

Abstract
The O2 and NO reactivity of a Cr(II) complex bearing a 12-membered tetraazamacrocyclic TMC ligand, [CrII(12-TMC)(Cl)]+ (1), and the NO reactivity of its peroxo derivative, [CrIV(12-TMC)(O2)(Cl)]+ (2), are described. By contrast to the previously reported Cr(III)-superoxo complex, [CrIII(14-TMC)(O2)(Cl)]+, a Cr(IV)-peroxo complex (2) is formed in the reaction of 1 and O2. Full spectroscopic and X-ray analysis reveals that 2 possesses a side-on η2-peroxo ligation. A quantitative reaction of 2 with NO affords a reduction in Cr oxidation state and production of a Cr(III)-nitrato complex, [CrIII(12-TMC)(NO3)(Cl)]+ (3). The latter is suggested to form via a Cr(III)-peroxynitrite intermediate. A Cr(II)-nitrosyl complex, [CrII(12-TMC)(NO)(Cl)]+ (4), derived from 1 andNO could also be synthesized; however, it does not react with O2.
Mononuclear metal-dioxygen species (M-O2), such as metal-peroxo and -superoxo complexes, are critical components of processes leading to practical substrate oxidative transformations.1 They are also invoked as key intermediates in enzymatic reactions incorporating oxygen atoms into newly biosynthesized molecules as well as in the deleterious reactions of biological oxidative stress and enzymatic detoxification reactions of reactive oxygen species.2 In biomimetic studies, synthetic analogues of the M-O2 intermediates have been intensively investigated to understand factors that control their geometric and electronic properties and chemical reactivity to form related active oxygen species or effect substrate oxidations.3
Recently, metal complexes with O2-derived ligands bearing N-tetramethylated cyclam (TMC)4 chelates have been the subject of intense scrutiny.5-8 In these studies, it has been demonstrated that the nature of the TMC supporting ligands plays important roles in regulating the stability, geometric and electronic structure, and reactivity of the dioxygen or reduced derivatives in metal-O2 complexes. One notable example is the finding that [Ni(O2)(n-TMC)]+ complexes vary depending on the ring size of the TMC ligands; a side-on Ni(III)-peroxo complex is formed with 12-TMC,9 whereas a 14-TMC ligand9 affords an end-on Ni(II)-superoxo complex (Scheme 1).6 These results demonstrate that the supporting TMC ligand modulates not only the binding mode of the O2 ligand (e.g., an end- on superoxo vs a side-on peroxo) but also the preferred oxidation state of the Ni ion (e.g., Ni2+ vs Ni3+).
Scheme 1.
Metal ion/nitric oxide (NO) interactions are of great interest, since nitric oxide plays important roles in physiological processes, including as a signaling agent and in the mammalian immune response.10 However, overproduction of NO can lead to toxicological processes via reactive nitrogen species (RNS) formation, including NO2 and peroxynitrite. One manner in which proper NO levels are maintained is when microbial or mammalian heme protein NO dioxygenases (including hemoglobins, Hbs) mediate the reaction of O2 (via FeIII - superoxo species) and NO to yield the biologically benign nitrate ion (NO3−).11,12 In biomimetic studies, mononuclear metal-O2 complexes, best described as metal-superoxo species, have been recently shown to react with NO, giving peroxynitrite (−OON=O; “normally” formed from NO plus O2•−)11,13 species or chemistry.14-16 However, to the best of our knowledge, the reaction of NO with metal-peroxo intermediates has been rarely investigated.
We now report the synthesis, spectroscopic characterization, and crystal structure of a side-on Cr(IV)-peroxo complex bearing a 12-TMC ligand, [CrIV(12-TMC)(O2)(Cl)]+; a crystal structure of an end-on Cr(III)-superoxo complex bearing a 14-TMC ligand, [CrIII(14-TMC)(O2)(Cl)]+,7 was reported previously (see Scheme 1). We also describe the NO dioxygenase chemistry of the Cr(IV)-peroxo complex, in which a Cr(III)-nitrato complex, [CrIII(12-TMC)(NO3)(Cl)]+, is formed quantitatively.
