

Abstract
Since its discovery as a src kinase substrate more than three decades ago, appreciation for the physiologic functions of annexin A2 and its associated proteins has increased dramatically. With its binding partner S100A10 (p11), A2 forms a cell surface complex that regulates generation of the primary fibrinolytic protease, plasmin, and is dynamically regulated in settings of hemostasis and thrombosis. In addition, the complex is transcriptionally upregulated in hypoxia and promotes pathologic neoangiogenesis in the tissues such as the retina. Dysregulation of both A2 and p11 has been reported in examples of rodent and human cancer. Intracellularly, A2 plays a critical role in endosomal repair in postarthroplastic osteolysis, and intracellular p11 regulates serotonin receptor activity in psychiatric mood disorders. In human studies, the A2 system contributes to the coagulopathy of acute promyelocytic leukemia, and is a target of high-titer autoantibodies in patients with antiphospholipid syndrome, cerebral thrombosis, and possibly preeclampsia. Polymorphisms in the human ANXA2 gene have been associated with stroke and avascular osteonecrosis of bone, two severe complications of sickle cell disease. Together, these new findings suggest that manipulation of the annexin A2/S100A10 system may offer promising new avenues for treatment of a spectrum of human disorders.
1. The Annexin A2/S100A10 System
1.1. Assembly of the (A2·p11)2 Complex
Annexin A2 (A2) belongs to the annexin family of Ca2+-regulated phospholipid binding proteins, which are expressed in plants, animals, and protists throughout the phylogenetic tree [1]. A2 is a 36-kilodalton protein produced by endothelial cells, monocytes, macrophages, trophoblast cells, and some tumor cells and exists both free in the cytoplasm and in association with intracellular and plasma membrane surfaces [2, 3]. The human ANXA2 gene consists of 13 exons distributed over 40 kb of genomic DNA on chromosome 15 (15q21) [4]. Among mammalian species for which A2 has been sequenced, identity is approximately 98% at the amino acid level.
When A2 is membrane associated, the tightly packed, alpha-helical 33-kDa core domain forms a disk whose convex face is associated with membrane phospholipid and whose concave face is oriented away from the membrane. Membrane binding is mediated by at least two potential Ca2+-binding “annexin” repeats, features common to all annexin family proteins [3]. While the core domains of the annexin proteins are relatively well conserved, the hydrophilic amino-terminal “tail” or “interaction” domains are highly variable and essentially unique to each family member.
Protein S100A10, also known as p11, is a well-described binding partner of A2 [7, 8]. As a member of the S100 family of proteins, p11 contains Ca2+-binding helix-loop-helix motifs and confers increased phospholipid binding affinity on A2. Typical S100 proteins undergo a conformational change upon Ca2+-binding that places helix III (HIII) in a perpendicular orientation relative to helix IV(HIV), thus forming a cleft that can accept associated target proteins [9]. This calcium activation rule, however, does not apply to p11, which has permanently assumed a “calcium-on” state, due to replacement of the bidentate E65 by S70, and the monodentate D56 with C61 [10]. The published crystal structure of p11 in complex with the N-terminal 13 amino acids of A2 suggests that the basic unit of p11 structure is a noncovalently linked homodimer, each component of which can bind the A2 tail peptide to form a heterotetramer [10]. Upon binding, the A2 tail peptide assumes an α-helical conformation that presents key hydrophobic residues (V3, I6, L7, and L10) within a cleft formed by loop L2 and helix HIV of one monomer and helix HI of the other. The C-terminal region of p11, particularly its hydrophobic residues within the C-terminal extension (Y85FVVHM90), such as Y85 and F86, contributes critical contact points for binding to A2 [10].
1.2. The (A2·p11)2 Complex and Fibrinolysis
The primary fibrinolytic protease, plasmin, is formed upon cleavage of plasminogen at a single peptide bond at position R560-V561 by either of two serine proteases, tissue plasminogen activator (tPA), produced by vascular endothelial cells, or urokinase (uPA) [11–13]. tPA-dependent plasminogen activation is dramatically accelerated in the presence of fibrin, and to a lesser extent by cell surface fibrinolytic receptors. uPAR is expressed by monocyte/macrophages, tumor cells, and activated endothelial cells [14, 15], while the (A2·p11)2 complex is expressed on both resting and activated endothelial cells [16, 17]. In addition, an interesting array of plasminogen-binding receptors, including α-enolase [18], TATA-box protein interacting protein (TIP49) [19], histone H2B [20], αMβ2 integrin [21], amphoterin [22], and Plg-RKT, have been identified on many cell types [23].
On cell surfaces, the (A2·p11)2 complex serves as an assembly site for plasminogen and tPA [16, 17, 25, 26]. Although it is clear that heterotetramer-mediated colocalization of activator and substrate accelerates plasmin generation, there are, interestingly, two main theories as to the exact site of interaction of plasminogen and tPA with components of the heterotetramer complex. While one group suggests p11 as the key ligand interaction site and annexin A2 as the molecule that anchors it to the plasma membrane [26], another proposes annexin A2, in complex with p11, as the ligand binding site [17]. A third group has suggested that, in the context of the cell surface and its proteolytic milieu, both annexin A2 and p11 may have exposed lysine residues that are accessible to the lysine binding “kringle” domains of both tPA and plasminogen [27]. Detailed evidence for each view is outlined in the works cited above.
Translocation of A2 to the outer leaflet of the plasma membrane of the endothelial cell is a key regulatory step governing vascular fibrinolysis [16, 17]. Although cell surface appearance of A2 has been linked to plasma membrane fusion of multivesicular bodies in NIH 3T3 fibroblasts [28], and as a consequence of membrane disruption upon exocytosis of secretory granules in chromaffin cells [29], it is not clear whether similar mechanisms apply to the endothelial cell. Endothelial cell translocation, which can occur within minutes, is initiated by several factors including heat stress, thrombin stimulation, and hypoxia [30–32] and is known to require the presence of adequate p11 as well as src kinase phosphorylation at Y23. A2 was originally identified as a src kinase substrate [33], and translocation is driven by activation of pp60src [31].
1.3. Regulation at the Cell Surface
Translocation of A2 to the cell surface is dependent upon the abundance of p11. In the endothelial cell, p11 is stabilized by A2, which, upon binding, masks a critical “degron,” or polyubiquitination site on p11. In the absence of sufficient A2, p11 is polyubiquitinated and targeted to the proteasome for degradation [34]. In AnxA2−/− mice, which demonstrate low to nondetectable p11 expression, treatment with bortezomib, a proteasome inhibitor, restored p11 expression, verifying its regulation via a proteasome-linked pathway in vivo. In nonendothelial cells, p11 may be stabilized by one or more of its other partner proteins, which include a number of transmembrane channels and membrane receptors, such as the tetrodotoxin-resistant sodium channel Nav1.8 [35], the two predomain K+ channel TASK-1 [36], the acid-sensing ion channel ASIC1a [37], the transient receptor potential channels TRP5 and TRP6 [38], and the 5HT-1B serotonin receptor [39].
Protein kinase C-(PKC-) mediated phosphorylation of S11 or S25 residues on A2 appears to represent an additional regulatory pathway. Serine phosphorylation within the tail domain of A2 dissociates the heterotetramer complex, preventing further translocation to the cell surface by allowing polyubiquitination of p11 and its degradation in the proteasome [40, 41]. This event appears to be initiated by plasmin, which, once generated, signals activation of conventional PKC and thus limits its own generation. This mechanism appears to require cleavage of A2 by plasmin as well as activation of toll-like receptor 4.
In this paper, we summarize evidence from both animal models and human studies on the in vivo functions of the annexin A2/S100A10 system. The concept of the “annexinopathy” was first proposed in 1999 [42], and expanded in several subsequent reviews [43–47]. Here, we focus exclusively on the growing body of evidence that annexin A2 and its partner protein p11 contribute to human health and disease.
2. In Vivo Animal Studies
2.1. Fibrin Balance and Models of Thrombosis
The AnxA2−/− mouse has been highly informative in investigating the role of the annexin A2 system in vascular homeostasis in vivo. Although A2-deficient mice display normal development, fertility, and lifespan, fibrin accumulation is evident in both intravascular and extravascular locations within the lungs, spleen, small intestine, liver, and kidney (Figure 1) [5]. Microvascular endothelial cells isolated from AnxA2−/− mice, moreover, lack the ability to support tPA-dependent plasmin generation in vitro, and arterial injury in vivo leads to an increased rate and severity of vascular occlusion in the AnxA2−/− mouse. Recently, fibrinolysis was also assessed in p11-null mice, which also displayed increased vascular fibrin, reduced clearance of thrombi, and impaired neovascularization of Matrigel thrombi [48].
Figure 1.

Fibrin accumulation in both AnxA2−/− and hyperhomocysteinemic mice. Kidney sections from AnxA2+/+ mice on normal chow (a and b), from AnxA2−/− mice on normal chow (c), or from AnxA2+/+ mice on a high methionine diet (d). Sections from heparin-treated, highly perfused animals were stained with a polyclonal rabbit antifibrin(ogen)-specific antibody using either immunoperoxidase plus diaminobenzidine (brown immunoreaction product) with hematoxylin counterstaining [5] (a and c), or biotinylated goat anti-rabbit IgG followed by Alexa 568-conjugated streptavidin (b and d); red staining represents Alexa 568, blue represents DAPI nuclear staining, and green is tissue autofluorescence [6].
