

Although both CTTNBP2 and CTTNBP2NL interact with cortactin and striatin/zinedin, CTTNBP2, but not CTTNBP2NL, is predominantly expressed in neurons and regulates dendritic spine distribution of cortactin and striatin/zinedin. The finding may be relevant to the association of CTTNBP2 with autism.
Abstract
Cortactin-binding protein 2 (CTTNBP2) interacts with cortactin to regulate cortactin mobility and control dendritic spine formation. CTTNBP2 has also been associated with autistic spectrum disorder. The regulation of dendritic spinogenesis could explain the association of CTTNBP2 with autism. Sequence comparison has indicated that CTTNBP2 N-terminal–like protein (CTTNBP2NL) is a CTTNBP2 homologue. To confirm the specific effect of CTTNBP2 on dendritic spinogenesis, here we investigate whether CTTNBP2NL has a similar function to CTTNBP2. Although both CTTNBP2 and CTTNBP2NL interact with cortactin, CTTNBP2NL is associated with stress fibers, whereas CTTNBP2 is distributed to the cortex and intracellular puncta. We also provide evidence that CTTNBP2, but not CTTNBP2NL, is predominantly expressed in the brain. CTTNBP2NL does not show any activity in the regulation of dendritic spinogenesis. In addition to spine morphology, CTTNBP2 is also found to regulate the synaptic distribution of striatin and zinedin (the regulatory B subunits of protein phosphatase 2A [PP2A]), which interact with CTTNBP2NL in HEK293 cells. The association between CTTNBP2 and striatin/zinedin suggests that CTTNBP2 targets the PP2A complex to dendritic spines. Thus we propose that the interactions of CTTNBP2 and cortactin and the PP2A complex regulate spine morphogenesis and synaptic signaling.
INTRODUCTION
Dendritic spines are tiny, actin-rich protrusions of ∼0.5–1 μm in width and 1–2 μm in length that extend from dendrites (Matus et al., 1982; Landis and Reese, 1983) and are the sites of most of the excitatory synapses in the mammalian CNS (Harris and Stevens, 1989). F-actin dynamics controls the morphology of dendritic spines (Penzes and Cahill, 2012). The signals derived from neurotransmitters also influence F-actin dynamics and alter the morphology or even the density of dendritic spines (Schubert and Dotti, 2007; Bosch and Hayashi, 2012). Many of the ubiquitously expressed actin-associated proteins, including cortactin, have been shown to regulate the morphology or density of dendritic spines. However, because dendritic spines are neuron-specific subcellular structures, it is possible that some of the neuron-specific proteins or signals control the formation of dendritic spines. Indeed, we recently showed that cortactin-binding protein 2 (CTTNBP2), a neuron-specific, F-actin–associated protein (Ohoka and Takai, 1998), regulates the formation and maintenance of dendritic spines (Chen and Hsueh, 2012).
CTTNBP2 is also known as CortBP2 (cortactin-binding protein 2) or CBP90 (cortactin-binding protein at ∼90 kDa; Ohoka and Takai, 1998). It has been associated with autistic spectrum disorder (Cheung et al., 2001; Iossifov et al., 2012), indicating a critical role for CTTNBP2 in neural development and function. Through alternative splicing, a single CTTNBP2 gene encodes three different transcripts, namely short (S), long (L), and intron forms (Chen and Hsueh, 2012). The results of immunoblotting and reverse transcription PCR analyses indicated that CTTNBP2-S is the major product of the CTTNBP2 gene in neurons (Chen and Hsueh, 2012). An analysis of an Expressed Sequence Tags database revealed that the CTTNBP2-L and CTTNBP2-intron transcripts are potentially expressed in nonneuronal tissues, although their expression profiles have not been confirmed at the protein level. Our previous study indicated that CTTNBP2-S proteins are stably localized to dendritic spines, where interactions through the C-terminal proline-rich domain regulate the mobility of cortactin (Chen and Hsueh, 2012). The interaction with cortactin is required for the function of CTTNBP2-S in dendritic spine formation because the CTTNBP2-S mutant, which no longer interacts with cortactin, is unable to rescue the spine defects resulting from CTTNBP2 knockdown (Chen and Hsueh, 2012). Thus CTTNBP2-S may control cortactin–F-actin cytoskeletons and regulate the formation and maintenance of dendritic spines in neurons.
Based on an amino acid sequence analysis, a CTTNBP2-S homologue, CTTNBP2 N-terminal–like (CTTNBPNL) protein, has also been identified (Chen and Hsueh, 2012). A proteomics study revealed that the striatin–protein phosphatase 2A (PP2A) protein complex associates with CTTNBP2NL in HEK293 cells (Goudreault et al., 2009). In addition to CTTNBP2NL, a trace amount of CTTNBP2 was also identified using mass spectrometry (Goudreault et al., 2009). In mammals, the striatin family contains three members—striatin, zinedin, and SG2NA—which function as PP2A B-type regulatory subunits (Benoist et al., 2006) that target the PP2A complex to specific cellular compartments or protein substrates and thus influence the substrate specificity of PP2A (Arroyo and Hahn, 2005). In neurons, all the members of the striatin family are highly concentrated at dendritic spines (Gaillard et al., 2006). PP2A activity has been shown to play roles in synaptic signaling (Chan and Sucher, 2001; Belmeguenai and Hansel, 2005; Mauna et al., 2011) and neurodegeneration (Tian and Wang, 2002); therefore striatins are potentially involved in the regulation of this activity through their influence on PP2A catalytic subunits. In addition, striatins also interact with mammalian sterile 20-like (Mst) kinases and many other proteins (Goudreault et al., 2009; Gordon et al., 2011). Thus striatins may form a large signaling complex containing phosphatases and kinases to regulate synaptic signaling.
Although the hetero-oligomerization of striatins is required for synaptic targeting (Gaillard et al., 2006) and all of the members of the striatin family are highly concentrated at dendritic spines, the mechanism underlying the targeting of striatins to the dendritic spines remains unclear. The CTTNBP2-S proteins are stably localized to dendritic spines (Chen and Hsueh, 2012). Therefore we examined whether CTTNBP2-S interacts with striatin family proteins in neurons and regulates their distribution to dendritic spines. Moreover, we also investigated the sequence and functional similarities, particularly in dendritic spinogenesis, between CTTNBP2NL and CTTNBP2-S. We unexpectedly found that CTTNBP2-S and CTTNBP2NL are different from each other in terms of tissue and subcellular distribution and function in dendritic spinogenesis.