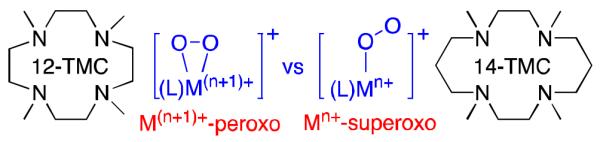
The starting material, [CrII(12-TMC)(Cl)]+ (1), was synthesized by reacting CrCl2 with 12-TMC ligand in CH3CN under Ar (see Experimental Section, Figure S2, and Tables S1 and S2 in Supporting Information (SI)). Bubbling O2 through a blue solution of 1 in CH3CN at −20 °C produced a purple intermediate 2, which persisted for several days at room temperature. The UV-vis spectrum of 2 shows a distinct absorption band at 532 nm (ε = 310 M−1 cm−1, Figure 1a). Electrospray ionization mass spectrum (ESI-MS) of 2 exhibits a prominent ion peak at a mass-to-charge ratio (m/z) of 346.9 (Figure 1b), whose mass and isotope distribution pattern correspond to [Cr(12-TMC)(O2)(Cl)]+ (calculated m/z of 347.1). When the reaction was carried out with isotopically labeled 18O2, a peak corresponding to [Cr(12-TMC)(18O2)(Cl)]+ (2-18O) appeared at m/z of 350.9 (calculated m/z of 351.1) (Figure 1b, inset). The observation of a four mass unit increase upon the substitution of 16O with 18O indicates that 2 contains an O2 unit. The resonance Raman (rRaman) spectrum of 2 was collected using 407-nm excitation in CH3CN at −20 °C. The sample of 2 prepared with 16O2 exhibits an isotope sensitive band at 865 cm−1, which shifts to 820 cm−1 when 2 is prepared with 18O2, consistent with its assignment as an O-O peroxide stretching vibration,17 also on the basis of the 16Δ - 18Δ value of 45 cm−1 (16Δ - 18Δ (calculated) = 49 cm−1) (Figure 1a, inset). This value is comparable to those recorded for spectroscopically and structurally characterized side-on metal-peroxo complexes bearing TMC ligands, such as [Fe(14-TMC)(O2)]+ (825 cm−1),8a [Co(12-TMC)(O2)]+ (902 cm−1),8b and [Ni(12-TMC)(O2)]+ (1002 cm−1).6b Moreover, 2 (1 mM) in CH3CN at 5 K reveals a silent EPR spectrum, consistent with the tetravalency assigned to Cr ion in 2.
Figure 1.
(a) UV-vis spectra of 1 (black) and 2 (red) in CH3CN at 0 °C. Inset; rRaman spectra of 2 prepared with 16O2 (red) and 18O2 (blue) and the difference spectrum (black). (b) ESI-MS of 2. Inset; isotope distribution patterns for 2-16O (red) and 2-18O (blue). The peak at m/z = 315.3 with asterisk was assigned to [CrII(12-TMC)(Cl)]+ (calculated m/z of 315.1).
