

Leukotrienes are bioactive signaling molecules derived from arachidonic acid that initiate and amplify innate immunity. A single structure, the leukotriene synthetic complex, on the nuclear membrane of neutrophils integrates and transduces extracellular signals to generate the chemotactic lipid LTB4.
Abstract
Leukotrienes (LTs) are lipid-signaling molecules derived from arachidonic acid (AA) that initiate and amplify inflammation. To initiate LT formation, the 5-lipoxygenase (5-LO) enzyme translocates to nuclear membranes, where it associates with its scaffold protein, 5-lipoxygenase–activating protein (FLAP), to form the core of the multiprotein LT synthetic complex. FLAP is considered to function by binding free AA and facilitating its use as a substrate by 5-LO to form the initial LT, LTA4. We used a combination of fluorescence lifetime imaging microscopy, cell biology, and biochemistry to identify discrete AA-dependent and AA-independent steps that occur on nuclear membranes to control the assembly of the LT synthetic complex in polymorphonuclear leukocytes. The association of AA with FLAP changes the configuration of the scaffold protein, enhances recruitment of membrane-associated 5-LO to form complexes with FLAP, and controls the closeness of this association. Granulocyte monocyte colony–stimulating factor provides a second AA-independent signal that controls the closeness of 5-LO and FLAP within complexes but not the number of complexes that are assembled. Our results demonstrate that the LT synthetic complex is a signal integrator that transduces extracellular signals to modulate the interaction of 5-LO and FLAP.
INTRODUCTION
The mammalian innate immune system is poised to respond quickly to bacterial infection and tissue injury, with polymorphonuclear leukocytes (PMNs) being the initial responders. Not only do PMNs provide the first line of host defense to bacteria, but they also amplify the inflammatory response by producing cytokines, chemokines, and the lipid-signaling molecule leukotriene (LT) B4 (Borgeat and Samuelsson, 1979; Scapini et al., 2001; Cloutier et al., 2009). LTB4 is a product of the 5-lipoxygenase (5-LO) pathway of arachidonic acid (AA) metabolism and initiates and amplifies the innate immune response by functioning as a highly potent chemoattractant for PMNs, monocytes, mast cells, and certain T-cell subsets (Ford-Hutchinson et al., 1980; Martin et al., 1989; Showell et al., 1995). Recently Afonso et al. (2012) showed that LTB4 plays a key role in controlling cellular responses to formyl peptide–mediated neutrophil polarization, resulting in augmented chemotaxis and an appropriate response to infectious and inflammatory challenges. Therefore cells must be able to both integrate extracellular signals to generate LTB4 and transduce these signals to yield a flexible and appropriate LTB4 response. How they achieve this is not known.
To prevent the unwanted initiation of inflammation and ensure that an inflammatory response is appropriately matched to a given challenge, cells have evolved a complex series of cellular and molecular controls to regulate LT synthesis (Figure 1). In resting cells, 5-LO, the initial enzyme of the LT pathway, resides in the cytosol and nucleoplasm (Rouzer and Samuelsson, 1987; Rouzer et al., 1989; Woods et al., 1993) and cytosolic phospholipase A2 (cPLA2) in the cytosol (Clark et al., 1991; Leslie, 2004). The substrate AA is esterified to phospholipids, and the generation of free AA by cPLA2 is required for the oxygenation of AA by 5-LO (Leslie, 2004). Upon cell activation, increased intracellular calcium triggers the translocation of cPLA2 to the endoplasmic reticulum (ER), outer nuclear membrane, and the Golgi (Glover et al., 1995; Evans et al., 2001; Leslie, 2004). In parallel, 5-LO translocates to the inner and outer nuclear membranes in a calcium-dependent process (Rouzer and Kargman, 1988; Rouzer et al., 1989; Woods et al., 1993; Kulkarni et al., 2002). The N-terminal C2 domain of cPLA2 (Nalefski et al., 1994; Evans et al., 2004) and C2-like domain of 5-LO (Kulkarni et al., 2002) mediate their association with specific membrane phospholipids. Although cPLA2 efficiently releases AA, the integral membrane protein 5-lipoxygenase–activating protein (FLAP) is required for the efficient conversion of endogenous AA to LTA4 (Dixon et al., 1990; Miller et al., 1990; Reid et al., 1990). X-ray crystallography identified FLAP as a trimer (Ferguson et al., 2007). FLAP contains four transmembrane helices that are connected by two cytosolic loops and one loop facing the lumen. FLAP is localized to the inner and outer nuclear membranes and also ER (Dixon et al., 1990; Miller et al., 1990). FLAP is considered to function as a scaffold protein that brings AA in sufficient proximity with 5-LO to allow efficient metabolism of AA.
FIGURE 1:
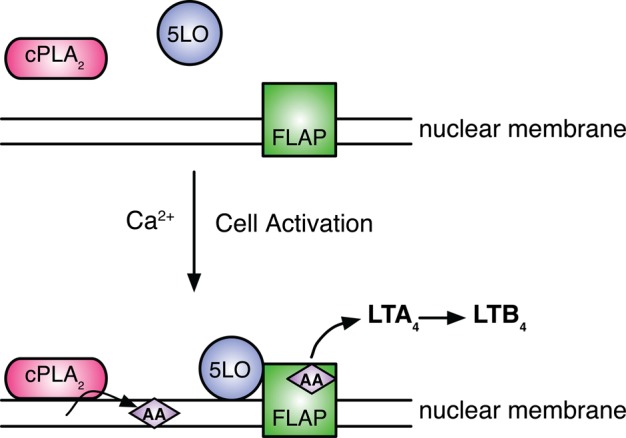
Assembly of the LT synthetic complex and the initiation of LT synthesis. Under resting conditions, cPLA2 and 5-LO reside in the cytoplasm of neutrophils. Transmembrane cell activation results in an increase in intracellular calcium leading to the translocation of 5-LO to the nuclear membrane, where it associates with FLAP. Concomitantly, cPLA2 traffics to the nuclear membrane/ER, where it releases free AA from phospholipids. 5-LO, FLAP, and AA assemble together at the correct time on the nuclear membrane, making up the core of the LT biosynthetic complex. LTA4 is subsequently converted to LTB4 by soluble LTA4 hydrolase.
Prior findings from our laboratory indicate that 5-LO directly interacts with FLAP in activated cells to form the core of a multiprotein LT synthetic complex (Mandal et al., 2008). To initiate LT synthesis, 5-LO converts AA to 5-hydroperoxyeicosatetraenoic acid, which is then converted to LTA4 (Rouzer et al., 1986). In some myeloid cell types such as mast cells, LTA4 is converted to LTC4 by the enzyme LTC4 synthase (Penrose et al., 1992; Lam et al., 1994). LTC4 synthase is an integral membrane protein that is tightly associated with FLAP in the outer nuclear membrane (Mandal et al., 2004). Alternatively LTA4 is subsequently converted to LTB4 by LTA4 hydrolase, which is present in the cytosol of PMNs (Radmark et al., 1984).
It is not known how multiple extracellular signals from cytokines, chemokines, lipid mediators, and C5a are integrated in the correct temporal and spatial order to assemble LT synthetic complexes and generate an appropriately graded LTB4 output. PMNs are a useful cell to address this question because they require two distinct signals for optimal LTB4 synthesis (Dahinden et al., 1988; Krump and Borgeat, 1994). The complement component C5a signals through its cognate G protein–coupled receptor (GPCR) to give an increase in intracellular calcium, leading to the translocation of 5-LO and cPLA2 to the nuclear envelope and the subsequent conversion of AA to a small but significant amount of LTB4 (Dahinden et al., 1988; Krump and Borgeat, 1994). Pretreatment, or “priming,” of PMNs with a cytokine such as granulocyte macrophage-colony–stimulating factor (GM-CSF) augments C5a-dependent LTB4 synthesis by threefold to fourfold, even though no LTB4 is produced in response to GM-CSF treatment alone (DiPersio et al., 1988; Krump and Borgeat, 1994; Brock et al., 1996). Because priming is not simply an additive effect, various mechanisms have been proposed to explain the process. Phosphorylation of cPLA2 at Ser-505 enhances its activity, leading to increased AA availability (McColl et al., 1991; Lin et al., 1993). Phosphorylation of 5-LO has been suggested to play a role in regulating activity (Werz et al., 2002), but these observations were made using nonphysiologic conditions. No previous studies have addressed the concept that discrete extracellular signals might alter macromolecular interactions in the LT synthetic complex.