In neurons, the short form is the major protein product of CTTNBP2; therefore CTTNBP2 is used to represent CTTNBP-S in this study. The notations BP2 and NL are used in the figures to represent the genes and encoded protein products of CTTNBP2 and CTTNBP2NL, respectively.
RESULTS
CTTNBP2 shares sequence similarity with CTTNBP2NL
Both CTTNBP2 and CTTNBP2NL were highly conserved among the human, rat, and mouse genomes. In addition, CTTNBP2 and CTTNBP2NL also shared amino acid sequence similarities, particularly in the N-terminal region, coiled-coil domain, and C-terminal proline-rich motifs. We observed ∼36% identity and ∼50% similarity between the mouse CTTNBP2 and CTTNBP2NL proteins (Figure 1). Previously we showed that the Pro540 and Pro543 residues of CTTNBP2 are critical for interaction with cortactin, because a proline-to-alanine substitution at these two residues dramatically reduced the interaction between CTTNBP2 and cortactin (Chen and Hsueh, 2012). These two proline residues and the flanking sequences are also conserved in CTTNBP2NL (Figure 1, gray box), suggesting an interaction between CTTNBP2NL and cortactin. Owing to the high sequence similarity, it has been suggested that CTTNBP2 and CTTNBP2NL are the same type of protein molecule (Goudreault et al., 2009).
FIGURE 1:

Amino acid sequence alignment of CTTNBP2NL and CTTNBP2. Mouse CTTNBP2NL and the short form of CTTNBP2 (CTTNBP2-s) were aligned using ClustalW2. Identical residues are indicated with asterisks. The black and gray boxes indicate the coiled-coil domain and the cortactin-interacting motif, respectively. The line above the CTTNBP2NL sequence indicates the sequence of polypeptides used to generate the anti-CTTNBP2NL antibody, and the underlined CTTNBP2 sequence represents the region (amino acids 498–625 ) used as the immunogen for the production of anti-CTTNBP2 antibodies.
CTTNBP2NL interacts with cortactin and targets cortactin to stress fibers
CTTNBP2NL also contains the proline-rich motif for interaction with cortactin; therefore we investigated whether, similar to CTTNBP2, CTTNBP2NL interacts with cortactin. We performed a coimmunoprecipitation experiment using COS cells cotransfected with Myc-cortactin and hemagglutinin (HA)-tagged CTTNBP2NL. The coprecipitation of Myc-tagged cortactin and HA-tagged CTTNBP2NL using an anti-Myc antibody (Figure 2A) indicated an interaction between CTTNBP2NL and cortactin. The precipitation was specific because nonimmune immunoglobulin G (IgG) precipitated neither HA-CTTNBP2NL nor Myc-cortactin (Figure 2A).
FIGURE 2:

Comparison of the interaction of CTTNBP2NL and CTTNBP2 with cortactin. (A) Coimmunoprecipitation of cortactin and CTTNBP2NL. Whole-cell extracts of COS cells cotransfected with Myc-tagged cortactin (Myc-Cort) and HA-tagged CTTNBP2NL (HA-NL) were precipitated using anti-Myc and nonimmune mouse IgG antibodies. Immunoblotting (IB) was performed to assess the presence of CTTNBP2NL and cortactin in the precipitates. (B) The overlapping distribution of cortactin (Cort) and F-actin at the cell cortex (arrowheads). COS cells were fixed and stained with anti-cortactin and phalloidin (to label F-actin). (C, D) COS cells were transfected with CTTNBP2NL and immunostained with anti-CTTNBP2NL antibody and (C) phalloidin and (D) anti-tubulin antibodies. Arrows indicate the untransfected COS cells, which did not show any obvious immunoreactivity of CTTNBP2NL. (E) CTTNBP2NL preferentially associates with actin stress fibers (arrows), whereas CTTNBP2 colocalizes with cortactin at the cell cortex (arrowheads). COS cells expressing HA-tagged CTTNBP2NL (top) or Myc-tagged CTTNBP2 (Myc-BP2; bottom) were stained with phalloidin, anti-cortactin, and anti-HA or anti-Myc, respectively. Scale bars, 10 μm.
A previous study indicated that cortactin is distributed in the cortex and intracellular puncta in chicken embryonic cells, rat aortic smooth muscle cells, and rat endothelial cells (Wu and Parsons, 1993). A similar subcellular distribution of cortactin was also observed in COS cells (Figure 2B). Our previous study showed that CTTNBP2 is not expressed in COS cells (Chen and Hsueh, 2012). Similarly, here, expression of endogenous CTTNBP2NL in COS cells was not detected (Figures 2, C and D, and 3A). We compared the distribution of CTTNBP2NL and CTTNBP2 in the cortactin-positive subcellular structures using COS cells transfected with HA-tagged CTTNBP2NL or Myc-tagged CTTNBP2. In contrast to the endogenous cortactin, only a trace amount of HA-CTTNBP2NL was distributed to the cell cortex. Phalloidin staining revealed more promising immunoreactivities of HA-tagged CTTNBP2NL along stress fibers (Figure 2E, top). Moreover, cortactin was distributed to the stress fibers in the presence of HA-tagged CTTNBP2NL (Figure 2E, top). These results suggest that CTTNBP2NL interacts with cortactin and redistributes this protein to stress fibers. This activity is quite different from that of CTTNBP2, because CTTNBP2 colocalized with cortactin at the cell cortex (Figure 2E, bottom). In addition, the presence of CTTNBP2 increased the number of cortactin-positive puncta, which were also CTTNBP2 positive (Figure 2E, bottom). Taken together, the results from the immunostaining analyses suggest interactions between cortactin and CTTNBP2NL and CTTNBP2. The presence of CTTNBP2 and CTTNBP2NL thus influences the subcellular distribution of cortactin.
FIGURE 3:

Expression of CTTNBP2 and CTTNBP2NL in mouse tissues. (A) Characterization of the anti-CTTNBP2NL antibody by immunoblotting. Whole-cell extracts of COS cells transfected with HA-tagged CTTNBP2NL or the control vector were immunoblotted with anti-CTTNBP2NL antibodies. (B, C) Lysates from various mouse organs were immunoblotted using anti-CTTNBP2NL (B) and anti-CTTNBP2 (C) antibodies. B, brain; H, heart; K, kidney; L, lung; Lv, liver; M, muscle; Sk, skin; Sp, spleen; T, testis. (D) Real-time PCR analysis of the actual copy numbers of CTTNBP2 and CTTNBP2NL transcripts in total RNA extracted from different regions of the adult mouse brain. Cb, cerebellum; Cx, cerebral cortex; Hp, hippocampus; St, striatum.
