

Myosin- and Rab-interacting protein is not a classic receptor for MyoVa on large, dense-core secretory granules (SGs), but it aids in PKA-dependent phosphorylation of MyoVa-associated proteins on SGs in endocrine and neuroendocrine cells.
Abstract
Myosin- and Rab-interacting protein (MyRIP), which belongs to the protein kinase A (PKA)–anchoring family, is implicated in hormone secretion. However, its mechanism of action is not fully elucidated. Here we investigate the role of MyRIP in myosin Va (MyoVa)-dependent secretory granule (SG) transport and secretion in pancreatic beta cells. These cells solely express the brain isoform of MyoVa (BR-MyoVa), which is a key motor protein in SG transport. In vitro pull-down, coimmunoprecipitation, and colocalization studies revealed that MyRIP does not interact with BR-MyoVa in glucose-stimulated pancreatic beta cells, suggesting that, contrary to previous notions, MyRIP does not link this motor protein to SGs. Glucose-stimulated insulin secretion is augmented by incretin hormones, which increase cAMP levels and leads to MyRIP phosphorylation, its interaction with BR-MyoVa, and phosphorylation of the BR-MyoVa receptor rabphilin-3A (Rph-3A). Rph-3A phosphorylation on Ser-234 was inhibited by small interfering RNA knockdown of MyRIP, which also reduced cAMP-mediated hormone secretion. Demonstrating the importance of this phosphorylation, nonphosphorylatable and phosphomimic Rph-3A mutants significantly altered hormone release when PKA was activated. These data suggest that MyRIP only forms a functional protein complex with BR-MyoVa on SGs when cAMP is elevated and under this condition facilitates phosphorylation of SG-associated proteins, which in turn can enhance secretion.
INTRODUCTION
Insulin is stored in large, dense-core secretory granules (SGs) that are released upon nutrient stimulation from pancreatic beta cells. When blood glucose is elevated, insulin is secreted in a biphasic manner. The first, rapid phase is due to the ATP-sensitive K+ (KATP+) channel–dependent (triggering) pathway, which increases [Ca2+]i. This is followed by a sustained second phase of secretion, which includes the KATP+ channel–dependent pathway because of the need for elevated [Ca2+]i and additional signals from KATP+ channel–independent (amplifying) pathways. The latter include stimulation of adenylyl cyclase activity and activation of protein kinase A (PKA) by incretin hormones such as glucagon-like peptide-1 (GLP-1; Bratanova-Tochkova et al., 2002; Seino and Shibasaki, 2005; Doyle and Egan, 2007). SGs released during the first phase are likely to be situated at the plasma membrane (Olofsson et al., 2002), but they might also be recruited from relatively distant areas of beta cells (Kasai et al., 2008). During the second phase new SGs need to be recruited from the central regions to the periphery of the cell, which occurs by directed SG transport. Our studies showed that SGs are transported from the cell center to the cell cortex primarily by the microtubule-based motor protein conventional kinesin (Varadi et al., 2002, 2003). The short-range movements in the cortical regions of the cell that carry SGs over the last few hundred nanometers to the cell surface involve myosin Va (MyoVa; Varadi et al., 2005; Ivarsson et al., 2005). Inhibition of MyoVa function by the expression of the dominant-negative–acting MyoVa-tail domain or small interfering RNA (siRNA) knockdown led to a significant decrease in the number of docked SGs and to a reduction in insulin release (Varadi et al., 2005).
Our recent work revealed that the SG-docking proteins granuphilin-a/b (Gran-a/b) and rabphilin-3A (Rph-3A) link the neuronal/brain isoform of MyoVa (BR-MyoVa) to SGs (Brozzi et al., 2012). These findings were rather unexpected because this was the first time that direct interaction between these C2-domain–containing proteins and MyoVa was demonstrated. Previous studies of the melanocyte isoform of MyoVa (MC-MyoVa) receptor melanophilin (Mlph; Fukuda and Kuroda, 2002) on melanosomes raised the possibility that in endocrine and neuroendocrine cells the complementary myosin- and Rab-interacting protein (MyRIP)/Slac2-c fulfils the same role. Although it is clear from previous studies that MyRIP is important for beta-cell function, its mechanism of action is controversial.
Knockdown of MyRIP protein level by siRNA significantly reduces stimulated hormone release (Waselle et al., 2003; Goehring et al., 2007). Furthermore, MyRIP partially localizes to insulin-containing SGs (Waselle et al., 2003; Goehring et al., 2007). It is not clear, however, whether MyRIP is able to interact with BR-MyoVa under physiological conditions. The majority of in vitro and in vivo studies investigated the MC-MyoVa isoform interaction with MyRIP, and very little is known about the interactions and role of BR-MyoVa, which is expressed in cells with key physiological functions (e.g., endocrine and neuroendocrine cells and neurons). These MyoVa variants contain different alternative exons in their medial tail domain (Seperack et al., 1995). MC-MyoVa contains exons D and F, both of which are important for cargo binding in melanocytes (Au and Huang, 2002). In contrast, the BR-MyoVa contains exon B but lacks both exons D and F (Seperack et al., 1995). In vitro binding assays revealed that BR-MyoVa interaction with MyRIP is negligible compared with that of MC-MyoVa (Fukuda and Kuroda, 2002), demonstrating that alternative splicing of MyoVa influences its ability to interact with MyRIP. One study showed that the affinity of MyRIP binding even to the MC-MyoVa variant is very low in vitro (∼15-fold less than the Mlph–MC-MyoVa interaction), and overexpression of MyRIP could not rescue melanosome transport in Mlph-knockout cells (Kuroda and Fukuda, 2005a). This study suggested that it is highly unlikely that interaction could occur between BR-MyoVa and MyRIP under physiological conditions unless MyRIP is either posttranslationally modified or forms a complex with other proteins to enhance its interaction with BR-MyoVa. In support of this finding, overexpression of the MyoVa-binding domain of MyRIP did not affect SG localization and hormone secretion from pancreatic beta cells (Waselle et al., 2003). Furthermore, overexpression of MyRIP containing a mutation A748P that has reduced binding activity to the MC-MyoVa tail domain did not change global insulin vesicle distribution (Mizuno et al., 2011). Moreover, in PC12 cells the control of SG exocytosis by MyRIP depends on its actin- and Rab27A-binding ability but is independent of interaction with MyoVa (Desnos et al., 2003). Our recent study of pancreatic beta cells also suggested that MyRIP is unlikely to behave as a classic receptor for MyoVa on SGs (Brozzi et al., 2012).
In striking contrast, another study found that the affinity of MyRIP to MC-MyoVa is similar to that of Mlph and that overexpression of MyRIP is able to restore melanosome transport in Mlph-knockout cells (Ramalho et al., 2009). It suggested that MyRIP and MyoVa might interact in cells in which both proteins are endogenously coexpressed (e.g., insulin-secreting beta cells and PC12 cells). In support of this study, an antibody that prevents the interaction of MyoVa with MyRIP has been shown to reduce the recruitment of insulin granules for release (Ivarsson et al., 2005). The main reason for these confounding results is that the functionally different MC-MyoVa and BR-MyoVa variants were used interchangeably. To better understand the role of MyRIP in MyoVa transport in endocrine, neuroendocrine cells, and neurons, it is essential to investigate endogenous BR-MyoVa and MyRIP in a cell type that solely expresses this MyoVa splice variant.
In addition to its potential MyoVa receptor function, MyRIP has been shown to operate as a PKA-anchoring protein (AKAP; Goehring et al., 2007). MyRIP colocalizes with the PKA regulatory domain (PKAR) and forms complexes with both regulatory (PKAR) and catalytic subunits (PKAC), and PKA activity is enriched in the MyRIP complexes in pancreatic beta cells (Goehring et al., 2007). Furthermore, MyRIP interacts with the exocyst complex, an evolutionarily conserved protein complex that controls protein trafficking and exocytosis (Novick et al., 1980; Munson and Novick, 2006; Bajjalieh, 2004; Tsuboi et al., 2005). Thus, MyRIP may mediate correct positioning of PKA at sites of exocytosis to synchronize SG transport in response to second-messenger and other cellular signals. However, no targets for MyRIP-facilitated PKA phosphorylation have been identified to date.
The following questions regarding MyRIP functions are unresolved: Does MyRIP functionally couple to BR-MyoVa? Are Rab27A–MyRIP–MyoVa and the MyRIP–PKA part of the same complex? If so, is there a target for PKA on this complex? The present study addresses these questions.
RESULTS
The neuronal/brain form of MyoVa is expressed in pancreatic beta cells
The schematic working model of the MyoVa–SG complex and the alternative exons of the MyoVa medial tail domain are shown in Figure 1, A and B. Both MIN6 insulinoma cells and primary rat islets of Langerhans express a single MyoVa splice variant, BR-MyoVa, which contains exon B but not exon D or F (Figure 1, B and C). In contrast, the neuroendocrine PC12 cells coexpress several MyoVa splice variants (Figure 1C), including the melanocyte-spliced form that contains exons D and F (MC-MyoVa).
