6:

Unscheduled genome replication in RbCKO retina. (A–D) Representative images of dissociated retinal cells 4 h after EdU injection from control (A, D) Six3-Cre+;Rb1lox/+, Rb1lox/lox and (B, D) RbCKO retinas stained with EdU (red) at P7 and P10. (E) Percentage of EdU+ cells from dissociated control (black) and RbCKO (red) retinas at P7 and P10 (****p < 0.0001, **p < 0.005, two-tailed t-test). (F) Dissociated RbCKO retinal cells were colabeled with EdU (right, yellow outlines ) and calbindin (white arrowhead) at P7. (G) Quantification of dual-positive cells in RbCKO at P7. DAPI staining is in blue. Shown is the mean ± SD of three independent experiments. Scale bars, A–D and F, 10 μm.
To determine whether RbCKO horizontal cells are capable of DNA synthesis during the postnatal period, we double labeled a subset of cells in RbCKO and control retina with EdU and calbindin at P7 and P10, 4 h post-EdU injection. EdU+ horizontal cells were absent in control retina at P7 and P10 (unpublished data). In contrast, RbCKO horizontal cells that were EdU+ after a 4-h pulse were observed at P7 (Figure 6, F and G; 6.2 ± 1.1%) and P10 (unpublished data). These data demonstrate that RbCKO horizontal cells are capable of synthesizing new DNA before changes in horizontal cell size are detected.
Rb1-deficient horizontal cells sustain persistent DNA damage
Several reports have shown that Rb1 deficiency leaves cells more susceptible to DNA damage (Manning and Dyson, 2011). To evaluate Rb1-deficient horizontal cells for the presence of DNA damage, we double-labeled sections with calbindin and γ-H2AX, a marker for DNA damage induced by double-strand breaks (Rogakou et al., 1998). Cells that contain DNA damage show multiple punctate foci of γ-H2AX staining (Rogakou et al., 1999), whereas cells that are apoptotic will have a pannuclear staining pattern (Marti et al., 2006). We found that only a few scattered calbindin-negative cells stained for γ-H2AX in a pannuclear pattern in control retinas (arrowheads in Figure 7, A, C, and E), consistent with naturally occurring cell death (Young, 1984). In contrast, both types of γ-H2AX staining patterns were readily observed in postnatal RbCKO retina (Figure 7, B, D, F, and H). We found that punctate γ-H2AX staining was the most prominent and easily detected pattern in RbCKO horizontal cells (yellow arrowheads in Figure 7, D, F, and H).
FIGURE 7:

Persistent DNA damage in Rb1-deficient horizontal cells. (A–H) Retinal sections were double labeled with γ-H2AX (red) and calbindin (green) at several time points postnatally. DAPI staining is in blue. Absence of colocalized markers in control (Six3-Cre+;Rb1lox/+, Rb1lox/+, Rb1lox/lox) retina (A, C, E, G). Sparse γ-H2AX staining was observed in a pannuclear pattern in control retina at early postnatal ages (A, C, E; large white arrowheads) and absent by P15 (G). RbCKO horizontal cells display multiple punctate foci of γ-H2AX staining as early as P7 (D; yellow arrowhead) that persist at later postnatal ages (F, H; yellow arrowheads). Representative dissociated cells colabeled with calbindin and γ-H2AX were examined for the presence and pattern of γ-H2AX foci staining in control (data not shown) and RbCKO retina (I–K). (L) Quantification of horizontal cells with different γ-H2AX foci staining patterns at three postnatal ages revealed a mild decrease in the staining pattern with few (two to six) γ-H2AX foci in the RbCKO retina from P10 to P15 (blue line, *p < 0.05, one-way ANOVA followed by Tukey's post hoc test). Shown is the mean ± SD of three independent experiments for each age and genotype. Scale bars, 10 μm.
To measure the frequency of horizontal cells that contain DNA damage, we scored and analyzed the γ-H2AX foci in individual calbindin-positive cells at P7, P10, and P15 from dissociated control and RbCKO retina (Figure 7, I–K). To ensure that the effects do not result from nonspecific nuclease activity from the Cre recombinase, controls containing Cre (Six3-Cre;Rblox/+) were used at P15. We found that 32.3 ± 5.4% of calbindin-positive horizontal cells were positive for γ-H2AX during the first 2 wk of postnatal development (Figure 7L). Staining in individual RbCKO horizontal cells ranged from relatively few (2–6) to many (>6) foci. Our data suggest a mild decrease in the number of horizontal cells, with few γ-H2AX foci between P10 and P15 in RbCKO retina (p < 0.05; Figure 7L). In Cre+ controls at P15, we observed a small fraction of horizontal cells that were positive for γ-H2AX, which contained either few (4/250) or many (1/250) foci. This small fraction of γ-H2AX+ cells in these controls suggests that the effects of nonspecific nuclease activity are minimal. Overall these analyses revealed that Rb1-deficient horizontal cells sustain persistent DNA damage during postnatal development.