CTTNBP2NL is expressed at low levels in the brain
Because CTTNBP2NL also interacts with cortactin, which regulates dendritic spinogenesis in neurons, we examined whether, similar to CTTNBP2, CTTNBP2NL controls cortactin mobility and regulates dendritic spinogenesis in neurons. We generated a CTTNBP2NL-specific antibody to investigate the expression and distribution of CTTNBP2NL. A synthetic peptide corresponding to the CTTNBP2NL-specific sequence (Figure 1) was used as an immunogen to minimize the cross-reactivity against CTTNBP2. With the use of cell lysates prepared from transfected COS cells, immunoblot analysis showed that our antibody recognized HA-tagged CTTNBP2NL proteins but did not show nonspecific signals in cells transfected with vector control (Figure 3A). Subsequently we investigated the expression of CTTNBP2NL proteins. The most promising CTTNBP2NL immunoreactivities were present in the skin; CTTNBP2NL proteins were also expressed in the lungs and spleen. However, CTTNBP2NL protein was barely detected in the brain (Figure 3B). This distribution pattern is distinct from that of CTTNBP2, which was specifically expressed in the brain (Figure 3C).
To confirm the low expression of CTTNBP2NL in the brain, we performed quantitative real time (RT)-PCR analysis. The copy numbers of CTTNBP2 and CTTNBP2NL transcripts in different mouse brain regions were determined. The CTTNBP2NL mRNA levels were ∼20- to 25-fold lower than those of CTTNBP2 in various brain regions, including the cerebral cortex, cerebellum, hippocampus, and striatum (Figure 3D). This result is consistent with the immunoblot data and suggests that in contrast to CTTNBP2, CTTNBP2NL is expressed at extremely low levels in the brain.
CTTNBP2, but not CTTNBP2NL, regulates dendritic spinogenesis in cultured hippocampal neurons
The low level of expression and distinct subcellular distribution of CTTNBP2NL suggest that CTTNBP2NL is unlikely to perform a function similar to CTTNBP2 in the regulation of dendritic spinogenesis. Next we constructed an artificial microRNA (miRNA)-expressing plasmid to knock down CTTNBP2NL expression and directly examine the effect of CTTNBP2NL on the regulation of dendritic spine density. We confirmed the knockdown efficiency of the CTTNBP2NL miRNA construct (NL-miR) in COS cells. Compared with the control vector (Ctrl-miR), NL-miR effectively reduced the protein levels of CTTNBP2NL, which was coexpressed in COS cells (Figure 4A, left). The effect of NL-miR was specific to CTTNBP2NL because it did not reduce CTTNBP2 expression (Figure 4A, right). Using NL-miR, we investigated the role of CTTNBP2NL in dendritic spinogenesis. Similar to observations in our previous study (Chen and Hsueh, 2012), the inhibition of endogenous CTTNBP2 expression using an artificial miRNA (BP2-miR) significantly reduced the number of dendritic spines in cultured hippocampal neurons (Figure 4C). However, the knockdown of CTTNBP2NL did not reduce the density of dendritic spines (Figure 4, B and C). Moreover, the overexpression of CTTNBP2NL did not rescue the spine defects in the CTTNBP2 knockdown (Figure 4C), suggesting that CTTNBP2NL is not functionally similar to CTTNBP2, at least in the regulation of dendritic spinogenesis.
FIGURE 4:
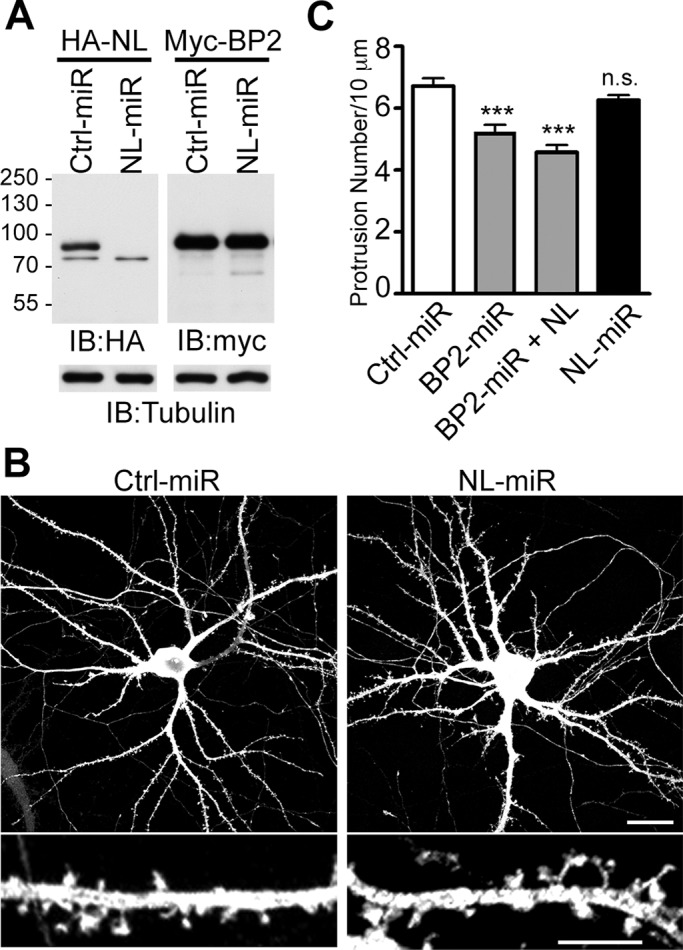
CTTNBP2NL does not significantly regulate the density of dendritic spines in cultured hippocampal neurons. (A) The knockdown efficiency of NL-miR. Whole-cell extracts of COS cells cotransfected with HA-tagged CTTNBP2NL or Myc-tagged CTTNBP2 and Ctrl-miR or NL-miR, as indicated, were immunoblotted with the anti-HA antibody or the anti-Myc antibody. Tubulin was used as an internal control. (B, C) The effect of CTTNBP2NL on dendritic spines. Rat hippocampal neurons were transfected at 12 DIV and analyzed at 18 DIV. (B) Representative images of Ctrl-miR– or NL-miR–transfected neurons. Bottom, a higher magnification of the dendrites. Scale bars, 20 μm (top) and 5 μm (bottom). (C) Quantification of the density of dendritic spines in neurons transfected with Ctrl-miR, BP2-miR, BP2-miR and CTTNBP2NL, or NL-miR. More than 20 dendrites from 10 neurons were analyzed for each group.