Interestingly, mice with diet-induced hyperhomocysteinemia share this phenotypic feature with the AnxA2−/− mouse (Figure 1) [6]. Homocysteine (HC) is a thiol-containing amino acid that is generated during the conversion of methionine to cysteine [50]. Elevated levels of circulating HC have been associated with both thrombotic and atherosclerotic vascular disease [51], although therapies that lower plasma HC have not been shown to reduce the risk of recurrent cardiovascular disease [52]. Pretreatment, but not cotreatment, of endothelial cells with HC blocks their ability to bind tPA and inhibits endothelial cell-related, tPA-dependent plasminogen activation [53]. Incubation of purified A2 with HC, moreover, interferes with its ability to bind tPA [54]. When wild type mice were subjected to diet-induced hyperhomocysteinemia, fibrin accumulated in multiple tissues (Figure 1(d)), and extracted A2 failed to support tPA binding or tPA-dependent plasmin generation, revealing that HC-induced blockade of the cell surface A2 pathway can occur in vivo.
The potential clinical utility of recombinant annexin A2 protein (rA2) in ischemic stroke has emerged from thrombosis models in rats. Animals were treated with low-dose tPA with or without rA2 at 2 or 4 hours following the initiation of focal embolic stroke [55]. Those receiving both agents had a significantly lower infarct size and greater cerebral blood flow compared to animals treated with low-dose tPA alone. In similar experiments, in which animals underwent middle cerebral artery embolization with autologous clot, pretreatment with rA2 not only improved blood flow but also reduced infarct size compared to saline-treated controls [56]. These findings are significant in view of reported neurotoxicity and cerebral hemorrhage associated with the use of tPA in the treatment of thrombotic stroke in humans [57–59]. Thus, rA2 or related agents may constitute a useful adjunct to tPA alone for the restoration of cerebral blow flow [60].
A2 or its analogs might also prove efficacious in the treatment of peripheral arterial occlusion. When carotid artery thrombosis was induced by adventitial application of FeCl3, administration of recombinant full length, but not truncated, A2 was associated with improved cerebral blood flow and reduced thrombus size in comparison with untreated control animals [61]. This treatment had no effect on bleeding time, prothrombin time, or activated partial thromboplastin time, indicating that global clotting parameters remained intact. Thus, A2 or its analogs may constitute a useful adjuvant to conventional thrombolytic treatment by reducing the effective dose of tPA, thereby limiting its potential toxicity.
2.2. A Model of Hypercholesterolemic Atherosclerosis
The fibrinolytic system appears to modulate the development of plaque-like vascular lesions in mouse models of atherosclerosis in a complex fashion. Mice deficient in both plasminogen and apolipoprotein E (ApoE), for example, display an enhanced tendency toward atherosclerosis compared to those lacking ApoE alone, suggesting that plasminogen protects against lesion formation [62]. On the other hand, when macrophages overexpressed uPA in ApoE−/− mice, plaque development was accelerated through a plasminogen-dependent pathway [63]. When ApoE deficiency is combined with global deficiency of either uPA or tPA, however, the predilection for early fatty streaks and advanced plaque development was similar to that seen in mice with isolated ApoE deficiency [64]. These data suggest that the fibrinolytic system acts at multiple levels in the regulation atherogenesis.
In order to determine whether blocking plasmin(ogen) binding to A2 on the surface of macrophages is an effective strategy to reduce the development of atherosclerosis, ApoE−/− mice were crossed with AnxA2−/− mice to generate double nulls (Figure 2). Following weaning, ApoE−/−AnxA2−/− mice were placed on a Western chow diet (30% fat) and sacrificed at 12 or 24 wks. Aortas were removed and evaluated for lesion development by en face Oil Red O staining and morphometry of histologic sections taken through the aortic root at the base of the heart. There was no difference in en face lesion area or lesion size in ApoE−/−AnxA2+/+ mice as compared to ApoE−/−AnxA2−/− mice. Therefore, we conclude that the redundant nature of plasmin(ogen)-binding sites on macrophages renders targeting a single binding site ineffective in modulating lesion development in this model system.
Figure 2.

Diet-induced atherogenesis in AnxA2+/+ and AnxA2−/− mice. AnxA2−/− mice on the C57Bl/6 background were backcrossed to ApoE−/− mice (Jackson Laboratories) to generate AnxA2−/−ApoE−/− double knockout mice. At 3 weeks of age, both AnxA2+/+ApoE−/− and AnxA2−/−ApoE−/− mice male and female mice were placed on a western diet, composed of 1% (wt/wt) adjusted calories from fat and 0.15% (wt/wt) cholesterol (TD88137, Harland Tekland Laboratory) ad libitum, for 12 (n = 14–20 mice/group) or 24 weeks (n = 17–23 mice/group). At 12 or 24 weeks (a and b, resp.), the animals were perfused with PBS for 20 minutes, whereupon the entire aorta from the heart to 5–10 mm below the bifurcation of the iliac arteries was removed and fixed in 4% paraformaldehyde. The aorta was evaluated for lesion development by en face Oil red O staining, and morphometry of digital images of the stained aortas was performed using Adobe Photoshop 7.0 software [24]. For aortic root analyses, hearts removed at 24 weeks (c) were fixed in 3% PFA and paraffin embedded. Serial sections, 10 microns in thickness, obtained from the region of the proximal aorta to the level of the aortic leaflet, were stained with hematoxylin and eosin. Digital microscopic images were analyzed using image analysis software (NIH Image 1.63). Lesion size for each mouse was calculated as the average lesion size in 10–15 sections over a distance of 200 to 300 microns in the aortic root. Representative lesions are shown.
2.3. Angiogenesis and Oxygen-Induced Retinopathy
Although embryonic vasculogenesis appears to be normal, AnxA2−/− mice display diminished neovascularization in several in vivo assays, including Matrigel implant, corneal pocket, and oxygen-induced retinopathy (OIR) models [5]. Mice with diet-induced hyperhomocysteinemia also display impaired corneal neoangiogenesis, which can be corrected upon intravenous injection of recombinant annexin A2 [6]. Microvascular endothelial cells from AnxA2−/− mice, as well as HC-treated human endothelial cells, moreover, migrate less efficiently in growth factor-enriched Matrigel. Together, these data suggest that absence of A2, or its modification by HC, leads to impairment of angiogenesis-related endothelial cell function.
Interestingly, annexin A2 is upregulated in OIR (Figure 3). In this model, newborn mouse pups are transitioned to room air after 5 days in a 75% oxygen environment, whereupon relative hypoxia initiates a robust vascular proliferative response in the retina [66]. The return to 21% oxygen also triggers A2 synthesis out of proportion to the increase in vascular endothelial cell abundance [32]. The retinal neoangiogenic response is inhibited by about 50% in the AnxA2−/− mouse. A2 expression is also increased in the endothelial cell under true hypoxia through the direct action of hypoxia-inducible factor-1 (HIF-1) with the A2 promoter. Electrophoretic mobility shift experiments, chromatin immunoprecipitation studies, and luciferase promoter reporter assays all indicate binding of HIF-1α and HIF-1β to a hypoxia-responsive element within the promoter region of the human A2 gene, leading to its activation.
Figure 3.
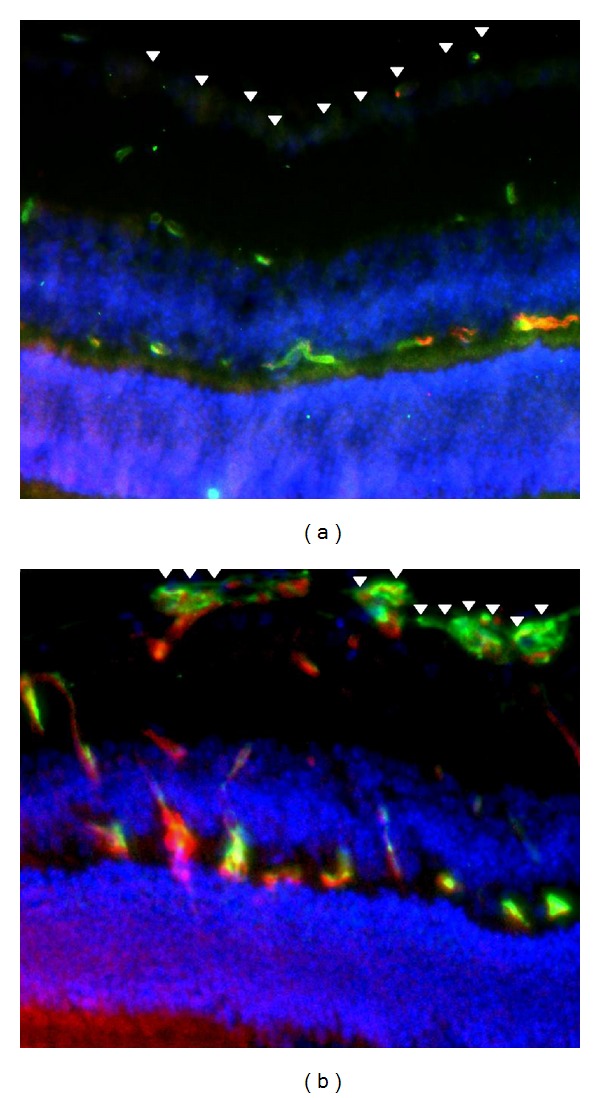
A2-expressing neovessels in oxygen-induced retinopathy in the mouse. Sections through retinas of wild type mice maintained in room air (a) or treated for five days with 75% oxygen to initiate oxygen-induced retinopathy (b) were stained with an endothelial cell-specific lectin (isolectin B4—green), 4′,6-diamidino-2-phenylindole for nuclei (DAPI—blue), and anti-A2 (red) as described [32]. Note A2-positive neovascular tufts arrayed at the inner limiting membrane of the oxygen-treated retina (b, arrowheads) versus control (a, arrowheads).