Although the steps leading to the assembly of the LT synthetic complex and LT synthesis are well established (Figure 1), two key questions remain regarding the assembly of 5-LO and FLAP on the nuclear envelope: 1) Is the association of 5-LO with the nuclear membrane sufficient to promote binding to FLAP, or are additional signals required to initiate this protein–protein interaction? 2) Does free AA released from phospholipids modulate LT synthetic complex formation? We used fluorescence lifetime imaging microscopy (FLIM) to define a multistep assembly process for formation of LT synthetic complexes on the nuclear envelope. We identified distinct AA-dependent and AA-independent steps that control the interaction of 5-LO and FLAP. Overall, we identify the LT synthetic complex as a regulatory structure that functions by integrating and transducing diverse extracellular signals to regulate the interaction of 5-LO with FLAP. These qualitative changes are directly associated with levels of the cellular LTB4 response to cell activation by combinations of C5a and GM-CSF.
RESULTS
Multiple signals regulate the assembly of the LT synthetic complex in PMNs
We sought to define the multiple potential steps in assembling the LT synthetic complex. We confirmed the priming effect of GM-CSF in mature PMNs isolated from mouse bone marrow. Stimulation of PMNs with 100 nM C5a for 5 min induced a small but significant amount of LTB4 generation (Figure 2). Preincubation with 1 ng/ml GM-CSF (∼70 pM) for 60 min augmented the generation of LTB4 by PMNs in response to stimulation with C5a by threefold to fourfold. Neither unstimulated cells nor cells only primed with GM-CSF generated detectable amounts of LTB4.
FIGURE 2:
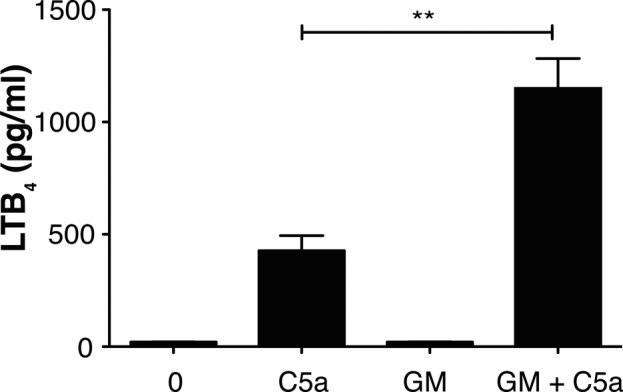
Priming and activation of PMNs for LTB4 production. PMNs were analyzed for LTB4 synthesis 5 min after stimulation with 100 nM C5a or after priming with 1 ng/ml GM-CSF for 60 min before the addition of C5a. Alternatively, cells were primed for 60 min with GM-CSF alone or left unstimulated (0) before analysis. Cells were centrifuged, and the supernatants were removed and analyzed by EIA for LTB4. Data are represented as mean ± SEM from four separate experiments. Data analysis was performed using one-way ANOVA followed by Bonferroni's multiple comparison test, where p < 0.05 is significant. Unpaired Student's t test was performed to obtain p values. **p = 0.0028 for C5a vs. GM-CSF + C5a. p < 0.05 for C5a and GM-CSF + C5a vs. unstimulated.
Because cPLA2 requires Ca2+ for its association with phospholipids in nuclear membranes (Glover et al., 1995; Evans et al., 2001, 2004), we tested whether the priming effect of GM-CSF could be explained by an increase in Ca2+ flux, potentially leading to an increase in membrane-associated cPLA2 molecules. Cells were primed with GM-CSF, followed by stimulation with C5a. Calcium flux was compared with that seen in cells stimulated with C5a alone. As shown in Table 1, both primed and unprimed PMNs showed a comparable increase in Ca2+ in response to C5a, indicating that differences in Ca2+ levels did not account for the increased LTB4 production with GM-CSF priming. Cells treated with ionomycin (positive control) showed a marked increase in Ca2+, whereas no change in Ca2+ flux was seen in unstimulated cells.
TABLE 1:
Calcium flux in stimulated PMNs.
| Condition | Δ[Ca2+]i, AU |
|---|---|
| Unstimulated | 0.1 ± 0.2 |
| C5a | 0.5 ± 0.1 |
| GM-CSF + C5a | 0.7 ± 0.2 |
| Ionomycin | 3.0 ± 0.4 |
Values represent mean ± SEM for three independent experiments. AU, arbitrary units.
We hypothesized that differences in LTB4 synthesis could ultimately be translated into discrete events at the level of membrane organization of the LT synthetic complex. To test this, we evaluated the relationship between 5-LO and FLAP on the nuclear membrane in response to cell stimulation by a combination of epifluorescence and FLIM. Aliquots of cells from each experimental condition were collected in parallel, fixed, permeabilized, and probed with antibodies (Abs) to the C-terminus of 5-LO and to the C-terminus of FLAP, which were detected with Alexa Fluor 488– and Alexa Fluor 594–conjugated secondary Abs, respectively. Because FLAP is an integral membrane protein that is present predominantly in the nuclear membrane of PMNs (Woods et al., 1993), we were able to distinguish nuclear membranes from the nucleoplasm by comparing 4′,6-diamidino-2-phenylindole (DAPI) staining to the localization of FLAP. Therefore an overall estimate of the nuclear membrane distribution of 5-LO could be made using FLAP and DAPI staining as references. Alexa Fluor fluorophores also served as the donor and acceptor fluorophores for FLIM analysis, allowing the same cells to be analyzed by epifluorescence and FLIM by alternating the imaging system interfacing with the microscope. Epifluorescence imaging of DAPI staining in control cells shows the nuclei that is clearly bordered by FLAP (Figure 3A). These results are supported by confocal microscopy (Supplemental Figure S1). After cellular activation by C5a alone or by C5a after priming with GM-CSF, 5-LO colocalized with FLAP on the nuclear envelope (Figure 3B).