CTTNBP2 interacts with striatins in neurons
CTTNBP2 is more abundant than CTTNBP2NL in neurons; therefore we investigated whether CTTNBP2 could replace the function of CTTNBP2NL in neurons in terms of the interactions with the striatin family proteins at dendritic spines. We used immunofluorescence staining to examine the distribution of CTTNBP2 and the striatin proteins in cultured hippocampal neurons. Two striatin family proteins were investigated here: striatin and zinedin. Consistent with the previous study (Gaillard et al., 2006), striatin and zinedin were distributed to dendritic spines, where they were colocalized with CTTNBP2 (Figure 5A). Coimmunoprecipitation using an anti-CTTNBP2 antibody in rat brain extracts also demonstrated the interaction of CTTNBP2 with striatin and zinedin (Figure 5, B and C). Moreover, another striatin-interacting protein, phocein (Baillat et al., 2001), was also present in the CTTNBP2 immunocomplex purified from rat brain (Figure 5C), suggesting that CTTNBP2 interacts with the striatin protein complex in neurons.
FIGURE 5:
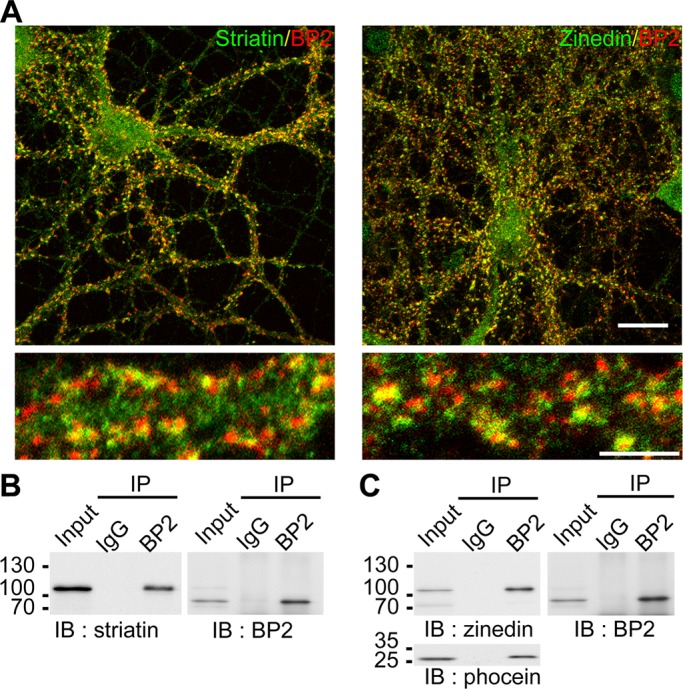
The association between CTTNBP2 and the striatin complex in neurons. (A) Colocalization of CTTNBP2 and striatin/zinedin in cultured hippocampal neurons. Rat hippocampal neurons were stained at 21 DIV using anti-CTTNBP2 and anti-striatin (left) or anti-zinedin (right) antibodies. Scale bars, 20 μm (top) and 5 μm (bottom). (B, C) Coimmunoprecipitation of CTTNBP2 and the striatin complex from rat brain lysate using anti-CTTNBP2 and nonimmune rabbit IgG antibodies. The precipitates were immunoblotted with anti-CTTNBP2, anti-striatin (B), anti-zinedin (C), or anti-phocein (C) antibodies.
The N-terminal region of CTTNBP2 associates with the N-terminal domain of striatin
We then investigated how CTTNBP2 interacts with striatin. The domain of CTTNBP2 required for the interaction with striatin was first studied using transfected HEK293T cells. HA-tagged full-length striatin and various Myc-tagged CTTNBP2 domain–deletion constructs (Figure 6A) were cotransfected into HEK293T cells. Coimmunoprecipitation using Myc-tagged antibody showed that the NC domain containing the N-terminal region and the coiled-coil domain of CTTNBP2 are sufficient for the interaction with striatin (Figure 6B). To identify the striatin domain that associates with CTTNBP2, we cotransfected Myc-tagged CTTNBP2 with HA-tagged striatin constructs (Figure 6C) into HEK293T cells. The N-terminal coiled-coil (N1) domain of striatin was shown to be the region associated with CTTNBP2 (Figure 6D). These analyses indicate that the N-terminal regions of CTTNBP2 and striatin are involved in the association between these two proteins.
FIGURE 6:

The N-terminal regions of CTTNBP2 and striatin (STN) are required for the association. (A, C) The full-length and domain deletion constructs used in coimmunoprecipitation for CTTNBP2 (A) and striatin (C). (B, D) HEK293T cells were cotransfected with various striatin and CTTNBP2 constructs, as indicated, and subjected to immunoprecipitation and immunoblotting with the antibodies as shown. Arrows point out the expected positions of the proteins in a SDS–PAGE. The asterisk indicates a potential dimer of the NC construct of CTTNBP2. IgH, IgG heavy chain.
CTTNBP2 is required for the synaptic distribution of striatin and zinedin
Our previous study indicated that CTTNBP2 stably resides at dendritic spines (Chen and Hsueh, 2012). We therefore investigated whether CTTNBP2 regulates the synaptic distribution of striatin and zinedin, using knockdown of endogenous CTTNBP2. Although a reduced number of dendritic spines was observed in the CTTNBP2 knockdown (Figure 4C), we could still find residual dendritic spines along the dendrites of some CTTNBP2-knockdown hippocampal neurons. We measured the immunoreactivities of striatin and zinedin in the residual dendritic spines. A line scan was performed to measure the signals radiating from the tips of the dendritic spines to the other edge of the dendritic shaft. Compared with Ctrl-miR, BP2-miR expression reduced the immunoreactivities of both striatin and zinedin in dendritic spines (Figure 7, A and B). The striatin signals in the control spines were at least twofold higher than those in the CTTNBP2-knockdown spines (Figure 7B, left). Similarly, zinedin expression in the control spines was also at least twofold higher than that in the CTTNBP2 knockdown spines (Figure 7B, right).
