

Abstract
Celiac disease (CD) is one of the most common diseases, resulting from both environmental (gluten) and genetic factors [human leukocyte antigen (HLA) and non-HLA genes]. The prevalence of CD has been estimated to approximate 0.5%-1% in different parts of the world. However, the population with diabetes, autoimmune disorder or relatives of CD individuals have even higher risk for the development of CD, at least in part, because of shared HLA typing. Gliadin gains access to the basal surface of the epithelium, and interact directly with the immune system, via both trans- and para-cellular routes. From a diagnostic perspective, symptoms may be viewed as either “typical” or “atypical”. In both positive serological screening results suggestive of CD, should lead to small bowel biopsy followed by a favourable clinical and serological response to the gluten-free diet (GFD) to confirm the diagnosis. Positive anti-tissue transglutaminase antibody or anti-endomysial antibody during the clinical course helps to confirm the diagnosis of CD because of their over 99% specificities when small bowel villous atrophy is present on biopsy. Currently, the only treatment available for CD individuals is a strict life-long GFD. A greater understanding of the pathogenesis of CD allows alternative future CD treatments to hydrolyse toxic gliadin peptide, prevent toxic gliadin peptide absorption, blockage of selective deamidation of specific glutamine residues by tissue, restore immune tolerance towards gluten, modulation of immune response to dietary gliadin, and restoration of intestinal architecture.
Keywords: Celiac disease, Demography, Diagnosis, Pathogenesis, Treatment
INTRODUCTION
Celiac disease (CD) is a life-long gluten-sensitive autoimmune disease of the small intestine affecting genetically susceptible individuals worldwide. CD individuals may present gastrointestinal symptoms, extraintestinal symptoms or no signs of symptoms. The classical symptoms include gastrointestinal-related symptoms such as diarrhea, steatorrhea and weight loss due to malabsorption. About 50% of CD patients present extraintestinal or atypical symptoms, such as anemia, osteoporosis, dermatitis herpetiformis, neurological problems and dental enamel hypoplasia[1-3]. The variable clinical picture of CD is due to having both genetical and immunological bases, with age of onset, extent of mucosal injury, dietary habits, and gender[4], affecting the clinical manifestation of the disease.
CD diagnosis is based on presence of predisposing genetic factor human leukocyte antigen (HLA) DQ2/8, with positive biopsy and serological antibodies upon gluten contained diet. The spectrum of CD may present in different forms[5]. The classical form may be diagnosed at any age of life and is often characterized by crypt hyperplasia and villous atrophy along with features of malabsorption. The atypical form is characterized by positive celiac serology, limited abnormalities of the small intestinal mucosa or no intestinal symptoms, but associated extraintestinal conditions such as osteoporosis, peripheral neuropathy, anemia and infertility. The latent form is defined by presence of predisposing gene HLA-DQ2 and/or HLA-DQ8, normal intestinal mucosa and, possible positive serology. Extraintestinal features and biopsies of the small bowel show alterations with gluten intake (i.e., gluten-sensitive). Rarely, usually after age 50 years[6], some that have initially responded to a gluten-free diet (GFD), develop recurrent symptoms and biopsy changes despite a GFD. This is the refractory form[7]. If no response to GFD was initially documented, however, the use of sprue-like intestinal disease or unclassified sprue has been used.
PREVALENCE
CD originally thought to almost exclusively affect white Europeans, is now known to be widely distributed worldwide[8]. Epidemiological studies conducted in areas supposedly free of CD, including Africa, the Middle East, Asia, and South America, show that the disease was previously underdiagnosed[9]. This provides evidence that CD is one of the most common genetic diseases, resulting from both environmental (gluten) and genetic (HLA and non-HLA genes) factors.
The world distribution of CD seems to have followed the mankind wheat consumption and the migratory flows. Man originally fed on meat, fruit and vegetables, with no exposer to gluten-containing cereals. It was only about 10 000 years ago in a small region called the “Fertile Crescent” of the Middle-East (including Anatolia (Southern Turkey), Lebanon, Syria, Palestine and Iraq) where wild wheat and barley grains successfully cultivated due to favorable environmental conditions. In the Fertile Crescent some tribes changed from nomadic to stable settlement style of living because land cultivation permitted food storage, and later migrated westwards to obtain new lands for cultivation. These persons spread through the Mediterranean area (Northern Africa, Southern Europe) and Central Europe. The expansion continued from 9000 to 4000 BC by which time the cultivation of wheat and barley had spread all over the Old Continent, also reaching Northern Europe (Ireland, Denmark and the Scandinavian countries). This expansion in farming was due to the diffusion of agricultural practices and replacement of local inhabitants by descendants from the Middle-East[10]. Hence, the European and North-African populations share genetic background with the peoples of Middle-East origin.
In the last few years a number of studies in different populations have been carried out using molecular genetics methods to identify genes causing CD. CD-predisposing genetic loci are CELIAC1 on chromosome 6, CELIAC2 on chromosome 5q31-33[11], CELIAC3 on chromosome 2q33[12], and CELIAC4 on chromosome 19p13.1[13]. PARD3 and MAGI2 tight junction genes associations have also been reported in Dutch CD or ulcerative colitis patients, suggesting a common intestinal defect in these two conditions[14]. Another gene expressed in major histocompatibility complex (MHC) I antigen presenting cell is HLA B8, was found to be associated with CD in Algeria[15], Iraq[16,17] and Turkey[18]. Moreover, atypical CD Saharawi patients were found to over-express the MHC class I chain-related gene A (MICA) allele 5.1[15], which have also been reported in Western countries[19]. Increased prevalence of HLA-A25 in Turkish children with CD was also reported, suggesting that this genotype is particularly encountered among this population[18], with no association described in Western countries.
Useful Background: Genes causing CD CELIAC1 on chromosome 6 (HLA-DQ2 and HLA-DQ8); CELIAC2 on chromosome 5q31-33[11]; CELIAC3 on chromosome 2q33 (containing T lymphocyte regulatory genes CD28, CTLA4 and ICOS)[12]; and CELIAC4 (myosin IXB gene, MYO9XB) on chromosome 19p13.1[13].
HLA genotype contributes to the genetic risk for CD at 30%-50%[20,21]. Non-HLA genes contribute more evidence to the CD genetic background than the HLA genes, but each by itself contributes only a modest to the disease development. Hence, it is reasonable to assume that the susceptibility to CD involves with polymorphic genes that influence the immune response to gluten, as shown for the HLA-linked genes[22].
Ninety percent of European patients with CD carry the HLA-DQ2 molecule, encoded either in cis on the HLA-DQA1*0501-DQB1*0201 haplotype (HLA-DQ2.5cis) or in trans on the HLA-DQA1*0505 DQB1*0301/DQA1*0201-DQB1*0202 haplotypes (HLA-DQ2.5trans). Approximately 5% express HLA-DQ8, encoded by HLA-DQA1*0301-DQB1*0302. The majority of the remainder carry the HLA-DQA1*0201-DQB1*0202 haplotype[20]. With genetic testing, DQ2 is almost synonymous with DQB1*02, a gene with two common alleles designated DQB1*0201 and DQB1*0202. The DQ2 frequency in Caucasian in Western Europe populations has been estimated at 20%-30%, and relatively high frequencies also occur in Northern and Western Africa, the Middle East and central Asia[23]. Thereafter, the overall frequency of DQ2 declines from West to East with low frequencies in populations in South-East Asia and the virtual absence of DQ2 in Japan (Table 1). DQ8 frequency has a worldwide distribution, whereas DQ2.5, is common in South and Central America; approximately 90% of Amerindian populations carry DQ8 and may display the celiac phenotype[24,25]. The frequency of DQ8 population is shown in Table 1.
Table 1.
Frequency of human leukocyte antigen-DQ2, encoded by human leukocyte antigen-DQB1*02 and human leukocyte antigen-DQ8, encoded by human leukocyte antigen-DQA1*0301-DQB1*0302
| < 5% | 5%-20% | 20% |
| HLA-DQ2 | ||
| Albania | Belarus Algeria | Algeria |
| Canada BC (Athabaskans) | Cameroon | Australia |
| Cook Islands | Congo | Belgium |
| Indonesia | Costa Rica | Central African Republic |
| Japan | China | Croatia |
| Jordan | Cuba | England |
| Papua New Guinea | Ecuador Africans | Equatorial Guinea Bioko Island |
| Philippines | France | Ethiopia |
| Samoa | India | Germany |
| Malaysia | Greece | |
| Mexico | Iran | |
| Poland | Ireland South | |
| Russia | Israel | |
| Singapore | Italy | |
| South Korea | Mongolia | |
| Spain | New Zealand | |
| Sri Lanka | Pakistan | |
| Sweden | Saudi Arabia | |
| Taiwan, China | Slovenia | |
| Thailand | Tunesia | |
| Turkey | United States | |
| Uganda | ||
| Ukraine | ||
| Vietnam | ||
| HLA-DQ8 | ||
| Australia | Algeria | Argentina |
| China | Belgium | Ecuador |
| Georgia | Brazil | Ethiopia |
| Greece | Canada BC (Athabaskans) | Mexico |
| North India | Croatia | Venezuela |
| Spain | England Caucasoid | |
| Uganda | France | |
| South India | ||
| Israel | ||
| Italy | ||
| Japan | ||
| Russia | ||
| South Korea | ||
| Tunisia | ||
| Turkey | ||
| Ukraine | ||
| United States | ||
| European American | ||
Estimates are based on studies included in a comprehensive Internet website[24]. In several countries, the frequency is not known. HLA: Human leukocyte antigen.