FIGURE 3:
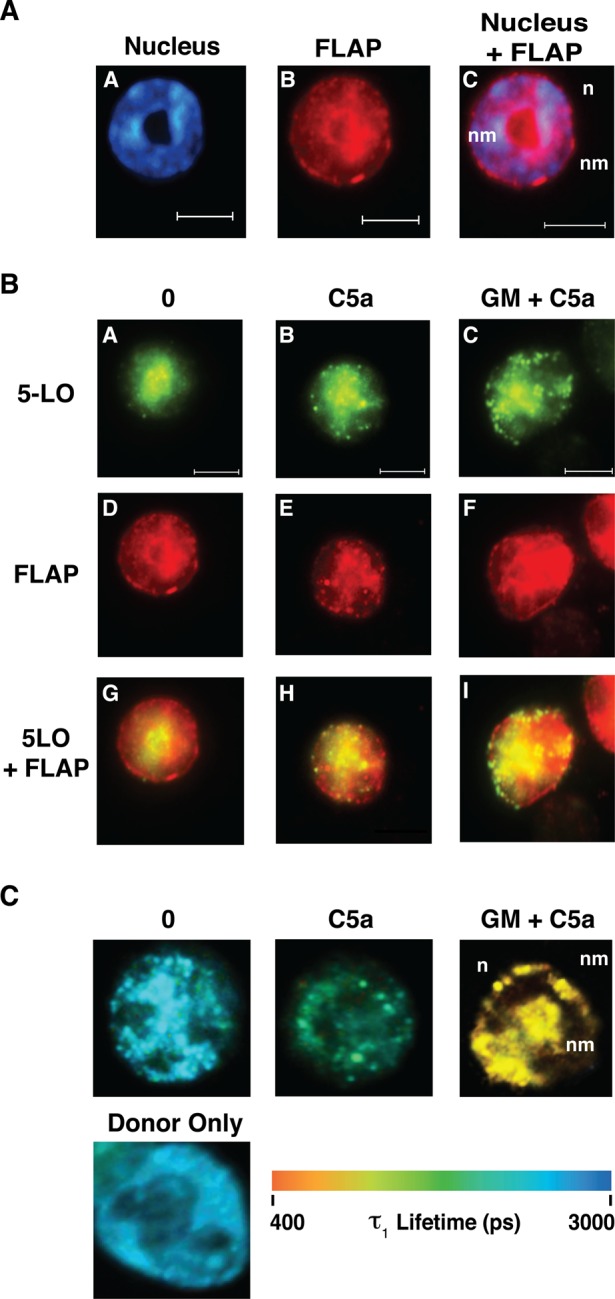
5-LO and FLAP interact on the nuclear membrane. Aliquots of cells from the groups studied in Figure 2 were collected for analysis. (A) Localization of FLAP and DNA. A representative unstimulated PMN is shown. The cell was costained with DAPI and anti-FLAP antibody followed by Alexa Fluor 594–conjugated secondary antibody. The cell was imaged using a filter cube for DAPI in A or TRITC in B. A digitally merged image is shown in C. Scale bar, 5 μM. n, nucleus; nm, nuclear membrane. (B) Redistribution of 5-LO in response to cell activation. PMNs were either activated with C5a (B, E, H) or primed with GM-CSF and then stimulated with C5a (C, F, I). The same cell shown in A is used as an unstimulated control in A, D, and G (of B). Cells were fixed, permeabilized, and then simultaneously probed with anti-5-LO (A, B, C) and anti-FLAP (D, E, F) primary antibodies. 5-LO was detected with Alexa Fluor 488–conjugated secondary antibody and FLAP with Alexa Fluor 594–conjugated secondary antibody. The cells were then analyzed with a filter set containing FITC or TRITC. In addition, the foregoing data were digitally merged (G, H, I). Scale bar, 5 μM. Cells shown are representative of at least four experiments. n, nucleus; nm, nuclear membrane. (C) Pseudocolor images of the FLIM analysis of the assembly of the LT synthetic complex. The cells shown are the same cells shown in B and are a representation of interacting fraction, τ1. The color scale for the τ1 lifetimes (picoseconds) ranging from 400 to 3000 is shown below the cellular images. 5-LO was identified with the donor fluorophore (Alexa Fluor 488) and FLAP with the acceptor fluorophore (Alexa Fluor 594). n, nucleus; nm, nuclear membrane. Analysis was performed using Becker and Hickl SPCImage software.
Cells were also analyzed by FLIM, a quantitative method for measuring Förster resonance energy transfer (FRET; Table 2 and Figure 3C). FLIM allows evaluation of the fluorescence lifetime of a donor molecule, in this case 5-LO, labeled with Alexa Fluor 488–conjugated secondary. After determining the donor fluorescence lifetime in the absence of the acceptor fluorophore (Figure 3C, Donor only), FRET between donor and acceptor was defined by the lifetime of interacting molecules (τ1), with a1(%) representing the fraction of interacting molecules. We found that a small percentage of 5-LO molecules interacted with FLAP on the nuclear membrane in unstimulated cells (Table 2), consistent with previously reported findings in unprimed RBL-2H3 cells (Mandal et al., 2008). After stimulation with C5a, a1(%) was not significantly different from that in unstimulated cells, but τ1 significantly decreased (Table 2 and Figure 3C), indicating increased proximity between 5-LO and FLAP. When cells were primed with GM-CSF before C5a stimulation, a further decrease in τ1 and a significant increase in a1(%) were observed, indicating a closer association and an increase in the number of interactions between 5-LO and FLAP. GM-CSF itself had no effect on τ1 (Supplemental Table S1).
TABLE 2:
FLIM analysis of 5-LO/FLAP interactions.
| Condition | τ1 (ps) | a1(%) |
|---|---|---|
| Donor only | 3000 ± 1 | 100 ± 0 |
| Unstimulated | 1920 ± 1 | 9.5 ± 1a |
| C5a | 1356 ± 74b | 12 ± 2a |
| GM-CSF + C5a | 826 ± 89b,c | 29 ± 5b,c |
| cPLA2 Inh + C5a | 1828 ± 89a | 12 ± 1a |
| cPLA2 Inh + GM-CSF + C5a | 1256 ± 187a,b | 9 ± 2a |
Values represent means ± SEM for n = 20 for each condition.
ap < 0.01 vs. GM-CSF + C5a.
bp < 0.001 vs. unstimulated cells.
cp < 0.0001 vs. C5a.
AA-dependent and AA-independent signals control the assembly of 5-LO and FLAP
Several studies equated the effect of GM-CSF or other cytokines to an increased availability of AA secondary to phosphorylation of cPLA2 (DiPersio et al., 1988), resulting in increased catalytic activity (McColl et al., 1991; Lin et al., 1993; Krump and Borgeat, 1994). We therefore hypothesized that a subtle rearrangement of the structure of the FLAP trimer occurs upon binding of AA and that this could bring a FLAP domain in closer apposition to the C-terminal catalytic domain of 5-LO; this would manifest as an AA-dependent decrease in τ1. To test this possibility, we first determined the effect of cPLA2 inhibitor (cPLA2 Inh) N-{(2S,4R)-4-(biphenyl-2-ylmethyl-isobutyl-amino)-1-[2-(2,4-difluorobenzoyl)-benzoyl]-pyrrolidin-2-ylmethyl}8–3-[4-(2,4-dioxothiazolidin-5-ylidenemethyl)-phenyl]acrylamide) on both LTB4 synthesis and the interaction between 5-LO and FLAP. Treatment of PMNs with cPLA2 Inh at concentrations between 100 nM and 10 μM before stimulation significantly inhibited LTB4 synthesis (Figure 4 and Supplemental Figure S2). Of note, the pyrrilidone cPLA2 inhibitor used in this study has been shown to have no detectable inhibition of purified, recombinant human sPLA2-IIA, group V, or group X sPLA2 at a concentration of 10 mM and no significant inhibition of either recombinant cytosolic PLA2-γ or the calcium-independent PLA2, iPLA2 (Ghomashchi et al., 2001; Bryant et al., 2011).
FIGURE 4:
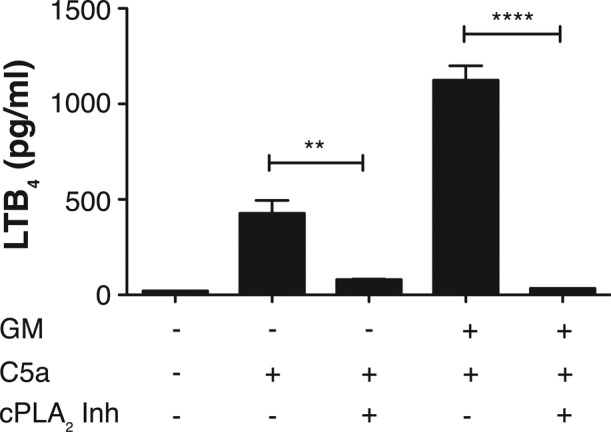
Inhibition of LTB4 synthesis by cPLA2 Inh. PMNs were analyzed for the synthesis of LTB4 5 min after stimulation with 100 nM C5a for or after priming with 1 ng/ml GM-CSF for 60 min before the addition of C5a. In parallel, cells were preincubated with 5 μM N-{(2S,4R)-4-(biphenyl-2-ylmethyl-isobutyl-amino)-1-[2-(2,4-difluorobenzoyl)-benzoyl]-pyrrolidin-2-ylmethyl}8–3-[4-(2,4-dioxothiazolidin-5-ylidenemethyl)-phenyl]acrylamide (cPLA2 Inh) for 60 min before activation with C5a for 5 min. In addition, cells were concomitantly exposed to cPLA2 Inh and GM-SCF for 60 min, followed by C5a stimulation for 5 min or left unstimulated (0) for control. Cells were centrifuged, and the supernatants were removed and analyzed by EIA for LTB4. Aliquots of cells were collected for analysis and shown in Figure 5. Data are represented as mean ± SEM from four separate experiments. Comparison of data were performed using one-way ANOVA followed by Bonferroni's multiple comparison test, where p < 0.05 is significant. Unpaired Student's t test was performed to obtain p values. **p = 0.0022 and ****p < 0.0001.