FIGURE 7:

Knockdown of CTTNBP2 reduces the dendritic spine distribution of striatin and zinedin. (A) Rat hippocampal neurons were transfected with Ctrl-miR or BP2-miR at 12 DIV and stained at 18 DIV using anti-striatin (left) or anti-zinedin (right) antibodies. The GFP signals highlight the dendritic morphology. (B) Quantitative analysis of striatin (left) or zinedin (right) distribution. In the higher magnification of a spine (inset), the line indicates the path for line scanning, which starts from the tip of the spine (set as 0) and crosses the dendritic shaft. The means ± SEM of the fluorescence intensities along the scanning path are provided. More than 30 spines from seven neurons were analyzed for each group. (C, D) Dendritic spine distribution of striatin and zinedin were rescued by expression of Myc-tagged BP2-rescue construct, which is resistant to BP2-miR. The experiment was carried as described in A and B, except that one more group of neurons cotransfected with BP2-miR and BP2-rescue was included for analysis. To make the DNA amounts equal, vector control was also included as indicated. Note that the expression levels of BP2-rescue construct are not identical among transfected neurons. One BP2-rescue lower-expressing cell and two BP2-rescue higher-expressing cells are shown in Cc. Scale bar, 5 μm.
Rescue experiments were also performed using a BP2-rescue construct that is resistant to BP2-miR (Chen and Hsueh, 2012). Compared with CTTNBP2 knockdown, expression of the BP2-rescue construct increased the amounts of both striatin and zinedin at the dendritic spines (Figure 7, C and D), supporting the conclusion that BP2 regulates the dendritic spine distribution of striatin and zinedin. However, we noticed that the rescue effect was partial. The means did not reach to the levels of neurons transfected with ctrl-miR (Figure 7D). We speculate that this partial rescue effect was due to ectopic dendritic distribution of the overexpressed BP2-rescue construct. It is difficult to precisely control the expression levels of exogenous genes in cells. In some of the neurons, the levels of BP2-rescue mutant proteins were moderate and those exogenous CTTNBP2 proteins still specifically targeted to dendritic spines (Figure 7Cc, left). In this case, the endogenous striatin proteins were clearly concentrated at dendritic spines (Figure 7Cc, left). However, in the majority of transfected neurons, the levels of BP2-rescue construct were too high and ectopically distributed to the dendritic shaft (Figure 7Cc, middle and right), which may have thus reduced the rescue effect on dendritic spine distribution of striatin and zinedin.
Glutamate stimulation induces the redistribution of striatin and zinedin
Our previous study indicated that CTTNBP2 is stably localized to dendritic spines after glutamate stimulation (Chen and Hsueh, 2012). In contrast, cortactin and F-actin are redistributed to the dendritic shaft after glutamate stimulation (Hering and Sheng, 2003; Chen and Hsueh, 2012). The redistribution of cortactin has been suggested to play a role in synaptic remodeling (Hering and Sheng, 2003). PP2A is also critical for neuronal plasticity; therefore we examined whether neural activation also induces the redistribution of striatin and zinedin. Indeed, N-methyl-d-aspartate (NMDA) treatment for 15 min reduced the immunoreactivities of striatin and zinedin in dendritic spines, because the colocalization of CTTNBP2 and striatin or zinedin was reduced (Figure 8A). This reduction was not due to protein degradation, because the total protein levels of striatin and zinedin after NMDA stimulation were comparable to those without NMDA treatment (Figure 8B).
FIGURE 8:

NMDA treatment induces the redistribution of striatin and zinedin. (A) At 18 DIV, cultured hippocampal neurons were stimulated with NMDA (100 μM) for 15 min, followed by staining with phalloidin, anti-CTTNBP2, and anti-striatin (top) or anti-zinedin (bottom) antibodies. (B) Using the same stimulation conditions as in A, we prepared whole-cell extracts of neurons for immunoblotting for striatin and zinedin, as indicated. Tubulin was used as an internal control. (C) An experiment similar to that performed as described in A was carried out, except that neurons were treated with 20 μM NMDA for 3 min. After 12 min of recovery, cells were harvested for immunostaining. (D) The total protein levels of striatin and zinedin in cultured hippocampal neurons were examined under the condition as described in C. Scale bar, 5 μm.
In the aforementioned experiment, NMDA at a concentration of 100 μM was added to stimulate cultured hippocampal neurons. A weaker stimulation condition was also applied to cultured hippocampal neurons. When the concentration of NMDA was reduced to 20 μM and the incubation time was shortened to 3 min, redistribution of F-actin to dendritic shaft still occurred in ∼40% of cultured neurons. In those cells, CTTNBP2 proteins were still concentrated at the dendritic spines, but the signals of striatin and zinedin at the dendritic spines were greatly reduced (Figure 8C). In agreement with the results of treatment with 100 μM NMDA, this low-dose treatment did not result in reduction of striatin or zinedin at the protein level (Figure 8D). Taken together, these results indicate that, similar to cortactin, striatin and zinedin were redistributed upon NMDA receptor (NMDAR) activation.
To further confirm the dissociation of CTTNBP2 and striatin upon NMDAR activation, we performed coimmunoprecipitation using CTTNBP2 and striatin antibodies. Compared with vehicle control, NMDA treatment reduced the protein amounts of striatin in the complex precipitated by CTTNBP2 antibody (Figure 9). Similarly, the presence of CTTNBP2 in the precipitates using striatin antibodies was also decreased upon NMDA stimulation, indicating that NMDAR activation weakens the association between CTTNBP2 and striatin.
FIGURE 9:
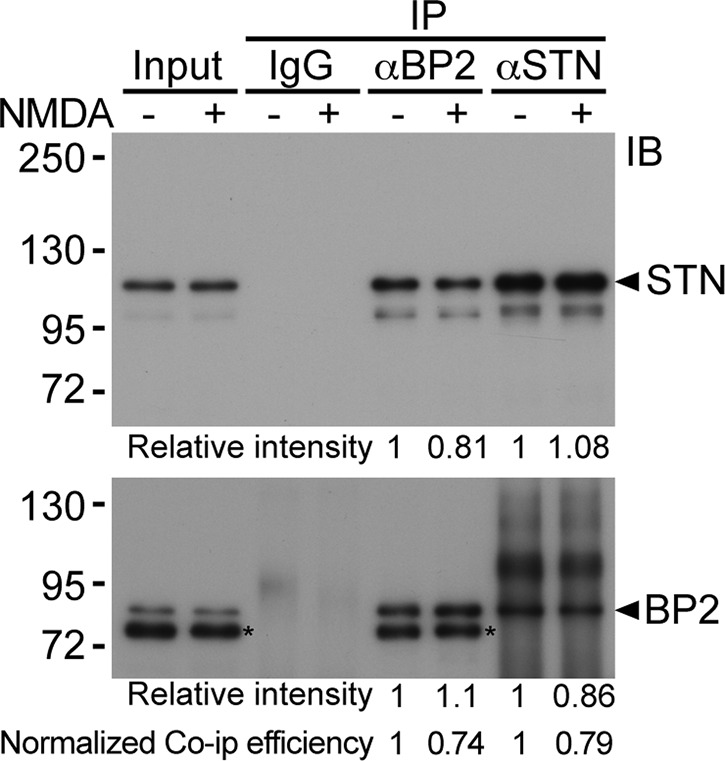
NMDA treatment reduces the association between CTTNBP2 and striatin (STN). Cultured hippocampal neurons were treated with 100 μM NMDA for 15 min and subjected to immunoprecipitation using CTTNBP2 and striatin antibody. Nonimmune IgG was used as a negative control. The associations of striatin with CTTNBP2 were analyzed by immunoblotting. The asterisks indicate unknown proteins recognized by CTTNBP2 antibody, which may be either degraded CTTNBP2 or cross-reactive protein species.