Rb1-deficient horizontal cells enter mitosis
Because Rb1-deficient horizontal cells were capable of undergoing unscheduled genome replication and sustained DNA damage, we wanted to further explore the mechanism associated with elevated ploidy and determine whether the abnormal cells were capable of entering mitosis. We first examined hematoxylin and eosin (H&E)–stained sections and found the presence of condensed chromatin and mitotic figures located in the sclerad portion of the INL in RbCKO retina at P10, P15, and P18 (Figure 8, A and C, and unpublished data). Double immunolabeling with calbindin and PH3 revealed that Rb1-deficient horizontal cells are mitotic at P10 and P15 (Figure 8, B and D).
FIGURE 8:

Deregulated cell cycle checkpoint in Rb1-deficient retina. H&E-stained sections show the presence of mitotic figures and condensed chromatin in the outer INL of RbCKO retina at P10 (A) and P15 (C). Sections double labeled with phospho–histone H3 Ser-10 (PH3, green) and calbindin (red) demonstrates that differentiated RbCKO horizontal cells are mitotic at P10 (B) and P15 (D). (E–G) Subset of dissociated RbCKO horizontal cells displays γ-H2AX foci (red) and DAPI (blue) patterns consistent with mitosis at several postnatal ages. (H–K) Dissociated RbCKO retinal cells were colabeled with γ-H2AX (red) and PH3 (green). (L) Quantification of the percentage of mitotic (PH3+) cells that are γ-H2AX+ in control (Six3-Cre+;Rb1lox/+, Rb1lox/+, Rb1lox/lox) and RbCKO at P7 (left) and P10 (right). (M, N) RbCKO horizontal cells labeled with calbindin (white arrowheads, far right) show both markers of genomic instability (γ-H2AX) and DNA synthesis (EdU) after a 4-h in vivo pulse of EdU. EdU+ cells are outlined in yellow in merged images to aid visibility. Shown is the mean ± SD of three independent experiments. Scale bars, A–N, 10 μm.
To assess whether Rb1-deficient horizontal cells with DNA damage are able to enter mitosis, we carefully examined dissociated cells double labeled with calbindin and γ-H2AX for the presence of condensed chromatin, a typical characteristic of mitotic cells, using DAPI stain. We compared the pattern of γ-H2AX staining foci with the pattern and intensity of the DAPI stain because mitotic cells are generally brighter than nonmitotic cells. We found rare examples of γ-H2AX foci arranged in a single linear position located at the center of the cell or in two parallel lines at opposite ends of the cell (Figure 8, E–G). These patterns are consistent with the arrangement of chromosomes at different phases of mitosis. Moreover, the DAPI staining in these same cells was brighter than for cells in the same field of view, consistent with condensation of DNA during mitosis, and did not show signs of nuclear fragmentation.
To quantify the extent to which mitotic Rb1-deficient retinal cells exhibit markers of DNA damage, we dissociated RbCKO and control retinas and processed them for γ-H2AX and PH3 double labeling (Figure 8, H–K). We then scored mitotic cells (PH3+) for the presence or absence of γ-H2AX foci labeling. Consistent with our previous data, no PH3+ cells were observed in control retinal cells (unpublished data). In contrast, we found that 10.9 ± 4.0% of mitotic cells in RbCKO retina contained γ-H2AX foci at P7 (Figure 8L, left), whereas at P10, only 0.34 ± 0.07% of mitotic cells had γ-H2AX foci present (Figure 8L, right). To measure the extent of calbindin-positive cells that were mitotic, we examined at least 100 calbindin-positive cells from four to seven RbCKO and control retinas at P7, P10, and P15. We found that the frequency of calbindin-positive cells that were PH3+ (± SD) in RbCKO was 2.0% (± 1.8), 3.3% (± 4.5), and 0.98% (± 1.0) at P7, P10, and P15, respectively (unpublished data). In contrast, no double-positive cells were observed in control retinas at the same ages (unpublished data). These data suggest that the percentage of mitotic horizontal cells is small and variable in the RbCKO retina. Taken together, these data suggest that there is selective vulnerability to DNA damage after Rb1 loss in the retina; most mitotic cells suffer genomic damage and subsequently undergo apoptosis, whereas a small percentage of horizontal cells enter into an endomitotic cycle, which may contribute to elevated ploidy/aneuploidy.