Although OIR-associated retinal neovascularization is impaired in the AnxA2−/− mouse, it can be reestablished upon treatment of AnxA2−/− mice with a subretinal injection of an A2-encoding adenovirus, which restores A2 expression [32]. In addition, neovascularization of the hyperoxia-treated AnxA2−/− retina can be repaired upon treatment with the defibrinating agent ancrod, which depletes fibrinogen, thereby preventing fibrin formation. Together, these findings provide a link between fibrin accumulation and diminished neoangiogenesis and imply that new therapeutic avenues for proliferative retinal vascular disorders, such as retinopathy of prematurity or diabetic retinopathy, could involve blockade of A2 in addition to inhibition of angiogenic growth factors.
2.4. Models of Cancer
In the central nervous system, glioblastomas, malignant tumors derived from glial cells, are usually highly aggressive and refractory to treatment due to the early development of widespread infiltrative loci [67]. Glioma-generated proteases, such as plasminogen activators and matrix metalloproteinases, contribute substantially to glioma cell invasion [67, 68]. High concentrations of annexin A2, similarly, are associated with the pseudopodia of invasive glioma cells [69], and knockdown of A2 reduces their migratory capacity in vitro [70]. In both mouse and rat in vivo models, stable knockdown of A2 expression in glioblastoma cells retarded overall tumor progression upon implantation of the cells into rodent brains; cellular invasion, proliferation, apoptosis, and angiogenesis were all inhibited [49]. Interestingly, when A2 expression was stably reduced by transfection of RNAi directed against A2 in rat GL261 glioma cells, tumor growth and invasiveness were reduced, even though p11 expression persisted; this result indicated that the contribution of A2 to tumor invasiveness was p11 independent (Figure 4). These data suggest that A2-directed treatment could offer a new therapeutic modality for human glioblastoma.
Figure 4.
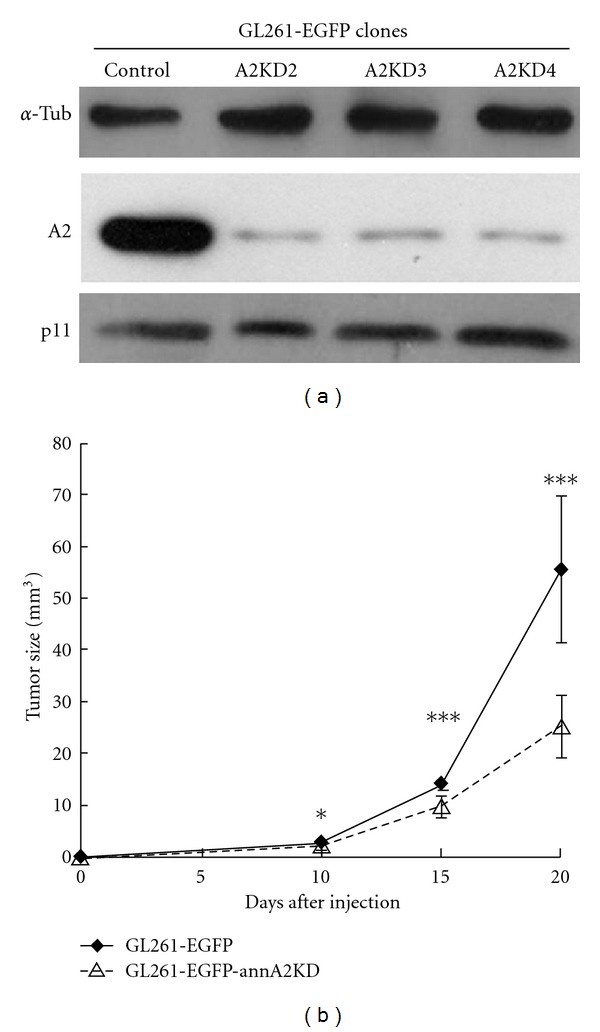
The glioblastoma-promoting effect of A2 is p11 independent. (a) When A2 expression in GL261 murine glioma cells was suppressed with siRNA, p11 expression remained intact. (b) Suppression of A2 expression significantly dampened the growth of intracranial gliomas derived from stereotactically implanted cells. Reproduced with permission [49].
In a xenograft model in which highly invasive and metastatic breast cancer cells were implanted into nude mice, both tumor growth and vascular density were blocked by administration of anti-A2 monoclonal antibody [71]. The tumor cells employed in this experiment expressed abundant A2, strongly supported tPA binding and tPA-dependent plasmin generation and exhibited plasmin- and A2-dependent cellular matrix invasion [72, 73]. These studies suggest that A2 may contribute to aggressive breast cancer cellular invasion and tumor angiogenesis through production of localized protease activity.
In a third in vivo model, growth of Lewis lung and T241 sarcoma tumors implanted into p11-deficient mice was markedly reduced compared to wild type controls [74]. Impaired tumor growth was correlated with diminished macrophage density within the tumors, and clodronate-mediated depletion of macrophages in wild type mice led to a similar reduction in tumor size. This study recapitulates the finding that thioglycollate-induced macrophage invasion into the peritoneum, and macrophage invasion of subcutaneous Matrigel plugs, were also impaired in the p11 knockout mouse [75]. A related study reveals that soluble (A2·p11)2 tetramer activates human and murine monocyte-derived macrophages, that this activation requires toll-like receptor 4 (TLR-4), and that the tetramer modulates cytokine production in the macrophage [76]. Thus, tumor infiltration by macrophages may in part be due to (A2·p11)2 tetramer signaling.
2.5. Aseptic Osteolysis
Arthroplastic surgery has revolutionized the treatment of arthritis and related rheumatologic disorders. Nevertheless, aseptic osteolysis, due to generation of wear debris particles (WDP), is an emerging problem that leads to failure of 10–30% of all joint replacements [77]. Recent investigations have thus focused on the mechanism by which WDP induce joint inflammation. Alkane polymers, 8–12 carbon atoms in length and derived from the breakdown of WDP, bind directly to toll-like receptors 1 and 2 and activate the downstream signaling pathway [78]. In addition, endocytosed WDP can induce endosomal membrane damage and disruption in phagocytic cells, and this process is associated with dramatic recruitment of cytoplasmic annexin A2 to the endosomal membrane (Figure 5). In the absence of A2, endosomal disruption leads to leakage of lysosomal cathepsins and H+ ions into the cytosol with subsequent activation of the NLRP inflammasome and an accelerated inflammatory response [65].
Figure 5.

Intracellular A2 redistributes to lysosomal and late endosomal membranes in dendritic cells upon endocytosis of wear debris particles (WDP). (a) Western blot analysis of A2 and p11 localization in late endosomes (LE) and lysosomes (LYS) isolated from WDP-treated dendritic cells. (b) Confocal analysis of untreated and 12 h WDP-treated dendritic cells stained with anti-LAMP1 (green) and anti-A2 (red) revealed well-delineated LAMP+ endosomal compartments with cytosolic distribution of A2 in control cells. Cells cultured with WDP for 12 h (b) demonstrated an extensive increase in the number and size of the endosomal compartments with the subsequent loss of the cytosolic distribution of A2. Bar corresponds to 10 mm. Reproduced with permission [65].
2.6. Neuropsychiatric Disease
A large body of work has focused on the role of p11 in neuropsychiatric function. p11 binds to both the serotonin 1B and serotonin 4 receptors, suggesting a role for p11 in regulation of mood [39]. p11-deleted mice show depression-like behavior, characterized by increased immobility in the tail suspension test, increased thigmotaxis, and decreased responsiveness to a sucrose reward. In wild type mice, adenovirus-mediated deletion of p11 specifically within the nucleus accumbens (NA) resulted in depressive behavior, indistinguishable from that seen in mice with global p11 deficiency; exogenous administration of p11 within the NA of p11-deleted mice restored normal behavior [79]. These data correlate with findings in human depression, in which p11 protein levels were reduced in the NA. Furthermore, reduced p11 mRNA levels in peripheral blood mononuclear cells may serve as a potential biomarker for patients at high risk of suicide [80]. These studies raise the possibility that some forms of human depression may be reversible by augmentation of p11 expression [81]. Indeed, commonly used anti-inflammatory drugs that attenuate the antidepressive effects of serotonin reuptake inhibitors may do so by inhibiting the effects of interferonγ, a known inducer of p11 [82, 83].
p11 also potentiates nociception. The tetrodotoxin-resistant sodium channel (Nav1.8/SNS), whose expression is restricted to sensory neurons, is the major pain perception receptor and is expressed in 85% of neurons emerging from the dorsal route ganglia. p11 binds to the amino terminus of the Nav1.8 protein and promotes its translocation to the plasma membrane to produce functional channels [35]. Deletion of p11 specifically in primary nociceptor sensory neurons was achieved using Nav1.8 promoter-directed Cre recombinase and led to a loss of tetrodotoxin-resistant sodium current density, and severe compromise of noxious coding in sensory neurons from the dorsal root horn [84]. Thus, directed p11 targeting may prove useful in the treatment of refractory pain disorders.