DISCUSSION
In this study, we compared the subcellular and tissue distribution, protein–protein interaction, and function of CTTNBP2 and CTTNBP2NL. The tissue distribution of CTTNBP2 and CTTNBPNL was different. The results of immunoblot and quantitative RT-PCR analyses indicate that CTTNBP2 was predominantly expressed in the brain. Similarly, the subcellular distribution of CTTNBP2 and CTTNBP2NL was also distinct. CTTNBP2 interacts with cortactin in the cell cortex and at intracellular puncta. In contrast, CTTNBP2NL targets and redistributes cortactin to the stress fibers. These differential interactions suggest that CTTNBP2 is involved in F-actin branching, whereas CTTNBP2NL associates with F-actin bundles. The difference in tissue distribution and distinct ability to associate with different microfilaments of the F-actin cytoskeleton might explain why CTTNBP2, but not CTTNBP2NL, is critical for dendritic spinogenesis. The heads of dendritic spines are supported by branched F-actin (Chen and Hsueh, 2012). Thus, suppressing the expression of cortactin, a factor that stabilizes F-actin branching, reduces the size of spine heads and eventually results in the withdrawal of dendritic spines (Hering and Sheng, 2003). CTTNBP2 regulates the mobility and synaptic distribution of cortactin (Chen and Hsueh, 2012) and is predominantly expressed in the brain, suggesting that CTTNBP2 regulates dendritic spinogenesis. In contrast, CTTNBP2NL is expressed at extremely low levels in the brain, which makes it virtually impossible for CTTNBP2NL to regulate dendritic spinogenesis. Indeed, the overexpression of CTTNBP2NL did not rescue the spinal defects in the CTTNBP2 knockdown, indicating that CTTNBP2 and CTTNBP2NL are functionally different, at least in the regulation of dendritic spinogenesis.
Although CTTNBP2 and CTTNBP2NL are different in terms of tissue and subcellular distribution, our data indicate that, similar to CTTNBP2NL, CTTNBP2 associates with members of the striatin family, namely striatin and zinedin. Moreover, CTTNBP2 is critical for the distribution of striatin and zinedin in dendritic spines. The members of the striatin family function as regulatory B subunits of the PP2A holoenzyme (Benoist et al., 2006); therefore it has been suggested that the striatin proteins target the PP2A complex to specific substrates or cellular compartments and thus determine the specificity of PP2A (Arroyo and Hahn, 2005). The role of CTTNBP2 in the regulation of the synaptic distribution of striatin and zinedin suggests that CTTNBP2 regulates synaptic signaling through PP2A. In the spleen, lungs, and skin, CTTNBP2NL, but not CTTNBP2, might regulate the distribution of the striatin–PP2A complex in cells. Given that CTTNBP2NL preferentially associates with stress fibers, the striatin–PP2A complex potentially modulates stress fibers through an association with CTTNBP2NL.
The interaction between CTTNBP2 and SG2Na, the third member of the striatin family, was not examined in this study because we lacked a reliable reagent to detect SG2Na in cells. However, the hetero-oligomerization of striatin, zinedin, and SG2Na has been previously demonstrated and is required for synaptic targeting of the striatin family (Gaillard et al., 2006). Thus it is reasonable to speculate that CTTNBP2 also associates with SG2Na at dendritic spines.
A previous proteomic study indicated the association between striatin family proteins and CTTNBP2NL/CTTNBP2 (Goudreault et al., 2009). In the present article, we provide evidence that the N-terminal coiled-coil domains of CTTNBP2 and striatin family proteins are involved in this association. Because CTTNBP2 controls the synaptic distribution of striatin family proteins, our study provides an explanation how the coiled-coil domain of striatin proteins is involved in synaptic targeting of the striatin family (Gaillard et al., 2006).
CTTNBP2 proteins are stably localized to dendritic spines (Chen and Hsueh, 2012), and they are involved in the synaptic targeting of cortactin and proteins of the striatin family. However, the mechanism underlying the targeting of CTTNBP2 to dendritic spines remains unknown. Further experiments concerning the stable localization of CTTNBP2 in dendritic spines are needed. The evidence suggests that interactions between CTTNBP2 and cortactin and the striatin-family proteins are regulated through neuronal activation. However, it is also unclear how neuronal activation disrupts these interactions. The striatin-family proteins also associate with Mst kinases; therefore it is possible that the phosphorylation of CTTNBP2, cortactin, or the striatin-family proteins regulates the interactions between these molecules. More investigations are required to examine this possibility.
Human genetic studies indicated an association between CTTNBP2 and autism (Cheung et al., 2001; Iossifov et al., 2012). Evidence revealed that abnormalities in synaptic structures and signaling are a cause of autistic spectrum disorder (Walsh et al., 2008). Our study reveals the roles of CTTNBP2 in the regulation of dendritic spinogenesis and the synaptic distribution of the PP2A complex in neurons. These results also explain, at least in part, how mutations in the CTTNBP2 gene result in autistic spectrum disorder.