To support the model that Rb1 loss overrides genomic checkpoints and promotes proliferation of horizontal cells containing DNA damage, we evaluated Rb1-deficient horizontal cells for the presence of γ-H2AX and EdU at P7. RbCKO mice and littermate controls received a single IP dose of EdU. After 4 h, the retinas were removed, dissociated, and triple labeled for calbindin, EdU, and γ-H2AX. Calbindin-positive horizontal cells in control retina were immunonegative for EdU and/or γ-H2AX (unpublished data). In contrast, we observed calbindin-positive horizontal cells that were immunopositive for both EdU and γ-H2AX (Figure 8, M and N). These data suggest that cell cycle checkpoints are compromised in Rb1-deficient horizontal cells.
Mitotic spindle assembly defects and abnormal centrosome number in a rare population of mitotic cells in Rb1-deficient retina
A number of studies reported that the loss of Rb1 results in supernumerary centrosomes and is associated with changes in ploidy (Manning and Dyson, 2011). To evaluate whether the loss of Rb1 leads to alterations in centrosome number, we examined the expression and distribution of γ-tubulin, a centrosomal marker (Muresan et al., 1993), in mitotic cells from dissociated retina. First, to ensure that the dissociation process would not disrupt or alter the distribution of centrosomes within the cell, we examined the expression and distribution of γ-tubulin in control and RbCKO PH3-labeled retinal progenitors at P0. We found that the overwhelming majority of mitotic cells in either genotype contained a single pair of centrosomes distributed in the central part of the nucleus at varying distances depending on the mitotic phase, and did not contain more than two centrosomes (unpublished data).
Next we evaluated whether there were centrosome abnormalities in mitotic (PH3+) cells at later stages of development in the RbCKO retina. We found that centrosomes were positioned abnormally within the nucleus and that mitotic Rb1-deficient retinal cells contained an aberrant number of centrosomes during a 2-wk postnatal time period (Supplemental Figure S5, A–R). For example, we observed centrosomes positioned in RbCKO retinal cells indicative of multipolar and unequal bipolar mitoses (Supplemental Figure S5, A–E, G–J, and L–R). In addition, some cells had misoriented, opposing centrosomes at the cleavage furrow (Supplemental Figure S5, F, J–L, and P–R). Next we measured the frequency of mitotic cells with more than two γ-tubulin foci and found an age-dependent increase in the number of mitotic cells that contained three centrosomes (P7 to P15; p < 0.05) but not in those that contained four centrosomes (p = 0.44; Supplemental Figure S5S). In rare cases, we observed RbCKO horizontal cells that contained three centrosomes (unpublished data). These data indicate that abnormalities in centrosome number and distribution observed between the first and second postnatal weeks are unlikely to play a role in the elevated ploidy observed in adult Rb1-deficient horizontal cells.
DISCUSSION
In this study we showed that the loss of Rb1 in early mouse retinal progenitors leads to the rapid degeneration of most retinal cell types, except horizontal cells, which can survive for months despite sustained DNA damage and abnormal genome content. Even though retinal horizontal cells exhibit markers of genomic instability and altered ploidy, both of which have been shown to be sufficient for tumorigenesis in various mouse tissues (Knudsen and Knudsen, 2008; Viatour and Sage, 2011), we did not observe retinal tumors in our model (unpublished data). This finding is consistent with earlier studies that showed that Rb1 loss alone is not sufficient to generate retinoblastoma in murine retina but requires inactivation of at least one other Rb family member, either p107 or p130 (Robanus-Maandag et al., 1998; Chen et al., 2004; Dannenberg et al., 2004; MacPherson et al., 2004; Zhang et al., 2004) or combined loss of Rb1 and p27 (Sangwan et al., 2012). Thus our data suggest that horizontal cells are an exceptionally robust cell type in the murine retina after Rb1 inactivation but that DNA damage and abnormal genome content in horizontal cells are not sufficient to initiate tumor formation in this model.
Earlier studies in the mouse suggested that horizontal cells were among several cell types that could represent a cell of origin for retinoblastoma (Robanus-Maandag et al., 1998; Chen et al., 2004; Dannenberg et al., 2004; Dyer and Bremner, 2005; Laurie et al., 2006; Ajioka et al., 2007; Johnson et al., 2007). However, our findings of genomic instability and altered ploidy in Rb1-deficient horizontal cells contrast sharply with a recent whole-genome sequencing study of human retinoblastoma, which showed remarkable genomic stability in these tumors (Zhang et al., 2012). These differences suggest that caution is warranted when comparing horizontal cell–derived tumors in mice with human retinoblastoma (Ajioka et al., 2007).