FIGURE 1:
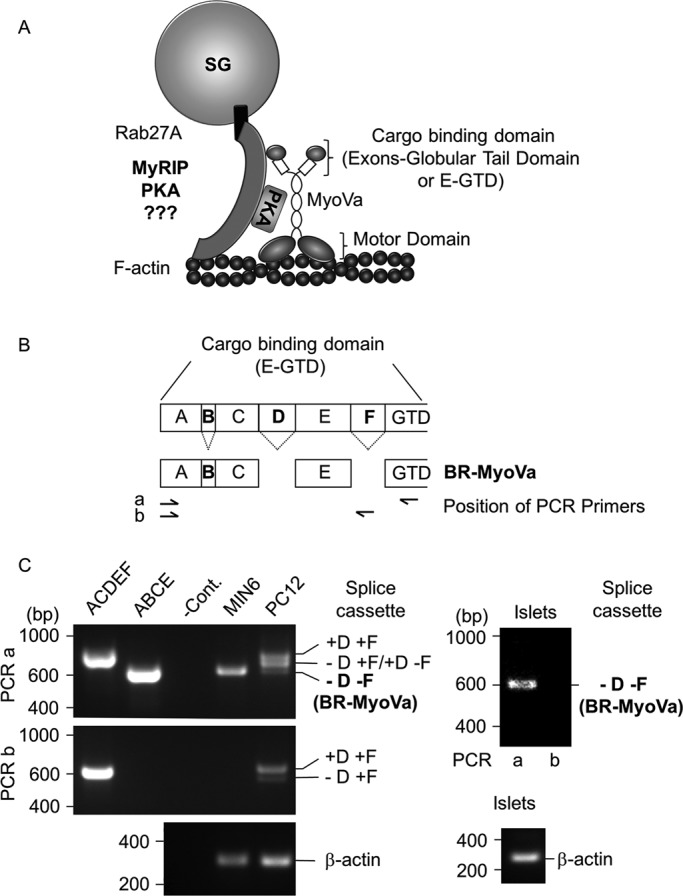
Schematic model of the MyoVa–SG complex and MyoVa expression in pancreatic beta cells. (A) The alternative exons ABCE and the globular tail domain (E-GTD) of MyoVa are involved in the interaction with SGs. Rab27 is an essential component of a protein complex that serves as the SG receptor for MyoVa. This complex is likely to include a Rab effector protein(s) such as MyRIP in pancreatic beta cells. Note that MyRIP is an AKAP (Goehring et al., 2007), hence the presence of PKA in the model. (B) Schematic representation of the different alternatively spliced forms of MyoVa and location of the primers used for PCR. The capital letters represent exons as described previously (Seperack et al., 1995; Brozzi et al., 2012). (C) Amplification of different alternative spliced forms of MyoVa by RT-PCR. Two sets of PCR primers were used: PCR a and b (positions are shown on B). Agarose gels show the MyoVa and β-actin amplification products from MIN6 beta cells, PC12 cells (left) and primary rat islets of Langerhans (right). Plasmids encoding exons ACDEF or ABCE were used as controls (left). PCR amplification in the absence of DNA template was used as negative control (–Cont.). Data are representative of three independent experiments.
MyRIP and BR-MyoVa are not part of the same protein complex, and they do not colocalize in beta cells at stimulatory glucose concentrations
To identify whether MyRIP interacts with BR-MyoVa in glucose-stimulated MIN6 cells (16.7 mM glucose), we generated glutathione S-transferase (GST)–tagged constructs encoding the most likely site for cargo interaction: exons ABCE with globular tail domain (E-GTD) and globular tail domain (GTD; Figures 1, A and B, and 2A), which were used for GST pull-down assays (Figure 2B). Although MyRIP and Rab3 are clearly detectable in the input (Figure 2B, lane 1), they never appeared in pull-down samples (Figure 2B, lanes 2–4). In contrast, under the same conditions the known interaction partner Rab27 (Brozzi et al., 2012) was equally detectable in the pull-down fractions (Figure 2B, lanes 2 and 3), which can link BR-MyoVa to SG via Gran-a, Gan-b, or Rph-3A (Brozzi et al., 2012).
FIGURE 2:

MyRIP and BR-MyoVa are present on insulin-containing vesicles, but they show limited colocalization, and they do not interact in glucose-stimulated MIN6 beta cells. (A) Schematic representation of the MyoVa constructs used for GST pull-down assays. GST–E-GTD contains exons ABCE expressed in pancreatic beta cells (Figure 1C, BR-MyoVa) and the globular tail domain. GST-GTD contains the globular tail domain alone. (B) Proteins eluted from beads coated with GST-GTD (lanes 2 and 5), GST–E-GTD (lanes 3 and 6), and GST (lanes 4 and 7) in the presence (lanes 2–4) or absence (lanes 5–7) of solubilized MIN6 cell homogenates (cells were stimulated with 16.7 mM glucose) were analyzed by 10% SDS–PAGE, followed by immunoblotting with anti-MyRIP (1:1000 dilution), anti-Rab27 (1:2000 dilution), and anti-Rab3 (1:1000 dilution) antibodies. Input represents 1/25 of the reaction mixture used for pull-down. The positions of the molecular mass markers are shown on the left. Data are representative of four independent experiments. (C) Glucose-stimulated MIN6 cells were permeabilized with digitonin before fixation (Varadi et al., 2006) and coimmunostained with a rabbit polyclonal anti-MyoVa (1:200 dilution) and a goat polyclonal anti-MyRIP (1:250 dilution) primary antibody and then visualized with an Alexa Fluor goat anti-rabbit 568 and an Alexa Fluor donkey anti-goat 568. Enlarged regions are shown on the fourth image, and arrows indicate noncolocalized regions on individual vesicles. Pearson's r as a measure for colocalization of MyoVa and MyRIP doubly labeled structures (200–500 nm), which corresponded closely with SGs, was 0.12 ± 0.11 in 256 cells (which indicates little or no association) in three independent experiments. Scale bars, 5 and 2 μm. (D) MyoVa (6-nm gold particles) and MyRIP (10-nm gold particles) were immunolocalized on cryosections of MIN6 cells. cyt., cytosol; PM, plasma membrane; SG, insulin-containing secretory granules. White and black arrowheads indicate MyoVa and MyRIP, respectively. Scale bar, 50 nm.
To test whether the lack of colocalization can be observed in situ in glucose-stimulated beta cells, we costained MIN6 cells for MyoVa and MyRIP. As demonstrated previously, both MyoVa (Varadi et al., 2005; Brozzi et al., 2012) and MyRIP (Waselle et al., 2003) partially colocalize with insulin, but staining is also observed in the cytosol. In contrast, MyRIP and MyoVa showed very little colocalization on insulin granules (Figure 2, C and D). Immunogold electron microscopy revealed that only 7% of SGs were colabeled with both antibodies (250 SGs on eight different sections from four different blocks). However, when both MyoVa and MyRIP localized onto the same SGs, the distance between the gold particles was >30 nm indicating that they are not part of the same protein complex even on these SGs (van Weering et al., 2010; Brozzi et al., 2012). Occasionally the gold particles were found within 10–30 nm in the cytosol (14 such clusters were observed on eight sections). These results indicate that the majority of MyoVa is not linked to insulin vesicles via MyRIP.
As an alternative approach and also to test whether the lack of interaction seen in GST pull down is not the result of using a truncated form of MyoVa, we performed immunoprecipitations (IPs) of native endogenous proteins. Whereas MyoVa could specifically be immunoprecipitated with the anti-MyoVa antibody (Figure 3A, Spec. IgG, first row) MyRIP was not detectable in the same sample (Figure 3A, Spec. IgG, second row). In contrast, kinesin, which is known to be in the same complex as MyoVa (Huang et al., 1999), was clearly detectable (Figure 3A, Spec. IgG, third row). PC12 cell homogenate was used as a positive control, which is known to contain several splice variants of MyoVa, including the MC-MyoVa (Figure 1C), and it has been shown that MyRIP can interact with this MyoVa variant (Fukuda and Kuroda, 2002; Desnos et al., 2003). In these cells MyRIP and MyoVa are part of the same complex because MyRIP was clearly visible in the samples immunoprecipitated with the anti-MyoVa antibody (Figure 3A, Spec. IgG, bottom row) but was absent in the IP samples obtained using preimmune immunoglobulins (Figure 3A, Cont. IgG, bottom row). In agreement with the result of the MyoVa IP in MIN6 cells, MyoVa was not present in the MyRIP immunocomplexes (Figure 3B, second row). As another control, IP was performed with the anti-Rab27 antibody. Rab27A is known to be in the same protein complex as MyRIP, MyoVa, or β-actin (Desnos et al., 2003; Brozzi et al., 2012), all of which could clearly be detected in samples immunoprecipitated with the Rab27-specific antibody but was absent in controls (Figure 3B). Note that Rab27A could not be detected in the MyRIP or MyoVa immunoprecipitates due to the presence of a nonspecific band (∼27 kDa) corresponding to immunoglobulin G (IgG) light chain. Cytochrome c was used as a negative control for IP (Figure 3, A and B). These data demonstrate that endogenous BR-MyoVa and MyRIP are not part of the same protein complexes, and they are differentially distributed in glucose-stimulated beta cells. Rab27A binds directly to MyRIP and via another Rab27A effector protein such as Gran-a, Gran-b, or Rph-3A to MyoVa. Our data also showed that the other splice variants of MyoVa that contain exon F or D or both can interact with MyRIP in PC12 cells.