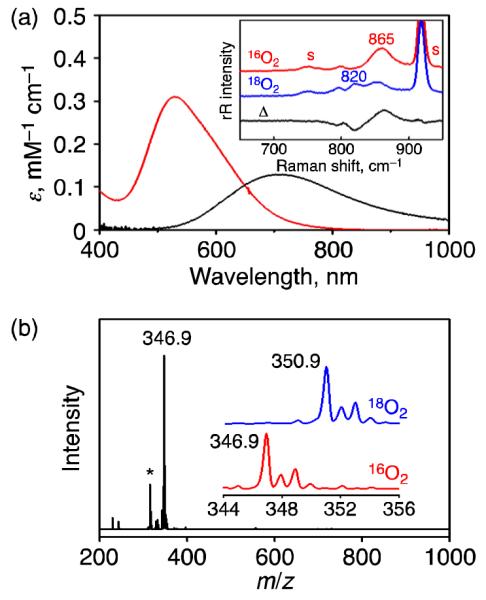
The X-ray crystal structure of 2 reveals that the Cr center is situated in a 7-coordinated environment with the 12-TMC, O2, and an aqua ligand (Figure 2a). Interestingly, it was found that the aqua ligand coordinates to the Cr center, and two chloride anions exist as counter anions in the crystal packing to formulate the crystal as [Cr(12-TMC)(O2)(OH2)]Cl2 (2′•Cl2), although the chloride ligand is found within the formula of 2 in ESI-MS measurements.18 Notably, the O-O moiety was found as side-on manner to the Cr center, and the O-O bond length in 2′ (1.394 Å) is longer than that of the end-on Cr(III)-superoxo complex, [CrIII(14-TMC)(O2)(Cl)]+ (1.231 Å).7a 2′ should be categorized as a metal-peroxo (O-O = ~1.4 - 1.5 Å) rather than a metal-superoxo complex (O-O = ~1.2 - 1.3 Å).17 The O-O bond length in 2′ is short; however, a similar O-O bond distance is found for a related nickel-peroxo complex (1.386 Å), ([NiIII(12-TMC)(O2)]+).6b In 2′, all four N-methyl groups are pointed syn to the peroxo moiety, which allows this ligand to be cis to the aqua ligand, whereas those in the Cr-superoxo complex, [CrIII(14-TMC)(O2)(Cl)]+, are oriented anti to the superoxo moiety.7a Crystallographic data and selected bond distances (Å) and angles (°) for 2′•Cl2 are summarized in SI, Tables S1 and S3.19
Figure 2.
ORTEP diagram of (a) [CrIV(12-TMC)(O2)(OH2)]2+ (2′) and (b) [CrIII(12-TMC)(NO3)(Cl)]+ (3) with 30% probability thermal ellipsoid. Hydrogen atoms are omitted for clarity. Gray, carbon; blue, nitrogen; red, oxygen; sky blue, chromium; orange, chlorine.
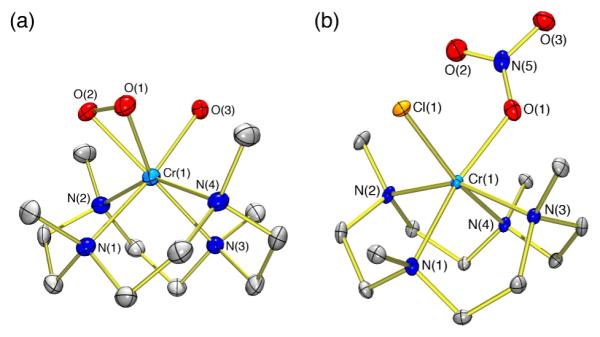
To further elaborate the chemistry of [CrIV(12-TMC)(O2)(Cl)]+ (2), we reacted it with nitric oxide (NO(g)). Complex 2 in CH3CN (2 mM) was bubbled with Ar for 30 min at 0 °C, and an excess amount of NO(g) was then added over the top of this solution. The UV-vis spectral changes observedreveal isosbestic points at 383, 447, and 605 nm (Figure 3a). In an ESI-MS measurement, the peak cluster centered at m/z of 377.0 is assigned to a Cr(III)-nitrato complex formulated as [CrIII(12-TMC)(NO3)(Cl)]+ (3) (calculated m/z of 377.1) (Figure 3b). An EPR spectrum of 3 reveals signals expected of a d3 chromium(III) (S = 3/2) (SI, Figure S3).20 Further, an X-ray crystal structure of 3 reveals that a nitrate anion coordinates to the chromium center bearing the 12-TMC ligand to give a six-coordinated configuration (Figure 2b).21 Selected bond distances (Å) and angles (°) are given in SI, Table S4.
Figure 3.
(a) UV-vis spectral changes of 2 (2 mM) upon addition of NO(g) (10 cm3, ~1 atm) into the head-space of an CH3CN solution at 0 °C under Ar (2, black line; 3, red line). (b) ESI-MS spectrum of 3. Inset; isotope distribution pattern for 3 at m/z of 377.1. The peaks at m/z of 350.1 and 332.1 with asterisk are assigned to [CrIII(12-TMC)(Cl2)]+ and [CrIII(12-TMC)(OH)(Cl)]+, respectively.