In the past, the prevalence of CD had been underestimated, but it is now regarded one of the most common genetic disorders in the West with 1% prevalence[26-28]. Interestingly, there is increased prevalence of CD amongst women compared to men with male:female ratio of 1:2.8[29]. This could be due to the finding that men with CD were diagnosed at an older age[30]. Indeed, there have been reported CD cases among immigrant children native of Eastern Europe, Northern, West and East Africa, the Middle East and Southern Asia, according to their acquisition of Western dietary practices (i.e., short period or lack of breast feeding and early weaning with a great amount of gluten intake)[31]. This suggests that many persons may have the genetic predisposition to CD but the clinical presentation only occurs when there is sufficient gluten in the diet.
Normal at-risk persons
In several parts of the world, the presence of the combination of antibody (serum tissue transglutaminase and endomysial autoantibodies) positivity and an HLA haplotype associated with CD is predictive of small-bowel abnormalities indicative of CD. For the majority of countries, the CD prevalence is unknown. Figure 1 shows a range of estimated normal at-risk CD prevalence in continents and nations around the globe. It must be noted that some studies report prevalence of CD based on serology, others on celiac compatible small bowel biopsies and a few on serology, biopsy and response to gluten challenge.
Figure 1.
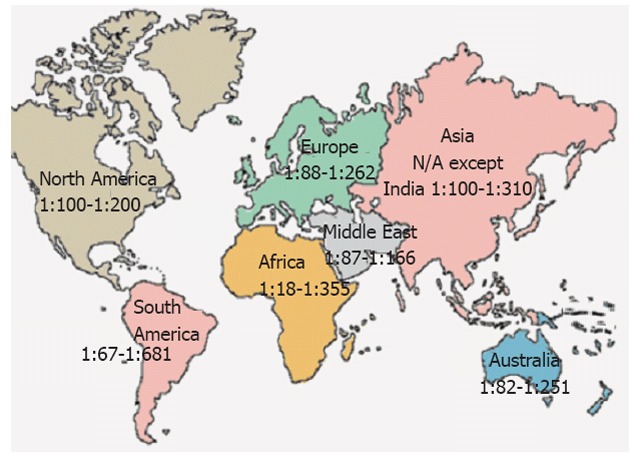
Prevalence of celiac disease worldwide. N/A: Not available.
North America
CD prevalence in North American and Europe were found to be similar in symptomatic patients and not-at-risk subjects. In the United States, CD is believed to affect 0.5%-1.0% of the general population[32]. The study by Fasano et al[28] on serum antibody and biopsy screening were performed for a total of 13 145 United States subjects: first-degree (n = 4508) and second-degree relatives of patients (n = 1275) with biopsy-proven CD, symptomatic patients (n = 3236) (with either gastrointestinal symptoms or a disorder associated with CD), and not-at-risk individuals (n = 4126). The overall CD prevalence is 1:133 in the not-at-risk groups, whereas in the at-risk group, the prevalence is 1:22 in first-degree relatives, 1:39 in second-degree relatives, and 1:56 in symptomatic patients[28].
South America
In South America, CD had been historically considered a rare disorder and the prevalence investigations have not been extensively studied. However, during the last few years studies in Brazil disclosed a prevalence of 1:681 in healthy blood donors[33] and 1:473 among adult outpatients attending a clinical laboratory for routine blood testing[34]. In an urban area of Argentina, the overall prevalence of CD, among 2000 adults from the general population (996 women; median age 29 year, range 16-79 year) was 1:167, with prevalence in women double that for men[35]. The high CD prevalence in Argentina could be correlated with HLA DQ8 (> 20%) in the Argentina population.
Europe
The overall prevalence of CD in Western populations is close to 1% (1:100) and may be higher in Northern European countries (Table 2)[36-38]. The Scandinavian countries, Ireland, and the United Kingdom population tended to show a higher prevalence of CD of approximately 1.0%-1.5%, although there also were studies that showed a lower prevalence in these countries. A study of small-intestinal biopsy obtained from healthy Dutch blood donors at Arnhem and Nijmegen Blood Donation Centers shows that the prevalence of CD-compatible biopsies of 1:330[39]. The prevalence of CD among 3654 children (age range, 7-16 years) in Finland was at least 1:99 based on serum autoantibodies and small-bowel abnormalities[40]. The prevalence of CD in Northern Spain in the general population was 1:389[41], 1:132 (0.75%) in Eastern Switzerland adolescents[42].
Table 2.
Prevalence of celiac disease in Europeans based on unselected population serological screenings[36-38] (adapted)
| Countries | Prevalence |
| Czechoslovakia | 0.193 |
| Estonia | 0.103 |
| Finland | 0.110 |
| Hungary | 0.101 |
| Ireland | 0.126 |
| Italy | 0.115 |
| Norway | 0.224 |
| Portugal | 0.135 |
| Spain | 0.124 |
| Sweden | 0.174 |
| Switzerland | 0.133 |
| Netherlands | 0.179 |
| United Kingdom | 0.111 |
Africa
In Northern African populations (including Morocco, Algeria, Tunisia, Libya and Egypt) higher incidences of 0.28%-5.6% of CD have recently been reported in the general population[43-46]. The prevalence of CD in asymptomatic Tunisian school children was estimated to be about 1:157, which is close to the European prevalence. In this respect, the highest world frequency of 16.4% is reported in the CD associated with Insulin Dependent Diabetes Mellitus[47], in Oran (Algeria). A recent serological screening in 2500 Tunisian healthy blood donors[45], showed similar that the prevalence of anti-endomysium antibodies in the general population of 1:355, to that of Europeans. Due to high wheat and barley consumption in the North American countries[45], as well as high frequency of CD predisposing DR3-DQ2 haplotypes in these populations[48-50], these high CD frequencies are not surprising.
Saharawi population in North Africa, who are of Arabian and Berberian origin, having a high degree of cognation and live as refugees in the Sahara desert (Algeria), has the highest prevalence of CD (5.6%) known in the world today[43,49]. This elevated prevalence in this population may be explained both by genetic factors: very high frequency of the DR3-DQ2 haplotype, and by environmental factors: changed of dietary habits in the last few decades. The reduced rates and duration of breast-feeding and increased consumption of gluten in early life as part of the staple diet, supplied by Western countries as humanitarian aids[51], may have played a role in this elevated CD prevalence. However, there are other unknown genetic and environmental factors that explain such a high frequency of CD in the Saharawi people, because there is a much lower prevalence of CD in Sardinia population with similar staple diet consumption and frequencies of DR3-DQ2[52].
Australia and New Zealand
Australia and New Zealand are the two countries having the highest proportion of individuals from Caucasian background, with high prevalence of HLA DQ2 and per capita wheat consumption of > 150 and 75-150 kg per person per year, respectively[23]. Only two prevalence studies have been carried out in these two countries. From a random population of 1064 adults in Christchurch, New Zealand (96% Caucasian), CD was confirmed histologically in all patients with positive serology giving an overall prevalence of 1:82 (1.2%)[53]. A larger study in 3011 adults from a large Caucasian community in Western Australia, revealed an overall prevalence of CD of 1:251 (0.4%) of the population[54].
Asia
CD is likely to be rare in Indonesia, South Korea, Philippines and many smaller Pacific islands because of their low wheat consumption and a low frequency of HLA-DQB1*02. In South-East Asia, HLA-DQB1*02 is often present in more than 5% of the population but CD is predicted to be rare, as staple diets are based on rice. In contrast, prevalence rates that are similar to those in Europe are likely to apply from Pakistan in the South to Kazakhstan in the North. Ancient migration patterns that determine the frequency of DQB1*02 would also predict more patients with CD in Western China than in Eastern China[23]. Interestingly, there is one report of CD in three adult descendants of Chinese and Japanese families who migrated to Canada[55].
In genetic studies of CD in India, the appearance of HLA associations is similar to those in Western countries with a frequency of HLA-DQB1*02 of close to 100%[56]. This association is more frequent in the population of Northern and North-Eastern India (16%-27%)[57], than in groups of adults in the Southern state of Tamil Nadu (9%-14%)[58]. The prevalence of CD in India is nearly the same as that in Western Caucasian populations[59]. In Punjab (North-west India) school children, CD frequency was estimated to be 0.3%[60]. This prevalence is probably an underestimation. A retrospective analysis of confirmed cases of CD between 1995 and 2000 in Dayanand Medical College and Hospital (Ludhiana, Punjab) from a total of 202 cases showed an initial of 10 positive cases with a significant growth rate of 79.43% annually with a trend equation increase of 15.49 cases/year[61]. These studies showed that CD is relatively common in Northern India where there has been a history of wheat cultivation from before 1000 BC[62]. Hence, the relative rarity of CD in Southern India reflects the effect of both genetic and environmental factors.
The prevalence in first-degree relatives of North Indian children with CD diagnosed as per the European Society for Pediatric Gastroenterology and Nutrition criteria is 4.4% of the first-degree relatives (85% positive for HLA DQ2/DQ8), which is 14 times higher than that of the general population[63]. There have been reports on clinical experience of biopsy-defined CD in 10 North Indian Immigrants or descendants born in Canada out of 14 Asians diagnosed since 1988 in a single Canadian teaching hospital[55]. Several studies, particularly from Northern and North-West India, have also documented the presence of CD in children presenting with chronic diarrhea[64].
Middle East
It seems likely that the prevalence of CD in the Middle East is similar to that of Europe[65]. CD is a relatively common cause of chronic diarrhea in Iran, Iraq and Kuwait and has been diagnosed in 2%-8% of patients with type 1 diabetes in Iran, Israel and Saudi Arabia[39]. Many of these countries have a per capita wheat consumption that ranks among the highest in the world (> 150 kg per person per year)[23]. Although only a limited number of genetic studies have been carried out, the population of countries such as Iran, Israel, Saudi Arabia and Turkey have a high frequency of HLA-DQB1*02. The prevalence of CD in adult blood donors in Iran, Israel, Syria, Turkey and Anatolia are 1:166[66], 1:157[67], 1:62[68], 1:87[69], 1:100[70], respectively. Similar prevalence rates were determined in surveys of Iranian children (1:165, 0.6%)[71], and Turkish children (1:115, 0.9%)[72].