FIGURE 3:

MyRIP and BR-MyoVa become part of the same protein complex only when insulin secretion is stimulated via activation of PKA but not glucose alone. (A) MIN6 or PC12 cell lysates were immunoprecipitated with anti–MyoVa polyclonal antibody (Spec. IgG) or rabbit IgG (Cont. IgG). Copurification of BR-MyoVa and MyRIP, kinesin 1, or cytochrome c (Cyt c) was assessed by immunoblotting using specific antibodies as indicated (anti-MyRIP, 1:1000 dilution; anti–kinesin 1, 1:2000 dilution; anti–Cyt c, 1:500 dilution). (B) Immunoprecipitation was performed with anti-MyRIP polyclonal antibody, and copurification of MyRIP and BR-MyoVa or Cyt c was assessed by immunoblotting using specific antibodies as indicated. Coimmunoprecipitation was performed as described for A but using a rabbit anti-Rab27a polyclonal antibody. For immunoblotting Rab27a was used at 1:2000 dilution and β-actin at 1:2000 dilution. (C) MIN6 cells were cultured at 3 mM glucose overnight and incubated in buffer containing 3 mM glucose for a further 1 h and then with 3 mM glucose or 16.7 mM glucose, 10 μM forskolin, and 1 mM IBMX for 60 min. Cell lysates were immunoprecipitated with anti-MyRIP polyclonal antibody (Spec. IgG) or goat IgG (Cont. IgG) and analyzed by immunoblot for the presence of MyRIP (anti-MyRIP, 1:1000 dilution). Coprecipitation of MyRIP and MyoVa with Rph-3A was assessed by immunoblot using specific antibodies as indicated (anti-MyoVa, 1:1000 dilution; anti–Rph-3A, 1:500 dilution). IP was also carried out with the anti-MyoVa antibody (bottom two rows). Data are representative of four independent experiments. Note that films for IP samples (Cont. IgG and Spec. IgG; shown on A–C) were exposed simultaneously, whereas longer exposure of the same blot was used to detect proteins in the homogenates. (D) MyoVa (6-nm gold particles) and MyRIP (10-nm gold particles) were immunolocalized on cryosections of MIN6 cells stimulated with IBMX and forskolin as for C. cyt., cytosol; SG, insulin-containing secretory granules. White and black arrowheads indicate MyoVa and MyRIP, respectively. Scale bar, 50 nm.
MyRIP and BR-MyoVa become part of the same complex when the cAMP-PKA pathway is activated
Because MyRIP functions as scaffolding protein for PKA in beta cells (Goehring et al., 2007), we investigated whether activation of cAMP-PKA would induce the formation of a complex between MyRIP and MyoVa. MIN6 cells were treated for 60 min with 1 mM 3-isobutyl-1-methylxanthine (IBMX; inhibitor of phosphodieseterases) and 10 μM forskolin (activator of adenylyl cyclases), which increase cellular cAMP in a concentration-dependent manner (Yajima et al., 1999; Chan et al., 2001). We performed immunoprecipitation of solubilized proteins using an anti-MyRIP antibody, which specifically precipitated endogenous MyRIP from both unstimulated (3 mM glucose) and stimulated (16.7 mM glucose) MIN6 cells (Figure 3C, first row). Similar to the result presented on Figure 3B, MyoVa was not part of the MyRIP immunocomplex in unstimulated or glucose-stimulated cells. However, MyoVa became part of the MyRIP complex after an increase in cAMP (Figure 3C, second row). Our recent study (Brozzi et al., 2012) demonstrated that the SG-associated protein Rph-3A directly interacts with MyoVa and colocalizes with MyoVa exclusively on SGs (Brozzi et al., 2012). We investigated whether MyRIP interacts with MyoVa alone (i.e., cytosolic MyoVa) or with the MyoVa-SG complex. Similar to MyoVa, Rph-3A was hardly detectable in the IP samples at nonstimulatory condition, but it was clearly present after activation of PKA (Figure 3C, third row). In agreement with the results discussed earlier, MyRIP was only detectable in immunoprecipitated samples obtained with the anti-MyoVa antibody when cAMP level was increased (Figure 3C, bottom two rows). To confirm our biochemical data, we performed immunogold electron microscopy, which revealed that MyRIP and MyoVa are colocalized on SGs more frequently after activation of the cAMP pathway than after stimulation with glucose alone (Figure 2D). Thirty-eight of SGs showed double labeling (238 SGs on 12 different sections from four different blocks). In most cases MyoVa and MyRIP labeling was observed on the SG periphery at a distance from 10 to 30 nm apart (84% of double-labeled SGs; Figure 3D). These data suggest that MyRIP forms a complex with BR-MyoVa on SGs when cAMP is elevated.
Rph-3A is expressed in pancreatic beta cells
Given that Rph-3A is a receptor for MyoVa on SGs, we investigated its expression in primary beta cells. In contrast to previous reports showing that Rph-3A expression was undetectable in INS-1 cells and primary islets of Langerhans (Inagaki et al., 1994; Regazzi et al., 1996), we found by reverse transcription (RT)-PCR that both MIN6 and INS-1 beta-cell lines and primary rat and mouse islets of Langerhans express Rph-3A mRNA (Figure 4A). This finding is in agreement with the transcriptional profiling of Rph-3A in fluorescence-activated cell sorting–purified mouse beta cells and mouse islets of Langerhans available in the EMBL-EBI database (www.ebi.ac.uk/gxa/experiment/E-TABM-877/ENSMUSG00000029608). RNA from spleen was used as a negative control in which Rph-3A was not detectable, whereas the PCR product for β-tubulin was clearly visible (Figure 4A). The corresponding proteins were also clearly detectable in MIN6 and INS-1 cells (Figure 4B). A high amount of Rph-3A protein is found in the brain (Inagaki et al., 1994), and thus we used a protein preparation from rat brain as a positive control on immunoblots. Proteins prepared from 200 rat islets of Langerhans were used for immunoblots, but the presence of Rph-3A was not clear in these samples, probably due to the small amount of total protein in the sample (compared with the cultured beta cells, for which 50 μg of protein was loaded onto the gel) and also to the relatively low amount of Rph-3A mRNA in primary rat and mouse beta cell samples (Figure 4A).
FIGURE 4:

The insulin vesicle–associated protein Rph-3A is expressed in primary rodent and human beta cells. (A) The agarose gel shows the mRNA expression of Rph-3A in MIN6 and INS-1 cells, in rat spleen, rat, and mouse islets of Langerhans. (B) Immunoblot shows Rph-3A protein in samples prepared from rat brain, INS1, C6, MIN6, and COS-7 cells. (C) Distribution of Rph-3A proteins was analyzed using immunocytochemistry in isolated primary rat beta cells. Cells were fixed, permeabilized, and coimmunostained with a guinea pig polyclonal anti-insulin (a, 1:250 dilution) and a rabbit polyclonal anti-Rph-3A (b, 1:10 dilution) primary antibody and then visualized with an Alexa Fluor goat anti-guinea pig 647 and an Alexa Fluor goat anti-rabbit 488 secondary antibody. Overlay of a and b is shown in image c. Scale bar; 5 μm; on the enlarged images, 2.5 μm. Pearson's r as a measure for colocalization of Rph-3A and insulin doubly labeled structures was 0.78 ± 0.11 in 21 cells (which indicates significant association) in three independent experiments. (D) Human pancreatic sections were immunostained using the same primary antibodies that are described in C. Scale bar, 10 μm. Data are representative of three independent experiments. Exoc. cells, exocrine cells.
In immunocytochemistry of isolated primary rat beta cells the Rph-3A antibody specifically labeled vesicle-like structures, which showed good colocalization with insulin (Figure 4C). The same staining pattern was observed when the Rph-3A antibody was used alone (unpublished data). To test that the Rph-3A staining in primary beta cells is specific, we labeled human pancreatic sections for Rph-3A or Rph-3A and insulin. Similar to the staining shown in the human protein atlas database (www.proteinatlas.org/ENSG00000089169/normal/pancreas), the insulin-containing cells were specifically labeled, whereas no staining was observed in the surrounding exocrine cells (Figure 4D). The same staining pattern was observed when the Rph-3A antibody was used alone (unpublished data). Note that isolated human islets were not available for RNA or protein extraction. These data suggest that both cultured and primary beta cells express Rph-3A, but its amount is somewhat lower in primary cells.
Rph-3A becomes phosphorylated after elevation of cAMP, which depends on the presence of MyRIP
Rph-3A is phosphorylated in vitro and in vivo in neurons by PKA, and the physiological phosphorylation site is the Ser-234 (S234) residue (Fykse et al., 1995; Lonart and Südhof, 1998, 2001). We next investigated whether Rph-3A is phosphorylated on Ser-234 in pancreatic beta -cells using a previously well-characterized antibody raised against a 12–amino acid Rph-3A synthetic peptide containing a central phosphorylated serine residue (Ser-234; Lonart and Südhof, 1998, 2001; Foletti et al., 2001). There is moderate phosphorylation in the presence of 3 or 16.7 mM glucose (Figure 5, A, lanes 1 and 2, and B). In contrast, elevation of cAMP by IBMX and forskolin significantly increased the amount of P-Rph-3A(S234) (Figure 5, A, lanes 3 and 4, and B). To test whether this phosphorylation of Rph-3A could occur under physiological conditions, we treated cells with the cAMP-elevating agent exendin-4, which displays biological properties similar to those of the naturally occurring incretin hormone GLP-1 (Göke et al., 1993). Exendin-4 also induced phosphorylation of Rph-3A, which was significantly higher than glucose alone (Figure 5, A, lane 5 vs. lane 2, and B). We observed 3.5-fold increase in Rph-3A phosphorylation, which, based on a previous study (Foletti et al., 2001), likely represents ∼30–40% phosphorylation of the total Rph-3A. We next investigated the cellular localization of Rph-3A and P-Rph-3A. In agreement with our immuno–electron microscopy studies (Brozzi et al., 2012), both Rph-3A and P-Rph-3A were found exclusively in the SG-enriched fraction and were not detectable in the supernatant that contains the cytosol (Figure 5C). In contrast, MyoVa was detectable in both pellet and supernatant. The distribution of Rph-3A did not change after PKA activation, suggesting that the phosphorylation of Rph-3A takes place when it is associated with SGs.