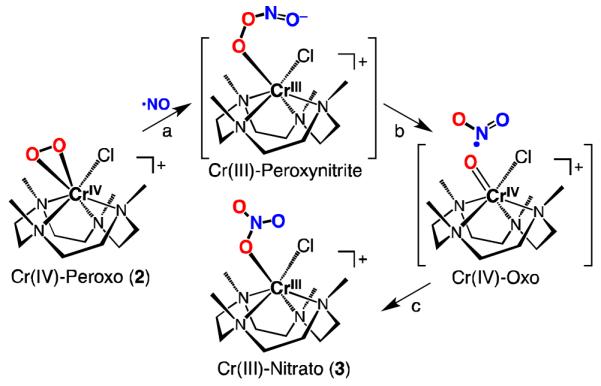
The yield of reaction is quantitative based on the UV-vis spectral change and the absorptivity of authentic complex 3 at 415 nm (ε = 180 M−1cm−1).22 According to the results, we propose a reaction mechanism as depicted in Scheme 2. As for the generally accepted course of reactions for Fe(III)-superoxo species with NO by Hbs,11,12b the first step should be the nucleophilic attack of [CrIV(O2)(12-TMC)(Cl)]+ (2) on NO. As the Cr-peroxo species has a one-electron further reduced O2 fragment (a peroxo character), formation of a Cr-peroxynitrite intermediate would result in metal reduction, giving (CrIII-(−OON=O)) (Scheme 2, step a). Although we have proposed the reaction between the Cr(IV)-peroxo species and NO, we cannot rule out the presence of an undetectable equilibrium between Cr(IV)-peroxo and Cr(III)-superoxo species in solution, and that the latter, present in tiny amounts, is what reacts with NO to give the Cr(III)-peroxynitrite intermediate. Qualitatively, we observe that increased amounts of NO lead to faster reactions, suggesting a direct interaction of nitric oxide with 2. Subsequent homolytic O-O bond cleavage of the peroxynitrite moiety would lead to a Cr(IV)-oxo species and nitrogen dioxide (NO2) (Scheme 2, step b); as suggested for many examples,10b,11,23c the latter would rebind to the Cr(IV)-oxo species to form the observed Cr(III)-nitrato complex (Scheme 2, step c). The lifetime of such a metal-bound peroxynitrite species is expected to be extremely short.23 The one isolated metal-peroxynitrite complex is [Co(CN)5(OONO)]3−;24 most recently, another well characterized (IR, isotope labeling, DFT calculations) CoIII-peroxynitrite complex has been confirmed.25 Thus, isolation of Cr(III)-nitrato complex 3 supports the intermediacy of a Cr(III)-peroxynitrite species. Even stronger evidence is that addition of 2,4-di-t-butylphenol to a solution of 2 (for which no reaction is observed) prior to NO addition, leads to a 93% yield of 2,4-di-t-butyl-6-nitrophenol, indicating the added phenol can trap the peroxynitrite intermediate.
Scheme 2.
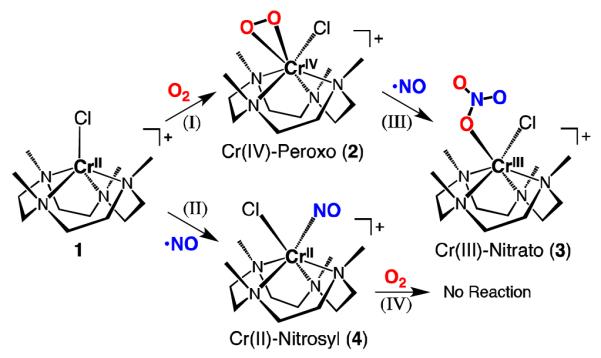
Supplemental experiments were performed with a Cr(II)-nitrosyl complex, [CrII(12-TMC)(NO)(Cl)]Cl (4•Cl),26 to complete a systematic study of [Cr(12-TMC)]2+ complex with O2 and NO. Reactions of metal-nitrosyl complexes with O2 have a considerable history, including with iron,14 cobalt,15 and copper16 systems, leading to nitrate or nitrite products through suggested peroxynitrite complex generation. Interestingly, different from the iron,14 cobalt,15 and copper16 complexes, the chromium complex 4 shows no reactivity toward O2 in acetonitrile at room temperature.