How does the altered ploidy state emerge in horizontal cells lacking Rb1? As a tumor suppressor gene, Rb1 is well known for its role in regulating cell proliferation; however, recent data from a variety of different in vivo and in vitro model systems suggest that Rb1 may also play a role in the maintenance of genome stability (Knudsen et al., 2006; Manning and Dyson, 2011; Coschi and Dick, 2012). It has been proposed that the loss of Rb1 disrupts the coordinated transitions through the cell cycle and that compromised cell cycle checkpoints culminate in genomic change (Knudsen et al., 2006; Manning and Dyson, 2011; Coschi and Dick, 2012). In the present study, we found that multiple cell cycle checkpoints are compromised in Rb1-deficient horizontal cells before changes in horizontal cell size and ploidy were detected. Rb1-deficient horizontal cells underwent proliferation, sustained persistent DNA damage, and entered mitosis at inappropriate stages of postnatal development from P7 to P15. In addition, abnormally positioned excess centrosomes were observed in a small population of Rb1-deficient mitotic cells during the same time period. Remarkably, horizontal cells in the RbCKO retina survived despite compromised cell cycle checkpoints and increased DNA content. Our findings suggest that Rb1 plays multiple roles in coordinating cell cycle progression to prevent aberrant ploidy in horizontal cells.
How might Rb1 mediate its effect on horizontal cell size and ploidy? Our data suggest that this effect is likely mediated through the transcription factor E2f1, since the size of the horizontal cells is rescued when we cross the RbCKO mouse onto an E2f1-null line. After Rb1 loss in the retina a subset of E2fs and known E2f target genes that regulate the cell cycle and apoptosis, including E2f1, is up-regulated (Chen et al., 2007). In agreement with these data, during the postnatal time period, when horizontal cells are progressing through the cell cycle, we found that E2f1 protein is up-regulated in RbCKO retina (Donovan and Corbo, unpublished observations). Two potential E2f1 target genes that may play a role in altered horizontal cell ploidy are mad2 and geminin. Mad2 has been shown to be elevated in human retinoblastoma tumors and implicated in chromosome instability (Hernando et al., 2004; Schvartzman et al., 2011) and prolonged DNA damage checkpoint arrest (Dotiwala et al., 2010). Geminin helps to prevent the relicensing of replication origins after they have fired (McGarry and Kirschner, 1998), is deregulated in Rb1-null cells, and contains E2F sites in the promoter region (Markey et al., 2004). Alternatively, other mechanisms involving chromatin modifications may contribute to genomic instability (Knudsen et al., 2006; Manning and Dyson, 2011).
MATERIALS AND METHODS
Mice
The Six3-Cre transgenic mouse was published previously (Furuta et al., 2000). Briefly, Cre recombinase is controlled by regulatory elements of the Six3 promoter and enables gene deletion in retinal progenitors as early as E9.5. In contrast to the Pax6-α-Cre line, which confines gene deletion to the peripheral retina as early as E10.5 (Marquardt et al., 2001), the Six3-Cre line leads to widespread gene inactivation throughout the retina. Rb1loxP/loxP mice were obtained from the National Cancer Institute Mouse Repository (Frederick, MD; strain number 01XC1), and E2f1−/− mice were purchased from the Jackson Laboratory (Bar Harbor, ME; stock #002785). The original background of the Rb1loxP/loxP mice carried the rd1 allele, which was removed by backcrossing to C57Bl/6 mice (Gimenez and Montoliu, 2001). Genotyping was performed as previously described for the Rb1loxP/loxP strain (Vooijs et al., 2002) and Six3-Cre transgenic mice (Furuta et al., 2000). Genotyping of E2f1−/− mice was performed according to protocols at the Jackson Laboratory (http://jaxmice.jax.org/strain/002785.html). Mice were maintained on a 12-h light/dark schedule at 22°C with free access to food and water. The health of the animals was regularly monitored. All studies were conducted in accordance with the Guide for the Care and Use of Laboratory Animals and the Animal Welfare Act and were approved by the Washington University in St. Louis Institutional Animal Care and Use Committee (approval no. 20110089).