FIGURE 5:

Rph-3A is phosphorylated on insulin vesicles and is dependent on the AKAP function of MyRIP. (A) Cells were incubated as described in Figure 3C. Changes in the PKA-dependent phosphorylation of Rph-3A on Ser-234 (S234) were detected by immunoblotting using a rabbit polyclonal anti-phosphor S234-Rph-3A antibody (1:500 dilution). To detect total Rph-3A, the same blots were reprobed with a mouse monoclonal anti–Rph-3A antibody (1:500 dilution). (B) Quantification of Rph-3A phosphorylation under the conditions described in A. Density of P-Rph-3A was normalized to total Rph-3A. Data are means ± SD Three independent experiments were carried out with three parallel samples for each condition (n = 9). (C) MIN6 cells were incubated as described in Figure 3C. SG-enriched pellet (Pellet) and supernatant (Sup.) were obtained after the centrifugation of the postnuclear supernatant (Hom.) at 10,000 × g for 20 min. (D) Silencing of MyRIP was carried out as described in Materials and Methods, and the amount of MyRIP was determined by immunoblotting using anti MyRIP antibody (1:1000 dilution, top lane). Phosphorylation of Rph-3A on S234 after MyRIP knockdown was detected on immunoblots using a rabbit polyclonal anti–P-Rph-3A antibody. Total Rph-3A and β-tubulin was detected in the same samples. (E) Relative amount of MyRIP normalized to α-tubulin. Data are means ± SD. Three independent experiments were carried out with four parallel samples for each condition (n = 48). (F) Relative density of P-Rph-3A normalized to total Rph-3A. Three independent experiments were carried out with four parallel samples for each condition (n = 12). (G) Insulin secretion from control (black bars) and MyRIP-silenced (white bars) MIN6 cells were measured. After siRNA or scrambled RNA transfection, cells were kept in culture for 3 d (see Materials and Methods) and then incubated overnight at 3 mM glucose. On the following day cells were equilibrated at 3 mM glucose-containing buffer for 1 h and then incubated at 3 mM glucose, 16.7 mM glucose, or in the presence of stimulatory concentration of glucose, IBMX, and forskolin (16.7 mM Glu, 1 mM IBMX, 10 μM forskolin) for 60 min. The total amount of insulin in the cells and the fraction released during the incubation was determined by enzyme-linked immunosorbent assay. Data are means ± SD. Three independent experiments were performed with four parallel samples for each condition (n = 12).
Rph-3A is part of the MyRIP–MyoVa–SG complex (Figure 3C) and is phosphorylated by PKA (Figure 5, A and B). This raises the possibility that the localized phosphorylation of Rph-3A might be mediated by the AKAP function of MyRIP. We therefore decided to reduce MyRIP protein level with siRNA (Figure 5, D, even lanes, and E and F) and investigated Rph-3A phosphorylation on Ser-234 using the same experimental protocol as shown in Figure 5A. Clear phosphorylation of Rph-3A on Ser-234 was observed in controls (Figure 5D, odd lanes), which was significantly reduced in the MyRIP siRNA samples (Figure 5, D, even lanes, and F). In contrast, the amounts of total Rph-3A and β-tubulin were unchanged. As demonstrated previously (Goehring et al., 2007), the MyRIP siRNA significantly reduced high-glucose-, forskolin-, and IBMX-induced insulin release (Figure 5G). However. it had no significant effect on glucose-stimulated hormone release (Figure 5G). It was not possible to specifically block MyRIP and PKA interaction with the Ht31 peptide (commonly used to block interaction between the AKAPs and PKA) because MyRIP is an atypical AKAP that contains two PKA-anchoring domains (Goehring et al., 2007). Furthermore, rescue experiments using siRNA-resistant forms of MyRIP that contain or lack the PKA-binding domain were inconclusive in beta cells (Goehring et al., 2007). Nevertheless, these data demonstrate that MyRIP is important for cAMP-regulated insulin secretion and aids phosphorylation of the SG-associated protein Rph-3A.
Rph-3A phosphorylation on Ser-234 is important for its interaction with 14-3-3 and for stimulated hormone secretion
It has been shown that P-Rph-3A interacts in vitro with 14-3-3 protein (Sun et al., 2003), which regulates the subcellular localization of its interaction partners. It is important for normal synaptic transmission (Broadie et al., 1997) and stimulation of catecholamine secretion in chromaffin cells (Morgan and Burgoyne, 1992). We investigated whether the same interaction can take place in MIN6 beta cells. Cells were incubated and IP was carried out as described for Figure 3C using an anti-14-3-3 antibody. This antibody specifically immunoprecipitated the 14-3-3 protein (Figure 6A, Spec. IgG), which was absent in the control samples (Figure 6A, Cont. IgG). Rph-3A was already detectable in the IP samples obtained from unstimulated cells using the anti-14-3-3 antibody (Figure 6A, second row, Spec. IgG). However, there was a significant 2.6-fold increase (p < 0.05) in Rph-3A in the immunoprecipitates after IBMX and forskolin treatment. As shown (Figure 5, A and B), under these conditions Rph-3A becomes phosphorylated (Figure 5A, third and fourth rows). These data suggest that the interaction between Rph-3A and 14-3-3 is increased after PKA-dependent phosphorylation of Rph-3A.
FIGURE 6:

Rph-3A phosphorylation at S234 is important in its interaction with 14-4-3 and cAMP-dependent secretion. (A) MIN6 cell lysates were immunoprecipitated with anti–14-3-3 polyclonal antibody (Spec. IgG) or rabbit IgG (Cont. IgG) and analyzed by immunoblotting for the presence of 14-3-3. Copurification of Rph-3A was assessed by immunoblotting using a specific antibody as indicated (top two rows). IP was carried out with the P-Rph-3A–specific antibody as described for 14-3-3. Note that films for IP samples (Cont. IgG and Spec. IgG) were exposed simultaneously; for Hom. longer exposure of the same blot was used. (B) MIN6 cells were transiently cotransfected with a plasmid encoding hGH and either empty vector (pcDNA3, Cont., black bars) or plasmids encoding full-length, wild type Rph-3A (WT, white bars) or phosphorylation mutant Rph-3A (S234A, gray bars) as indicated. After 3 d in culture, cells were incubated for 60 min under basal conditions (3 mM Glu) or in the presence of stimulatory concentration of glucose, IBMX, and forskolin (16.7 mM Glu, 1 mM IBMX, 10 μM forskolin). The total amount of hGH expressed in the cells and the fraction released during the incubation was determined by enzyme-linked immunosorbent assay. Data are means ± SD (n = 9). Three independent experiments were performed with three parallel samples for each condition. (C) Cells were incubated as described in B, but the transfection was carried out using the WT (white bars) and phosphomimic Rph-3A (S234D, black bars, or S234E, gray bars). Four independent experiments were performed with three parallel samples for each condition. Data are means ± SD (n = 12). The transfection efficiencies were very similar for all constructs, and the total hGH content was not significantly different between these samples.
We next investigated the functional importance of Rph-3A phosphorylation in stimulated hormone release. First, Ser-234–Rph-3A was replaced by Ala (S234A–Rph-3A) using a standard site-directed mutagenesis approach. Hormone release was measured from empty vector, full-length Rph-3A wild-type (WT)–, or S234A–Rph-3A-mutant–expressing MIN6 cells cotransfected with plasmid encoding human growth hormone (hGH; Coppola et al., 2002; Varadi et al., 2002, 2005). Consistent with previous studies (Joberty et al., 1999; Brozzi et al., 2012), overexpression of Rph-3A WT enhanced stimulated secretion. In contrast, the S234A mutation nearly completely abolished this effect (Figure 6B). We also generated phosphomimic mutants by substituting serine with aspartate (S234D) or glutamate (S234E). Both of these mutants significantly enhanced hormone release induced by high glucose, forskolin, and IBMX compared with wild-type Rph-3A (Figure 6C). In contrast, they had no effect on glucose-stimulated hormone release (Figure 6C). These data suggest that Rph-3A phosphorylation on Ser-234 is important for cAMP-regulated hormone release.
MyRIP becomes phosphorylated after PKA activation
To find other potential PKA phosphorylation targets on the MyoVa–SG complex, we probed anti-MyRIP antibody–immunoprecipitated samples with an anti–phospho Ser/Thr PKA substrate antibody. Although numerous proteins become phosphorylated after PKA activation (Figure 7A, lane 2 vs. lane 1), one very dominant band of ∼98 kDa (Pho-98) appeared consistently in the IP samples (Figure 7A, lane 5 vs. lane 3). To test that the phosphorylation of this protein requires the presence of MyRIP, cells were incubated with MyRIP siRNA as described previously (Figure 5D) and then immunoprecipitated with the anti-MyRIP antibody. There was a 3.05 ± 0.07–fold (p < 0.003) reduction in the phosphorylation of this 98-kDa protein in the MyRIP siRNA–treated samples under stimulatory conditions (Figure 7B, lane 7 vs. lane 3). Of interest, this 98-kDa protein was recognized as MyRIP on the same blots (Figure 7B, right). An identical result was obtained when the total-cell homogenates of the MyRIP siRNA–treated cells were probed with the anti–phospho Ser/Thr PKA substrate antibody. Numerous phosphorylated bands appeared after IBMX and forskolin stimulation, as shown in Figure 7A (first two lanes). However, a clear 2.01 ± 0.013–fold (p < 0.0025) reduction in the phosphorylation of the 98-kDa protein was observed in the MyRIP siRNA–treated samples (Figure 7C, lane 6 vs. lane 4). This 98-kDa phosphorylated band was again recognized by the MyRIP antibody. Despite the correct molecular weight and the dependence on the AKAP function of MyRIP, there is a remote possibility that the identified 98-kDa phosphorylated protein may represent another, unidentified interaction partner. To eliminate this possibility, we used a pretreatment (Moult et al., 2006; Gladding et al., 2009) that completely dissociated MyRIP and its associated proteins before immunoprecipitation. This showed that the immunoprecipitated MyRIP remains phosphorylated under these conditions (Figure 7D), confirming that indeed MyRIP is the identified 98-kDa protein. Similar to Mlph, MyRIP is heavily phosphorylated on serine and threonine (Passeron et al., 2004), and 12 potential PKA phosphorylation sites (e.g., S296, S363, S470) have been identified by NetPhosK software. These data suggest that MyRIP is phosphorylated after PKA activation, which likely aids its interaction with BR-MyoVa.