In summary, we have successfully constructed a systematic study of the [CrII(12-TMC)]2+ complex with O2 and NO. The reactions performed are summarized in Scheme 3, where theat starting Cr(II) complex 1 reacts with O2 or NO to produce a Cr(IV)-peroxo (2) (reaction I) or Cr(II)-nitrosyl complex (4) (reaction II), respectively. Complex 2 was isolated and characterized as a side-on Cr(IV)-peroxo complex which notably is very stable in acetonitrile even at room temperature. It is notable that for all TMC ligated transition metal-peroxo complexes,5 2 is the first to be formed from O2 reaction with a reduced metal ion, rather than via use of hydrogen peroxide as the origin of the final peroxide fragment. The Cr(IV)-peroxo complex (2) reacts with NO to give a Cr(III)-nitrato complex (3) (reaction III), which is most likely formed through a Cr(III)-peroxynitrite intermediate and involving atypical metal ion reduction chemistry. Different from many other metalnitrosyl complexes,14-16 the Cr(II)-nitrosyl complex (4) did not react with O2 (reaction IV). We are continuing our efforts to systematically characterize TMC chelated redox-active first row transition metal complexes, and their O2 and/or peroxide and/or O2/NO derived chemistry, that as further seen here, which is strongly influenced by the TMC ring size.
Scheme 3.
Supplementary Material
ACKNOWLEDGMENT
The authors gratefully acknowledge research support of this work by the NRF of Korea through CRI, WCU (R31-2008-000-10010-0), and GRL (2010-00353) Programs (W.N.), the Ministry of Education, Culture, Sports, Science and Technology of Japan through the Global COE program and Priority Area (No. 22018026) (T.O.), and the USA National Institutes of Health (K.D.K.).
Footnotes
ASSOCIATED CONTENT
Supporting Information. Experimental section, crystallographic data, crystal structure of 1, UV-vis spectrum and ESI-MS of 4•Cl, IR spectrum of 4•Cl, and EPR spectra of 3 and 4. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).Sheldon RA, Kochi JK. Metal-Catalyzed Oxidations of Organic Compounds. Academic Press; New York: 1981. [Google Scholar]
- (2).(a) Solomon EI, Brunold TC, Davis MI, Kemsley JN, Lee S-K, Lehnert N, Neese F, Skulan AJ, Yang Y-S, Zhou J. Chem. Rev. 2000;100:235–349. doi: 10.1021/cr9900275. [DOI] [PubMed] [Google Scholar]; (b) Costas M, Mehn MP, Jensen MP, Que L., Jr Chem. Rev. 2004;104:939–986. doi: 10.1021/cr020628n. [DOI] [PubMed] [Google Scholar]; (c) Bollinger JM, Jr., Krebs C. Curr. Opin. Chem. Biol. 2007;11:151–158. doi: 10.1016/j.cbpa.2007.02.037. [DOI] [PubMed] [Google Scholar]; (d) Kovaleva EG, Lipscomb JD. Science. 2007;316:453–457. doi: 10.1126/science.1134697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).(a) Mirica LM, Ottenwaelder X, Stack TDP. Chem. Rev. 2004;104:1013–1045. doi: 10.1021/cr020632z. [DOI] [PubMed] [Google Scholar]; (b) Bakac A. Coord. Chem. Rev. 2006;250:2046–2058. [Google Scholar]; (c) Cramer CJ, Tolman WB. Acc. Chem. Res. 2007;40:601–608. doi: 10.1021/ar700008c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Kieber-Emmons MT, Riordan CG. Acc. Chem. Res. 2007;40:618–625. doi: 10.1021/ar700043n. [DOI] [PubMed] [Google Scholar]; (e) Himes RA, Karlin KD. Curr. Opin. Chem. Biol. 2009;13:119–131. doi: 10.1016/j.cbpa.2009.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Yao S, Driess M. Acc. Chem. Res. 2012;45:276–287. doi: 10.1021/ar200156r. [DOI] [PubMed] [Google Scholar]