3. Lessons from the Clinic
3.1. Acute Promyelocytic Leukemia
At diagnosis, acute promyelocytic leukemia (APL) is commonly associated with life-threatening hemorrhage [85]. In APL, clonal expansion of immature promyelocytes harboring a balanced chromosomal translocation (t(15; 17)(q22–24; q12–21)) gives rise to the transcriptionally active promyelocytic leukemia-retinoic acid receptor α (PML-RARα) fusion protein. APL frequently responds to differentiation therapy with all-trans retinoic acid (ATRA), which triggers degradation of PML-RARα [86]. While disseminated intravascular coagulation promotes coagulopathy in APL, consumption of the plasmin inhibitor alpha2-antiplasmin and development of a hyperfibrinolytic state due to excessive plasmin generation is also a primary driver.
High-level expression of annexin A2 occurs specifically in APL blast cells (Figure 6). A2 was detected in blast cells recovered from 6 of 6 APL patients, all of whom had evidence of hyperfibrinolysis, as evidenced by elevated circulating fibrin degradation products and D-dimer and depletion of plasma fibrinogen [87]. NB4 cells, which carry the t(15; 17) translocation and express the PML-RARα fusion protein, displayed steady state A2 mRNA levels that were approximately 10-fold higher than those found on leukemia cells that lacked the fusion protein. Treatment of NB4 cells with the retinoic acid receptor ligand, all-trans retinoic acid (ATRA), attenuated A2 expression in a time frame associated with clinical resolution of bleeding [87]. Intracranial bleeding, an unusually frequent problem in APL, may be due to the relatively high level expression of the A2 system on cerebral microvascular endothelial cells compared to those of other vascular beds [85]. Elevated expression of p11 in NB4 cells was also recently demonstrated and shown to respond to treatment with ATRA [88].
Figure 6.
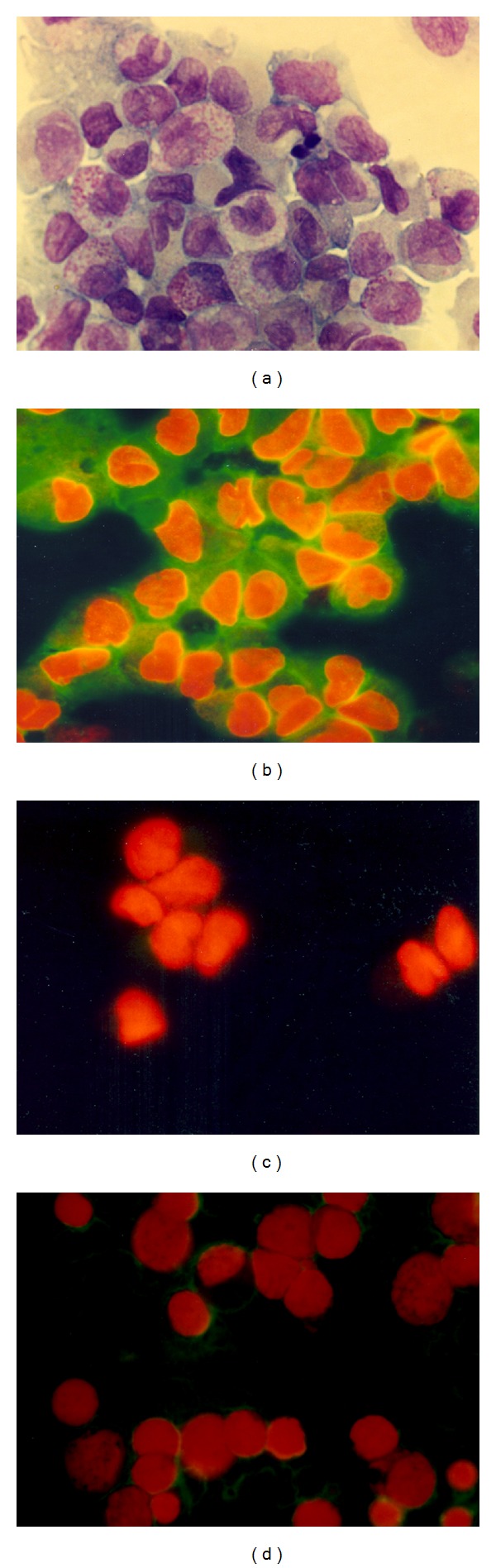
Expression of A2 is specifically elevated in acute promyelocytic leukemia cells. (a) Wright-Giemsa stain of human acute promyelocytic leukemia (APL) cells (M3). (b) APL cells stained with anti-A2 antibody (green fluorescence) and counterstained with ethidium bromide to visualize nuclei. (c) APL cells stained with nonimmune IgG and ethidium bromide. (d) Undifferentiated leukemia cells (M0) stained with anti-A2 antibody and ethidium bromide.
In a second clinical study, a cohort of 26 patients were studied prospectively and found to have enhanced fibrinolysis at diagnosis, despite normal tPA levels and increased PAI-1 [89]. APL cells harvested from these subjects expressed 3-fold higher levels of A2, and their rate of tPA-dependent plasmin generation was similarly elevated over that seen in the presence of M1, M2, M4, or acute lymphoblastic leukemia cells. Both elevated A2 expression in blast cells and hyperfibrinolytic hemorrhage corrected in 23 patients upon treatment with differentiation therapy, consisting of all-trans retinoic acid (ATRA) or ATRA plus arsenic trioxide. This study confirms the role of the A2 system in fibrinolytic bleeding in patients with APL.
3.2. Solid Tumors
Expression levels of annexin A2 have been examined in a variety of human malignancies. In some, such as renal cell [90–92], gastric [93], prostate [94], pancreatic [95], breast [72] carcinoma, and osteogenic sarcoma [96], increased expression levels appear to correlate with higher histologic grade and/or development of distant metastases. In human glioblastoma, A2 expression correlates with histologic grade and CNS dissemination [49, 97–99]. In human breast cancer, A2 appears to be associated with the surface of invasive, malignant cells, but not normal ductal or acinar epithelial cells, and expression correlated with neoangiogenic activity [73]. Proteomic profiling of colorectal cancer, moreover, revealed differentially increased expression of A2 in tumors that had progressed to lymph node metastases versus localized tumors [100]. Primary multiple myeloma cells harvested from a cohort of patients displayed 10-fold higher cell surface A2 expression than that observed on normal plasma cells; silencing of A2 in related cell lines suppressed expression of proangiogenic genes [101]. A2 and a related A2-binding “receptor” has been reported to promote myeloma cell adhesion and growth in the bone marrow [102]. On the other hand, A2 expression in oral squamous cell carcinoma [103] or sinonasal adenocarcinoma [104] was found to be inversely related to histopathologic grade. These studies suggest that expression levels of A2 may have prognostic value in malignancy, but would need to be validated for each specific tumor.
Expression of p11 in distinct tumor types has been less extensively studied. High levels of both p11 and A2 were found in 100% of anaplastic thyroid carcinomas, and correlated with their aggressive behavior [105]. In a comprehensive study of S100 gene expression in over 300 primary breast cancers, both p11 (S100A10) and S100A11 were selectively upregulated in basal versus nonbasal breast cancer subtypes, but did not predict overall survival [106]. Among 62 cases of human esophageal squamous cell carcinoma, 11 of 12 S100 genes, including p11, were downregulated, based on reverse transcription-polymerase chain reaction assays [107]. p11 transcripts have also been reported to be increased in both renal cell and gastric carcinomas [92, 108, 109]. Further studies may define p11 expression as a viable biomarker or prognostic indicator in selected tumors.
3.3. Antiphospholipid Syndrome
Antiphospholipid syndrome (APS) is a major cause of acquired thrombophilia [110]. It is characterized by thrombosis and recurrent fetal loss in association with circulating antiphospholipid antibodies. The latter are distinct, often coexisting antibodies directed against either β2-glycoprotein I or other intravascular proteins, which may be found in complex with anionic membrane phospholipids [111]. A2 has been identified as a prominent target of autoantibodies arising specifically in patients with APS with severe thrombosis and/or pregnancy morbidity [112, 113]. In vitro, patient-derived antiannexin A2 antibodies blocked endothelial surface tPA-dependent plasmin generation, and also “activated” cultured endothelial cells, inciting them to express elevated levels of the prothrombotic agent, tissue factor [112]. Other groups have noted that A2 can serve as a binding site for β(2)-glycoprotein I [114] in APS and can initiate A2-dependent endothelial cell activation [115]. Together, these data implicate A2 in the pathogenesis of APS-associated thrombosis through several possible mechanisms [116].
3.4. Cerebral Thrombosis
Cerebral venous thrombosis is a rare disorder of unknown etiology that mainly affects children and young adults [117]. Among a cohort of 40 consecutive patients studied 2 to 6 months following the index thrombotic event, 12.5% were found to have high titer anti-A2 antibodies compared to 2.1% in healthy subjects [118]. Thus, anti-A2 may define a new subset of individuals with immune-mediated thrombosis, play a role in the pathogenesis of this disorder, and/or offer novel therapeutic targets.