In parallel, we analyzed cells from each of these populations by both epifluorescence and FLIM. Inhibition of AA release by cPLA2 Inh (5 μM) had no obvious effect on the movement of 5-LO to the nuclear membrane in cells either stimulated directly with C5a or primed with GM-CSF before stimulation with C5a (Figure 5A), suggesting that 5-LO movement to the nuclear membrane is independent of AA release. We next analyzed the interaction between 5-LO and FLAP in the presence and absence of cPLA2 inhibition. There was a small but quantifiable percentage of 5-LO that interacts with FLAP on the nuclear membrane of unstimulated cells (Table 2). In addition, τ1 values remained unchanged upon exposure to GM-CSF alone and cPLA2 Inh alone (Supplemental Table S1). Also, as noted, cells stimulated with C5a exhibited a decreased τ1 compared with unstimulated controls (Table 2). However, when C5a-stimulated cells were pretreated with cPLA2 Inh, the τ1 values were similar to those of unstimulated cells. These results strongly support a role for AA in regulating the closeness between 5-LO and FLAP.
FIGURE 5:
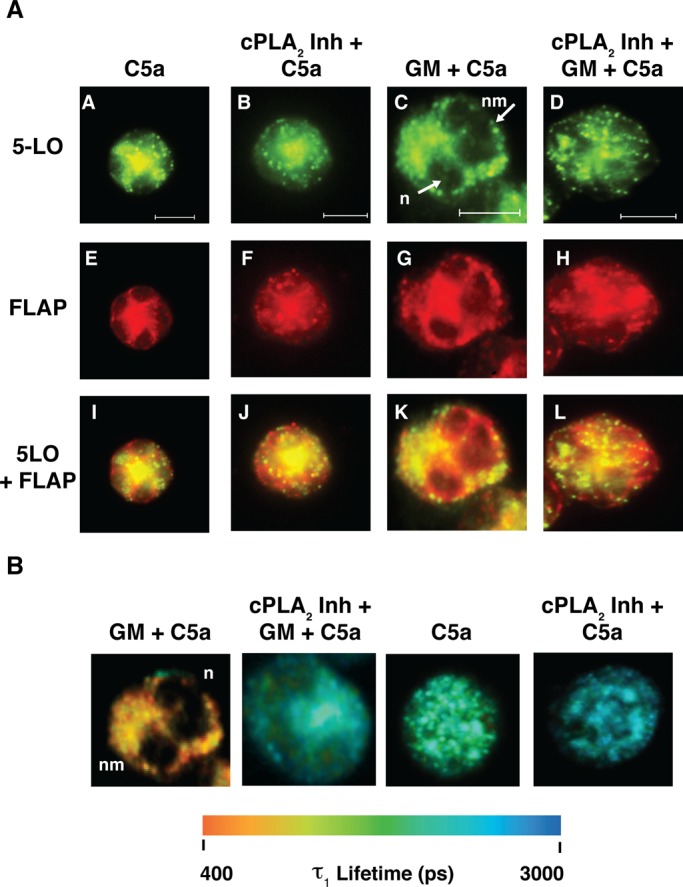
AA controls the interaction of 5-LO and FLAP. (A) Targeting of 5-LO to the nuclear membrane. PMNs were either activated with C5a (A, E, I), or primed with GM-CSF and then stimulated with C5a (C, G, K). In addition, cells were preincubated with cPLA2 Inh and then stimulated with C5a (B, F, J) or coincubated with cPLA2 Inh and GM-CSF, followed by C5a stimulation (D, H, L). Cells were fixed, permeabilized, and then simultaneously probed with anti-5-LO (A–D) and anti-FLAP (E–H) primary antibodies. 5-LO was detected with Alexa Fluor 488–conjugated secondary antibody (donor fluorophore) and FLAP with Alexa Fluor 594–conjugated secondary antibody (acceptor fluorophore). The cells were imaged using a filter set containing FITC and TRITC. In addition, the foregoing acquired data were digitally merged. (I–L). Cells shown are representative of at least three experiments. Scale bar, 5 μM. n, nucleus; nm, nuclear membrane. (B) Pseudocolor images of the FLIM analysis of the assembly of the LT synthetic complex. The cells shown are the same cells shown in A and are a representation of interacting fraction, τ1. The color scale for the τ1 lifetimes (picoseconds) ranging from 400 to 3000 is shown below the cellular images. 5-LO was identified with the donor fluorophore (Alexa Fluor 488) and FLAP with the acceptor fluorophore (Alexa Fluor 594). n, nucleus; nm, nuclear membrane. Analysis was performed using Becker and Hickl SPCImage software.
A distinct response was observed in cells primed with GM-CSF before C5a stimulation (Table 2 and Figure 5B). In the presence or absence of cPLA2 Inh, τ1 decreased relative to values in unstimulated cells. However, cPLA2 inhibition altered the interaction between FLAP and 5-LO, with τ1 approaching those of cells treated with C5a alone. This suggests that in addition to the AA-dependent effect observed with C5a, an AA-independent, GM-CSF–dependent process regulates the relationship of 5-LO to FLAP. When cPLA2 was inhibited, a1(%) values returned to baseline, indicating a role for AA in controlling the number of LT synthetic complexes formed on nuclear membranes.
Two of the major intracellular signaling pathways that are engaged by the activation of the GM-CSF receptor are p38 mitogen-activated protein kinase (MAPK) and extracellular-regulated kinase (ERK; Hercus et al., 2009), each of which has been linked to posttranslational modification of enzymes regulating LT synthesis. Both p38 MAPK and ERK have been shown to play redundant roles in the phosphorylation and regulation of cPLA2 (Geijsen et al., 2000). Furthermore, pharmacological inhibition of ERK has been shown to inhibit LT formation in response to f-Met-Leu-Phe (fMLP; Werz et al., 2002), potentially, in part, by blocking phosphorylation of 5-LO. Inhibition of ERK with 10 μM U0126 completely eliminated LTB4 generation in response to combinations of GM-CSF and C5a (Supplemental Figure S3). When aliquots of the same cells were analyzed for FLIM, the low τ1 values seen in stimulated cells returned to those seen in unstimulated control cells, whereas a1(%) levels remained elevated when compared with unstimulated controls (Table 3). When the same experiment was performed in the presence of the cPLA2 inhibitor, in addition to τ1, the a1(%) values returned those seen in unstimulated cells. These data indicate a role for ERK in controlling the qualitative association of 5-LO and FLAP that is independent of AA.
TABLE 3:
FLIM analysis of 5-LO/FLAP interactions.
| Condition | τ1 (ps) | a1(%) |
|---|---|---|
| Donor only | 2900 ± 4 | 100 ± 0 |
| Unstimulated | 1899 ± 30 | 11 ± 9 |
| GM-CSF + C5a | 562 ± 32 | 49 ± 11 |
| 10 μM SB203580 + GM-CSF + C5a | 1635 ± 81 | 32 ± 27 |
| 10 μM U0126 + GM-CSF + C5a | 2003 ± 29 | 28 ± 19 |
| 5 μM cPLA2 Inh + GM-CSF + C5a | 1492 ± 33 | 11 ± 9 |
| 10 μM SB203580 + 5 μM cPLA2 Inh + GM-CSF + C5a | 2001 ± 80 | 6 ± 3 |
| 10 μM U0126 + 5 μM cPLA2 Inh + GM-CSF + C5a | 1960 ± 91 | 9 ± 7 |
Values represent means ± SEM from two separate experiments, where n = 15 for each condition.
When p38 MAPK was inhibited in cells by 10 μM SB203580, LTB4 production was inhibited by ∼80% in response to the combination of GM-CSF and C5a (Supplemental Figure S3). When compared with stimulated cells, an increase in τ1 was observed; however, there was no change in a1(%) (Table 3). With the inclusion of the cPLA2 inhibitor, a further increase in τ1 was observed, matching what was seen unstimulated controls. This indicates a role for p38 MAPK independent of AA in regulating the interaction of 5-LO and FLAP. The mechanism by which ERK and p38 function in this process remains to be determined.