The prevalence of CD is approximately 0.5%-1% in all parts of the world, except for populations with very low and very high intake of gluten in their diet.
High at-risk persons
In the general celiac population (without classical CD symptoms, e.g., diarrhea or weight loss), there are high risk groups that may have higher CD prevalence rates (Table 3). Among factors that denote a higher risk for CD, the most important factor is familial history of biopsy proven CD with an estimate of 20% or more of first-degree relatives having CD[73]. Some authors observed a higher prevalence in CD siblings as compared to parents[74-76]. A study in Swedish youth (< 20 years old) diagnosed with type 1 diabetes (T1D) confirmed the low prevalence (0.7%) of diagnosed symptomatic CD at initial onset of clinical diagnosis, but document by screening an increasing prevalence of silent CD during a 5-year follow-up to reached an overall prevalence of 10%[77]. Thus, the prevalence of an association with CD in high risk groups may increase over time.
Table 3.
High risk populations for celiac disease[73] (adapted)
| Relatives, especially first-degree |
| Anemia, especially iron deficiency |
| Osteopenic bone disease |
| Insulin-dependent diabetes (type 1), especially children |
| Liver disorders, especially Autoimmune hepatitis and primary biliary cirrhosis |
| Genetic disorders, including down and Turner’s syndrome |
| Autoimmune endocrinopathy, especially thyroid disease |
| Skin disorders, particularly dermatitis herpetiformis |
| Neurological disorders, including ataxia, seizures, myasthenia gravis |
| Others, including immunoglobulin A nephropathy |
The overall prevalence of CD is highly dependent on the HLA DQ2/DQ8 typing and gluten consumption. The population with positive HLA typing for celiac have high chances of developing celiac symptoms when on high gluten consumption. However, the population with diabetes, autoimmune disorder or relatives of CD individuals) have even higher risk for the development of CD, since they share the same HLA typing.
PATHOGENESIS
CD is an intestinal enteropathy triggered by the ingestion of gliadin and of other related prolamins in genetically predisposed individuals[22,78]. Wheat and related species such as barley and rye also induce CD[79]. A small minority of CD patients also react to oat[80]. Gliadin peptides exert damaging effects since they are resistant to gastrointestinal enzymes[81], they have amino acid sequences that are specific for HLA-DQ2, which is a class II major histocompatibility complex, they also have preferred glutamine residues for tissue transglutaminase (tTG)-mediated deamidation[82], and lastly, they affect intestinal permeability[83]. Hence the pathogenesis of CD is dependent on genetic and environmental factors. The environmental factor is mainly ingestion of gluten, while several genes contribute to the genetic predisposition[13]. CD commonly appears in early childhood, with severe symptoms including chronic diarrhea, abdominal distension, and failure to thrive. In many patients, symptoms may not develop until later in life, when the disease symptoms include fatigue, diarrhea, and weight loss due to malabsorption, anemia, and neurological symptoms (Table 4). Celiac disease is a life-long disorder, and if untreated, it is associated with increased morbidity and mortality[84,85].
Table 4.
Possible clinical manifestations of celiac disease[8] (printed with permission)
| Typical symptoms | Atypical symptoms | Associated conditions |
| Chronic diarrhea | Secondary to malabsorption | Possible gluten dependent |
| Failure to thrive | Sideropenic anemia | IDDM |
| Abdominal distention | Short stature | Autoimmue thyroiditis |
| Osteopenia | Autoimmune hepatitis | |
| Recurrent abortions | Sjogren syndrome | |
| Hepatic steatosis | Addison disease | |
| Recurrent abdominal pain | Autoimmune atrophic gastritis | |
| Gaseousness | Autoimmune emocytopenic diseases | |
| Independent of malabsorption | Gluten independent | |
| Dermatitis herpetiformis | Down syndrome | |
| Dental enamel hypoplasia | Turner syndrome | |
| Ataxia | Williams syndrome | |
| Alopecia | Congenital heart defects | |
| Primary biliary cirrhosis | IgA deficiency | |
| Isolated hypertransaminasemia | ||
| Recurrent aphthous stomatitis | ||
| Myasthenia gravis | ||
| Recurrent pericarditis | ||
| Psoriasis | ||
| Polyneuropathy | ||
| Epilepsy | ||
| Vasculitis | ||
| Dilatative cardiomyopathy | ||
| Hypo/hyperthyroidism |
IgA: Immunoglobulin A; IDDM: Insulin dependent diabetes mellitus.
Possible triggers
Genetic predisposition association (HLA, MYO9B), exogenous trigger (gluten), pro-autoimmune genetic background, viral infections, tissue damage, early termination of breastfeeding and gender contribute to the development of CD (Table 5)[86].
Table 5.
The most important factors contributing to the development of celiac disease[86] (printed with permission)
| Factors contributing to the onset of celiac disease | Mechanism |
| Gluten | Elicit T cell responses |
| Induces cytokine production and intestinal lesion | |
| Age of introduction of gluten | Weak gut immune during early childhood |
| HLA-DQ2 or HLA-DQ8 | Gluten presentation |
| MYO9Bo | Increased permeability of the intestine |
| Pro-autoimmune genetic background | Shift in Th1/Th2 balance towards Th1 |
| Viral infections | Defect in generation of active tolerance (e.g., regulatory T cells) |
| IFN production | |
| Tissue damage | Tissue damage |
| Increased level of tTG | |
| Danger signals | |
| Early termination of breastfeeding | Decreased protection against infections |
| Gender | Hormone-related pro-autoimmune status |
Th1: T helper 1; Th2: T helper 2; tTG: Tissue transglutaminase; HLA: Human leukocyte antigen; IFN: Interferon.
Apart from introduction of gluten during the first year of life, infectious agents may play a role in development of CD. Several studies have implicated infections with Adenovirus type 12[87-89], hepatitis C virus[90,91], Campylobacter jejuni[92], Giardia lamblia[93], Rotavirus[94] and Enterovirus infection[95] as triggers for the development of CD. The immunologic response in persons genetically susceptible CD may be triggered due shared viral sequence of 8 to 12 amino acids with the toxic gliadin fraction[89]. Other factors such as timing of gluten ingestion and breast feeding cessation may involve in the pathogenesis and disease development of CD[96]. Some initiating factors, such as gluten overload, gastric surgery “unmasking”, giving up smoking, and infections can also trigger the disease, which can become apparent in an abrupt manner[97,98].
Prolamin trigger
Gluten is a protein that appears in wheat, barley, rye and oat, compositing of prolamin and glutelin. The majority of the proteins in food responsible for the immune reaction in CD are the prolamins. Prolamins is found in several grains, such as wheat (gliadin), barley (hordein) and rye (secalin), corn (zein) and as a minor protein, avenin in oats. Because of their high glutamine content and specific sequence patterns, prolamins are resistant to gastrointestinal proteolytic enzymes and are excellent substrates for deamidation by tissue transglutaminase.
The incomplete gastrointestinal digestion of gluten leads to the appearance of gluten-derived gliadin peptides such as 33mer (LQLQPFPQPQLPYPQPLPYPQPQL PYPQPQPF) with a variety of characteristics[81]. It contains overlapping T-cell epitopes, and its deamidated form is a potent T-cell stimulator, generating the glutamic acid residues essential for binding to HLA-DQ2 in celiac patients[99]. The ingestion of prolamins from wheat, barley, rye and possibly oats causes histological changes in the small intestine mucosa of celiac patients, leading to a malabsorption syndrome[100]. Clinical symptoms of an autoimmune attack after ingestion of the gluten containing food include digestive symptoms and skin reactions.
Gliadin peptides cause stimulation of the innate and adaptive immune system[83,101,102]. The prototype of peptides effective on innate response is peptide 31-43/49, which has been proved both in vitro and in vivo to be toxic for CD patients[103,104]. Peptide 31-43 (p31-43) stimulates the synthesis and release of interleukin (IL)-15, a proinflammatory cytokine, that promotes the adaptive immune response[101], involving CD4+ T cells that recognize several deamidated gliadin peptides[82]. Unlike p31-43 which is not immunogenic for T cells, peptide 57-68 (p57-68), which binds to HLA-DQ2/8 molecules, is one of the dominant epitopes recognized by T cells isolated from the intestine of CD patients[82]. The so-called toxic peptides, of which p31-43 is probably the most fully studied, modulate the small-intestinal mucosal biology via an innate immune mechanism.
Time of trigger
Several studies related the rise in childhood CD to infant feeding practices[96,105]. Consumption of wheat, barley, and rye in the first 3 mo children have significantly increased the risk of developing CD-associated autoantibodies, as compared with exposure during first 4 to 6 mo[105,106].
Although CD can be diagnosed at any time of life, it is present mostly in either early childhood (between 9 and 24 mo) or in the third or fourth decade of life[8,85,107,108]. In contrast to the 1/1 sex ratio in children, in adulthood it is diagnosed twice in females. Interestingly, celiac disease is also becoming more frequently recognized in the elderly, and in this population, a 1/1 sex ratio has also been noted[109]. Although the “classical” gastrointestinal malabsorption syndrome characterized by diarrhea, steatorrhoea, weight loss, fatigue, and anemia may occur in severe cases, most patients have a milder symptoms such as abdominal discomfort, bloating, indigestion, or non-gastrointestinal symptoms (or no symptoms at all)[8,85,107,108]. Mäki et al[2] reported a shift of 5-6 years of age at diagnosis in Finland, with less than 50% of new cases presenting typical gastrointestinal symptoms. In England[110], Scotland[111], Canada[112], and the United States[28], reports have also shown that almost 50% of newly diagnosed CD patients do not present with gastrointestinal symptoms.