- (4).Barefield EK. Coord. Chem. Rev. 2010;254:1607–1627. [Google Scholar]
- (5).Cho J, Sarangi R, Nam W. Acc. Chem. Res. 2012;45:1321–1330. doi: 10.1021/ar3000019. and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).(a) Kieber-Emmons MT, Annaraj J, Seo MS, Van Heuvelen KM, Tosha T, Kitagawa T, Brunold TC, Nam W, Riordan CG. J. Am. Chem. Soc. 2006;128:14230–14231. doi: 10.1021/ja0644879. [DOI] [PubMed] [Google Scholar]; (b) Cho J, Sarangi R, Annaraj J, Kim SY, Kubo M, Ogura T, Solomon EI, Nam W. Nature Chem. 2009;1:568–572. doi: 10.1038/nchem.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).(a) Cho J, Woo J, Nam W. J. Am. Chem. Soc. 2010;132:5958–5959. doi: 10.1021/ja1015926. [DOI] [PubMed] [Google Scholar]; (b) Cho J, Woo J, Han JE, Kubo M, Ogura T, Nam W. Chem. Sci. 2011;2:2057–2062. [Google Scholar]
- (8).(a) Cho J, Jeon S, Wilson SA, Liu LV, Kang EA, Braymer JJ, Lim MH, Hedman B, Hodgson KO, Valentine JS, Solomon EI, Nam W. Nature. 2011;478:502–505. doi: 10.1038/nature10535. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Cho J, Sarangi R, Kang HY, Lee JY, Kubo M, Ogura T, Solomon EI, Nam W. J. Am. Chem. Soc. 2010;132:16977–16986. doi: 10.1021/ja107177m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Annaraj J, Cho J, Lee Y-M, Kim SY, Latifi R, de Visser SP, Nam W. Angew. Chem. Int. Ed. 2009;48:4150–4153. doi: 10.1002/anie.200900118. [DOI] [PubMed] [Google Scholar]; (d) Seo MS, Kim JY, Annaraj J, Kim Y, Lee Y-M, Kim S-J, Kim J, Nam W. Angew. Chem. Int. Ed. 2007;46:377–380. doi: 10.1002/anie.200603414. [DOI] [PubMed] [Google Scholar]
- (9).Abbreviations used: 12-TMC, 1,4,7,10-tetramethyl-1,4,7,10-tetraazacyclododecane; 14-TMC, 1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetradecane.
- (10).(a) Richter-Addo GB, Legzdins P, Burstyn J. Chem. Rev. 2002;102:857–859. doi: 10.1021/cr010188k. [DOI] [PubMed] [Google Scholar]; (b) Ford PC, Lorkovic IM. Chem. Rev. 2002;102:993–1017. doi: 10.1021/cr0000271. [DOI] [PubMed] [Google Scholar]; (c) McCleverty JA. Chem. Rev. 2004;104:403–418. doi: 10.1021/cr020623q. [DOI] [PubMed] [Google Scholar]; (d) Fry NL, Mascharak PK. Acc. Chem. Res. 2011;44:289–298. doi: 10.1021/ar100155t. [DOI] [PubMed] [Google Scholar]; (e) Berto TC, Speelman AL, Zheng S, Lehnert N. Coord. Chem. Rev. 2012 doi.org/10.1016/j.ccr.2012.05.007. [Google Scholar]
- (11).Schopfer MP, Wang J, Karlin KD. Inorg. Chem. 2010;49:6267–6282. doi: 10.1021/ic100033y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).(a) Ouellet H, Ouellet Y, Richard C, Labarre M, Wittenberg B, Wittenberg J, Guertin M. Proc. Natl. Acad. Sci. USA. 2002;99:5902–5907. doi: 10.1073/pnas.092017799. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gardner PR, Gardner AM, Brashear WT, Suzuki T, Hvitved AN, Setchell KDR, Olson JS. J. Inorg. Biochem. 2006;100:542–550. doi: 10.1016/j.jinorgbio.2005.12.012. [DOI] [PubMed] [Google Scholar]
- (13).Blough NV, Zafiriou OC. Inorg. Chem. 1985;24:3502–3504. [Google Scholar]