AA controls the relationship between the N- and C-terminal domains of FLAP
We postulated that AA could function to promote the interaction of 5-LO with FLAP by binding to the scaffold protein and causing a change in the molecular arrangement of FLAP. To monitor this effect, we used FLIM to monitor interactions between the N- and C-termini of FLAP. The N-terminus of FLAP (amino acids 1–19) was identified with Alexa Fluor 488–conjugated secondary antibody (donor fluorophore) and the C-terminus of FLAP (amino acids 148–161) detected with secondary antibody conjugated to Alexa Fluor 594 (acceptor fluorophore). This approach allowed the detection of potential changes in the relationship between the domains of individual monomers, between monomers of a given trimeric FLAP molecule, or between monomers of adjacent trimers. The relatively low values of a1(%) in all samples (Table 4) suggest that the majority of interactions detected between N- and C-termini are not within individual monomers but between either adjacent trimeric FLAP molecules or, possibly, between different FLAP trimers. The most likely reason for this is steric considerations of the relationships between Abs and that the size of FLAP affected analysis of the domains of individual monomers.
TABLE 4:
FLIM between the N- and C- termini of FLAP.
| Condition | τ1 (ps) | a1(%) |
|---|---|---|
| Unstimulated | 543 ± 27 | 32 ± 2 |
| C5a | 865 ± 49a | 16 ± 4b |
| GM-CSF + C5a | 522 ± 34 | 18 ± 2b |
| cPLA2 Inh + C5a | 622 ± 30 | 41 ± 2 |
| cPLA2 Inh + GM-CSF + C5a | 568 ± 27 | 41 ± 2 |
FLIM between N-terminus (amino acids 1–19) and C-terminus (amino acids 148–161) is represented by values ± SEM of n = 20 for each experimental condition.
ap < 0.001 vs. all other τ1 conditions.
bp < 0.001 vs. unstimulated a1 condition.
In unstimulated cells, the τ1 value was 543 ± 27 ps (Table 4). When PMNs were stimulated with C5a, τ1 increased, indicating a significant change in the relationship of the N- and C-termini; this change was not observed when cells were primed with GM-CSF before C5a stimulation. However, the percentage of interacting N-and C-termini decreased after C5a stimulation in both unprimed and primed cells. When cPLA2 was inhibited, τ1 values were not statistically different from those in unstimulated cells; furthermore, a1(%) increased to levels seen in unstimulated cells. Although the mechanism is unclear, these results indicate that AA affects the molecular configuration of FLAP. In control experiments using either 100 nM or 5 μM, cPLA2 Inh blocked LTB4 synthesis in response to GM-CSF and C5a (Supplemental Figure S2), but had no effect on the interaction between the N- and C-termini of FLAP (Supplemental Table S2).
DISCUSSION
LT biosynthesis in PMNs requires the dynamic construction of macromolecular complexes on the nuclear membrane. Although prior studies focused on the discrete step of 5-LO translocation to the nuclear envelope and the use of AA as a substrate for generating leukotrienes through the activation of cPLA2 (Nahas et al., 1996), these studies did not address how LT synthesis is organized on the nuclear envelope. We report here that multistep assembly and organization of the LT synthetic complex in isolated murine PMNs involves processes that are both dependent on and independent of AA release. The flexibility of this process and response to different signals supports a role for the LT synthetic complex as the regulator of the qualitative interaction between 5-LO and FLAP, a relationship that is likely to be reflected in the output of LTA4.
To elucidate whether assembly of the LT synthetic complex on the nuclear envelope of PMNs was altered by different signals, we exploited the concept of “priming,” in which a signal by a transmembrane cytokine receptor, such as the GM-CSF receptor, synergistically augments LTB4 synthesis in concert with a GPCR agonist such as C5a (DiPersio et al., 1988). As expected, priming by GM-CSF increased LTB4 synthesis threefold to fourfold even though there was no difference in calcium flux compared with unprimed, stimulated cells.
The additional amplification of LTB4 synthesis imparted by GM-CSF priming has two effects. First, it amplifies the AA-driven assembly of the LT synthetic complex, regulating an increase in the percentage of nuclear membrane–associated 5-LO molecules interacting with FLAP. This is distinct from the effect seen by stimulation with C5a alone. GM-CSF also provides a signal that drives the closeness of the association of 5-LO and FLAP (Figures 3 and 6 and Table 2). The mechanism by which GM-CSF exerts these two distinct activities may be related to the phosphorylation of cPLA2 and 5-LO. Priming by GM-CSF stimulates the MAPK pathway, which can phosphorylate cPLA2 (Geijsen et al., 2000). This would augment the release of AA from membrane phospholipids. Phosphorylation of 5-LO can affect its intracellular trafficking; whether it directly affects the association of 5-LO and FLAP on the nuclear envelope, however, is not known. Both ERK and p38 MAPK inhibition alter the interaction of 5-LO and FLAP independent of AA (Table 3). However, whether this is secondary to phosphorylation of 5-LO or the posttranslational modification of a yet-to-be-determined complex member is unknown.
FIGURE 6:
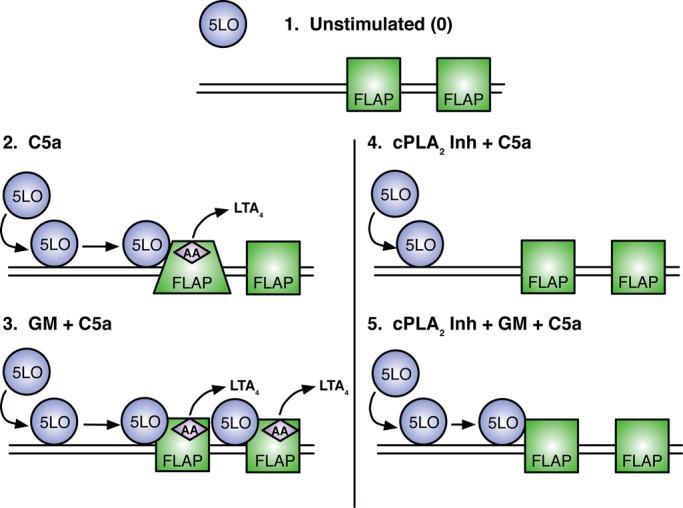
The multistep assembly of the LT synthetic complex. 1) Under resting conditions 5-LO resides in the cytoplasm and FLAP within the nuclear membrane. No LTs are made under this condition. 2) C5a-induced cell activation, followed by a rise in intracellular calcium, redistributes 5-LO to the nuclear membrane in close proximity with FLAP. AA is cleaved from phospholipids and interacts with FLAP; modest amounts of LTA4 are generated. Change in FLAP domains by FLIM are illustrated by the trapezoidal FLAP. 3) Priming cells with GM-CSF followed by C5a activation results in increased 5-LO translocation to the nuclear membrane, tighter interactions with FLAP, and robust LTA4 production. 4) AA controls the assembly of the LT synthetic complex. In the presence of cPLA2 Inh, 5-LO translocates to the nuclear membrane after C5a stimulation but does not show an increase in either the percentage of molecules interacting with FLAP or the “closeness” of its interaction. No LTs are made. 5) Priming with GM-CSF controls the interaction of 5-LO and FLAP by AA-dependent and AA-independent mechanisms. Cells treated with cPLA2 Inh and primed with GM-CSF followed by C5a activation show an increase in the closeness of 5-LO and FLAP but not in the percentage of membrane-associated molecules interacting with FLAP. No LTs are produced.
The ability of cells to qualitatively modify the association of 5-LO with FLAP in response to discrete signals has the potential to provide the flexibility to generate the appropriate amount of LTB4 in different biological settings. This can be illustrated by considering the diverse signaling responses of the GM-CSF receptor (Hercus et al., 2009). At low (femtomole) levels of GM-CSF, the receptor is exclusively phosphorylated at βc Ser-585, engaging PI3 kinase and Akt pathways that do not lead to posttranslational modification of either cPLA2 or 5-LO, and are not known to prime PMNs for augmented LTB4 synthesis. This would potentially correspond to a setting of early or low-level tissue inflammation or injury, and the generation of LTB4 would be determined solely by the AA levels induced by C5a, fMLP, or chemokines. In settings of more severe inflammation or infection, higher levels of GM-CSF would be generated. This would lead to exclusive phosphorylation of βc Tyr-577 and extinction of Ser-585 phosphorylation. This engages the MAP kinase pathway, allowing for the phosphorylation of cPLA2 and increased release of free AA. In turn, this would lead to the incorporation of a higher percentage of nuclear membrane–associated 5-LO into LT synthetic complexes with increased closeness between 5-LO and FLAP. Phosphorylation of 5-LO would have an additional effect by increasing the closeness of its association with FLAP. The net effect of these changes would be the generation of more LTB4 and the recruitment of more PMNs and other LTB4-responsive cells to the inflamed tissue. The levels of GM-CSF used in this study to prime PMNs are within this higher range.