Tissue processing and immunohistochemistry
Retinas were rapidly dissected, fixed in 4% paraformaldehyde overnight at 4°C, and processed for paraffin sectioning. Paraffin sections were cut at 4-μm thickness through the optic nerve and stained with H&E. Subsequent immunostaining was performed on paraffin sections at the same retinal eccentricity for all genotypes and ages or on dissociated retinal cells. Briefly, paraffin sections were dewaxed in xylenes and then rehydrated through a graded ethanol series to water. Slides were then incubated in 0.05 M Tris, pH 8.0, with 0.1% glycine for 2 h at room temperature. Antigen retrieval was performed on all antibodies by boiling paraffin sections in 0.01 M citrate buffer, pH 6.0, for 10 min. Dissociated cells, prepared as previously described (Morrow et al., 1998), and antigen-retrieved sections were incubated in blocking solution (2% normal donkey serum, 0.5% Triton X-100 in phosphate-buffered saline [PBS]) for 1 h, followed by an overnight incubation at 4°C with primary antibodies diluted in the same blocking solution. A list of antibodies, including the manufacturer, catalogue number, and dilution used for single- or double-labeling experiments on dissociated cells and tissue sections, is given in Supplemental Table S1. Fluorescently conjugated secondary antibodies directed against the appropriate species of primary antibodies were used at 1:1000 (Alexa Fluor 488 donkey anti-rabbit, A-21206; Alexa Fluor 555 donkey anti-mouse, A-31570). Tissue or cells were counterstained with DAPI (D9542; Sigma Aldrich, St. Louis, MO). To quantify the frequency of immunostained dissociated cells, 100–500 immunopositive cells were scored per age per genotype from three independent experiments. Control tissue was processed from age-matched littermates and included the following genotypes: Rb1lox/+, Rb1lox/lox, Six3-Cre+;Rb1+/+, and Six3-Cre+;Rb1lox/+.
In vivo labeling of S-phase cells
To label cells in active DNA synthesis/S phase, the Click-iT Alexa Fluor 647 EdU Imaging Kit was used (C10340; Life Technologies, Carlsbad, CA). Cells were first processed for immunostaining using 1× PBS for washes in between steps, followed by EdU labeling. Briefly, control and RbCKO mice were given an IP injection of EdU (25 μg/g). Retinas were harvested 4 h after the injection, dissociated, and fixed in 4% paraformaldehyde for 1 h at 4°C. The cells were washed and incubated in blocking solution for 1 h at room temperature, followed by an overnight incubation at 4°C with primary antibodies. The following day, the cells were washed and incubated with appropriate secondary antibodies (as described) for 1 h at room temperature. Next the cells were washed, refixed for 10 min with 4% paraformaldehyde, and washed again with 3% bovine serum albumin in 1× PBS. EdU labeling was performed according to the manufacturer's instructions (Life Technologies). The cells were counterstained with DAPI.
Statistical analysis
Data are plotted with the mean and SD. To compare two groups, statistical analyses were performed by using a two-tailed t test. To compare three or more groups, statistical analyses were performed using one-way analysis of variance (ANOVA) followed by Tukey's multiple comparison post hoc test. A p < 0.05 was taken as statistically significant. GraphPad Prism software, version 5.04 for Windows (GraphPad Software, San Diego, CA), was used to generate all of the graphs and carry out statistical analyses.
Morphometric analysis of nuclear area in the INL
Paraffin sections were dewaxed in xylenes and rehydrated through a graded ethanol series to PBS and stained with SYTOX Green fluorescent nuclear counterstain (S7020; Invitrogen),. From 10 to 12 fields encompassing the INL were randomly chosen for RbCKO and control at P26 and P139 (∼4.5 mo). Images were acquired with an ORCA-ER camera (Hamamatsu, Hamamatsu, Japan) on a spinning disk confocal microscope (BX61WI; Olympus, Tokyo, Japan). The outline of each nucleus in the INL was traced onto a transparency. The traced images were scanned, and the areas of each outlined nucleus were measured using ImageJ software (National Institutes of Health, Bethesda, MD). In the RbCKO retina 1154 nuclei were evaluated at P26, and 609 nuclei were analyzed at 4.5 mo. In control retina, 1699 nuclei were evaluated at P26, and 1322 nuclei were analyzed at 4.5 mo. A histogram of the frequency of nuclear area in the INL was plotted for each genotype at both time points.
Cytofluorometric DNA analysis
To measure DNA content in horizontal cells, dissociated (asynchronous) retinal cells from adult (P35) control and RbCKO retina were immunostained with calbindin and counterstained with DAPI. Images of calbindin-positive and -negative cells were acquired along with the corresponding DAPI image. To obtain optimal light levels during the exposure, a neutral density filter was used. Exposure times were adjusted to achieve a maximum pixel value close to 3000. The same exposure settings were used to capture all of the images. The images were then analyzed using ImageJ to calculate the integrated density measurement (IDM = area of the DAPI-stained nuclei × mean gray value) for individual calbindin-positive cells. The mean gray value is the average gray value of all the pixels in the selection (area) divided by the total number of pixels. A total of 27 RbCKO and 20 control calbindin-positive horizontal cells were analyzed from three independent experiments. Raw IDM values in RbCKO calbindin-positive and -negative cells were normalized to the mean IDM of control calbindin-positive cells, which represents a 2N, diploid population. The data were graphed in a vertical scatter plot with the mean and SD.