GENETICS
Genetics play a strong role in CD, indicated by the high disease concordance in monozygotic twin[113]. The CD prevalence rose to 17.6% in sisters, 10.8% in brothers, and 3.4% in parents[21]. CD is associated with HLA alleles as well as more than 250 other MHC and non-MHC genes. The main genetic factor is the given HLA-DQ genes, i.e., the genes encoding DQ2 or DQ8 in the HLA complex on 6p21. Approximately 95% of celiac have a DQ2 comprised of DQB1*302 and DQA1*03. A small number of individuals lacking either of those heterodimers have DQB1*02 and DQA1*05 alone[20,114]. Gene dosage also affect CD susceptibility; individuals with the heterodimer comprised of DQB1*02 and DQA1*05 and most of the remaining 5% have a DQ8 heterodimer. Homozygous individuals who carry DQB1*02 and DQA1*05 in cis on both chromosomes have a great risk developing complicated forms of CD[115]. A significant higher risk for CD of 1:7 for DQ2 and DQ8 individuals and 1:2518 for subjects lacking all predisposing factors have been determined[21].
It is found that 30% of the Caucasian populations carry HLA-DQ2 and most will eat wheat, while only 1 in 100 will develop disease[116]. The remaining susceptibility is thought to be due to a combination of genetic and environmental factors (Figure 2).
Figure 2.
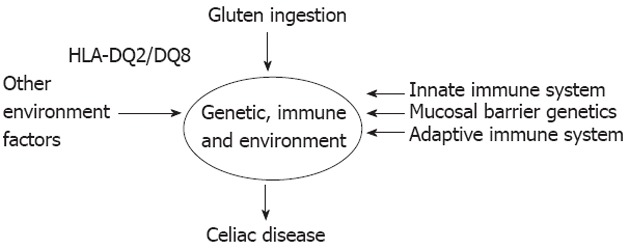
Factors necessary for celiac disease development[96] (adapted). HLA: Human leukocyte antigen.
HLA-DQ2.5 carriage is necessary for disease development, but it is not sufficient by itself. A combination of other genetic factors influencing the mucosal barrier, the adaptive and the innate immune system also impact the likelihood of disease development. Wheat ingestion is a known environment factor that is necessary for disease development but on top of this, a number of factors such as timing of gluten ingestion and breast feeding cessation may influence disease development[96].
Studies using twins, which are assumed to share environmental factors, have estimated the percentage of non-HLA genetic variants which predispose to disease as approximately 60%[117]. To date a large list of variants have been suggested to predispose to CD through a combination of linkage and association studies, a large number of variants, however, do not stand up to further scrutiny. Only those that have been validated with convincing evidence in multiple populations are mentioned here.
In CD, like many common diseases, this genome wide linkage approach has been fairly unsuccessful at locating variants. Linkage was found to various regions including 5q and 19p, however, the only genomic region that was replicated with some reliability in other populations was 2q33, a region that contains the CTLA4[118], ICOS and CD28 genes[117]. CTLA4 is an excellent candidate gene for involvement in CD not only due to its integral involvement in the suppression of immune responses but also because it has been implicated as a genetic variant that increases susceptibility to T1D.
The prevalence of CD in patients diagnosed with T1D has been estimated at up to 15% in children and 6% in adults[119]. The reason for this association has never been fully elucidated, but common mechanisms within the pathogenesis and genetics of the two conditions may provide some insights. IL-21 region displays CD associations to T1D[120], rheumatoid arthritis[121], Grave’s disease[122] and psoriatic arthritis[123]; but genetic involvement in all these conditions is not currently understood. There is possibility of shared genetic susceptibility to autoimmunity through IL-2, IL-21 locus, both inside and outside of the HLA region, with almost no function identified thus far[116]. Like the studies associated with the HLA-DQ2.5 variant, further identification of the causal variant and its function will provide a unique insight into CD and other autoimmune disease biology.
Immune function
CD is an autoimmune disease associated with the genetic predisposition HLA and tTG autoantigen. tTG is a calcium dependent enzyme that plays a crucial role in CD pathogenesis[124]. tTG mediates ordered and specific deamidation of gliadins, creating an epitope that binds efficiently to DQ2 and is recognized by gut-derived T cells[125]. During gluten consumption, these tTG autoantibodies are produced by the mucosa of the small intestine, and detected in patients’ serum but disappear slowly from the patient’s circulation on a GFD[126]. Extraintestinal CD symptoms may be associated with immunoglobulin A (IgA) deposits on extracellular tTG in the liver, kidney, lymph nodes and muscles of CD patients[126].
The toxic peptides, such as the 19-mer, trigger an innate immune response[101], characterized by the production of IL-15 by epithelial cells and lamina propria dendritic cells[127]. There is some evidence that this response is a generalized response in all individuals, but is amplified in CD patients (possibly due to a lower threshold to IL-15) who only get disease as a result of adaptive immune system involvement[128]. IL-15 affects the epithelial barrier, both by increasing the permeability through disruption of the tight junctions[129,130] and acting on intraepithelial lymphocytes (IELs) promoting interferon γ (IFN-γ) production as well as a potent cytotoxic activity particularly by NKG2D+ cells[131,132]. Therefore, immunoadaptive peptides, like the 33-mer, can now reach the lamina propria, where they are presented by dendritic cells to gluten-specific T cells[133,134].
Other autoantigens that are normally “cryptic” can be unmasked and cause a self-aggressive immunologic response following the gliadin-initiated inflammatory process[8]. In fact, persistent stimulation by some proinflammatory cytokines (IFN-γ and tumor necrosis factor α) can cause further processing of autoantigens and their presentation to T lymphocytes by antigen-presenting cells. The mucosa is expanded by increased numbers of lymphoid cells both in the intraepithelial compartment, in which there is an increase in γδ T cells, and in the lamina propria, which is expanded by lymphocytes and plasma cells. The intestinal crypts are elongated because of an increase in dividing epithelial cells, and villi are shortened or even completely absent because of rapid loss of mature epithelial cells from the villus tip.
Intestinal epithelium function
Intestinal epithelium plays a central role in CD disease pathogenesis. It modulates the intestinal immune system that is acutely altered by gliadin. This indicates that gliadin can gain access to the basal surface of the epithelium, and therefore interact directly with the immune system, via both trans- and paracellular routes of absorption (Figure 3).
Figure 3.

Mechanisms of mucosal damage in celiac disease[80] (adapted). Gliadin peptides crosses the enterocyte by paracellular tight junctions (TJ) as a consequence of increased release of zonulin leading to impaired mucosal integrity upon 19 mer gliadin binding to chemokine (C-X-C motif) receptor 3 (CXCR3) receptor, or via transcytosis, or retrotranscytosis of secretory immunoglobulin A (IgA) through transferrin receptor CD71. Tissue transglutaminase (tTG) deamidates or crosslinks 33 mer gliadin which is then recognized by human leukocyte antigen (HLA)-DQ2 or -DQ8 molecules of antigen presenting cell (APC). APC presents the toxic peptide to CD4+ T cells. Activated gluten-reactive CD4+ T-cells produce high levels of pro-inflammatory cytokines. T helper 1 (Th1) cytokines promote increased cytotoxicity of intraepithelial lymphocytes (IELs) and natural killer (NK) T cells which cause apoptotic death of enterocytes by the Fas/Fas ligand (FasL) system, or interleukin 15 (IL-15)-induced perforin/granzyme and homodimeric natural killer-activating receptor-major histocompatibility-classIchain-related gene A comple (NKG2D–MICA) signaling pathways. The production of T-helper2 (Th2) cytokines activate and induce clonal expansion of B cells, which differentiate into (antigliadin and anti-tTG) antibody secreting plasma cells. Interaction between with the extracellular tTG and anti-tTG-autoantibody may induce epithelial damage. TCR: T cell receptor.
Retrotranscytosis
The protected retrotransport of secretory IgA into the intestinal lumen via the transferrin receptor CD71, allows the entry of intact and thus harmful gliadin peptides into the intestinal mucosa by a transcellular route. The overexpression of the transferring receptor CD71 in patients with active CD by transportation of gliadin across the intestinal mucosa through retrotranscytosis of secretory immunoglobulin-gliadin complexes is shown in Figure 4[135,136].
Figure 4.
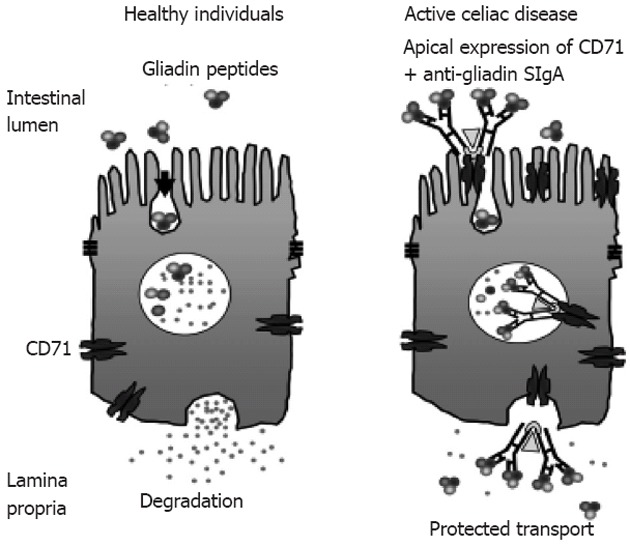
CD71 receptor-mediated transport of immunoglobulin A-gliadin complexes in celiac disease[136] (adapted). Gliadin bound to apically expressed CD71 receptor in active celiac disease individual allows protected transport of gliadin into the lamina propria. SIgA: Secretory immunoglobulin A.
Transcytosis of α2-gliadin-33mer (an important trigger of CD) by apical-to-basal is stimulated by IFN-γ, which is a key cytokine involved in CD immunopathogenesis[137].
Paracellular route
There have been recent hypothesis associated with non-digested gliadin absorption in the intestinal lumen during the early event in CD pathogenesis by stimulation of the innate and adaptive immune system[83,101,102]. Zonulins provide information on the regulation of intercellular tight junctions (TJs) and increased intestinal permeability[138-142]. It is released by the enterocyte upon apical exposure to α-gliadin digests[129,143]. Lammers et al[143] have identified that MyD88 induces release of zonulin upon gliadin binding to CXCR3 on enterocytes, as a result inducing greater epithelial permeability and subsequent paracellular gliadin passage to the gut mucosa.