3.5. Preeclampsia and Pregnancy Loss
Both A2 and p11 are expressed on the brush border of the placental syncytiotrophoblast [119]. In 60 patients with preeclampsia, A2 mRNA and protein levels in placenta were significantly reduced compared to those in placentas from 30 matched normal pregnancy controls [120]. High titer anti-A2 antibodies, moreover, were detected more frequently in sera from subjects in the pre-eclamptic group and associated with increased placental vascular thrombosis. Impaired local fibrinolytic function due to blunted A2 expression may contribute to the pathogenesis of pre-eclampsia and maternal and perinatal infant morbidity.
3.6. Sickle Hemoglobinopathy
Sickle hemoglobinopathy arises from a point mutation within the 6th codon of the human β-globin chain [121]. Polymerization of abnormal hemoglobin under deoxygenating conditions induces erythrocyte shape change and non deformability, which leads to vascular occlusion, impaired vasodilatation, distal ischemia, and endothelial cell activation with adhesion of leukocytes. In children, sickle cell disease is complicated by stroke in 6–8% of patients. A recent analysis of 108 single nucleotide polymorphisms in 39 candidate genes revealed that variations in the annexin A2 (ANXA2) gene, among several others, were associated with increased risk of stroke [122]. A second independent study linked ANXA2 polymorphisms to increased risk for stroke in sickle cell disease [123], while additional ANXA2 SNPs have been associated with avascular necrosis of bone (osteonecrosis) in sickle hemoglobinopathy [124]. These data suggest that annexin A2 may represent a significant modifier gene that shapes the clinical expression and natural history of sickle cell disease.
References
- 1.Moss SE, Morgan RO. The annexins. Genome Biology. 2004;5(4, article 219) doi: 10.1186/gb-2004-5-4-219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gerke V, Creutz CE, Moss SE. Annexins: linking Ca2+ signalling to membrane dynamics. Nature Reviews Molecular Cell Biology. 2005;6(6):449–461. doi: 10.1038/nrm1661. [DOI] [PubMed] [Google Scholar]
- 3.Gerke V, Moss SE. Annexins: from structure to function. Physiological Reviews. 2002;82(2):331–371. doi: 10.1152/physrev.00030.2001. [DOI] [PubMed] [Google Scholar]
- 4.Spano F, Raugei G, Palla E, Colella C, Melli M. Characterization of the human lipocortin-2-encoding multigene family: its structure suggests the existence of a short amino acid unit undergoing duplication. Gene. 1990;95(2):243–251. doi: 10.1016/0378-1119(90)90367-z. [DOI] [PubMed] [Google Scholar]
- 5.Ling Q, Jacovina AT, Deora A, et al. Annexin II regulates fibrin homeostasis and neoangiogenesis in vivo. The Journal of Clinical Investigation. 2004;113(1):38–48. doi: 10.1172/JCI200419684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jacovina AT, Deora AB, Ling Q, et al. Homocysteine inhibits neoangiogenesis in mice through blockade of annexin A2-dependent fibrinolysis. The Journal of Clinical Investigation. 2009;119(11):3384–3394. doi: 10.1172/JCI39591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Waisman DM. Annexin II tetramer: structure and function. Molecular and Cellular Biochemistry. 1995;149-150:301–322. doi: 10.1007/BF01076592. [DOI] [PubMed] [Google Scholar]
- 8.Rescher U, Gerke V. S100A10/p11: family, friends and functions. Pflugers Archiv European Journal of Physiology. 2008;455(4):575–582. doi: 10.1007/s00424-007-0313-4. [DOI] [PubMed] [Google Scholar]
- 9.Donato R. S100: a multigenic family of calcium-modulated proteins of the EF-hand type with intracellular and extracellular functional roles. International Journal of Biochemistry and Cell Biology. 2001;33(7):637–668. doi: 10.1016/s1357-2725(01)00046-2. [DOI] [PubMed] [Google Scholar]
- 10.Réty S, Sopkova J, Renouard M, et al. The crystal structure of a complex of p11 with the annexin II N-terminal peptide. Nature Structural Biology. 1999;6(1):89–95. doi: 10.1038/4965. [DOI] [PubMed] [Google Scholar]
- 11.Cesarman-Maus G, Hajjar KA. Molecular mechanisms of fibrinolysis. British Journal of Haematology. 2005;129(3):307–321. doi: 10.1111/j.1365-2141.2005.05444.x. [DOI] [PubMed] [Google Scholar]
- 12.Hajjar KA. The molecular basis of fibrinolysis. In: Orkin SH, Nathan DG, Ginsburg D, Look AT, Fisher DE, Lux SE, editors. Nathan and Oski's Hematology of Infancy and Childhood. Philadelphia, Pa, USA: Saunders Elsevier; 2009. pp. 1425–1447. [Google Scholar]
- 13.Hajjar KA, Ruan J. Fibrinolysis and thrombolysis. In: Kaushansky K, Lichtman MA, Beutler E, Kipps TJ, Seligsohn U, Prchal JT, editors. Williams Hematology. New York, NY, USA: McGraw-Hill; 2010. pp. 2219–2246. [Google Scholar]
- 14.Blasi F, Carmeliet P. uPAR: a versatile signalling orchestrator. Nature Reviews Molecular Cell Biology. 2002;3(12):932–943. doi: 10.1038/nrm977. [DOI] [PubMed] [Google Scholar]
- 15.Blasi F, Sidenius N. The urokinase receptor: focused cell surface proteolysis, cell adhesion and signaling. FEBS Letters. 2010;584(9):1923–1930. doi: 10.1016/j.febslet.2009.12.039. [DOI] [PubMed] [Google Scholar]
- 16.Dassah M, Deora AB, He K, Hajjar KA. The endothelial cell annexin A2 system and vascular fibrinolysis. General Physiology and Biophysics. 2009;28:F20–F28. [PMC free article] [PubMed] [Google Scholar]
- 17.Flood EC, Hajjar KA. The annexin A2 system and vascular homeostasis. Vascular Pharmacology. 2011;54(3–6):59–67. doi: 10.1016/j.vph.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miles LA, Dahlberg CM, Plescia J, Felez J, Kato K, Plow EF. Role of cell-surface lysines in plasminogen binding to cells: identification of α-enolase as a candidate plasminogen receptor. Biochemistry. 1991;30(6):1682–1691. doi: 10.1021/bi00220a034. [DOI] [PubMed] [Google Scholar]
- 19.Hawley SB, Tamura TA, Miles LA. Purification, cloning, and characterization of a profibrinolytic plasminogen-binding protein, TIP49a. The Journal of Biological Chemistry. 2001;276(1):179–186. doi: 10.1074/jbc.M004919200. [DOI] [PubMed] [Google Scholar]
- 20.Herren T, Burke TA, Das R, Plow EF. Identification of histone H2B as a regulated plasminogen receptor. Biochemistry. 2006;45(31):9463–9474. doi: 10.1021/bi060756w. [DOI] [PubMed] [Google Scholar]
- 21.Pluskota E, Soloviev DA, Bdeir K, Cines DB, Plow EF. Integrin αMβ2 Orchestrates and Accelerates Plasminogen Activation and Fibrinolysis by Neutrophils. The Journal of Biological Chemistry. 2004;279(17):18063–18072. doi: 10.1074/jbc.M310462200. [DOI] [PubMed] [Google Scholar]
- 22.Parkkinen J, Rauvala H. Interactions of plasminogen and tissue plasminogen activator (t-PA) with amphoterin: enhancement of t-PA-catalyzed plasminogen activation by amphoterin. The Journal of Biological Chemistry. 1991;266(25):16730–16735. [PubMed] [Google Scholar]
- 23.Andronicos NM, Chen EI, Baik N, et al. Proteomics-based discovery of a novel, structurally unique, and developmentally regulated plasminogen receptor, Plg-RKT, a major regulator of cell surface plasminogen activation. Blood. 2010;115(7):1319–1330. doi: 10.1182/blood-2008-11-188938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kraemer R, Baker PJ, Kent KC, et al. Decreased neurotrophin TrkB receptor expression reduces lesion size in the apolipoprotein E-null mutant mouse. Circulation. 2005;112(23):3644–3653. doi: 10.1161/CIRCULATIONAHA.105.587980. [DOI] [PubMed] [Google Scholar]
- 25.Kwon M, MacLeod TJ, Zhang Y, Waisman DM. S100A10, annexin A2, and annexin A2 heterotetramer as candidate plasminogen receptors. Frontiers in Bioscience. 2005;10(1):300–325. doi: 10.2741/1529. [DOI] [PubMed] [Google Scholar]
- 26.Madureira PA, Surette AP, Phipps KD, Taboski MAS, Miller VA, Waisman DM. The role of the annexin A2 heterotetramer in vascular fibrinolysis. Blood. 2011;118(18):4789–4797. doi: 10.1182/blood-2011-06-334672. [DOI] [PubMed] [Google Scholar]