FIGURE 7:

MyRIP becomes phosphorylated after activation of the cAMP pathway. (A) Solubilized MIN6 cell proteins obtained under the same conditions as described in Figure 3C were immunoprecipitated with the anti-MyRIP polyclonal antibody (Spec. IgG) or goat IgG (Cont. IgG). Copurification of phosphorylated proteins on Ser/Thr residues was assessed by immunoblotting using the anti–phospho Ser/Thr PKA substrate antibody. N/S, IgG-related nonspecific band; Pho-98, 98-kDa phosphorylated protein. Note that films for IP samples (Cont. IgG and Spec. IgG) were exposed simultaneously, but for Hom. longer exposure of the same blot was used. (B) Same immunoprecipitation as described in A of MyRIP siRNA–silenced MIN6 cell homogenates (same silencing as described in Figure 5D). Blots were probed with antibodies as indicated. (C) Phosphorylation of the 98-kDa protein in untreated (lanes 1 and 2), control siRNA–treated (lanes 3 and 4), or MyRIP siRNA–silenced (lanes 5 and 6) MIN6 cells. Proteins were separated on 10% SDS gels and run for several hours to obtain good separation of proteins, and immunoblots were probed with the anti–phospho-Ser/Thr PKA substrate or the anti-MyRIP antibody. (D) MyRIP and its associated proteins were solubilized and dissociated in a modified RIPA buffer at 100°C before MyRIP immunoprecipitation, which was carried out as described in Figure 3B. MyRIP phosphorylation on Ser/Thr residues was then assessed by immunoblotting using the anti–phospho Ser/Thr PKA substrate antibody. Three independent experiments were performed with three parallel samples for each conditions (n = 9).
DISCUSSION
MyRIP as a receptor for BR-MyoVa
Our study using in vitro and in vivo approaches showed that MyRIP does not interact or colocalize with the BR variant of MyoVa at basal or stimulatory glucose concentrations in beta cells (Figures 2 and 3). In contrast, it can interact with other variants of MyoVa (i.e., MC-MyoVa; Figure 1A) in unstimulated PC12 cells (Figure 3A), which has previously been demonstrated in this cell type (Desnos et al., 2003). Interaction between MC-MyoVa and MyRIP has been investigated extensively. An earlier study showed that the affinity of MyRIP to MC-MyoVa (Figure 1A) is very low, and it might occur only under physiological conditions, where the endogenous MC-MyoVa level is very high (Kuroda and Fukuda, 2005a). In contrast, another study showed that MyRIP affinity to MC-MyoVa is comparable to that of Mlph to MC-MyoVa (Ramalho et al., 2009), and the likely explanation for the difference between this and the previous studies (Kuroda and Fukuda, 2005a, b) is that different cell systems were used and different expression levels achieved. Interaction between BR-MyoVa and MyRIP was investigated previously using an in vitro binding assay with a very high amount of proteins (Kuroda and Fukuda, 2005a, b). In agreement with our results (Figures 2 and 3), these two proteins showed negligible interaction (Kuroda and Fukuda, 2005a, b), suggesting that interaction cannot occur between BR-MyoVa and MyRIP under physiological conditions unless MyRIP is either posttranslationally modified or forms a complex with other proteins to enhance its interaction with BR-MyoVa.
Induction of MyRIP and BR-MyoVa interaction
We next investigated whether interaction between MyRIP and BR-MyoVa can be induced under physiological conditions when hormone secretion is stimulated in pancreatic beta cells. It has been demonstrated that siRNA or short hairpin RNA knockdown of MyRIP reduced depolarization-stimulated (Ivarsson et al., 2005), KCl-stimulated (Mizuno et al., 2011), or high-glucose-, IBMX-, and forskolin-stimulated (Waselle et al., 2003; Goehring et al., 2007) hormone release from pancreatic beta cells. siRNA knockdown or expression of the dominant-negative-acting globular tail domain of BR-MyoVa reduced depolarization- and glucose-stimulated insulin secretion (Varadi et al., 2005). Therefore both MyRIP and BR-MyoVa result in similar effects on exocytosis. However, these two proteins are not part of the same protein complex in the presence of basal or stimulatory glucose concentrations (Figures 2 and 3), suggesting that their roles in SG transport and secretion under these conditions are independent of each other.
Although the main physiological trigger of insulin secretion is glucose, its effect is greatly augmented by incretin hormones (Doyle and Egan, 2007), which account for 60% of the insulin secretory response after oral glucose load (Nauck et al., 1986). The incretin hormones (glucose-dependent insulinotropic peptide and GLP-1) potentiate insulin secretion through cAMP signaling (Supplemental Figure S1). cAMP action in insulin secretion is mediated through PKA-dependent (Supplemental Figure S1) and PKA-independent mechanisms, the latter involving Epac2 (Renström et al., 1997; Eliasson et al., 2003; Seino and Shibasaki, 2005). There is evidence that MyRIP knockdown by siRNA reduces cAMP-regulated hormone release (Waselle et al., 2003; Goehring et al., 2007). Therefore we investigated MyRIP and BR-MyoVa interaction when cAMP was increased (Supplemental Figure S1). Under this condition MyRIP and BR-MyoVa became part of the same protein complex on SGs (Figure 3, C and D), which is likely to be facilitated by phosphorylation of MyRIP (Figure 7). In our recent study we demonstrated that Rph-3A links BR-MyoVa to SGs in endocrine and neuroendocrine cells (Brozzi et al., 2012), and, surprisingly, Rph-3A remains associated with MyoVa-SG even after activation of PKA (Figures 3C and 5C), suggesting that MyRIP does not replace Rph-3A in this complex. This suggests that MyRIP's role is most likely to target PKA to the MyoVa–SG complex.
Target of PKA-dependent phosphorylation on the BR-MyoVa–SG complex
In our previous study we identified Rph-3A as a link between Rab27 and MyoVa on SGs (Brozzi et al., 2012). Rph-3A is phosphorylated by Ca2+/CaM- and cAMP-dependent kinases in vitro (Fykse et al., 1995) and in mossy fiber nerve terminals of the hippocampus in vivo (Lonart and Südhof, 1998). Rph-3A is physiologically phosphorylated on Ser-234 mediated by PKA (Lonart and Südhof, 2001). This phosphorylation altered Rph-3A affinity to membranes (Foletti et al., 2001) and occurred in different regions of the hippocampus (Lonart and Südhof, 1998). The phosphorylated Rph-3A, similar to another Rab3a-binding protein, RIM, is able to interact with 14-3-3 protein, which functions through its ability to bind specific phosphoserine sites (Sun et al., 2003).
Similar to neurons, Rph-3A is phosphorylated on Ser-234 by PKA (Figure 5, A and C). This site is phosphorylated after IBMX and forskolin treatment and, more important, by the physiologically relevant exendin-4 (Figure 5A and Supplemental Figure S1). It has been estimated that 75–80% of Rph-3A is phosphorylated after high-K+ stimulation in rat brain slices (Foletti et al., 2001). High-K+ stimulation produced a approximately sixfold to eightfold increase in phosphorylation compared with unstimulated basal levels in brain slices (Foletti et al., 2001). Under these conditions not only PKA but also Ca2+-dependent kinases would be activated. The maximal increase in phosphorylation after PKA activation was 2.5- to 3.5-fold (Foletti et al., 2001), which is similar to our findings (Figure 5B). Therefore it is estimated that a maximum of ∼30–40% of Rph-3A becomes phosphorylated on SGs (Figure 5B). Phosphorylation of Rph-3A is dependent on the presence of MyRIP, because knockdown of MyRIP with an siRNA that reduces IBMX- and forskolin-induced insulin release from pancreatic beta cells (Figure 5G; Goehring et al., 2007) also reduced Rph-3A phosphorylation on Ser-234, whereas the amount of total Rph-3A was unchanged (Figure 5, D and F, and Supplemental Figure S1). Overexpression of the S234A-Rph-3A mutant failed to increase stimulated secretion (Figure 6B). In contrast, the phosphomimic mutants of Rph-3A at position Ser-234D/E significantly enhanced hormone release (Figure 6C), suggesting that Ser-234 phosphorylation is important for Rph-3A function in insulin secretion (Supplemental Figure S1). The Ser-234D/E mutants elevated hormone release only when cAMP was elevated (Figure 6C) and similarly to MyRIP (Figure 5G) had no effect on glucose-stimulated hormone release (Figure 6C). There is evidence that Rph-3A phosphorylation is influenced by its interaction with other proteins (i.e., Rab-3a) and by its correct localization at the site of exocytosis (Foletti et al., 2001). In agreement, we showed that MyRIP influences Rph-3A phosphorylation (Figure 5D) when MyRIP becomes part of the MyoVa–Rph-3A–vesicle complex after PKA activation (Figure 3, C and D). Under this condition MyRIP, Rph-3A, and other, yet-unidentified proteins on the MyoVa–SG complex become phosphorylated (Figure 7). It has also been demonstrated that MyRIP and MyoVa mediate the retention/correct localization of secretory granules at the cell periphery (Mizuno et al., 2011; Huet et al., 2012). Thus the correct assembly and localization of the MyRIP–MyoVa–Rph-3A–vesicle complex is necessary for hormone release, and mere phosphorylation of one of the components (i.e., Rph-3A) is not sufficient to enhance secretion in the absence of PKA.