After binding to its surface receptor, gluten is internalized[144] and subsequently triggers a series of intracellular events including phospholipase C and Protein kinase Ca activation and actin polymerization, which lead to the opening of TJs[139,145] through Zot/Zonulin receptor (Figure 5).
Figure 5.
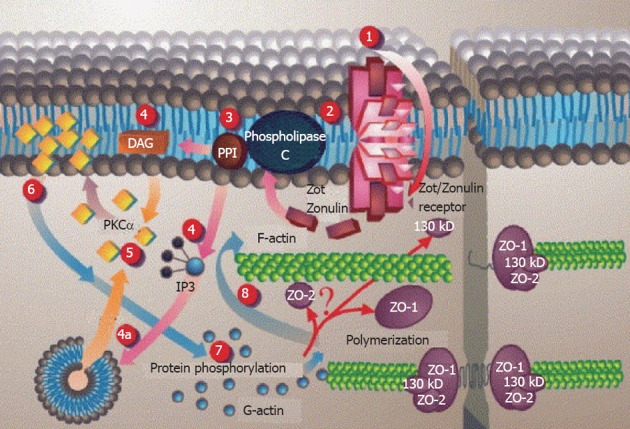
Proposed Zot intracellular signal mediated opening of intestinal tight junctions[146] (printed with permission). 1: Zot interacts with a specific Zot/Zonulin intestinal surface receptor; 2: Leading to protein internalization; 3: Activation of phospholipase C; 4: Hydrolyzes phosphatidyl inositol to release inositol 1,4,5-tris phosphate (PPI-3) and diacylglycerol (DAG), either via DAG or (4a) through the release of intracellular Ca2+ via PPI-3; 5: Protein kinase C alpha (PKCα) is then activated; 6: Membrane-associated, activated PKCα catalyzes the phosphorylation of target protein(s); 7: With subsequent polymerization of soluble G-actin in F-actin; 8: This polymerization causes the rearrangement of the tight junctions (TJ) filaments and displacement of proteins [including zonula occludens-1 (ZO-1)]. As a result, intestinal TJ becomes loosened. IP3: Inositol trisphosphate.
Other pathways
There are several pathways including cellular signals that may be involved in the mucosal damage in CD. Deamidation of gluten peptides by tissue tTG reinforces presentation of gluten peptides by HLA-DQ2 or HLA-DQ8 molecules of plasmacytoid dendritic cells (pDCs) to T cells, which activate gluten-reactive Th1 cells and produce high levels of proinflammatory cytokines. IL-21 is overproduced in the mucosa of CD patients, where it helps sustain T-bet expression and IFN-γ production[146]. Th1 cytokines promote increased cytotoxicity of IELs and natural killer (NK) T cells which cause apoptotic death of enterocytes by the Fas/Fas ligand system, or IL-15-induced perforin/granzyme and NKG2D-MICA signaling pathways. IFN-α released by activated pDCs perpetuates the inflammatory reaction by inducing Th1 cells to produce IFN-γ. IL-21 and IL-15 produced by DCs and intraepithelial cells also inhibit transforming growth factor beta signaling and regulatory T cells (Tregs) function. Additionally, the production of Th2 cytokines, Th2 cells drives the activation and clonal expansion and differentiate of B cells into plasma cells secreting anti-gliadin and anti-tissue transglutaminase antibodies[147], which interact with extracellular tTG, and may induce epithelial damage.
Hence in CD, there is impaired suppressor activity of Tregs. This defect in Tregs function could play a role in the pathogenesis of CD and in CD autoimmunity[148].
DIAGNOSIS
Approach to initial CD diagnosis
In 1970, the European Society of Paediatric Gastroenterology laid down criteria for the diagnosis of CD in children, entailing three biopsies of an initial flat mucosa in the upper small intestine, restoration of the mucosa to normal on a GFD, and a deterioration of the mucosa after gluten challenge[149]. Given the current availability of serological tests being highly sensitive and specific, the European Society of Paediatric Gastroenterology, Hepatology, and Nutrition has proposed a revised CD diagnostic protocol[150]. Based on this protocol, if the symptoms (either “classical” or “atypical”) and serological tests are suggestive of CD, small bowel biopsy followed by a favourable clinical and serological response to the GFD is now considered sufficient to definitely confirm the diagnosis. In asymptomatic patients improvement in mucosal appearance may be required to confirm the diagnosis, but in majority symptomatic patients, continual abnormality of mucosa at the second biopsy is more likely to indicate slow /partial mucosal recovery[151]. This may also reflect that the site of re-biopsy (proximal small intestine) is often the last site to improve.
The current approach to evaluating CD has been modified by the advent of highly sensitive and specific serological tests. An algorithm for diagnosing CD is given in Figure 6. Assays for IgA anti-tissue transglutaminase (TGA) and IgA anti-endomysial (EMA) have both the highest specificities and sensitivities, and are therefore regarded as being superior serological screening tools for diagnosis of CD[152].
Figure 6.
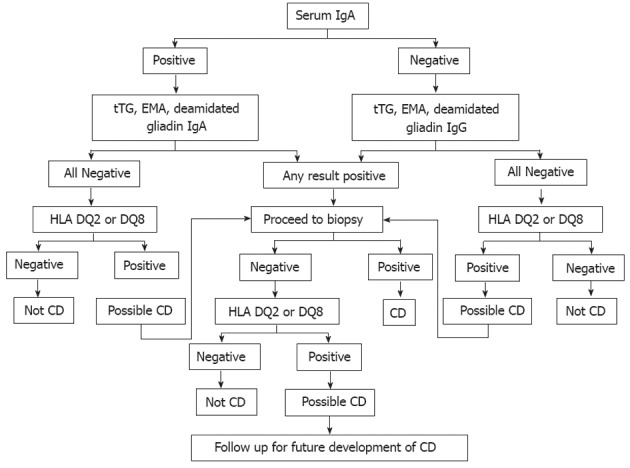
Celiac disease diagnostic testing algorithm (adapted from Mayo Medical Laboratories, Mayo Foundation for Medical Education and Research). IgA: Immunoglobulin A; IgG: Immunoglobulin G; tTG: Tissue transglutaminase; EMA: Endomysial; HLA: Human leukocyte antigen; CD: Celiac disease.
Initial CD evaluation is based on a combination of positive CD-specific serological tests, histological findings in the intestinal biopsy, CD-predisposing gene encoding HLA DQ2 or DQ8, family and medical history of CD, and clinical or histological response to GFD[26,80]. However, CD diagnosis can be challenging in some non-responsive patients to GFD[7]. Practically all patients with CD carry HLA-DQ2 or HLA-DQ8. Thus the absence of these gene pairs reflects a very high negative predictive value for CD and should prompt consideration of other causes of small bowel-related symptoms and pathological changes[153,154]. Positive TGA or EMA at initial diagnosis of CD or at any time in the clinical course of the disease helps to confirm the diagnosis of CD because of their excellent specificities of over 99% when small bowel villous atrophy is present on biopsy[155].
However, false positive serological assays may also occur[156], in liver disease and small-bowel inflammation[157], so documentation of gluten sensitivity is important. A combination of biopsy and serological antibody can also be used to support diagnosis to reduce false positive results. A validated subjective Celiac Dietary Adherence Test, a patient-completed tool, can also be used in conjunction with biological markers to assess dietary adherence and disease activity in individuals with CD[158].
Diagnosis of refractory CD
The influence of noncompliance to a GFD and the substantial number of patients being undiagnosed are of greatest concern, as these factors could possibly contribute to the refractory form of CD and to the development of malignancies. These patients’ CD symptoms do not revert on GFD. The first evaluation step of a potential RCD case is to confirm correct initial diagnosis of CD[7]. Sometimes neglected in this determination is the documentation of an initial and convincing response to a GFD, i.e., demonstration that the disease was truly a “gluten-sensitive” small bowel disorder. Otherwise, it may be difficult to ascertain if CD was initially present. More precise terms in this clinical setting include “sprue-like intestinal disease” or “unclassified sprue”.
Some patients with developed RCD are likely to have negative TGA and EMA[6,159], demonstrating that negative CD specific serology does not exclude the diagnosis of CD. A family history of CD in first-degree relatives (especially siblings) further supports the diagnosis of CD in patients, having 14% positive tTG test and 10% positive EMA with an estimated prevalence of 11% where 54% had “silent” disease, most with severe intestinal villous atrophy[76]. The diagnosis of CD by histological findings or clinical improvement after GFD without confirmation with other diagnostic criteria may not be entirely reliable because CD is just one of many causes of villous atrophy. Clinical response to GFD or exacerbation after gluten re-introduction have low sensitivity of 59% and specificity of 92% for CD[160], which account to a positive likelihood ratio of 7.37 (means that individuals with positive histology upon gluten re-introduction are 7.37 times more likely to have CD than those with negative histology upon gluten re-introduction) and a negative likelihood ratio of 0.44 (means that individuals with positive histology upon gluten re-introduction are 0.44 times less likely to lack CD than those with negative histology upon gluten re-introduction). Thus, a critical review of prior tests and villous histology is crucial to determine the accuracy of a prior diagnosis of CD. Ideally, documented normalization of biopsies after a GFD and then demonstration of recurrent symptoms with histological relapse best defines refractory CD (RCD). Obviously, this is not always possible.
RCD is believed to affect approximately 5% of patients with CD. It is subdivided into types I and II, with normal and aberrant (expressing cytoplasmic) CD3, but lacking surface expression of the T-cell markers CD3, CD4, CD8[161], and the T-cell receptor, intraepithelial T lymphocytes in the small intestinal mucosa, respectively[162]. Enteropathy-associated T-cell lymphoma (EATL) occurs in more than half of patients with RCD II within 4-6 years after RCD II diagnosis, and is the main cause of death in this group of patients[76,163]. RCD type 1 only rarely evolves into EATL. Recent data indicate a relative risk for patients with (untreated) CD to develop EATL[164,165].