- 27.Das R, Burke T, Plow EF. Histone H2B as a functionally important plasminogen receptor on macrophages. Blood. 2007;110(10):3763–3772. doi: 10.1182/blood-2007-03-079392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Valapala M, Vishwanatha JK. Lipid raft endocytosis and exosomal transport facilitate extracellular trafficking of annexin A2. The Journal of Biological Chemistry. 2011;286(35):30911–30925. doi: 10.1074/jbc.M111.271155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Faure AV, Migné C, Devilliers G, Ayala-Sanmartin J. Annexin 2 “secretion” accompanying exocytosis of chromaffin cells: possible mechanisms of annexin release. Experimental Cell Research. 2002;276(1):79–89. doi: 10.1006/excr.2002.5512. [DOI] [PubMed] [Google Scholar]
- 30.Peterson EA, Sutherland MR, Nesheim ME, Pryzdial ELG. Thrombin induces endothelial cell-surface exposure of the plasminogen receptor annexin 2. Journal of Cell Science. 2003;116(12):2399–2408. doi: 10.1242/jcs.00434. [DOI] [PubMed] [Google Scholar]
- 31.Deora AB, Kreitzer G, Jacovina AT, Hajjar KA. An annexin 2 phosphorylation switch mediates p11-dependent translocation of annexin 2 to the cell surface. The Journal of Biological Chemistry. 2004;279(42):43411–43418. doi: 10.1074/jbc.M408078200. [DOI] [PubMed] [Google Scholar]
- 32.Huang B, Deora AB, He K-L, et al. Hypoxia-inducible factor-1 drives annexin A2 system-mediated perivascular fibrin clearance in oxygen-induced retinopathy in mice. Blood. 2011;118(10):2918–2929. doi: 10.1182/blood-2011-03-341214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Erikson E, Erikson RL. Identification of a cellular protein substrate phosphorylated by the avian sarcoma virus-transforming gene product. Cell. 1980;21(3):829–836. doi: 10.1016/0092-8674(80)90446-8. [DOI] [PubMed] [Google Scholar]
- 34.He KL, Deora AB, Xiong H, et al. Endothelial cell annexin A2 regulates polyubiquitination and degradation of its binding partner S100A10/p11. The Journal of Biological Chemistry. 2008;283(28):19192–19200. doi: 10.1074/jbc.M800100200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Okuse K, Malik-Hall M, Baker MD, et al. Annexin II light chain regulates sensory neuron-specific sodium channel expression. Nature. 2002;417(6889):653–656. doi: 10.1038/nature00781. [DOI] [PubMed] [Google Scholar]
- 36.Girard C, Tinel N, Terrenoire C, Romey G, Lazdunski M, Borsotto M. p11, an annexin II subunit, an auxiliary protein associated with the background K+ channel, TASK-1. The EMBO Journal. 2002;21(17):4439–4448. doi: 10.1093/emboj/cdf469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Donier E, Rugiero F, Okuse K, Wood JN. Annexin II light chain p11 promotes functional expression of acid-sensing ion channel ASIC1a. The Journal of Biological Chemistry. 2005;280(46):38666–38672. doi: 10.1074/jbc.M505981200. [DOI] [PubMed] [Google Scholar]
- 38.Van de Graaf SFJ, Hoenderop JGJ, Gkika D, et al. Functional expression of the epithelial Ca2+ channels (TRPV5 and TRPV6) requires association of the S100A10-annexin 2 complex. The EMBO Journal. 2003;22(7):1478–1487. doi: 10.1093/emboj/cdg162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Svenningsson P, Chergui K, Rachleff I, et al. Alterations in 5-HT1B receptor function by p11 in depression-like states. Science. 2006;311(5757):77–80. doi: 10.1126/science.1117571. [DOI] [PubMed] [Google Scholar]
- 40.He KL, Sui G, Xiong H, et al. Feedback regulation of endothelial cell surface plasmin generation by PKC-dependent phosphorylation of annexin A2. The Journal of Biological Chemistry. 2011;286(17):15428–15439. doi: 10.1074/jbc.M110.185058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jost M, Gerke V. Mapping of a regulatory important site for protein kinase C phosphorylation in the N-terminal domain of annexin II. Biochimica et Biophysica Acta. 1996;1313(3):283–289. doi: 10.1016/0167-4889(96)00101-2. [DOI] [PubMed] [Google Scholar]
- 42.Rand JH. ‘Annexinopathies’—a new class of diseases. The New England Journal of Medicine. 1999;340(13):1035–1036. doi: 10.1056/NEJM199904013401310. [DOI] [PubMed] [Google Scholar]
- 43.Rand JH. The annexinopathies: a new category of diseases. Biochimica et Biophysica Acta. 2000;1498(2-3):169–173. doi: 10.1016/s0167-4889(00)00093-8. [DOI] [PubMed] [Google Scholar]
- 44.Hayes MJ, Moss SE. Annexins and disease. Biochemical and Biophysical Research Communications. 2004;322(4):1166–1170. doi: 10.1016/j.bbrc.2004.07.124. [DOI] [PubMed] [Google Scholar]
- 45.Hayes MJ, Longbottom RE, Evans MA, Moss SE. Annexinopathies. Sub-Cellular biochemistry. 2007;45:1–28. doi: 10.1007/978-1-4020-6191-2_1. [DOI] [PubMed] [Google Scholar]
- 46.Fatimathas L, Moss SE. Annexins as disease modifiers. Histology and Histopathology. 2010;25(4):527–532. doi: 10.14670/HH-25.527. [DOI] [PubMed] [Google Scholar]
- 47.Heizmann CW, Ackermann GE, Galichet A. Pathologies involving the S100 proteins and RAGE. Sub-cellular Biochemistry. 2007;45:93–138. doi: 10.1007/978-1-4020-6191-2_5. [DOI] [PubMed] [Google Scholar]
- 48.Surette AP, Madureira PA, Phipps KD, Miller VA, Svenningsson P, Waisman DM. Regulation of fibrinolysis by S100A10 in vivo. Blood. 2011;118(11):3172–3181. doi: 10.1182/blood-2011-05-353482. [DOI] [PubMed] [Google Scholar]
- 49.Zhai H, Acharya S, Gravanis I, et al. Annexin A2 promotes tumor progression in glioma cells. The Journal of Neuroscience. 2011;31:14346–14360. doi: 10.1523/JNEUROSCI.3299-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Selhub J. Homocysteine metabolism. Annual Review of Nutrition. 1999;19:217–246. doi: 10.1146/annurev.nutr.19.1.217. [DOI] [PubMed] [Google Scholar]
- 51.Humphrey LL, Fu R, Rogers K, Freeman M, Helfand M. Homocysteine level and coronary heart disease incidence: a systematic review and meta-analysis. Mayo Clinic Proceedings. 2008;83(11):1203–1212. doi: 10.4065/83.11.1203. [DOI] [PubMed] [Google Scholar]
- 52.Bønaa KH, Njølstad I, Ueland PM, et al. Homocysteine lowering and cardiovascular events after acute myocardial infarction. The New England Journal of Medicine. 2006;354(15):1578–1588. doi: 10.1056/NEJMoa055227. [DOI] [PubMed] [Google Scholar]
- 53.Hajjar KA. Homocysteine-induced modulation of tissue plasminogen activator binding to its endothelial cell membrane receptor. The Journal of Clinical Investigation. 1993;91(6):2873–2879. doi: 10.1172/JCI116532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hajjar KA, Mauri L, Jacovina AT, et al. Tissue plasminogen activator binding to the annexin II tail domain: direct modulation by homocysteine. The Journal of Biological Chemistry. 1998;273(16):9987–9993. doi: 10.1074/jbc.273.16.9987. [DOI] [PubMed] [Google Scholar]
- 55.Zhu H, Fan X, Yu Z, et al. Annexin A2 combined with low-dose tPA improves thrombolytic therapy in a rat model of focal embolic stroke. Journal of Cerebral Blood Flow and Metabolism. 2010;30(6):1137–1146. doi: 10.1038/jcbfm.2009.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tanaka Y, Ishii H, Hiraoka M, et al. Efficacy of recombinant annexin 2 for fibrinolytic therapy in a rat embolic stroke model: a magnetic resonance imaging study. Brain Research. 2007;1165(1):135–143. doi: 10.1016/j.brainres.2007.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yepes M, Sandkvist M, Moore EG, Bugge TH, Strickland DK, Lawrence DA. Tissue-type plasminogen activator induces opening of the blood-brain barrier via the LDL receptor-related protein. The Journal of Clinical Investigation. 2003;112(10):1533–1540. doi: 10.1172/JCI19212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang X, Tsuji K, Lee SR, et al. Mechanisms of hemorrhagic transformation after tissue plasminogen activator reperfusion therapy for ischemic stroke. Stroke. 2004;35(11):2726–2730. doi: 10.1161/01.STR.0000143219.16695.af. [DOI] [PubMed] [Google Scholar]
- 59.Kaur J, Zhao Z, Klein GM, Lo EH, Buchan AM. The neurotoxicity of tissue plasminogen activator? Journal of Cerebral Blood Flow and Metabolism. 2004;24(9):945–963. doi: 10.1097/01.WCB.0000137868.50767.E8. [DOI] [PubMed] [Google Scholar]
- 60.Fan X, Yu Z, Liu J, et al. Annexin A2: a tissue plasminogen activator amplifier for thrombolytic stroke therapy. Stroke. 2010;41(10):S54–S58. doi: 10.1161/STROKEAHA.110.596106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ishii H, Yoshida M, Hiraoka M, et al. Recombinant annexin II modulates impaired fibrinolytic activity in vitro and in rat carotid artery. Circulation Research. 2001;89(12):1240–1245. doi: 10.1161/hh2401.101066. [DOI] [PubMed] [Google Scholar]