MyRIP siRNA reduced cAMP-mediated hormone secretion but not glucose-stimulated secretion, suggesting that 1) MyRIP is crucial for cAMP-mediated hormone secretion, 2) Rph-3A phosphorylation can enhance hormone secretion but only when PKA is activated, and 3) the MyRIP–MyoVa complex is formed.
These findings suggest that MyRIP as an AKAP protein is involved in GLP-1–mediated signaling. Only a few AKAPs have been implicated in PKA activation in the context of GLP-1 signaling, such as AKAP18 (Fraser et al., 1998) and AKAP 79 (Nauert et al., 2003), and this is the first experimental evidence showing MyRIP's role as an AKAP in incretin-mediated signaling. Furthermore, this is the first study to identify a direct phosphorylation target for MyRIP-PKA. It is not clear whether Rph-3A is the only phosphorylation target on the MyoVa–SG or perhaps on the closely located soluble N-ethylmaleimide–sensitive factor attachment protein receptor complex. It is well known that Rph-3A interacts with SNAP-25 (Supplemental Figure S1), and thus it could be a potential target. SNAP-25 has multiple phosphorylation sites, and only the Ser-187 site, which is phosphorylated by PKC, has been investigated in primary islets (Gonelle-Gispert et al., 2002). The in vivo phosphorylation site for PKA on SNAP-25 is Thr-138 (Hepp et al., 2002), which regulates releasable vesicle pool sizes in chromaffin cells (Nagy et al., 2004), but its role is not known in beta cells. Most recently snapin, which can interact with SNAP-25, has been shown to be phosphorylated on Ser-50 by PKA after incretin-mediated signaling (Song et al., 2011). Future work will need to identify other targets for the GLP-1–MyRIP pathway. Given that cAMP-dependent phosphorylation increased the frequency of granule movement in beta cells (Yu et al., 2000), our data indicate that MyRIP functionally links insulin vesicle transport and GLP-1 signaling.
MATERIALS AND METHODS
Materials
The following antibodies were used: goat polyclonal anti-MyRIP and mouse monoclonal anti–kinesin 1 (Abcam, Cambridge, United Kingdom), rabbit polyclonal anti–myosin Va, rabbit anti–cytochrome c, anti–phospho-Ser/Thr PKA substrate, and rabbit polyclonal anti–protein kinase A catalytic subunit antibodies (Cell Signaling, Danvers, MA), rabbit polyclonal anti-Rab27, rabbit polyclonal anti-Rab3, and rabbit polyclonal anti–Rph-3A (Synaptic Systems, Göttingen, Germany), mouse monoclonal anti–β-actin (Sigma, Poole, United Kingdom), mouse monoclonal anti–rabphilin-3A (BD Transduction Laboratories, Swindon, United Kingdom), and (phosphor S234) rabbit polyclonal Rph-3A antibodies (Abnova, Heidelberg, Germany). The horseradish peroxidase–coupled secondary antibodies, glutathione Sepharose, and benzamidine Sepharose beads were obtained from GE HealthCare (Chalfont St Giles, United Kingdom). The fluorophore-coupled secondary antibodies were obtained from Invitrogen (Paisley, United Kingdom). Donkey anti–rabbit or anti–mouse immunoglobulins G (IgGs) conjugated to 6- or 15-nm gold particles and acetylated bovine serum albumin were from Aurion (Wageningen, Netherlands). Nonidet P-40 and protease inhibitor cocktail tablets were obtained from Roche Diagnostics (Burgess Hill, United Kingdom). IPTG, protein G Sepharose beads, Coomassie brilliant blue colloidal, and collagenase were obtained from Sigma (Poole, United Kingdom). MyRIP siGENOME SMART pool siRNA was purchased from Dharmacon Thermo Scientific (Fisher Scientific UK, United Kingdom). QuikChange II Site-Directed Mutagenesis Kit was from Agilent Technologies UK (Stockport, United Kingdom). Human islets of Langerhans sections were a gift from K. Gillespie (Diabetes and Metabolism Unit, University of Bristol, Bristol, United Kingdom).
Cell culture and transient transfection
MIN6 cells (passages 29–35) were cultured as described previously (Varadi et al., 2006). Cells were grown at 37°C in 5% CO2 and humidified atmosphere and at 50% confluency were transfected with the indicated DNA plasmids using Lipofectamine (Invitrogen) according to manufacturer's instructions.
Isolation of rodent islets of Langerhans, RNA preparation, and RT-PCR
Isolation of rat and mouse islets was performed as previously described (Varadi et al., 2005). The study was conducted in accordance with the Principles of Laboratory Care. Total RNA was isolated using TRI Reagent (Sigma; Varadi et al., 1996). Reverse transcription and PCR amplification using GoTaq polymerase were performed according to manufacturer's recommendations (Promega, Southampton, United Kingdom). Primers flanking the MyoVa tail domain corresponding to nucleotides 3660–3678 (forward), 4183–4208 (reverse), and 4352–5372 (reverse) of the sequence (NM_010864) were used. The sequences were verified by sequencing. The GST-tagged myosin Va constructs were generated and pull-down assays carried out as described previously (Brozzi et al., 2012).
Coimmunoprecipitation
MIN6 cell homogenates were prepared as described earlier for GST pull-down experiments (Brozzi et al., 2012) except that the pellet was resuspended in ice-cold buffer B (20 mM Tris, pH 7.5, containing 150 mM NaCl, 2 mM MgCl2, 1 μM CaCl2, 0.5 mM ATP, 1% [vol/vol] Triton X-100, 50 mM NaF, 1 mM Na glycerophosphate, 0.5 mg/ml Pefabloc, and protease inhibitor cocktail tablets). Proteins (800 μg) were incubated with 20 μl of protein G Sepharose beads for 1 h at 4°C and clarified by centrifugation at 1000 × g for 5 min at 4°C. The resulting supernatant, specific antibody, or corresponding preimmune immunoglobulin (1 μg) was then mixed and incubated overnight at 4°C and then with protein G Sepharose (20 μl) for 1 h. After five washing steps with buffer B, beads and associated proteins were resuspended in SDS–PAGE buffer and analyzed by immunoblotting.
The dissociation of MyRIP and its associated proteins was carried out using a previously established protocol (Moult et al., 2006; Gladding et al., 2009). In brief, MIN6 cell homogenates were incubated in a modified radioimmunoprecipitation assay (RIPA) buffer (1% [vol/vol] Triton X-100, 1% [wt/vol] SDS, 0.4 [wt/vol] sodium deoxycholate, 2 mM EDTA, 150 mM NaCl, 50 mM Tris-HCl, pH 7.4) in the presence of protease and phosphatase inhibitors at 100°C for 5 min. SDS was diluted out by adding 950 μl of RIPA buffer lacking SDS, transferred into preactivated membranes (size 1, molecular weight, 12,000–14,000 Da; Medicell International, London, United Kingdom) and dialyzed against 4 × 1 l of 0.1% SDS and 1% Triton X-100 containing lysis buffer overnight at 4°C. MyRIP was immunoprecipitated as described.
Immunoblotting, subcellular fractionation, immuno–electron microscopy, immunocytochemistry, and imaging
These were performed as described previously (Varadi et al., 2004, Brozzi et al., 2012). Imaging was performed using a wild-field microscope (Nikon TD300; Nikon, Melville, NY) and a confocal microscope (UltraView Real Time Confocal microscope; PerkinElmer Life Sciences, Boston, MA). On immunogold electron microscopy all SGs were counted manually in a visual field. Gold particles at the SG periphery (within 30 nm of the SG membrane) were counted, and two proteins were counted as colocalizing when distance between the gold particles detecting the respective proteins was <30 nm (van Weering et al., 2010; Brozzi et al., 2012).
siRNA knockdown of MyRIP expression
Cells were transfected with 70 nM MyRIP siGENOME SMART pool siRNA or BLOCK-iT Fluorescent Oligo as RNAi Transfection Control (Invitrogen) using Lipofectamine (Invitrogen) according to manufacturer's instructions. At 36 h posttransfection, cells were cultured in media containing 3 mM glucose overnight and then incubated in modified 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES)–balanced Krebs-Ringer bicarbonate (KRB) buffer containing 3 mM glucose for 1 h and then in KRB buffer supplemented with 3 mM or 16.7 mM glucose, 10 μM forskolin, and 1 mM IBMX, or 16.7 mM glucose and 10 μM exendin-4 for 60 min.