DIAGNOSTIC TESTS
Serological tests
HLA typing: The contribution of HLA type to the genetic risk for CD has been variously estimated at 30%-50%[20,21]. Many of the polymorphic genes are involved in susceptibility to CD encode products that influence the immune response upon gluten ingestion, as shown for the HLA-linked genes[22]. Although Non-HLA genes contribute more than HLA genes to the genetic background of CD, each of them adds only a minor contribution to the disease development.
There is strong association between CD and the presence of HLA DQA1*0501-DQB1*02 (DQ2) and DQA1*0301-DQB1 [0302 (DQ8) haplotypes. Approximately 90% to 95% of patients with CD carry DQ2 and those patients that are negative for HLA-DQ2 are usually positive for HLA-DQ8[166,167], indicating a strong genetic risk for the disease[100]. Several studies also have confirmed that the absence of HLA-DQ2, HLA-DQ8, or both virtually excludes the diagnosis of CD[168-170]. However, the modest sensitivity (HLA-DQ2, 70%-99.8%; HLA-DQ8, 1.6%-38%) and specificity (HLA-DQ2, 69%-77%; HLA-DQ8, 77%-85%) of the test means that a positive result is not sufficient to diagnose the disease[having a low positive predictive values (HLA-DQ2, 6.3-18; HLA-DQ8, 0.28-8.1) and likelihood ratios (HLA-DQ2, 2.25-4.33; HLA-DQ8, 0.07-2.53)][171]. Even the presence of HLA-DQ2 or HLADQ8 in patients with positive serologic test results is strongly suggestive but not pathognomonic for CD. Antibody screening to identify participants with preclinical CD may be reduced by preselecting HLA risk group from the large populations with long-term follow-up for CD[172]. Hence HLA-DQ genotyping could be included in the algorithm of selecting large populations prospectively screened for CD.
Antibody level: Several serum antibodies have been used to initially evaluate patients with suspected CD, monitor adherence and response to GFD, and screen asymptomatic individuals. Anti-gliadin antibodies (AGA) detection has low sensitivity and specificity, leading to high false-positive rate in patients[173]. Recent reports of deamidated gliadin peptide AGA (DGP-AGA) have suggested a much improved accuracy[174]. The sensitivity and specificity for IgA DGP-AGA is 84.3% and 79.8%, whereas for IgG DGP-AGA the sensitivity and specificity are 82.3% and 98.9%, respectively[175]. As shown in Table 6, EMA and TGA have been found to be superior to AGA and gives highest sensitivity and specificity of greater than 95% when used in combination[173,176,177]. EMA testing, however, produces a subjective and highly observer-dependent result, whereas TGA testing is quantitative.
Table 6.
Operating characteristics of serological markers to detect the celiac disease in adults[178] (adapted)
| Serological tests | Sensitivity | Specificity | Predictive value | Likelihood ratio | ||
| 95% CI (%) | 95% CI (%) | Positive | Negative | Positive | Negative | |
| IgG AGA | 57-78 | 71-87 | 0.2-0.9 | 0.4-0.9 | 1.96-6 | 0.25-0.61 |
| IgA AGA | 55-100 | 71-100 | 0.3-1.0 | 0.7-1.0 | 1.89-∞ | 0-0.63 |
| IgA EMA | 86-100 | 98-100 | 0.98-1.0 | 0.8-0.95 | 43-∞ | 0-0.14 |
| IgA TGA | 77-100 | 91-100 | > 0.9 | > 0.95 | 8.55-∞ | 0-0.25 |
| IgA TGA and EMA | 98-100 | 98-100 | > 0.9 | > 0.95 | 49-∞ | 0-0.02 |
IgG: Immunoglobulin G; IgA: Immunoglobulin A; AGA: Anti-gliadin antibodies; EMA: Endomysial; TGA: Transglutaminase.
Small intestinal mucosal biopsy
Histopathological analysis: Although the diagnosis of CD can be suspected on clinical or laboratory grounds, or as a result of serological tests, histology of the proximal small intestinal mucosa is still the diagnostic gold standard and must always be performed. Small intestinal histopathology of CD biopsy samples are characterized by typical architectural abnormalities. Marsh[178] has pioneered the theory of a sequence of progression of the CD lesion in the small intestinal mucosa.
According to the modified Marsh classification: normal mucosa is classified Marsh 0, intraepithelial lymphocytosis as Marsh I, intraepithelial lymphocytosis and crypt hyperplasia as Marsh II, and intraepithelial lymphocytosis, crypt hyperplasia and villous atrophy as Marsh III[179]. Later the Marsh-Oberhuger system was developed, where stage 3 was split into three sub stages (a, b and c)[178,180]. The Marsh-Oberhuber classification was based on a 6-stage grading, namely (1) type 1 infiltrative lesions, characterized by normal mucosal architecture with an increased number of IELs; (2) type 2 hyperplastic lesions, characterized by an increase in crypt depth without villous flattening; (3) type 3a, 3b, and 3c destructive lesion, characterized by mild, marked, and complete villous flattening, respectively; and (4) type 4 hypoplastic lesions, characterized by villous atrophy with normal crypt height and IEL count.
Considering the broad spectrum of lesions possibly present in CD, the Marsh-Oberhuber system is undoubtedly valid under optimal clinical conditions, but the considerable number of diagnostic categories involved makes it prone to a low inter-observer and intra-observer agreement.
False-positive and false negative test results may occur due to patchy mucosal damage, inter-observer variability, low-grade histopathological abnormalities and technical limitations. Hence, reliance on standard histological findings alone may result in failure to diagnose CD[181]. Several other limitations may be evident in high-volume, service-oriented laboratories with limited attention to quality control. Poorly oriented biopsies fixed in the endoscopy suite may be prone to difficult interpretation. Inter-observer variation in pathological interpretation may occur, especially if there is limited access to a pathologist with expertise focused on interpretation of small intestinal biopsies. Some patients with low-grade histopathological abnormalities (Marsh I/Marsh II) can present with gluten-dependent symptoms or disorders before overt villous atrophy occurs. Furthermore, some patients with isolated intraepithelial lymphocytosis (Marsh I), who are not clinically suspected of having CD, may develop CD during follow-up[182]. Although the mucosal changes in CD are thought to develop gradually, whether minor mucosal lesions in asymptomatic patients indicate CD in an early stage is not yet clear[183].
In case of strong clinical suspicion of CD, duodenal biopsy must be performed regardless of serological analysis[184]; in cases of low suspicion of disease or screening, duodenal biopsy probably only needs to be performed in seropositive patients. Hence, the new system for routine use of simplified grading system with uniform diagnosis and increase validity of the pathologic diagnosis of CD was developed by using only three categories (A, B1 or B2) with A representing normal villous with lymphocytic infiltration and B1 and B2 representing partial and complete villous atrophy, respectively[185]. The new proposed grading system classified the CD lesions into non-atrophic (grade A) and atrophic (grade B)[186]. Grade A was characterized by the isolated increase of IELs (> 25/100 enterocytes)[187], whereas grade B was split into B1, in which the villous/crypt ratio is less than 3/1, with still detectable villi, and B2, in which the villi are no longer detectable. A comparison between the Marsh-Oberhuber and the new grading criteria is shown in Figure 7. Figure 7C represents pictures of the grades proposed in the new histologic grading criteria.
Figure 7.

A comparison between the Marsh classification for celiac disease. 1: Marsh-Oberhuber; 2: Grading system for celiac disease, and the new grading system; 3: Representative pictures of the grades A (original magnification, 20×; insert, 60×), B1 (20×), and B2 (20×), proposed in the new grading system. An alternative classification may simply describe “mild”, “moderate” or “severe (flat)” architectural changes[186] (printed with permission).
Recently, quantitative measurements of villous height, apical and basal villous widths, and crypt length (morphometry) have been used to determine changes in duodenal morphology, particularly after the introduction of a GFD, in correlation with Marsh grade, self-reported adherence to GFD, and changes in serology. GFD resulted in increase in villous area and a progressive decrease in crypt length, with a plateau after 6-12 mo and mean villous area half that of control subjects[188].
Intraepithelial lymphocyte: The presence of aberrant IELs appears to be a reliable prognostic marker to differentiate between RCD typeIand type II patients, with characteristic normal and aberrant IELs, respectively. IELs are considered aberrant when there is cytoplasmic CD3 expression, but no expression of surface CD3, CD4 and CD8 T-cell markers[189,190]. The current methods for double CD3/CD8 T cell receptor clonal from intestinal tissue can be done by immunohistochemistry, polymerase chain reaction or flow cytometry[161,162,189]. The presence of these IELs is associated with a significant increase in EATL development[161,163,191,192]. Increased IEL’s may be used to support or exclude diagnosis of CD, and may be useful for follow up as mentioned in Table 7[193].
Table 7.
Factors that support the diagnosis of celiac disease in patients with an increased density of intraepithelial lymphocytes but no villous shortening[194] (printed with permission)
| Family history of celiac disease | 15% of first-degree relatives are affected |
| Concomitant autoimmune conditions | Risk of coeliac disease approximately 5-fold |
| Increased density of γδ+ IELs | Sensitivity 0.84, specificity 0.91 |
| Increased density of villous tip IELs | Sensitivity 0.84, specificity 0.95 |
| HLA DQ2 or DQ8 | High sensitivity, low specificity |
| Negative predictive value high | |
| Gluten dependence | Should be ascertained by gluten challenge or gluten-free diet |
IELs: Intraepithelial lymphocytes; HLA: Human leukocyte antigen.