Supplementary Material
Acknowledgments
We thank Belinda McMahan for assistance with paraffin sectioning, Anton Berns for the RbloxP/loxP mice, and Connie Myers for help with confocal microscopy and ImageJ. This work was supported by National Institutes of Health, National Eye Institute, Grant EY018825 (to J.C.C.) and Institutional Vision Science Training Grant 5-T32-EY13360-10 (to S.L.D.). This work was further supported by The Hope Center for Neurologic Diseases (to J.C.C.) and The Children's Discovery Institute (to J.C.C.).
Abbreviation used:
- IDM
integrated density measurement
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-04-0293) on September 26, 2012.
REFERENCES
- Ajioka I, Martins RA, Bayazitov IT, Donovan S, Johnson DA, Frase S, Cicero SA, Boyd K, Zakharenko SS, Dyer MA. Differentiated horizontal interneurons clonally expand to form metastatic retinoblastoma in mice. Cell. 2007;131:378–390. doi: 10.1016/j.cell.2007.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogenmann E, Lochrie MA, Simon MI. Cone cell-specific genes expressed in retinoblastoma. Science. 1988;240:76–78. doi: 10.1126/science.2451289. [DOI] [PubMed] [Google Scholar]
- Bourgo RJ, Ehmer U, Sage J, Knudsen ES. RB deletion disrupts coordination between DNA replication licensing and mitotic entry in vivo. Mol Biol Cell. 2011;22:931–939. doi: 10.1091/mbc.E10-11-0895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Livne-bar I, Vanderluit JL, Slack RS, Agochiya M, Bremner R. Cell-specific effects of RB or RB/p107 loss on retinal development implicate an intrinsically death-resistant cell-of-origin in retinoblastoma. Cancer Cell. 2004;5:539–551. doi: 10.1016/j.ccr.2004.05.025. [DOI] [PubMed] [Google Scholar]
- Chen D, Opavsky R, Pacal M, Tanimoto N, Wenzel P, Seeliger MW, Leone G, Bremner R. Rb-mediated neuronal differentiation through cell-cycle-independent regulation of E2f3a. PLoS Biol. 2007;5:e179. doi: 10.1371/journal.pbio.0050179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke AR, Maandag ER, van Roon M, van der Lugt NM, van der Valk M, Hooper ML, Berns A, te Riele H. Requirement for a functional Rb-1 gene in murine development. Nature. 1992;359:328–330. doi: 10.1038/359328a0. [DOI] [PubMed] [Google Scholar]
- Corson TW, Gallie BL. One hit, two hits, three hits, more? Genomic changes in the development of retinoblastoma. Genes Chromosomes Cancer. 2007;46:617–634. doi: 10.1002/gcc.20457. [DOI] [PubMed] [Google Scholar]
- Coschi CH, Dick FA. Chromosome instability and deregulated proliferation: an unavoidable duo. Cell Mol Life Sci. 2012;69:2009–2024. doi: 10.1007/s00018-011-0910-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dannenberg JH, Schuijff L, Dekker M, van der Valk M, te Riele H. Tissue-specific tumor suppressor activity of retinoblastoma gene homologs p107 and p130. Genes Dev. 2004;18:2952–2962. doi: 10.1101/gad.322004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davoli T, de Lange T. The causes and consequences of polyploidy in normal development and cancer. Annu Rev Cell Dev Biol. 2011;27:585–610. doi: 10.1146/annurev-cellbio-092910-154234. [DOI] [PubMed] [Google Scholar]
- Donovan SL, Schweers B, Martins R, Johnson D, Dyer MA. Compensation by tumor suppressor genes during retinal development in mice and humans. BMC Biol. 2006;4:14. doi: 10.1186/1741-7007-4-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dotiwala F, Harrison JC, Jain S, Sugawara N, Haber JE. Mad2 prolongs DNA damage checkpoint arrest caused by a double-strand break via a centromere-dependent mechanism. Curr Biol. 2010;20:328–332. doi: 10.1016/j.cub.2009.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyer MA, Bremner R. The search for the retinoblastoma cell of origin. Nat Rev Cancer. 2005;5:91–101. doi: 10.1038/nrc1545. [DOI] [PubMed] [Google Scholar]
- Furuta Y, Lagutin O, Hogan BL, Oliver GC. Retina- and ventral forebrain-specific Cre recombinase activity in transgenic mice. Genesis. 2000;26:130–132. [PubMed] [Google Scholar]
- Ganguly A, Shields CL. Differential gene expression profile of retinoblastoma compared to normal retina. Mol Vis. 2010;16:1292–1303. [PMC free article] [PubMed] [Google Scholar]