Human growth hormone release
MIN6 cells were seeded at a density of (4–60) × 105/ml and were cultured overnight. Cells were cotransfected with 0.5 μg of hGH-encoding plasmid together with 1 μg of wild-type, full-length Rph-3A, Rph-3A (S234A, S234D, S234E) pcDNA3 constructs, or empty vector using Lipofectamine. Two days after transfection cells were cultured at 3 mM glucose overnight and then washed two times for 15 min in 20 mM HEPES (pH 7.4), 128 mM NaCl, 5 mM KCl, 1 mM MgCl2, 2.7 mM CaCl2, and 3 mM glucose at 37°C. To stimulate secretion, cells were incubated in the same buffer containing 3 or 16.7 mM glucose, 1 mM IBMX, and 10 μM forskolin (16.7 mM Glu, IBMX, forskolin) at 37°C for 1 h. At the end of the incubation, the supernatants were removed and the cells lysed with 500 μl of 0.5% (vol/vol) Triton X-100. The hGH assay was carried out using a colorimetric sandwich enzyme-linked immunosorbent assay method according to the manufacturer's instructions (Roche Diagnostics).
Statistical analysis
Data are presented as mean ± SD for the number of observations given, and statistical significance was calculated using a two-tailed Student's t test. Every experiment was performed at least three times unless otherwise stated in the figure legend.
Supplementary Material
Acknowledgments
Work in the Varadi laboratory was supported by a grant from the Biotechnology and Biological Sciences Research Council UK and by the Wellcome Trust. S.L. was a Marie Curie Intra-European Postdoctoral Fellow. E. Molnar is supported by the Medical Research Council UK (Grant G0601509) and the Biotechnology and Biological Sciences Research Council UK (Grants BB/F011326/1, BB/J015938/1).
Abbreviations used:
- AKAP
PKA-anchoring protein
- BR-MyoVa
brain isoform of myosin Va
- GLP-1
glucagon-like peptide-1
- MC-MyoVa
melanocyte isoform of myosin Va
- MyRIP
myosin- and Rab-interacting protein
- PKA
protein kinase A
- Rph-3A
rabphilin-3A
- SG
secretory granule
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-05-0369) on September 19, 2012.
*These authors contributed equally to this work.
F.B, S.L., F.D., K.H., and S.R. carried out the experiments and analyzed the raw data. R.R. and E.M. contributed reagents and experimental suggestions. A.V. and E.M wrote the manuscript. All authors discussed the results and implications and commented on the manuscript at all stages. A.V. supervised the project.
REFERENCES
- Au JS-Y, Huang J-D. A tissue-specific exon of myosin Va is responsible for selective cargo binding in melanocytes. Cell Motil Cytoskeleton. 2002;53:89–102. doi: 10.1002/cm.10061. [DOI] [PubMed] [Google Scholar]
- Bajjalieh S. Trafficking in cell fate. Nat Genet. 2004;36:216–217. doi: 10.1038/ng0304-216. [DOI] [PubMed] [Google Scholar]
- Bratanova-Tochkova TK, Cheng H, Daniel S, Gunawardana S, Liu YJ, Mulvaney-Musa J, Schermerhorn T, Straub SG, Yajima H, Sharp GW. Triggering and augmentation mechanisms, granule pools, and biphasic insulin secretion. Diabetes. 2002;51(Suppl 1):S83–S90. doi: 10.2337/diabetes.51.2007.s83. [DOI] [PubMed] [Google Scholar]
- Broadie K, Rushton E, Skoulakis EM, Davis RL. Leonardo, a Drosophila 14-3-3 protein involved in learning, regulates presynaptic function. Neuron. 1997;19:391–402. doi: 10.1016/s0896-6273(00)80948-4. [DOI] [PubMed] [Google Scholar]
- Brozzi F, Diraison F, Lajus S, Rajatileka S, Philips T, Regazzi R, Fukuda M, Verkade P, Molnár E, Váradi A. Molecular mechanism of myosin Va recruitment to dense core secretory granules. Traffic. 2012;13:54–69. doi: 10.1111/j.1600-0854.2011.01301.x. [DOI] [PubMed] [Google Scholar]
- Chan SLF, Mourtada M, Morgan NG. Characterization of a KATP channel–independent pathway involved in potentiation of insulin secretion by efaroxan. Diabetes. 2001;50:340–347. doi: 10.2337/diabetes.50.2.340. [DOI] [PubMed] [Google Scholar]
- Coppola T, Frantz C, Perret-Menoud V, Gattesco S, Hirling H, Regazzi R. Pancreatic beta-cell protein granulophilin binds Rab3 and Munc-18 and controls exocytosis. Mol Biol Cell. 2002;13:1906–1915. doi: 10.1091/mbc.02-02-0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desnos C, et al. Rab27A and its effector MyRIP link secretory granules to F-actin and control their motion towards release sites. J Cell Biol. 2003;163:559–570. doi: 10.1083/jcb.200302157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle ME, Egan JM. Mechanisms of action of glucagon-like peptide 1 in the pancreas. Pharmacol Ther. 2007;113:546–593. doi: 10.1016/j.pharmthera.2006.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliasson L, et al. SUR1 regulates PKA-independent cAMP-induced granule priming in mouse pancreatic B-cells. J Gen Physiol. 2003;121:181–197. doi: 10.1085/jgp.20028707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foletti DL, Blitzer JT, Scheller RH. Physiological modulation of rabphilin phosphorylation. J Neurosci. 2001;21:5473–5483. doi: 10.1523/JNEUROSCI.21-15-05473.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser ID, Tavalin SJ, Lester LB, Langeberg LK, Westphal AM, Dean RA, Marrion NV, Scott JD. A novel lipid-anchored A-kinase anchoring protein facilitates cAMP-responsive membrane events. EMBO J. 1998;17:2261–2272. doi: 10.1093/emboj/17.8.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda M, Kuroda TS. Slac2-c (synaptotagmin-like protein homologue lacking C2 domains-c), a novel linker protein that interacts with Rab27, myosin Va/VIIa, and actin. J Biol Chem. 2002;277:43096–43103. doi: 10.1074/jbc.M203862200. [DOI] [PubMed] [Google Scholar]
- Fykse EM, Li C, Südhof TC. Phosphorylation of rabphilin-3A by Ca2+/calmodulin- and cAMP-dependent protein kinase in vitro. J Neurosci. 1995;15:2385–2395. doi: 10.1523/JNEUROSCI.15-03-02385.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gladding CM, Collett VJ, Jia Z, Bashir ZI, Collingridge GL, Molnár E. Tyrosine dephosphorylation regulates AMPAR internalisation in mGluR-LTD. Mol Cell Neurosci. 2009;40:267–279. doi: 10.1016/j.mcn.2008.10.014. [DOI] [PubMed] [Google Scholar]
- Goehring AS, Pedroja BS, Hinke SA, Langeberg LK, Scott JD. MyRIP anchors protein kinase A to the exocyst complex. J Biol Chem. 2007;282:33155–33167. doi: 10.1074/jbc.M705167200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Göke R, Fehmann HC, Linn T, Schmidt H, Krause M, Eng J, Göke B. Exendin-4 is a high potency agonist and truncated exendin-(9–39)-amide an antagonist at the glucagon-like peptide 1-(7–36)-amide receptor of insulin-secreting beta-cells. J Biol Chem. 1993;268:19650–19655. [PubMed] [Google Scholar]
- Gonelle-Gispert C, Costa M, Takahashi M, Sadoul K, Halban P. Phosphorylation of SNAP-25 on serine-187 is induced by secretagogues in insulin-secreting cells, but is not correlated with insulin secretion. Biochem J. 2002;368:223–232. doi: 10.1042/BJ20020896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hepp R, Cabaniols J-P, Roche PA. Differential phosphorylation of SNAP-25 in vivo by protein kinase C and protein kinase A. FEBS Lett. 2002;532:52–56. doi: 10.1016/s0014-5793(02)03629-3. [DOI] [PubMed] [Google Scholar]
- Huang JD, Brady ST, Richards BW, Stenolen D, Resau JH, Copeland NG, Jenkins NA. Direct interaction of microtubule- and actin-based transport motors. Nature. 1999;397:267–270. doi: 10.1038/16722. [DOI] [PubMed] [Google Scholar]
- Huet S, Fanget I, Jouannot O, Meireles P, Zeiske T, Larochette N, Drachen F, Desnos C. Myrip couples the capture of secretory granules by the actin-rich cell cortex and their attachment to the plasma membrane. J Neurosci. 2012;32:2564–2577. doi: 10.1523/JNEUROSCI.2724-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki N, Mizuta M, Seino S. Cloning of a mouse rabphilin-3A expressed in hormone-secreting cells. J Biochem. 1994;116:239–242. doi: 10.1093/oxfordjournals.jbchem.a124512. [DOI] [PubMed] [Google Scholar]
- Ivarsson R, Jing X, Waselle L, Regazzi R, Renström E. Myosin 5a controls insulin granule recruitment during late-phase secretion. Traffic. 2005;6:1027–1035. doi: 10.1111/j.1600-0854.2005.00342.x. [DOI] [PubMed] [Google Scholar]
- Joberty G, Stabila PF, Coppola T, Macara IG, Regazzi R. High affinity Rab3 binding is dispensable for rabphilin-dependent potentiation of stimulated secretion. J Cell Sci. 1999;112:3579–3587. doi: 10.1242/jcs.112.20.3579. [DOI] [PubMed] [Google Scholar]