- 62.Xiao Q, Danton MJS, Witte DP, et al. Plasminogen deficiency accelerates vessel wall disease in mice predisposed to atherosclerosis. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(19):10335–10340. doi: 10.1073/pnas.94.19.10335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kremen M, Krishnan R, Emery I, et al. Plasminogen mediates the atherogenic effects of macrophage-expressed urokinase and accelerates atherosclerosis in apoE-knockout mice. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(44):17109–17114. doi: 10.1073/pnas.0808650105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Carmeliet P, Moons L, Lijnen R, et al. Urokinase-generated plasmin activates matrix metalloproteinases during aneurysm formation. Nature Genetics. 1997;17(4):439–444. doi: 10.1038/ng1297-439. [DOI] [PubMed] [Google Scholar]
- 65.Scharf B, Clement CC, Wu X-X, et al. Annexin A2 binds to endosomes following organelle destabilization by particulate wear debris. Nature Communications. 2012;3, article 755 doi: 10.1038/ncomms1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Smith LEH, Wesolowski E, McLellan A, et al. Oxygen-induced retinopathy in the mouse. Investigative Ophthalmology and Visual Science. 1994;35(1):101–111. [PubMed] [Google Scholar]
- 67.Rao JS. Molecular mechanisms of glioma invasiveness: the role of proteases. Nature Reviews Cancer. 2003;3(7):489–501. doi: 10.1038/nrc1121. [DOI] [PubMed] [Google Scholar]
- 68.Tsatas D, Kaye AH. The role of the plasminogen activation cascade in glioma cell invasion: a review. Journal of Clinical Neuroscience. 2003;10(2):139–145. doi: 10.1016/s0967-5868(02)00328-4. [DOI] [PubMed] [Google Scholar]
- 69.Beckner ME, Chen X, An J, Day BW, Pollack IF. Proteomic characterization of harvested pseudopodia with differential gel electrophoresis and specific antibodies. Laboratory Investigation. 2005;85(3):316–327. doi: 10.1038/labinvest.3700239. [DOI] [PubMed] [Google Scholar]
- 70.Tatenhorst L, Rescher U, Gerke V, Paulus W. Knockdown of annexin 2 decreases migration of human glioma cells in vitro. Neuropathology and Applied Neurobiology. 2006;32(3):271–277. doi: 10.1111/j.1365-2990.2006.00720.x. [DOI] [PubMed] [Google Scholar]
- 71.Sharma M, Blackman MR, Sharma MC. Antibody-directed neutralization of annexin II (ANX II) inhibits neoangiogenesis and human breast tumor growth in a xenograft model. Experimental and Molecular Pathology. 2012;92(1):175–184. doi: 10.1016/j.yexmp.2011.10.003. [DOI] [PubMed] [Google Scholar]
- 72.Sharma MR, Koltowski L, Ownbey RT, Tuszynski GP, Sharma MC. Angiogenesis-associated protein annexin II in breast cancer: selective expression in invasive breast cancer and contribution to tumor invasion and progression. Experimental and Molecular Pathology. 2006;81(2):146–156. doi: 10.1016/j.yexmp.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 73.Sharma M, Ownbey RT, Sharma MC. Breast cancer cell surface annexin II induces cell migration and neoangiogenesis via tPA dependent plasmin generation. Experimental and Molecular Pathology. 2010;88(2):278–286. doi: 10.1016/j.yexmp.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 74.Phipps KD, Surette AP, O'Connell PA, Waisman DM. Plasminogen receptor S100A10 is essential for the migration of tumor-promoting macrophages into tumor sites. Cancer Research. 2011;71(21):6676–6683. doi: 10.1158/0008-5472.CAN-11-1748. [DOI] [PubMed] [Google Scholar]
- 75.O’Connell PA, Surette AP, Liwski RS, Svenningsson P, Waisman DM. S100A10 regulates plasminogen-dependent macrophage invasion. Blood. 2010;116(7):1136–1146. doi: 10.1182/blood-2010-01-264754. [DOI] [PubMed] [Google Scholar]
- 76.Swisher JFA, Burton N, Bacot SM, Vogel SN, Feldman GM. Annexin A2 tetramer activates human and murine macrophages through TLR4. Blood. 2010;115(3):549–558. doi: 10.1182/blood-2009-06-226944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cobelli N, Scharf B, Crisi GM, Hardin J, Santambrogio L. Mediators of the inflammatory response to joint replacement devices. Nature Reviews Rheumatology. 2011;7(10):600–608. doi: 10.1038/nrrheum.2011.128. [DOI] [PubMed] [Google Scholar]
- 78.Maitra R, Clement CC, Scharf B, et al. Endosomal damage and TLR2 mediated inflammasome activation by alkane particles in the generation of aseptic osteolysis. Molecular Immunology. 2009;47(2-3):175–184. doi: 10.1016/j.molimm.2009.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Alexander B, Warner-Schmidt J, Eriksson T, et al. Reversal of depressed behaviors in mice by p11 gene therapy in the nucleus accumbens. Science Translational Medicine. 2010;2(54) doi: 10.1126/scitranslmed.3001079.54ra76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhang L, Su TP, Choi K, et al. P11 (S100A10) as a potential biomarker of psychiatric patients at risk of suicide. Journal of Psychiatric Research. 2011;45(4):435–441. doi: 10.1016/j.jpsychires.2010.08.012. [DOI] [PubMed] [Google Scholar]
- 81.Chen G, Twyman R, Manji HK. p11 and gene therapy for severe psychiatric disorders: a practical goal? Science Translational Medicine. 2010;2(54) doi: 10.1126/scitranslmed.3001754.54ps51 [DOI] [PubMed] [Google Scholar]
- 82.Huang XL, Pawliczak R, Yao XL, et al. Interferon-γ induces p11 gene and protein expression in human epithelial cells through interferon-γ-activated sequences in the p11 promoter. The Journal of Biological Chemistry. 2003;278(11):9298–9308. doi: 10.1074/jbc.m212704200. [DOI] [PubMed] [Google Scholar]
- 83.Warner-Schmidt JL, Vanover KE, Chen EY, Marshall JJ, Greengard P. Antidepressant effects of selective serotonin reuptake inhibitors (SSRIs) are attenuated by antiinflammatory drugs in mice and humans. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(22):9262–9267. doi: 10.1073/pnas.1104836108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Foulkes T, Nassar MA, Lane T, et al. Deletion of annexin 2 light chain p11 in nociceptors causes deficits in somatosensory coding and pain behavior. The Journal of Neuroscience. 2006;26(41):10499–10507. doi: 10.1523/JNEUROSCI.1997-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Stein E, McMahon B, Kwaan H, Altman JK, Frankfurt O, Tallman MS. The coagulopathy of acute promyelocytic leukaemia revisited. Best Practice and Research. 2009;22(1):153–163. doi: 10.1016/j.beha.2008.12.007. [DOI] [PubMed] [Google Scholar]
- 86.Nasr R, Guillemin MC, Ferhi O, et al. Eradication of acute promyelocytic leukemia-initiating cells through PML-RARA degradation. Nature Medicine. 2008;14(12):1333–1342. doi: 10.1038/nm.1891. [DOI] [PubMed] [Google Scholar]
- 87.Menell JS, Cesarman GM, Jacovina AT, McLaughlin MA, Lev EA, Hajjar KA. Annexin II and bleeding in acute promyelocytic leukemia. The New England Journal of Medicine. 1999;340(13):994–1004. doi: 10.1056/NEJM199904013401303. [DOI] [PubMed] [Google Scholar]
- 88.O’Connell PA, Madureira PA, Berman JN, Liwski RS, Waisman DM. Regulation of S100A10 by the PML-RAR-α oncoprotein. Blood. 2011;117(15):4095–4105. doi: 10.1182/blood-2010-07-298851. [DOI] [PubMed] [Google Scholar]
- 89.Liu Y, Wang Z, Jiang M, et al. The expression of annexin II and its role in the fibrinolytic activity in acute promyelocytic leukemia. Leukemia Research. 2011;35(7):879–884. doi: 10.1016/j.leukres.2010.11.008. [DOI] [PubMed] [Google Scholar]
- 90.Ohno Y, Izumi M, Kawamura T, Nishimura T, Mukai K, Tachibana M. Annexin II represents metastatic potential in clear-cell renal cell carcinoma. British Journal of Cancer. 2009;101(2):287–294. doi: 10.1038/sj.bjc.6605128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zimmermann U, Woenckhaus C, Pietschmann S, et al. Expression of annexin II in conventional renal cell carcinoma is correlated with Fuhrman grade and clinical outcome. Virchows Archiv. 2004;445(4):368–373. doi: 10.1007/s00428-004-1103-4. [DOI] [PubMed] [Google Scholar]