It has been shown that ligands of scaffold proteins impart an allosteric effect on the scaffold protein's ability to recruit a second ligand (Good et al., 2011). We found that AA has a direct effect on the interaction of the N- and C- termini of FLAP. Furthermore, in the presence of the cPLA2 Inh, primed and unprimed cells treated with C5a exhibited statistically decreased LTB4 production. We also found that in cells exposed to the cPLA2 Inh and C5a, τ1 values significantly decreased close to those observed in unstimulated cells. Similarly, cells that were incubated with the cPLA2 Inh, primed, and treated with C5a have significantly reduced τ1 values compared with cells primed and treated without the inhibitor. Taken together, these results imply that AA governs the closeness of the association of 5-LO and FLAP.
The use of AA as a means of modulating the LT synthetic complex yields increased flexibility in terms of regulating LT synthesis. Studies have shown that the regulation of the reacylation of free AA is a key step in LT synthesis in PMNs, especially under the conditions of priming (Zarini et al., 2006). Therefore it will be important to understand the regulation of the transacylase enzymes.
In addition to the studies indicating that LTB4 dramatically amplified formyl peptide–mediated neutrophil chemotaxis and cytoskeletal rearrangement by acting on signaling pathways upstream of actin polymerization (Afonso et al., 2012), C5a, fMLP, and IL-8 all stimulate PMNs to generate LTB4, and all have LTB4 synthesis amplified by priming (Dahinden et al., 1988; DiPersio et al., 1988; Krump and Borgeat, 1994; Afonso et al., 2012). Therefore the observations that AA stimulates the assembly of the LT synthetic complex in response to C5a likely represent a general mechanism for transducing and amplifying signaling by chemoattractants.
MATERIALS AND METHODS
Mice
C57BL/6 mice 8–12 wk old were obtained from Jackson Labs (Bar Harbor, ME) and maintained under specific-pathogen–free conditions at the animal facilities at the Massachusetts General Hospital Charlestown Facility. Experimental manipulations were performed in accordance with Institutional Animal Care and Use Committee recommendations at Massachusetts General Hospital.
Reagents
Poly-l-lysine solution was purchased from Sigma-Aldrich (St. Louis, MO). Recombinant mouse GM-CSF was obtained from Millipore (Billerica, MA), and C5a was from R&D Systems (Minneapolis, MN). Vectashield with DAPI was purchased from Vector Laboratories (Burlingame, CA). Anti–5-LO monoclonal antibody was from BD Biosciences (San Diego, CA); rabbit and goat polyclonal anti-ALOX5AP/FLAP antibodies were purchased from Thermo Scientific (Rockford, IL) and Santa Cruz Biotechnology (Santa Cruz, CA), respectively. Alexa Fluor 488–conjugated donkey anti-mouse and donkey anti-goat and Alexa Fluor 594–conjugated donkey anti-rabbit secondary antibodies were purchased from Life Technologies (Grand Island, NY). N-{(2S,4R)-4-(biphenyl-2-ylmethyl-isobutyl-amino)-1-[2-(2,4-difluorobenzoyl)-benzoyl]-pyrrolidin-2-ylmethyl}8-3-[4-(2,4-dioxothiazolidin-5-ylidenemethyl)-phenyl]acrylamide (cPLA2 Inh) was obtained from Calbiochem/EMD4 Biosciences (San Diego, CA). Fura-2 AM was from Life Technologies. The p38 inhibitor SB203580, and the ERK (MEK1 and MEK2) kinase inhibitor U0126 were purchased from Enzo Life Sciences (Farmingdale, NY) and Cell Signaling Technology (Danvers, MA), respectively.
Purification and activation of mouse neutrophils
PMNs were purified from bone marrow of 8- to 12-wk-old mice by density gradient centrifugation with Histopaque 1077 and 1119 (Sigma-Aldrich) as the separation media (Feng et al., 2010). For each experimental condition, 1.0 × 106 PMNs were suspended in 300 μl of DMEM (phenol red free) containing 1.5 mM calcium and magnesium. For priming, PMNs were incubated with 1 ng/ml GM-CSF for 60 min at 37°C in an atmosphere of 5% CO2; PMNs for control and C5a-only conditions were incubated in media alone for 60 min at 37°C. PMNs were then stimulated with 100 nM C5a for 5 min at 37°C, centrifuged, and the supernatants removed and kept on ice for subsequent LTB4 analysis. To inhibit cPLA2 activity, cells were incubated with 5 μM cPLA2 Inh for 60 min at 37°C in the presence or absence of GM-CSF, followed by stimulation with C5a for 5 min. To inhibit p38 and ERK MAPK activity, cells were first incubated with 10 μM SB203580 or 10 μM U0126 for 5 min, followed by concomitant incubation of 5 μM cPLA2 Inh and 1 ng/ml GM-CSF for 40 min at 37°C in an atmosphere of 5% CO2.
Cell pellets were resuspended in 400 μl of 4% paraformaldehyde and transferred to eight-well Millicell EZ slides (EMD Millipore) that were precoated with poly-l-lysine (per manufacturer's instructions). The cells were allowed to adhere for 10 min, washed with phosphate-buffered saline (PBS), and permeabilized for 5 min at room temperature (RT) with 0.1% Triton X-100 in PBS. The slides were washed in PBS and blocked with 5% fetal bovine serum in PBS for 30 min at RT. Primary antibodies were diluted in blocking solution overnight at 4°C. For 5-LO/FLAP interaction studies, anti–5-LO antibody (1:100) and Thermo Scientific (Waltham, MA) anti-FLAP antibody (1:100) were used; for FLAP domain interaction studies, anti–FLAP N-terminus (1:50; Santa Cruz Biotechnology) and anti–FLAP C-terminus (1:100; Thermo Scientific) were applied. The next day, slides were washed and incubated with Alexa Fluor 488– and Alexa Fluor 594–conjugated secondary antibodies (1:1000 in blocking buffer) for 60 min at RT. Slides were then washed and mounted with Vectashield containing DAPI using a #1.5 glass coverslip. Slides were sealed with nail polish and stored at 4°C until use.
Time-correlated single-photon counting FLIM analysis
Protein interactions were defined by time-correlated single-photon counting FLIM with instrument modifications noted in the following (Mandal et al., 2004, 2008). The baseline lifetimes of Alexa Fluor 488 (donor fluorophore) were calculated by single-exponential-decay fitting of fluorescence emission in the absence of Alexa Fluor 594 (acceptor fluorophore). For samples stained for both donor and acceptor, lifetimes were fit to a biexponential decay with lifetime of one component fixed to the donor-only lifetime. The lifetime for the interacting component, τ1, as well as fractional contributions for the percentage of interacting fluorophores, a1(%), and the noninteracting component were determined. At least four separate experiments were performed, with the reported n the total number of cells analyzed, and within each cell at least five different pixels were used to determine the mean value.
Intracellular calcium measurements
Purified bone marrow PMNs were counted and loaded with the fluorescent Ca2+ indicator Fura-2 AM (2 μM for 30 min at 37°C) and transferred into a precoated poly-l-lysine 96-well plate. For cells primed with GM-CSF, Fura-2 AM was included during the priming and/or inhibitor incubation periods. Cells were then washed twice with HBSS (20 mM HEPES, 115 mM NaCl, 5.4 mM KCl, 2 mM CaCl2, 0.8 mM MgCl2, 13.8 mM glucose, pH 7.4). The plate was placed on an inverted Zeiss Axiovert 200 microscope (Zeiss, Jena, Germany), and cells were focused using a 40× objective. Cells were then activated for 5 min with C5a, PBS containing 1.5 mM calcium/magnesium for unstimulated controls, or 2 μM ionomycin as a positive control. After excitation at 340 and 380 nm, AxioCamMR and Illuminator N HBO 103 were used to collect data. AxioVision software 4.3.0 was used to analyze the data. Data from single cells and cell clusters were normalized, and the change in 340/380 ratio in arbitrary units was documented. Prism, version 5, software (GraphPad, La Jolla, CA) was used to graph data.