- Kasai K, Fujita T, Gomi H, Izumi T. Docking is not a prerequisite but a temporal constraint for fusion of secretory granules. Traffic. 2008;9:1191–1203. doi: 10.1111/j.1600-0854.2008.00744.x. [DOI] [PubMed] [Google Scholar]
- Kuroda TS, Fukuda M. Functional analysis of Slac2-c/MyRIP as a linker protein between melanosomes and myosin VIIa. J Biol Chem. 2005a;280:28015–28022. doi: 10.1074/jbc.M501465200. [DOI] [PubMed] [Google Scholar]
- Kuroda TS, Fukuda M. Identification and biochemical analysis of Slac2-c/MyRIP as a Rab27A-, myosin Va/VIIa-, and actin-binding protein. Methods Enzymol. 2005b;403:431–444. doi: 10.1016/S0076-6879(05)03038-7. [DOI] [PubMed] [Google Scholar]
- Lonart G, Südhof TC. Region-specific phosphorylation of rabphilin in mossy fiber nerve terminals of the hippocampus. J Neurosci. 1998;18:634–640. doi: 10.1523/JNEUROSCI.18-02-00634.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonart G, Südhof TC. Characterization of rabphilin phosphorylation using phosphor-specific antibodies. Neuropharmacology. 2001;41:643–649. doi: 10.1016/s0028-3908(01)00126-5. [DOI] [PubMed] [Google Scholar]
- Mizuno K, Ramalho JS, Izumi T. Exophilin8 transiently clusters insulin granules at the actin-rich cell cortex prior to exocytosis. Mol Biol Cell. 2011;22:1716–1726. doi: 10.1091/mbc.E10-05-0404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan A, Burgoyne RD. Exo1 and Exo2 proteins stimulate calcium-dependent exocytosis in permeabilized adrenal chromaffin cells. Nature. 1992;355:833–836. doi: 10.1038/355833a0. [DOI] [PubMed] [Google Scholar]
- Moult PR, Gladding CM, Sanderson T, Fitzjohn SM, Bashir ZI, Molnár E, Collingridge GL. Tyrosine phosphatases regulate AMPA receptor trafficking during metabotropic glutamate receptor-mediated long-term depression. J Neurosci. 2006;26:2544–2554. doi: 10.1523/JNEUROSCI.4322-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munson M, Novick P. The exocyst defrocked, a framework of rods revealed. Nat Struct Mol Biol. 2006;13:577–581. doi: 10.1038/nsmb1097. [DOI] [PubMed] [Google Scholar]
- Nagy G, Reim K, Matti U, Brose N, Binz T, Rettig J, Neher E, Sørensen JB. Regulation of releasable vesicle pool sizes by protein kinase A-dependent phosphorylation of SNAP-25. Neuron. 2004;41:351–365. doi: 10.1016/s0896-6273(04)00038-8. [DOI] [PubMed] [Google Scholar]
- Nauck MA, Homberger E, Siegel EG, Allen RC, Eaton RP, Ebert R, Creutzfeldt W. Incretin effects of increasing glucose loads in man calculated from venous insulin and C-peptide responses. J Clin Endocrinol Metab. 1986;63:492–498. doi: 10.1210/jcem-63-2-492. [DOI] [PubMed] [Google Scholar]
- Nauert JB, Rigas JD, Lester LB. Identification of an IQGAP1/AKAP79 complex in beta-cells. J Cell Biochem. 2003;90:97–108. doi: 10.1002/jcb.10604. [DOI] [PubMed] [Google Scholar]
- Novick P, Field C, Schekman R. Identification of 23 complementation groups required for post-translational events in the yeast secretory pathway. Cell. 1980;21:205–215. doi: 10.1016/0092-8674(80)90128-2. [DOI] [PubMed] [Google Scholar]
- Olofsson CS, Göpel SO, Barg S, Galvanovskis J, Ma X, Salehi A, Rorsman P, Eliasson L. Fast insulin secretion reflects exocytosis of docked granules in mouse pancreatic B-cells. Pflugers Arch. 2002;444:43–51. doi: 10.1007/s00424-002-0781-5. [DOI] [PubMed] [Google Scholar]
- Passeron T, Bahadoran P, Bertolotto C, Chiaverini C, Buscà R, Valony G, Bille K, Ortonne J-P, Ballotti R. Cyclic AMP promotes a peripheral distribution of melanosomes and stimulates melanophilin/Slac2-a and actin association. FASEB J. 2004;18:989–991. doi: 10.1096/fj.03-1240fje. [DOI] [PubMed] [Google Scholar]
- Ramalho JS, Lopes VS, Tarafder AK, Seabra MC, Hume AN. Myrip uses distinct domains in the cellular activation of myosin VA and myosin VIIA in melanosome transport. Pigment Cell Melanoma Res. 2009;22:461–473. doi: 10.1111/j.1755-148X.2009.00567.x. [DOI] [PubMed] [Google Scholar]
- Regazzi R, Ravazzola M, Iezzi M, Lang J, Zahraoui A, Andereggen E, Morel P, Takai Y, Wollheim CB. Expression, localization and functional role of small GTPases of the Rab3 family in insulin-secreting cells. J Cell Sci. 1996;109:2265–2273. doi: 10.1242/jcs.109.9.2265. [DOI] [PubMed] [Google Scholar]
- Renström E, Eliasson L, Rorsman P. Protein kinase A-dependent and -independent stimulation of exocytosis by cAMP in mouse pancreatic B-cells. J Physiol. 1997;502:105–118. doi: 10.1111/j.1469-7793.1997.105bl.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seino S, Shibasaki T. PKA-dependent and PKA-independent pathways for cAMP-regulated exocytosis. Physiol Rev. 2005;85:1303–1342. doi: 10.1152/physrev.00001.2005. [DOI] [PubMed] [Google Scholar]
- Seperack PK, Mercer JA, Strobel MC, Copeland NG, Jenkins NA. Retroviral sequences located within an intron of the dilute gene alter dilute expression in a tissue-specific manner. EMBO J. 1995;14:2326–2332. doi: 10.1002/j.1460-2075.1995.tb07227.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song WJ, et al. Snapin mediates incretin action and augments glucose-dependent insulin secretion. Cell Metab. 2011;13:308–319. doi: 10.1016/j.cmet.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Bittner MA, Holz RW. Rim, a component of the presynaptic active zone and modulator of exocytosis, binds 14-3-3 through its N terminus. J Biol Chem. 2003;278:38301–38309. doi: 10.1074/jbc.M212801200. [DOI] [PubMed] [Google Scholar]
- Tsuboi T, Ravier MA, Xie H, Ewart MA, Gould GW, Baldwin SA, Rutter GA. Mammalian exocyst complex is required for the docking step of insulin vesicle exocytosis. J Biol Chem. 2005;280:25565–25570. doi: 10.1074/jbc.M501674200. [DOI] [PubMed] [Google Scholar]
- van Weering JRT, Brown E, Sharp TH, Mantell J, Cullen PJ, Verkade P. Intracellular membrane traffic at high resolution. Methods Cell Biol. 2010;96:619–648. doi: 10.1016/S0091-679X(10)96026-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varadi A, Ainscow EK, Allan VJ, Rutter GA. Involvement of conventional kinesin in glucose-stimulated secretory granule movements and exocytosis in clonal pancreatic beta-cells. J Cell Sci. 2002;115:4177–4189. doi: 10.1242/jcs.00083. [DOI] [PubMed] [Google Scholar]
- Varadi A, Grant A, McCormack M, Nicolson T, Magistri M, Mitchell KJ, Halestrap AP, Yuan H, Schwappach B, Rutter GA. Intracellular ATP-sensitive K+ channels in mouse pancreatic beta cells: against a role in organelle cation homeostasis. Diabetologia. 2006;49:1567–1577. doi: 10.1007/s00125-006-0257-9. [DOI] [PubMed] [Google Scholar]
- Varadi A, Johnson-Cadwell LI, Cirulli V, Yoon Y, Allan VJ, Rutter GA. Cytoplasmic dynein regulates the subcellular distribution of mitochondria by controlling the recruitment of the fission factor dynamin-related protein-1. J Cell Sci. 2004;117:4389–440. doi: 10.1242/jcs.01299. [DOI] [PubMed] [Google Scholar]
- Váradi A, Molnár E, Ashcroft SJ. A unique combination of plasma membrane Ca2+-ATPase isoforms is expressed in islets of Langerhans and pancreatic beta-cell lines. Biochem J. 1996;314:663–669. doi: 10.1042/bj3140663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varadi A, Tsuboi T, Johnson-Cadwell LI, Allan VJ, Rutter GA. Kinesin I and cytoplasmic dynein orchestrate glucose-stimulated insulin-containing vesicle movements in clonal MIN6 beta-cells. Biochem Biophys Res Commun. 2003;311:272–282. doi: 10.1016/j.bbrc.2003.09.208. [DOI] [PubMed] [Google Scholar]
- Varadi A, Tsuboi T, Rutter GA. Myosin Va transports dense core secretory vesicles in pancreatic MIN6 beta-cells. Mol Biol Cell. 2005;16:2670–2680. doi: 10.1091/mbc.E04-11-1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waselle L, Coppola T, Fukuda M, Iezzi M, El-Amraoui A, Petit C, Regazzi R. Involvement of the Rab27 binding protein Slac2c/MyRIP in insulin exocytosis. Mol Biol Cell. 2003;14:4103–4113. doi: 10.1091/mbc.E03-01-0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yajima H, Komatsu M, Shermerhorn T, Aizawa T, Kaneko T, Nagai M, Sharp GW, Hashizume K. cAMP enhances insulin secretion by an action on the ATP-sensitive K+ channel-independent pathway of glucose signaling in rat pancreatic islets. Diabetes. 1999;48:1006–1012. doi: 10.2337/diabetes.48.5.1006. [DOI] [PubMed] [Google Scholar]
- Yu W, Niwa T, Fukasawa T, Hidaka H, Senda T, Sasaki Y, Niki I. Synergism of protein kinase A, protein kinase C, and myosin light-chain kinase in the secretory cascade of the pancreatic β-cell. Diabetes. 2000;49:945–952. doi: 10.2337/diabetes.49.6.945. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.