MATERIALS AND METHODS
Antibodies
The following antibodies were used in this study: rabbit polyclonal anti-cortactin (H-191; Santa Cruz Biotechnology, Santa Cruz, CA); mouse monoclonal anti-striatin (sc-136084; Santa Cruz Biotechnology); mouse monoclonal anti-phocein (C-4; Santa Cruz Biotechnology); rabbit polyclonal anti-striatin (NB110-74571; Novus Biologicals, Littleton, CO); mouse monoclonal anti-zinedin (K88/64; UC Davis/NIH NeuroMab Facility, University of California, Davis, CA); mouse monoclonal anti-HA (12CA5; Roche Applied Science); rat monoclonal anti-HA (3F10; Roche Applied Science, Indianapolis, IN); mouse monoclonal anti-Myc (9B11; Cell Signaling Technology, Beverly, MA); rabbit polyclonal anti–green fluorescent protein (GFP; A-6455; Invitrogen, Carlsbad, CA); chicken polyclonal anti-GFP (ab13970; Abcam, Cambridge, MA); mouse monoclonal anti-VCP (BD Biosciences, San Diego, CA); and mouse monoclonal anti–β-tubulin (TUB 2.1; Sigma-Aldrich, St. Louis, MO). The rabbit polyclonal anti-CTTNBP2 antibody was generated as previously described (Chen and Hsueh, 2012). The rabbit polyclonal anti-CTTNBP2NL antibody was produced at GenScript (Piscataway, NJ) using a synthetic peptide (TSDSSTENQGPPRE, amino acids 363–376 of CTTNBP2NL). The specific antibody was subsequently purified on a peptide-conjugated affinity column.
Plasmids
The plasmid pGW1-CMV-Myc-cortactin (Hering and Sheng, 2003) was a gift from Morgan Sheng (Howard Hughes Medical Institute and Massachusetts Institute of Technology, Cambridge, MA; current affiliation, Genentech, South San Francisco, CA). The constructs pGW1-CMV-Myc-CTTNBP2, pcDNA6.2-GW/EmGFP-miR-CTTNBP2 (BP2-miR), and pcDNA6.2-GW/EmGFP-neg control (Ctrl-miR) were constructed as previously described (Chen and Hsueh, 2012). To construct the CTTNBP2NL expression plasmid, we amplified the coding sequence from IMAGE 3498078 and cloned it into a pGW1-CMV vector containing a HA tag. For the knockdown of CTTNBP2NL, pcDNA6.2-GW/EmGFP-miR-CTTNBP2NL (NL-miR) was constructed using base pairs 741–761 of the CTTNBP2NL coding sequence. Full-length HA-striatin construct was a gift from David C. Pallas (Department of Biochemistry and Winship Cancer Institute, Emory University School of Medicine, Atlanta, GA). Striatin full-length and truncated mutants were further subcloned into pGW1-CMV vector containing a HA tag.
Real-time PCR
Different regions of the adult mouse brain were dissected, and total RNA was extracted using TRIzol (Invitrogen). Complementary DNA synthesis was performed using 3 μg of total RNA per reaction in the Transcriptor First Strand cDNA Synthesis Kit (Roche) with an oligo(dT)18 primer. Quantitative PCR was performed using Universal ProbeLibrary probes (UPL; Roche) and a LightCycler480 system (Roche). The primers and probes were designed using the Roche Universal ProbeLibrary Assay Design Center. For CTTNBP2, the probe was UPL #4, and the primers were BP2#4L, 5′-AGCCAGAGACCTGGTGATTG-3′, and BP2#4R, 5′-AAACCTCCCATACCGTTCCT-3′. For CTTNBP2NL, the probe was UPL #33, and the primers were NL#33L, 5′-AGTGCAAGAACATGCAGGAG-3′, and NL#33R, 5′-CTTCAAGGTCGAGGATCACC-3′. Each sample was assayed in triplicate. The PCR conditions were set as follows: denaturation at 95°C for 10 min, 45 cycles of 10-s denaturation at 95°C for 10 s, and annealing at 60°C for 30 s, followed by extension at 72°C for 1 s and a final cooling step at 40°C for 30 s. The plasmid pGW1-CMV-Myc-CTTNBP2 or pGW1-CMV-HA-CTTNBP2NL was 10-fold serially diluted (from 2.5 pg to 2.5 fg/μl of reaction) and assayed as standards in the same 96-well plate. The precise copy number of the specific cDNA molecules in each sample was subsequently calculated using standard curve fitting.
Multiple-tissue immunoblotting and immunoprecipitation
Adult mouse tissues were dissected and lysed in lysis buffer (10 mM Tris, pH 7.4, 320 mM sucrose, 2 mM dithiothreitol, 2 μg/ml leupeptin, 2 μg/ml pepstatin-A, 2 μg/ml aprotinin, 1 mM tosylphenylalanylchloromethane, and 2 mM phenylmethylsulfonyl fluoride) containing 1% Triton X-100. After centrifugation at 12,000 × g at 4°C for 10 min, the soluble fractions were collected and subjected to immunoblotting. For the immunoprecipitation from rat brain, the soluble synaptosome fraction was obtained and immunoprecipitated as previously described (Chen and Hsueh, 2012).
Neuronal cultures, immunostaining, and morphometry
Rat hippocampal neurons from embryonic day 18 to 19 embryos were dissociated and cultured as previously described (Chen and Hsueh, 2012). The transfection of neurons was performed at 12 d in vitro (DIV) using the calcium phosphate precipitation method. The transfection of COS cells was conducted with Lipo2000 reagent (Invitrogen). For imaging, COS cells were trypsinized at 1 d after transfection and replated on poly-l-lysine (0.1 mg/ml)–coated glass coverslips, followed by incubation for 4 h at 37°C before fixation. For immunostaining, the cells were fixed with 4% paraformaldehyde and 4% sucrose in phosphate-buffered saline (PBS), followed by permeabilization with 0.2% Triton X-100 in PBS. After blocking with 10% bovine serum albumin, the cells were incubated with primary antibodies diluted in PBS containing 3% bovine serum albumin at 4°C overnight. After PBS washes, the cells were incubated with secondary antibodies conjugated with Alexa Fluor 488, 555, and/or 647 (Invitrogen) for 2 h. The images were acquired using a confocal microscope (LSM700; Carl Zeiss, Jena, Germany) equipped with a 63×/numerical aperture 1.4 oil objective lens (Plan-Apochromat; Carl Zeiss) and Zen 2009 (Carl Zeiss) acquisition and analysis software. The quantitation of spine density was performed using ImageJ, version 1.45 (National Institutes of Health, Bethesda, MD). The density was manually quantitated along a 20-μm dendrite starting at 20 μm away from the soma. Image acquisition and quantitation were blindly performed to minimize the effect of bias. The statistical analysis of spine density was performed using a one-way analysis of variance and Tukey's post hoc test with Prism 5.0 (GraphPad Software, La Jolla, CA).