Microscopy equipment and settings
Experimental conditions were all performed at room temperature. A Plan APO VC 60× oil DC N2 objective with a numerical aperture of 1.4 was used in all microscopy experiments. A Nikon Ti-E inverted microscope (Nikon, Melville, NY) was used for all epifluorescence and FLIM. Nikon Elements 3.10 imaging software was used to collect epifluorescence data. The filter cube set contained 3 filters: 1) DAPI with excitation range of 340–380 nm, emission range of 435–485 nm, and mirror at 400 nm. 2) Fluorescein isothiocyanate (FITC) with excitation range of 405–495 nm, emission range of 515–555 nm, and mirror at 505 nm. 3) Tetramethylrhodamine isothiocyanate (TRITC) with excitation range of 528–553 nm, emission range of 590–650 nm, and mirror at 565 nm. Brightness and contrast were applied to equally to all images. All images were normalized to equal gamma levels. For FLIM acquisition, Becker and Hickl (Berlin, Germany) BDL-488-SMC Picosecond Diode Laser and both long-pass (HQ500LP) and bandpass (HQ435/50) emission filters were used in combination with a hybrid detector (HPM-100-40 GaAsP Hybrid Detector integrated with a Hamamatsu [Hamamatsu, Japan] R10467-40 hybrid photomultiplier tube). Becker and Hickl SPCM software with DCC was used to acquire FLIM data, and SPCImage 3.0 (Becker and Hickl) software was used for FLIM analysis. Data were imported into Photoshop CS5, version 12.1 x64, InDesign CS5.5, version 7.5, and Illustrator CS5, version 15.1.0 software (Adobe, San Jose, CA), where overall brightness and contrast were adjusted.
Enzyme immunoassay analysis
Cell supernatants were analyzed for LTB4 with enzyme immunoassay (EIA; Cayman Chemical; Ann Arbor, MI) according to manufacturer's instructions.
Statistics
For EIA LTB4 studies, an unpaired two-tailed Student's t test (±SEM) was performed to obtain p values. For FLIM studies, data were analyzed using one-way analysis of variance (ANOVA) followed by Bonferroni's correction for posttest corrections. p < 0.05 was considered significant.
Supplementary Material
Acknowledgments
We thank Kristin White of Massachusetts General Hospital for her assistance in confocal microscopy. This work was supported by National Institutes of Health Grants R01 AI068871 (R.J.S. and A.M.B.), K01 DK089145-01A1 (A.M.B.), R01 ARRA supplement AI068871-04S1 (M.V.T., C.A.V., R.J.S.), 1S10RR027931 (R.J.S.), 1R01HL097796 (R.J.S., A.M.B., R.A.P.), and T32 DK007540-22 (A.M.B. and M.V.T.).
Abbreviations used:
- AA
arachidonic acid
- cPLA2 Inh
cytosolic phospholipase A2 inhibitor
- FLAP
5-lipoxygenase–activating protein
- FLIM
fluorescence lifetime imaging microscopy
- 5-LO
5-lipoxygenase
- LT
leukotriene
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-06-0489) on September 26, 2012.
A.M.B., C.A.V., M.V.T., and R.J.S. planned experiments, analyzed data, and wrote the manuscript. A.M.B., C.A.V., and M.V.T. performed experiments, and R.A.P. reviewed the manuscript. All authors read the manuscript and provided input.
REFERENCES
- Afonso PV, Janka-Junttila M, Lee YJ, McCann CP, Oliver CM, Aamer KA, Losert W, Cicerone MT, Parent CA. LTB(4) is a signal-relay molecule during neutrophil chemotaxis. Dev Cell. 2012;22:1079–1091. doi: 10.1016/j.devcel.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgeat P, Samuelsson B. Arachidonic acid metabolism in polymorphonuclear leukocytes: unstable intermediate in formation of dihydroxy acids. Proc Natl Acad Sci USA. 1979;76:3213–3217. doi: 10.1073/pnas.76.7.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brock TG, McNish RW, Coffey MJ, Ojo TC, Phare SM, Peters-Golden M. Effects of granulocyte-macrophage colony-stimulating factor on eicosanoid production by mononuclear phagocytes. J Immunol. 1996;156:2522–2527. [PubMed] [Google Scholar]
- Bryant KJ, et al. A bifunctional role for group IIA secreted phospholipase A2 in human rheumatoid fibroblast-like synoviocyte arachidonic acid metabolism. J Biol Chem. 2011;286:2492–2503. doi: 10.1074/jbc.M110.123927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark JD, Lin LL, Kriz RW, Ramesha CS, Sultzman LA, Lin AY, Milona N, Knopf JL. A novel arachidonic acid-selective cytosolic PLA2 contains a Ca(2+)-dependent translocation domain with homology to PKC and GAP. Cell. 1991;65:1043–1051. doi: 10.1016/0092-8674(91)90556-e. [DOI] [PubMed] [Google Scholar]
- Cloutier A, Guindi C, Larivee P, Dubois CM, Amrani A, McDonald PP. Inflammatory cytokine production by human neutrophils involves C/EBP transcription factors. J Immunol. 2009;182:563–571. doi: 10.4049/jimmunol.182.1.563. [DOI] [PubMed] [Google Scholar]
- Dahinden CA, Zingg J, Maly FE, de Weck AL. Leukotriene production in human neutrophils primed by recombinant human granulocyte/macrophage colony-stimulating factor and stimulated with the complement component C5A and FMLP as second signals. J Exp Med. 1988;167:1281–1295. doi: 10.1084/jem.167.4.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiPersio JF, Billing P, Williams R, Gasson JC. Human granulocyte-macrophage colony-stimulating factor and other cytokines prime human neutrophils for enhanced arachidonic acid release and leukotriene B4 synthesis. J Immunol. 1988;140:4315–4322. [PubMed] [Google Scholar]
- Dixon RA, Diehl RE, Opas E, Rands E, Vickers PJ, Evans JF, Gillard JW, Miller DK. Requirement of a 5-lipoxygenase-activating protein for leukotriene synthesis. Nature. 1990;343:282–284. doi: 10.1038/343282a0. [DOI] [PubMed] [Google Scholar]
- Evans JH, Gerber SH, Murray D, Leslie CC. The calcium binding loops of the cytosolic phospholipase A2 C2 domain specify targeting to Golgi and ER in live cells. Mol Biol Cell. 2004;15:371–383. doi: 10.1091/mbc.E03-05-0338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans JH, Spencer DM, Zweifach A, Leslie CC. Intracellular calcium signals regulating cytosolic phospholipase A2 translocation to internal membranes. J Biol Chem. 2001;276:30150–30160. doi: 10.1074/jbc.M100943200. [DOI] [PubMed] [Google Scholar]
- Feng Y, Zou L, Si R, Nagasaka Y, Chao W. Bone marrow MyD88 signaling modulates neutrophil function and ischemic myocardial injury. Am J Physiol Cell Physiol. 2010;299:C760–769. doi: 10.1152/ajpcell.00155.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson AD, et al. Crystal structure of inhibitor-bound human 5-lipoxygenase-activating protein. Science. 2007;317:510–512. doi: 10.1126/science.1144346. [DOI] [PubMed] [Google Scholar]
- Ford-Hutchinson AW, Bray MA, Doig MV, Shipley ME, Smith MJ. Leukotriene B, a potent chemokinetic and aggregating substance released from polymorphonuclear leukocytes. Nature. 1980;286:264–265. doi: 10.1038/286264a0. [DOI] [PubMed] [Google Scholar]
- Geijsen N, Dijkers PF, Lammers JJ, Koenderman L, Coffer PJ. Cytokine-mediated cPLA(2) phosphorylation is regulated by multiple MAPK family members. FEBS Lett. 2000;471:83–88. doi: 10.1016/s0014-5793(00)01373-9. [DOI] [PubMed] [Google Scholar]
- Ghomashchi F, Stewart A, Hefner Y, Ramanadham S, Turk J, Leslie CC, Gelb MH. A pyrrolidine-based specific inhibitor of cytosolic phospholipase A(2)alpha blocks arachidonic acid release in a variety of mammalian cells. Biochim Biophys Acta. 2001;1513:160–166. doi: 10.1016/s0005-2736(01)00349-2. [DOI] [PubMed] [Google Scholar]