In 95% of non-refractory CD and control patients, the highest percentage of aberrant T-cells in duodenal biopsy specimens is in agreement with the cut-off of the % T cells which are aberrant. Such a cut-off has been previously suggested in the RCD group based on the clinical observation that none of the RCD patients with less than 20% aberrant T-cells eventually developed EATL[163]. Clonal T-cell population can be found in the intestinal mucosa of RCD patients, which relates to the development of EATL[161,194]. Immunophenotyping using flow cytometry[162], gives significant higher negative predictive value and sensitivity (both 100%) for aberrant T-cells were found with regard to EATL development in RCD, when compared to clonality in a duodenal biopsy specimen (75% and 78%, respectively). The positive predictive values (59%) and likelihood ratios (1.85) of these tests for EATL development in RCD are almost comparable.
Aberrant T-cells is quantified by flow cytometry is well suited to identify RCD patients at risk for EATL as it has a higher predictive value and sensitivity than T-cell clonality analysis of duodenal biopsy specimens. A cut-off value of 20% appears reliable for early risk stratification[159], and targeted therapeutic options in RCD patients[6,27,195]. This is particularly important since once overt T-cell lymphoma has developed, treatment outcome and survival are very poor[159,196]. Additionally, quantification of aberrant T-cells is useful for the subsequent follow-up of treated RCD II patients[27].
Useful background for the diagnosis of CD: The HLA class II molecules DQ2 and DQ8 are required for but are not sufficient for the development of CD: 50% of Americans are positive of one of those molecules, but only 1% develop CD. Negative HLA DQ2 or DQ8 may rule out CD as a cause of the enteropathy; IgA TGA serology is > 95% sensitive for CD, especially when there is a high titre, but false positive tests can occur; Anti-gliadin antibodies have a relationship high false negative rate, and have been replaced by IgG DGP assays that appear to have a sensitivity compared to TGA; The endoscopic features of CD (scalloping of mucosal folds, less prominent folds, fissules, and a nodular/mosaic pattern) are 59% sensitive but 92% specific for CD. For example, other small bowel disorders, including Crohn disease in the duodenum, may cause mucosal scalloping and other endoscopic features of CD.
TREATMENT
Existing treatment
GFD: Currently, the only effective treatment available for CD individuals is a strict life-long GFD[197]. In reality, total avoidance of gluten intake is extremely difficult, due to hidden gluten from food contamination[198]. For safety purposes, United States Food and Drug Administration has set the limit (August 2011) of < 20 ppm gluten (equivalent to 10 ppm gliadin) for gluten-free foods. The total daily consumption of gluten-free foods must be taken into account as it may exceed the tolerable limit of each celiac individual. It has been estimated that the threshold of prolonged gluten ingestion in some CD individuals may be lower than 50 mg/d[199]. However, some CD individuals can conceivably be more sensitive. The presence of hidden gliadin in contaminated food products represents an imminent risk for celiac consumers, because of long-term effect of regular ingestion of small amounts of gliadin[200], on causing positive tTG and characteristic small bowel biopsy.
Gluten modification: Approaches to modify dietary gluten have been made to render gliadin non-toxic, since it is a non-invasive approach to CD patients. This approach has been less appealing due to loss of baking characteristic, public refusal for genetically modified crops, contamination of genetically modified crops with gluten contained crops grown nearby and heterogeneous uncharacterised immunostimulatory epitopes in gluten, and difference among patients response to epitopes and gluten levels[201].
A greater understanding of the pathogenesis of CD allows alternative future treatments to be designed. A number of preliminary studies have been published that illustrate from a conceptual perspective future possible approaches that may be pursued in more detail (Table 8).
Table 8.
Future therapeutic approach for celiac disease treatment
| Mechanism | Therapeutic agent | Stage of study | |
| Hydrolysis of toxic gliadin | ALV003 | Glutenenases and endoprotease | Phase II |
| AN-PEP | Prolyl endopeptidase | Phase II | |
| Lactobacilli | Discovery | ||
| VSL#3 | Lyophilised bacteria, including Bifidobacteria, Lactobacilli and Streptococcus salivarius | Discovery | |
| Prevention of gliadin absorption | Larazotide | Hexapeptide derived from zonula occludens toxin of Vibrio cholera | Phase II |
| Synthetic polymer poly (hydroxyethylmethacrylate-co-styrene sulfonate) | Discovery | ||
| Anti-gliadin IgY | Discovery | ||
| tTG2 inhibitor | Dihydroisoxazoles | Discovery | |
| Cinnamoyltriazole | Discovery | ||
| Aryl β-aminoethyl ketones | Discovery | ||
| Peptide vaccination | Nexvax2 | Three deamidated peptides derived from wheat α-gliadin, ω-gliadin and β-hordein | PhaseI |
| Human hookworm (Necator americanus) inoculation | Phase II | ||
| Modulate immune response | HLA-DQ2 blocker | Discovery | |
| Interleukin blocker | Discovery | ||
| NKG2D antagonist | Discovery | ||
| Restore intestinal architecture | R-spondin-1 | Discovery |
tTG2: Tissue transglutaminase 2; PEP: Prolyl endopeptidases; NKG2D: Homodimeric natural killer-activating receptor; HLA: Human leukocyte antigen; IgY: Immunoglobulin Y.
FUTURE TREATMENT APPROACHES
Hydrolysis of toxic gliadin peptide
Prolyl endopeptidases: Prolyl endopeptidases (PEPs) are endoproteolytic enzymes expressed in micro-organisms and plants. These enzymes cleave proline-rich gluten to smaller peptides that are ready for digestion by intestinal brush-border enzymes (aminopeptidases and carboxypeptidases). Limited efficiency was found, since PEP required 3 h preincubation with gluten containing foods to achieve full detoxification of peptides and to prevent intestinal transport of active gluten fragments[202]. This is unlikely to be achieved by co-administration of PEP and gluten-containing diet.
A two-stages cross-over phase II clinical trial was performed using asymptomatic CD patients eating, a slice of bread daily and a slice of bread pre-treated with PEP daily[203]. After 2 wk of PEP treated gluten challenge, majority of patients did not develop malabsorption, measured by faecal fat excretion and D-xylose malabsorption tests. The tests likely lacked the necessary sensitivity to assess minor malabsorption resulting from active CD, since no histological confirmation was performed to determine deterioration in the Marsh grading[201]. When PEPs were consumed as jam spread on a slice of gluten-containing bread by CD patients, villous blunting was seen in small bowel biopsy histological evaluation in most patients[204]. Further studies are needed to determine the appropriate dose of enzyme and time of administration relative to the quantity of ingested gluten.
ALV003: ALV003, a mixture of two glutenases, an endoprotease from germinating barley and PEP, was pretreated with wheat flour and tested in CD patients[205]. Symptoms typically associated with gluten ingestion were not significantly reduced by ALV003 pre-treatment, but ALV003 abolished immune responses induced by gluten in CD patients. A randomized controlled phase IIa clinical trial has been performed where CD patients received either ALV003 or placebo daily for 6 wk at the time of 2 g gluten contained bread. This proof-of-concept study demonstrated that ALV003 can attenuate gluten-induced small intestinal mucosal injury in CD patients[206]. After six weeks period, biopsies proved lower small intestinal mucosal injury in patients treated with ALV003 than placebo-treated patients despite of persistent intestinal inflammation in many patients on a strict GFD. Placebo-treated patients were found to have suffered more adverse events, most commonly including abdominal distention, flatulence, eructation, abdominal pain and diarrhea[206].
Lactobacilli: Lactobacilli added to sourdough for fermentation are able to lyse the proline-/glutamine-rich gluten peptides and thus decrease immunotoxicity[207-210]. A mixture of fermented wheat flour with oat, millet and buckwheat allows sourdough bread to retain its baking characteristics. A pilot study in patients with CD suggested that this bread was well tolerated[209]. However, these patients were challenged for only 2 d, which is clearly not sufficient to draw any firm conclusions. Hence, another 60-d diet of fully hydrolyzed wheat flour with sourdough lactobacilli and fungal proteases (8 ppm residual gluten; n = 5) was further studied. The pretreated flour was rendered non-toxic by serological, morphometrical, and immunohistochemical analysis[211]. A larger group of subjects in the trial and palatability of digested flour baked products needs to be taken into consideration.
VSL#3: VSL#3 is a probiotic containing lyophilised bacteria, including bifidobacteria (Bifidobacterium longum, Bifidobacterium infantis and Bifidobacterium breve), lactobacilli (Lactobacillus acidophilus, Lactobacillus casei, Lactobacillus delbrueckii subsp., Lactobacillus bulgaricus and Lactobacillus plantarum) and Streptococcus salivarius subsp., Thermophiles. It is used to hydrolyse gliadin peptides in pre-treated flour and tested for efficacy in rat intestinal cell line and celiac jejunal biopsies[207]. VSL#3 pre-digested gliadins did not show an increase of the infiltration of CD3+ intraepithelial lymphocytes and caused a less pronounced effect on intestinal mucosa permeability (determined by lower F-actin rearrangement and zonulin release). Hence, VSL#3 may have importance during food processing to produce pre-digested gluten-free products.
Prevention of toxic gliadin peptide absorption
Larazotide: Larazotide (AT-1001, Alba Therapeutics, Baltimore, MA), is a synthetic hexapeptide derived from Zonula Occludens toxin of Vibrio cholera[212]. It is used to inhibit the opening of tight junctions of the small intestine epithelial cells. Clinical trial phase I in CD patients suggested that Larazotide therapy is well tolerated by patients and reduces intestinal barrier dysfunction, proinflammatory cytokine production, and gastrointestinal symptoms in CD individuals after gluten exposure[213]. Encouraging results were obtained from a 6-wk phase IIb trial in terms of symptoms and antibody titers[214], showing larazotide acetate a promising drug candidate. This drug inhibits the paracellular route of gliadin absorption through tight junctions, which is not the only mechanism of gliadin absorption. Indeed, gliadin may gain access to the mucosa through transcellular pathways in addition to paracellular route[135,137]. Hence, this strategy might be best exploited in combination with other treatments.