Acknowledgments
We thank Morgan Sheng and David Pallas for DNA constructs. This work was supported through grants from the Academia Sinica (AS-100-TP-B09) and National Science Council (NSC 100-2321-B-001-022 and 101-2321-B-001-010) to Y.P.H.
Abbreviations used:
- CTTNBP2
cortactin-binding protein 2
- CTTNBP2NL
cortactin-binding protein 2 N-terminal–like protein
- PP2A
protein phosphatase 2A
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-05-0365) on September 26, 2012.
*Present address: Margaret M. Dyson Vision Research Institute, Department of Ophthalmology, Weill Medical College of Cornell University, New York, NY 10065.
REFERENCES
- Arroyo JD, Hahn WC. Involvement of PP2A in viral and cellular transformation. Oncogene. 2005;24:7746–7755. doi: 10.1038/sj.onc.1209038. [DOI] [PubMed] [Google Scholar]
- Baillat G, Moqrich A, Castets F, Baude A, Bailly Y, Benmerah A, Monneron A. Molecular cloning and characterization of phocein, a protein found from the Golgi complex to dendritic spines. Mol Biol Cell. 2001;12:663–673. doi: 10.1091/mbc.12.3.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belmeguenai A, Hansel C. A role for protein phosphatases 1, 2A, and 2B in cerebellar long-term potentiation. J Neurosci. 2005;25:10768–10772. doi: 10.1523/JNEUROSCI.2876-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benoist M, Gaillard S, Castets F. The striatin family: a new signaling platform in dendritic spines. J Physiol Paris. 2006;99:146–153. doi: 10.1016/j.jphysparis.2005.12.006. [DOI] [PubMed] [Google Scholar]
- Bosch M, Hayashi Y. Structural plasticity of dendritic spines. Curr Opin Neurobiol. 2012;22:383–388. doi: 10.1016/j.conb.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan SF, Sucher NJ. An NMDA receptor signaling complex with protein phosphatase 2A. J Neurosci. 2001;21:7985–7992. doi: 10.1523/JNEUROSCI.21-20-07985.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YK, Hsueh YP. Cortactin-binding protein 2 modulates the mobility of cortactin and regulates dendritic spine formation and maintenance. J Neurosci. 2012;32:1043–1055. doi: 10.1523/JNEUROSCI.4405-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung J, Petek E, Nakabayashi K, Tsui LC, Vincent JB, Scherer SW. Identification of the human cortactin-binding protein-2 gene from the autism candidate region at 7q31. Genomics. 2001;78:7–11. doi: 10.1006/geno.2001.6651. [DOI] [PubMed] [Google Scholar]
- Gaillard S, Bailly Y, Benoist M, Rakitina T, Kessler JP, Fronzaroli-Molinieres L, Dargent B, Castets F. Targeting of proteins of the striatin family to dendritic spines: role of the coiled-coil domain. Traffic. 2006;7:74–84. doi: 10.1111/j.1600-0854.2005.00363.x. [DOI] [PubMed] [Google Scholar]
- Gordon J, Hwang J, Carrier KJ, Jones CA, Kern QL, Moreno CS, Karas RH, Pallas DC. Protein phosphatase 2a (PP2A) binds within the oligomerization domain of striatin and regulates the phosphorylation and activation of the mammalian Ste20-Like kinase Mst3. BMC Biochem. 2011;12:54. doi: 10.1186/1471-2091-12-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goudreault M, et al. A PP2A phosphatase high density interaction network identifies a novel striatin-interacting phosphatase and kinase complex linked to the cerebral cavernous malformation 3 (CCM3) protein. Mol Cell Proteomics. 2009;8:157–171. doi: 10.1074/mcp.M800266-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris KM, Stevens JK. Dendritic spines of CA 1 pyramidal cells in the rat hippocampus: serial electron microscopy with reference to their biophysical characteristics. J Neurosci. 1989;9:2982–2997. doi: 10.1523/JNEUROSCI.09-08-02982.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hering H, Sheng M. Activity-dependent redistribution and essential role of cortactin in dendritic spine morphogenesis. J Neurosci. 2003;23:11759–11769. doi: 10.1523/JNEUROSCI.23-37-11759.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iossifov I, et al. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012;74:285–299. doi: 10.1016/j.neuron.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landis DM, Reese TS. Cytoplasmic organization in cerebellar dendritic spines. J Cell Biol. 1983;97:1169–1178. doi: 10.1083/jcb.97.4.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matus A, Ackermann M, Pehling G, Byers HR, Fujiwara K. High actin concentrations in brain dendritic spines and postsynaptic densities. Proc Natl Acad Sci USA. 1982;79:7590–7594. doi: 10.1073/pnas.79.23.7590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauna JC, Miyamae T, Pulli B, Thiels E. Protein phosphatases 1 and 2A are both required for long-term depression and associated dephosphorylation of cAMP response element binding protein in hippocampal area CA1 in vivo. Hippocampus. 2011;21:1093–1104. doi: 10.1002/hipo.20823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohoka Y, Takai Y. Isolation and characterization of cortactin isoforms and a novel cortactin-binding protein, CBP90. Genes Cells. 1998;3:603–612. doi: 10.1046/j.1365-2443.1998.00216.x. [DOI] [PubMed] [Google Scholar]
- Penzes P, Cahill ME. Deconstructing signal transduction pathways that regulate the actin cytoskeleton in dendritic spines. Cytoskeleton (Hoboken) 2012;69:426–441. doi: 10.1002/cm.21015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert V, Dotti CG. Transmitting on actin: synaptic control of dendritic architecture. J Cell Sci. 2007;120:205–212. doi: 10.1242/jcs.03337. [DOI] [PubMed] [Google Scholar]
- Tian Q, Wang J. Role of serine/threonine protein phosphatase in Alzheimer's disease. NeuroSignals. 2002;11:262–269. doi: 10.1159/000067425. [DOI] [PubMed] [Google Scholar]
- Walsh CA, Morrow EM, Rubenstein JL. Autism and brain development. Cell. 2008;135:396–400. doi: 10.1016/j.cell.2008.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Parsons JT. Cortactin, an 80/85-kilodalton pp60src substrate, is a filamentous actin-binding protein enriched in the cell cortex. J Cell Biol. 1993;120:1417–1426. doi: 10.1083/jcb.120.6.1417. [DOI] [PMC free article] [PubMed] [Google Scholar]