- 92.Domoto T, Miyama Y, Suzuki H, et al. Evaluation of S100A10, annexin II and B-FABP expression as markers for renal cell carcinoma. Cancer Science. 2007;98(1):77–82. doi: 10.1111/j.1349-7006.2006.00355.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Emoto K, Sawada H, Yamada Y, et al. Annexin II overexpression is correlated with poor prognosis in human gastric carcinoma. Anticancer Research. 2001;21(2):1339–1346. [PubMed] [Google Scholar]
- 94.Banerjee AG, Liu J, Yuan Y, et al. Expression of biomarkers modulating prostate cancer angiogenesis: differential expression of annexin II in prostate carcinomas from India and USA. Molecular Cancer. 2003;2, article 34 doi: 10.1186/1476-4598-2-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Esposito I, Penzel R, Chaib-Harrireche M, et al. Tenascin C and annexin II expression in the process of pancreatic carcinogenesis. Journal of Pathology. 2006;208(5):673–685. doi: 10.1002/path.1935. [DOI] [PubMed] [Google Scholar]
- 96.Mintz MB, Sowers R, Brown KM, et al. An expression signature classifies chemotherapy-resistant pediatric osteosarcoma. Cancer Research. 2005;65(5):1748–1754. doi: 10.1158/0008-5472.CAN-04-2463. [DOI] [PubMed] [Google Scholar]
- 97.Reeves SA, Chavez-Kappel C, Davis R, Rosenblum M, Israel MA. Developmental regulation of annexin II (Lipocortin 2) in human brain and expression in high grade glioma. Cancer Research. 1992;52(24):6871–6876. [PubMed] [Google Scholar]
- 98.Nygaard SJT, Haugland HK, Kristoffersen EK, Lund-Johansen M, Laerum OD, Tysnes OB. Expression of annexin II in glioma cell lines and in brain tumor biopsies. Journal of Neuro-Oncology. 1998;38(1):11–18. doi: 10.1023/a:1005953000523. [DOI] [PubMed] [Google Scholar]
- 99.Roseman BJ, Bollen A, Hsu J, Lamborn K, Israel MA. Annexin II marks astrocytic brain tumors of high histologic grade. Oncology Research. 1994;6(12):561–567. [PubMed] [Google Scholar]
- 100.Pei H, Zhu H, Zeng S, et al. Proteome analysis and tissue microarray for profiling protein markers associated with lymph node metastasis in colorectal cancer. Journal of Proteome Research. 2007;6(7):2495–2501. doi: 10.1021/pr060644r. [DOI] [PubMed] [Google Scholar]
- 101.Bao H, Jiang M, Zhu M, Sheng F, Ruan J, Ruan C. Overexpression of Annexin II affects the proliferation, apoptosis, invasion and production of proangiogenic factors in multiple myeloma. International Journal of Hematology. 2009;90(2):177–185. doi: 10.1007/s12185-009-0356-8. [DOI] [PubMed] [Google Scholar]
- 102.D'Souza S, Kurihara N, Shiozawa Y, et al. Annexin II interactions with the annexin II receptor enhance multiple myeloma cell adhesion and growth in the bone marrow microenvironment. Blood. 2012;119(8):1888–1896. doi: 10.1182/blood-2011-11-393348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Rodrigo JP, Lequerica-Fernández P, Rosado P, Allonca E, García-Pedrero JM, De Vicente JC. Clinical significance of annexin A2 downregulation in oral squamous cell carcinoma. Head and Neck. 2011;33(12):1708–1714. doi: 10.1002/hed.21661. [DOI] [PubMed] [Google Scholar]
- 104.Rodrigo JP, García-Pedrero JM, Llorente JL, et al. Down-regulation of annexin A1 and A2 protein expression in intestinal-type sinonasal adenocarcinomas. Human Pathology. 2011;42(1):88–94. doi: 10.1016/j.humpath.2010.05.017. [DOI] [PubMed] [Google Scholar]
- 105.Ito Y, Arai K, Nozawa R, et al. S100A10 expression in thyroid neoplasms originating from the follicular epithelium: contribution to the aggressive characteristic of anaplastic carcinoma. Anticancer Research. 2007;27(4):2679–2683. [PubMed] [Google Scholar]
- 106.McKiernan E, McDermott EW, Evoy D, Crown J, Duffy MJ. The role of S100 genes in breast cancer progression. Tumour Biology. 2011;32(3):441–450. doi: 10.1007/s13277-010-0137-2. [DOI] [PubMed] [Google Scholar]
- 107.Ji J, Zhao L, Wang X, et al. Differential expression of S100 gene family in human esophageal squamous cell carcinoma. Journal of Cancer Research and Clinical Oncology. 2004;130(8):480–486. doi: 10.1007/s00432-004-0555-x. [DOI] [PubMed] [Google Scholar]
- 108.Teratani T, Watanabe T, Kuwahara F, et al. Induced transcriptional expression of calcium-binding protein S100A1 and S100A10 genes in human renal cell carcinoma. Cancer Letters. 2002;175(1):71–77. doi: 10.1016/s0304-3835(01)00724-8. [DOI] [PubMed] [Google Scholar]
- 109.El-Rifai W, Moskaluk CA, Abdrabbo MK, et al. Gastric cancers overexpress S100A calcium-binding proteins. Cancer Research. 2002;62(23):6823–6826. [PubMed] [Google Scholar]
- 110.Cockrell E, Espinola RG, McCrae KR. Annexin A2: biology and relevance to the antiphospholipid syndrome. Lupus. 2008;17(10):943–951. doi: 10.1177/0961203308095329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Cohen D, Berger SP, Steup-Beekman GM, Bloemenkamp KWM, Bajema IM. Diagnosis and management of the antiphospholipid syndrome. BMJ. 2010;340(7756):1125–1132. doi: 10.1136/bmj.c2541. [DOI] [PubMed] [Google Scholar]
- 112.Cesarman-Maus G, Ríos-Luna NP, Deora AB, et al. Autoantibodies against the fibrinolytic receptor, annexin 2, in antiphospholipid syndrome. Blood. 2006;107(11):4375–4382. doi: 10.1182/blood-2005-07-2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ao W, Zheng H, Chen XW, Shen Y, Yang CD. Anti-annexin II antibody is associated with thrombosis and/or pregnancy morbidity in antiphospholipid syndrome and systemic lupus erythematosus with thrombosis. Rheumatology International. 2011;31(7):865–869. doi: 10.1007/s00296-010-1379-4. [DOI] [PubMed] [Google Scholar]
- 114.Zhang J, McCrae KR. Annexin A2 mediates endothelial cell activation by antiphospholipid/anti- β2 glycoprotein I antibodies. Blood. 2005;105(5):1964–1969. doi: 10.1182/blood-2004-05-1708. [DOI] [PubMed] [Google Scholar]
- 115.Allen KL, Fonseca FV, Betapudi V, Willard B, Zhang J, McCrae KR. A novel pathway for human endothelial cell activation by antiphospholipid/anti-β2 glycoprotein I antibodies. Blood. 2012;119(3):884–893. doi: 10.1182/blood-2011-03-344671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Krone KA, Allen KL, McCrae KR. Impaired fibrinolysis in the antiphospholipid syndrome. Current Rheumatology Reports. 2010;12(1):53–57. doi: 10.1007/s11926-009-0075-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Jackson BF, Porcher FK, Zapton DT, Losek JD. Cerebral sinovenous thrombosis in children: diagnosis and treatment. Pediatric Emergency Care. 2011;27(9):874–880. doi: 10.1097/PEC.0b013e31822c9ccc. [DOI] [PubMed] [Google Scholar]
- 118.Cesarman-Maus G, Cantú-Brito C, Barinagarrementeria F, et al. Autoantibodies against the fibrinolytic receptor, annexin A2, in cerebral venous thrombosis. Stroke. 2011;42(2):501–503. doi: 10.1161/STROKEAHA.110.592121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kaczan-Bourgois D, Salles JP, Hullin F, et al. Increased content of Annexin II (p36) and p11 in human placenta brush-border membrane vesicles during syncytiotrophoblast maturation and differentiation. Placenta. 1996;17(8):669–676. doi: 10.1016/s0143-4004(96)80017-8. [DOI] [PubMed] [Google Scholar]
- 120.Xin H, Zhang Y, Wang H, Sun S. Alterations of profibrinolytic receptor annexin A2 in pre-eclampsia: a possible role in placental thrombin formation. Thrombosis Research. 2012;129(5):563–567. doi: 10.1016/j.thromres.2011.07.039. [DOI] [PubMed] [Google Scholar]
- 121.Heeney M, Dover GJ. Sickle cell disease. In: Orkin SH, Nathan DG, Ginsburg D, Look AT, Fisher DE, Lux SE, editors. Hematology of Infancy and Childhood. Philadelphia, Pa, USA: Saunders Elsevier; 2009. pp. 949–941. [Google Scholar]
- 122.Sebastiani P, Ramoni MF, Nolan V, Baldwin CT, Steinberg MH. Genetic dissection and prognostic modeling of overt stroke in sickle cell anemia. Nature Genetics. 2005;37(4):435–440. doi: 10.1038/ng1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Flanagan JM, Frohlich DM, Howard TA, et al. Genetic predictors for stroke in children with sickle cell anemia. Blood. 2011;117(24):6681–6684. doi: 10.1182/blood-2011-01-332205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Baldwin C, Nolan VG, Wyszynski DF, et al. Association of klotho, bone morphogenic protein 6, and annexin A2 polymorphisms with sickle cell osteonecrosis. Blood. 2005;106(1):372–375. doi: 10.1182/blood-2005-02-0548. [DOI] [PMC free article] [PubMed] [Google Scholar]