- Gimenez E, Montoliu L. A simple polymerase chain reaction assay for genotyping the retinal degeneration mutation (Pdeb(rd1)) in FVB/N-derived transgenic mice. Lab Anim. 2001;35:153–156. doi: 10.1258/0023677011911525. [DOI] [PubMed] [Google Scholar]
- Glubrecht DD, Kim JH, Russell L, Bamforth JS, Godbout R. Differential CRX and OTX2 expression in human retina and retinoblastoma. J Neurochem. 2009;111:250–263. doi: 10.1111/j.1471-4159.2009.06322.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haverkamp S, Wassle H. Immunocytochemical analysis of the mouse retina. J Comp Neurol. 2000;424:1–23. [PubMed] [Google Scholar]
- Hernando E, et al. Rb inactivation promotes genomic instability by uncoupling cell cycle progression from mitotic control. Nature. 2004;430:797–802. doi: 10.1038/nature02820. [DOI] [PubMed] [Google Scholar]
- Hurwitz RL, Bogenmann E, Font RL, Holcombe V, Clark D. Expression of the functional cone phototransduction cascade in retinoblastoma. J Clin Invest. 1990;85:1872–1878. doi: 10.1172/JCI114648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacks T, Fazeli A, Schmitt EM, Bronson RT, Goodell MA, Weinberg RA. Effects of an Rb mutation in the mouse. Nature. 1992;359:295–300. doi: 10.1038/359295a0. [DOI] [PubMed] [Google Scholar]
- Johnson DA, Zhang J, Frase S, Wilson M, Rodriguez-Galindo C, Dyer MA. Neuronal differentiation and synaptogenesis in retinoblastoma. Cancer Res. 2007;67:2701–2711. doi: 10.1158/0008-5472.CAN-06-3754. [DOI] [PubMed] [Google Scholar]
- Knudsen ES, Knudsen KE. Tailoring to RB: tumour suppressor status and therapeutic response. Nat Rev Cancer. 2008;8:714–724. doi: 10.1038/nrc2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudsen ES, Sexton CR, Mayhew CN. Role of the retinoblastoma tumor suppressor in the maintenance of genome integrity. Curr Mol Med. 2006;6:749–757. doi: 10.2174/1566524010606070749. [DOI] [PubMed] [Google Scholar]
- Laurie NA, et al. Inactivation of the p53 pathway in retinoblastoma. Nature. 2006;444:61–66. doi: 10.1038/nature05194. [DOI] [PubMed] [Google Scholar]
- Lee EY, Chang CY, Hu N, Wang YC, Lai CC, Herrup K, Lee WH, Bradley A. Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature. 1992;359:288–294. doi: 10.1038/359288a0. [DOI] [PubMed] [Google Scholar]
- Maandag EC, van der Valk M, Vlaar M, Feltkamp C, O'Brien J, van Roon M, van der Lugt N, Berns A, te Riele H. Developmental rescue of an embryonic-lethal mutation in the retinoblastoma gene in chimeric mice. EMBO J. 1994;13:4260–4268. doi: 10.1002/j.1460-2075.1994.tb06746.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacPherson D, Sage J, Kim T, Ho D, McLaughlin ME, Jacks T. Cell type-specific effects of Rb deletion in the murine retina. Genes Dev. 2004;18:1681–1694. doi: 10.1101/gad.1203304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning AL, Dyson NJ. pRB, a tumor suppressor with a stabilizing presence. Trends Cell Biol. 2011;21:433–441. doi: 10.1016/j.tcb.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino S, Hoogervoorst D, Brandner S, Berns A. Rb and p107 are required for normal cerebellar development and granule cell survival but not for Purkinje cell persistence. Development. 2003;130:3359–3368. doi: 10.1242/dev.00553. [DOI] [PubMed] [Google Scholar]
- Markey M, Siddiqui H, Knudsen ES. Geminin is targeted for repression by the retinoblastoma tumor suppressor pathway through intragenic E2F sites. J Biol Chem. 2004;279:29255–29262. doi: 10.1074/jbc.M313482200. [DOI] [PubMed] [Google Scholar]
- Marquardt T, Ashery-Padan R, Andrejewski N, Scardigli R, Guillemot F, Gruss P. Pax6 is required for the multipotent state of retinal progenitor cells. Cell. 2001;105:43–55. doi: 10.1016/s0092-8674(01)00295-1. [DOI] [PubMed] [Google Scholar]
- Marti TM, Hefner E, Feeney L, Natale V, Cleaver JE. H2AX phosphorylation within the G1 phase after UV irradiation depends on nucleotide excision repair and not DNA double-strand breaks. Proc Natl Acad Sci USA. 2006;103:9891–9896. doi: 10.1073/pnas.0603779103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayhew CN, Bosco EE, Fox SR, Okaya T, Tarapore P, Schwemberger SJ, Babcock GF, Lentsch AB, Fukasawa K, Knudsen ES. Liver-specific pRB loss results in ectopic cell cycle entry and aberrant ploidy. Cancer Res. 2005;65:4568–4577. doi: 10.1158/0008-5472.CAN-04-4221. [DOI] [PubMed] [Google Scholar]