- (14).(a) Kennedy MC, Antholine WE, Li W, Mao Q, Petering DH. Inorg. Chim. Acta. 1995;240:535–540. [Google Scholar]; (b) Roncaroli F, Videla M, Slep LD, Olabe JA. Coord. Chem. Rev. 2007;251:1903–1930. [Google Scholar]
- (15).Clarkson SG, Basolo F. Inorg. Chem. 1973;12:1528–1534. [Google Scholar]
- (16).(a) Maiti D, Lee D-H, Narducci Sarjeant AA, Pau MYM, Solomon EI, Gaoutchenova K, Sundermeyer J, Karlin KD. J. Am. Chem. Soc. 2008;130:6700–6701. doi: 10.1021/ja801540e. [DOI] [PubMed] [Google Scholar]; (b) Park GY, Deepalatha S, Puiu SC, Lee D-H, Mondal B, Narducci Sarjeant AA, del Rio D, Pau MY,M, Solomon EI, Karlin KD. J. Biol. Inorg. Chem. 2009;14:1301–1311. doi: 10.1007/s00775-009-0575-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Cramer CJ, Tolman WB, Theopold KH, Rheingold AL. Proc. Natl. Acad. Sci. U.S.A. 2003;100:3635–3640. doi: 10.1073/pnas.0535926100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).In terms of the nature of 2′•Cl2 in solution, the ESI-MS of the crystals was measured and found to be identical to that of 2 generated in situ in the reaction of 1 and O2. This result suggests the chloride anion instead of the aqua ligand coordinates to the Cr center in solution. In support of this supposition is an experiment carried out where 2.2 equiv of Ag-triflate was added to a solution of complex 2. ESI-MS analysis revealed the dominant ion present to be [CrIV(12-TMC)(O )(Cl)]+ 2 indicating one chloride still binds to the chromium ion and Ag+ only removed the Cl− counter-anion. The aqua ligand undoubtedly originates from residual solvent water, and it coordinates to the Cr center only in the crystal structure, probably due to preferred crystal packing.
- (19).For a recent review of literature of a variety of chromium peroxo complex structures, most also possessing oxo ligands: see Sergienko VS. Crystallography Reports. 2007;52:639–646.
- (20).(a) Hempel JC, Morgan LO, Lewis WB. Inorg. Chem. 1970;9:2064–2072. [Google Scholar]; (b) Ueki S, Yamauchi J. Inorg. Chim. Acta. 2002;338:13–18. [Google Scholar]
- (21).In the crystal, another nitrate anion was found as a counter anion instead of chloride; the formula of the crystal was given as [Cr(12-TMC)(NO3)(Cl)]NO3. See SI, crystallographic data and Table S1.
- (22).The crystal of [Cr(12-TMC)(NO3)(Cl)]NO3 was used as an authentic sample for the calculation of the reaction yield (ε = 180 M−1cm−1 at 415 nm).
- (23).(a) Goldstein S, Lind J, Merényi G. Chem. Rev. 2005;105:2457–2470. doi: 10.1021/cr0307087. [DOI] [PubMed] [Google Scholar]; (b) Yukl ET, de Vries S, Moënne-Loccoz P. J. Am. Chem. Soc. 2009;131:7234–7235. doi: 10.1021/ja9026924. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kurtikyan TS, Ford PC. Chem. Commun. 2010;46:8570–8572. doi: 10.1039/c0cc02665d. [DOI] [PubMed] [Google Scholar]
- (24).Wick PK, Kissner R, Koppenol WH. Helv. Chim. Acta. 2000;83:748–754. [Google Scholar]
- (25).Kurtikyan TS, Eksuzyan SR, Hayrapetyan VA, Martirosyan GG, Hovhannisyan GS, Goodwin JA. J. Am. Chem. Soc. 2012;134:13861–13870. doi: 10.1021/ja305774v. [DOI] [PubMed] [Google Scholar]
- (26).4•Cl was synthesized by reacting 1 with •NO(g) and characterized by UV-vis, EPR, ESI-MS, IR, and elemental analysis (see SI, Experimental Section, Figures S4 - S6).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.