- Glover S, de Carvalho MS, Bayburt T, Jonas M, Chi E, Leslie CC, Gelb MH. Translocation of the 85-kDa phospholipase A2 from cytosol to the nuclear envelope in rat basophilic leukemia cells stimulated with calcium ionophore or IgE/antigen. J Biol Chem. 1995;270:15359–15367. doi: 10.1074/jbc.270.25.15359. [DOI] [PubMed] [Google Scholar]
- Good MC, Zalatan JG, Lim WA. Scaffold proteins: hubs for controlling the flow of cellular information. Science. 2011;332:680–686. doi: 10.1126/science.1198701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hercus TR, Thomas D, Guthridge MA, Ekert PG, King-Scott J, Parker MW, Lopez AF. The granulocyte-macrophage colony-stimulating factor receptor: linking its structure to cell signaling and its role in disease. Blood. 2009;114:1289–1298. doi: 10.1182/blood-2008-12-164004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krump E, Borgeat P. Kinetics of 5-lipoxygenase activation, arachidonic acid release, and leukotriene synthesis in human neutrophils: effects of granulocyte-macrophage colony-stimulating factor. Biochim Biophys Acta. 1994;1213:135–139. doi: 10.1016/0005-2760(94)90019-1. [DOI] [PubMed] [Google Scholar]
- Kulkarni S, Das S, Funk CD, Murray D, Cho W. Molecular basis of the specific subcellular localization of the C2-like domain of 5-lipoxygenase. J Biol Chem. 2002;277:13167–13174. doi: 10.1074/jbc.M112393200. [DOI] [PubMed] [Google Scholar]
- Lam BK, Penrose JF, Freeman GJ, Austen KF. Expression cloning of a cDNA for human leukotriene C4 synthase, an integral membrane protein conjugating reduced glutathione to leukotriene A4. Proc Natl Acad Sci USA. 1994;91:7663–7667. doi: 10.1073/pnas.91.16.7663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie CC. Regulation of the specific release of arachidonic acid by cytosolic phospholipase A2. Prostaglandins Leukot Essent Fatty Acids. 2004;70:373–376. doi: 10.1016/j.plefa.2003.12.012. [DOI] [PubMed] [Google Scholar]
- Lin LL, Wartmann M, Lin AY, Knopf JL, Seth A, Davis RJ. cPLA2 is phosphorylated and activated by MAP kinase. Cell. 1993;72:269–278. doi: 10.1016/0092-8674(93)90666-e. [DOI] [PubMed] [Google Scholar]
- Mandal AK, et al. The nuclear membrane organization of leukotriene synthesis. Proc Natl Acad Sci USA. 2008;105:20434–20439. doi: 10.1073/pnas.0808211106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal AK, et al. The membrane organization of leukotriene synthesis. Proc Natl Acad Sci USA. 2004;101:6587–6592. doi: 10.1073/pnas.0308523101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin TR, Pistorese BP, Chi EY, Goodman RB, Matthay MA. Effects of leukotriene B4 in the human lung. Recruitment of neutrophils into the alveolar spaces without a change in protein permeability. J Clin Invest. 1989;84:1609–1619. doi: 10.1172/JCI114338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McColl SR, Krump E, Naccache PH, Poubelle PE, Braquet P, Braquet M, Borgeat P. Granulocyte-macrophage colony-stimulating factor increases the synthesis of leukotriene B4 by human neutrophils in response to platelet-activating factor. Enhancement of both arachidonic acid availability and 5-lipoxygenase activation. J Immunol. 1991;146:1204–1211. [PubMed] [Google Scholar]
- Miller DK, et al. Identification and isolation of a membrane protein necessary for leukotriene production. Nature. 1990;343:278–281. doi: 10.1038/343278a0. [DOI] [PubMed] [Google Scholar]
- Nahas N, Waterman WH, Sha'afi RI. Granulocyte-macrophage colony-stimulating factor (GM-CSF) promotes phosphorylation and an increase in the activity of cytosolic phospholipase A2 in human neutrophils. Biochem J. 1996;313:503–508. doi: 10.1042/bj3130503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalefski EA, Sultzman LA, Martin DM, Kriz RW, Towler PS, Knopf JL, Clark JD. Delineation of two functionally distinct domains of cytosolic phospholipase A2, a regulatory Ca(2+)-dependent lipid-binding domain and a Ca(2+)-independent catalytic domain. J Biol Chem. 1994;269:18239–18249. [PubMed] [Google Scholar]
- Penrose JF, Gagnon L, Goppelt-Struebe M, Myers P, Lam BK, Jack RM, Austen KF, Soberman RJ. Purification of human leukotriene C4 synthase. Proc Natl Acad Sci USA. 1992;89:11603–11606. doi: 10.1073/pnas.89.23.11603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radmark O, Shimizu T, Jornvall H, Samuelsson B. Leukotriene A4 hydrolase in human leukocytes. Purification and properties. J Biol Chem. 1984;259:12339–12345. [PubMed] [Google Scholar]
- Reid GK, Kargman S, Vickers PJ, Mancini JA, Leveille C, Ethier D, Miller DK, Gillard JW, Dixon RA, Evans JF. Correlation between expression of 5-lipoxygenase-activating protein, 5-lipoxygenase, and cellular leukotriene synthesis. J Biol Chem. 1990;265:19818–19823. [PubMed] [Google Scholar]
- Rouzer CA, Bennett CD, Diehl RE, Jones RE, Kargman S, Rands E, Dixon RA. Cloning and expression of human leukocyte 5-lipoxygenase. Adv Prostaglandin Thromboxane Leukot Res. 1989;19:474–477. [PubMed] [Google Scholar]
- Rouzer CA, Kargman S. Translocation of 5-lipoxygenase to the membrane in human leukocytes challenged with ionophore A23187. J Biol Chem. 1988;263:10980–10988. [PubMed] [Google Scholar]
- Rouzer CA, Matsumoto T, Shimizu T, Samuelsson B. The human leukocyte 5-lipoxygenase: enzyme purification, multi-component regulatory system, and LTA4 synthase activity. Adv Prostaglandin Thromboxane Leukot Res. 1986;16:3–16. [PubMed] [Google Scholar]
- Rouzer CA, Samuelsson B. Reversible, calcium-dependent membrane association of human leukocyte 5-lipoxygenase. Proc Natl Acad Sci USA. 1987;84:7393–7397. doi: 10.1073/pnas.84.21.7393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scapini P, Laudanna C, Pinardi C, Allavena P, Mantovani A, Sozzani S, Cassatella MA. Neutrophils produce biologically active macrophage inflammatory protein-3alpha (MIP-3alpha)/CCL20 and MIP-3beta/CCL19. Eur J Immunol. 2001;31:1981–1988. doi: 10.1002/1521-4141(200107)31:7<1981::aid-immu1981>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Showell HJ, et al. The in vitro and in vivo pharmacologic activity of the potent and selective leukotriene B4 receptor antagonist CP-105696. J Pharmacol Exp Ther. 1995;273:176–184. [PubMed] [Google Scholar]
- Werz O, Burkert E, Fischer L, Szellas D, Dishart D, Samuelsson B, Radmark O, Steinhilber D. Extracellular signal-regulated kinases phosphorylate 5-lipoxygenase and stimulate 5-lipoxygenase product formation in leukocytes. FASEB J. 2002;16:1441–1443. doi: 10.1096/fj.01-0909fje. [DOI] [PubMed] [Google Scholar]
- Woods JW, Evans JF, Ethier D, Scott S, Vickers PJ, Hearn L, Heibein JA, Charleson S, Singer II. 5-lipoxygenase and 5-lipoxygenase-activating protein are localized in the nuclear envelope of activated human leukocytes. J Exp Med. 1993;178:1935–1946. doi: 10.1084/jem.178.6.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarini S, Gijon MA, Folco G, Murphy RC. Effect of arachidonic acid reacylation on leukotriene biosynthesis in human neutrophils stimulated with granulocyte-macrophage colony-stimulating factor and formyl-methionyl-leucyl-phenylalanine. J Biol Chem. 2006;281:10134–10142. doi: 10.1074/jbc.M510783200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.