- McGarry TJ, Kirschner MW. Geminin, an inhibitor of DNA replication, is degraded during mitosis. Cell. 1998;93:1043–1053. doi: 10.1016/s0092-8674(00)81209-x. [DOI] [PubMed] [Google Scholar]
- Morrow EM, Belliveau MJ, Cepko CL. Two phases of rod photoreceptor differentiation during rat retinal development. J Neurosci. 1998;18:3738–3748. doi: 10.1523/JNEUROSCI.18-10-03738.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munier FL, Balmer A, van Melle G, Gailloud C. Radial asymmetry in the topography of retinoblastoma. Clues to the cell of origin. Ophthalmic Genet. 1994;15:101–106. doi: 10.3109/13816819409057835. [DOI] [PubMed] [Google Scholar]
- Muresan V, Joshi HC, Besharse JC. Gamma-tubulin in differentiated cell types: localization in the vicinity of basal bodies in retinal photoreceptors and ciliated epithelia. J Cell Sci. 1993;104:1229–1237. doi: 10.1242/jcs.104.4.1229. [DOI] [PubMed] [Google Scholar]
- Robanus-Maandag E, Dekker M, van der Valk M, Carrozza ML, Jeanny JC, Dannenberg JH, Berns A, te Riele H. p107 is a suppressor of retinoblastoma development in pRb-deficient mice. Genes Dev. 1998;12:1599–1609. doi: 10.1101/gad.12.11.1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogakou EP, Boon C, Redon C, Bonner WM. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol. 1999;146:905–916. doi: 10.1083/jcb.146.5.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- Sangwan M, McCurdy SR, Livne-Bar I, Ahmad M, Wrana JL, Chen D, Bremner R. Established and new mouse models reveal E2f1 and Cdk2 dependency of retinoblastoma, and expose effective strategies to block tumor initiation. Oncogene. 2012 doi: 10.1038/onc.2011.654. doi:10.1038/onc.2011.654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santagata S, Maire CL, Idbaih A, Geffers L, Correll M, Holton K, Quackenbush J, Ligon KL. CRX is a diagnostic marker of retinal and pineal lineage tumors. PLoS One. 2009;4:e7932. doi: 10.1371/journal.pone.0007932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schvartzman JM, Duijf PH, Sotillo R, Coker C, Benezra R. Mad2 is a critical mediator of the chromosome instability observed upon Rb and p53 pathway inhibition. Cancer Cell. 2011;19:701–714. doi: 10.1016/j.ccr.2011.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viatour P, Sage J. Newly identified aspects of tumor suppression by RB. Dis Model Mech. 2011;4:581–585. doi: 10.1242/dmm.008060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vooijs M, te Riele H, van der Valk M, Berns A. Tumor formation in mice with somatic inactivation of the retinoblastoma gene in interphotoreceptor retinol binding protein-expressing cells. Oncogene. 2002;21:4635–4645. doi: 10.1038/sj.onc.1205575. [DOI] [PubMed] [Google Scholar]
- Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323–330. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- Williams BO, Schmitt EM, Remington L, Bronson RT, Albert DM, Weinberg RA, Jacks T. Extensive contribution of Rb-deficient cells to adult chimeric mice with limited histopathological consequences. EMBO J. 1994;13:4251–4259. doi: 10.1002/j.1460-2075.1994.tb06745.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu XL, Fang Y, Lee TC, Forrest D, Gregory-Evans C, Almeida D, Liu A, Jhanwar SC, Abramson DH, Cobrinik D. Retinoblastoma has properties of a cone precursor tumor and depends upon cone-specific MDM2 signaling. Cell. 2009;137:1018–1031. doi: 10.1016/j.cell.2009.03.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young RW. Cell death during differentiation of the retina in the mouse. J Comp Neurol. 1984;229:362–373. doi: 10.1002/cne.902290307. [DOI] [PubMed] [Google Scholar]
- Zacksenhaus E, Jiang Z, Chung D, Marth JD, Phillips RA, Gallie BL. pRb controls proliferation, differentiation, and death of skeletal muscle cells and other lineages during embryogenesis. Genes Dev. 1996;10:3051–3064. doi: 10.1101/gad.10.23.3051. [DOI] [PubMed] [Google Scholar]
- Zhang J, et al. A novel retinoblastoma therapy from genomic and epigenetic analyses. Nature. 2012;481:329–334. doi: 10.1038/nature10733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Schweers B, Dyer MA. The first knockout mouse model of retinoblastoma. Cell Cycle. 2004;3:952–959. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.