Synthetic polymer poly (hydroxyethylmethacrylate-co-styrene sulfonate): Poly (hydroxyethylmethacrylate-co-styrene sulfonate) [P (HEMA-co-SS)] forms supra-molecular particles upon gliadin complexation in gastric and intestinal conditions[215,216], and deteriorates gliadin’s effect on epithelial cells[217]. This complexation decreases the effect of gastrointestinal (GI) digestive enzymes on gliadin absorption, and thus the formation of immunogenic peptides is reduced. Gluten-sensitive HLA-HCD4/DQ8 mice co-administered with P (HEMA-co-SS) showed attenuated gliadin-induced changes in permeability and inflammation[217]. Low side effect, cost and possibility to be taken, occasionally with gluten-containing food, makes it an attractive option. Further investigation of the mechanisms of action and its interaction with human tissues is required before its efficacy is investigated in human trials[218].
Anti-gliadin egg yolk antibody: Oral antibody passive immunotherapy may be of value due to the advantages of reduced cost, ease of administration, and potential to treat localized conditions in the gastrointestinal tract[219]. Among antibodies, chicken egg yolk immunoglobulin (IgY), is ideal for passive immunotherapy, as it may be readily obtained in large quantities from egg yolk, presenting a more cost-effective, convenient, and hygienic alternative to mammalian antibodies. Oral immunotherapeutic IgY is a promising alternative to neutralize gliadin in the GI tract and prevention it from absorption[220]. Mannitol contained antibody preparation is highly resistant against GI enzymes and proved to effectively neutralized gliadin under simulated GI conditions in the presence of food. In vivo study; BALB/c mice fed with IgY formulation and gliadin ratio of 1:5 (w/w), demonstrated that gliadin absorption in the gastrointestinal tract was minimal at < 1%[221]. Further investigations in CD patients is requires to prove its efficacy and determine dosing regimen of antibody relative to the amount of gliadin ingestion.
Blockage of selective deamidation of specific glutamine residues by tissue transglutaminase 2 inhibitor
Transglutaminases (a family of eight enzymes) have diverse functions in human and are involved in several biological and pathological processes[222]. tTG2 is an enzyme that has a pro-inflammatory effect and increases the immunostimulatory epitopes present in the lamina propria of the small intestine. Blockage of tTG2 may be a promising approach to inhibit the inflammatory process upon gluten ingestion. There are two essential classes of tTG2 inhibitors; irreversible and reversible inhibitors[223]. Irreversible inhibitors form a stable covalent bond with this enzyme, and thus prevent deamidation of gliadin peptides[223,224]. Reversible inhibitors are more desirable to minimize possible side effects. These include aldehyde-bearing tTG modulators[225], cinnamoyl triazole derivatives[226], and the highly specific modified peptide targeting the active cysteine site of tTG2[227]. Since a few gluten T-cell epitopes can be recognized without being deamidated by tTG2[228,229], this approach will not inhibit the innate response[101], or the immune response induced by non-deamidated peptides[82]. To be able to use tTG2 inhibitors clinically, it is critical to design highly specific inhibitors, since all human tTG share high sequence homology.
Vaccine application to restore immune tolerance towards gluten
Autoimmune enteropathy in CD has been proposed to be due to impairment of immunoregulatory mechanisms that controls oral tolerance. Systematic peptide mapping of T-cell was performed to determine gliadin reactive epitopes recognized by approximately 90% of CD patients. A clinical trial phase I study has been initiated as Nexvax2® (Nexpep Pty, Ltd., Australia) peptide vaccine-containing mixture of immunotoxic α- and ω-gliadins and B-hordein[230].
Engineered Lactococcus lactis secreting a DQ8-re-stricted gliadin peptide administered orally[231], or recombinant α-gliadin in HLA-DQ8 administered intranasally in transgenic mouse model[232], have been studied to modulate immune response to gluten. However, it is difficult to appreciate how the vaccine or the intranasal peptide administration can modulate the Tr1 response. More work is needed to assess the effect of these therapies on the spectrum of gluten peptides presented to the gut.
Dermal inoculation of human hookworm (Necator americanus) has also been used to modulate the immune response to gluten[233]. A phase II trial with CD patients suggested that hookworm infection on its own may not obviate the necessity for a restricted diet in CD, but appears to be safe and might impact on immune pathology[234]. Here in, hookworm infection is expected to reduce gluten sensitivity and immune reactivity.
Modulation of immune response to dietary gliadin
HLA-DQ blocker: HLA-DQ blocker is used to block the binding sites of HLA-DQ2 or DQ8 for it to be unrecognized by T cells as well to block the presentation of the antigen. This is not a new concept that was developed without much success to treat type 1 diabetes mellitus and rheumatoid arthritis, due to difficulties in effective drug delivery[235,236]. By amino-acid substitution of gliadin T-cell stimulatory sequence, the epitope can be converted to an agonist or antagonist, abolishing the inflammatory cascade[237]. IFN-γ production by peripheral blood lymphocytes was prevented when either an alanine or lysine amino acid was substituted through the immunodominant α-gliadin peptide, corresponding to the peptide’s anchor to the HLA-DQ cleft[238]. To develop this as a new therapeutic agent, more studies need to be performed, looking at the mass T-cell action of the gut towards these modified peptides.
Interleukin blocker: Modulation of cytokine production has been evaluated for the treatment of several autoimmune diseases, although their side effects may be severe. Modulation of proinflammatory IL-15 and anti-inflammatory IL-10 cytokines has been suggested to influence the immune balance between tolerance and autoimmunity[127,239-241]. Blocking IL-15 may promote maintenance of epithelial integrity, limit epithelial destruction, leading to decreased passage of dietary gliadin.
NKG2D antagonists: MICA molecules, strongly expressed on active CD epithelial cell surface upon gliadin challenge[132], interact with the NKG2D-activating receptor on human natural killer cells and CD8 T cells, leading to villous atrophy due to an IEL-mediated damage to enterocytes[131,132]. Thus, NKG2D antagonists[131] and anti-NKG2D antibodies[242], have been proposed as therapeutics in CD.
Restoration of intestinal architecture by R-spondin-1
R-spondin-1 is an intestinal mitogen, shown to stimulate crypt cell growth, accelerate mucosal regeneration and restore intestinal architecture in mouse models of colitis[243]. This agent has yet to be tested in human to be considered as a therapeutic agent in CD.
CONCLUSION
CD has been kept in the dark for decades with very little known about what is a relatively common medical condition. It is only recently that we have greater understanding of its prevalence, diagnosis and pathogenesis, which has supported the development of new therapeutic approaches to treat CD. There are several future directions to follow to treat CD, which if successful will supplement or even replace the current only effective treatment, the use of a GFD. A greater understanding of the pathogenesis of CD allows alternative future CD treatments to hydrolyse toxic gliadin peptide, prevent toxic gliadin peptide absorption, blockage of selective deamidation of specific glutamine residues by tissue, restore immune tolerance towards gluten, modulation of immune response to dietary gliadin, and restoration of intestinal architecture (Table 9).
Table 9.
Key points from recent findings
| Cause |
| Environmental (gluten) and genetic factors (HLA and non-HLA genes) |
| Prevalence |
| 0.5%-1% worldwide in normal at-risk population |
| Higher risk in the population with diabetes, autoimmune disorder or relatives of CD individuals |
| Pathogenesis |
| Gliadin gains access via trans- and para-cellular routes to the basal surface of the epithelium, and interact directly with the immune system |
| Types of CD symptoms: “typical” or “atypical” |
| Diagnosis |
| Positive serological (TGA or EMA) screening results suggestive of CD, should lead to small bowel biopsy followed by a favourable clinical and serological response to the GFD to confirm the diagnosis |
| Current treatment |
| Strict life-long GFD |
| Alternative future CD treatments strategies |
| Hydrolysis of toxic gliadin peptide |
| Prevention of toxic gliadin peptide absorption |
| Blockage of deamidation of specific glutamine residues by tissue |
| Restoration of immune tolerance towards gluten |
| Modulation of immune response to dietary gliadin |
| Restoration of intestinal architecture |
HLA: Human leukocyte antigen; CD: Celiac disease; EMA: Endomysial; TGA: Transglutaminase; GFD: Gluten-free diet.
Footnotes
Peer reviewers: Khaled Jadallah, MD, Assistant Professor of Medicine, Consultant, Department of Internal Medicine, King Abdullah University Hospital, Jordan University of Science and Technology, Irbid 22110, Jordan; Cuong D Tran, PhD, Gastroenterology Unit, Children, Youth and Women's Health Service, University of Adelaide, 72 King William Rd, North Adelaide, SA 5006, Australia
S- Editor Gou SX L- Editor A E- Editor Li JY
References
- 1.Rampertab SD, Pooran N, Brar P, Singh P, Green PH. Trends in the presentation of celiac disease. Am J Med. 2006;119:355.e9–355.14. doi: 10.1016/j.amjmed.2005.08.044. [DOI] [PubMed] [Google Scholar]
- 2.Mäki M, Kallonen K, Lähdeaho ML, Visakorpi JK. Changing pattern of childhood coeliac disease in Finland. Acta Paediatr Scand. 1988;77:408–412. doi: 10.1111/j.1651-2227.1988.tb10668.x. [DOI] [PubMed] [Google Scholar]
- 3.Tursi A, Brandimarte G, Giorgetti G, Gigliobianco A, Lombardi D, Gasbarrini G. Low prevalence of antigliadin and anti-endomysium antibodies in subclinical/silent celiac disease. Am J Gastroenterol. 2001;96:1507–1510. doi: 10.1111/j.1572-0241.2001.03744.x. [DOI] [PubMed] [Google Scholar]
- 4.Bai D, Brar P, Holleran S, Ramakrishnan R, Green PH. Effect of gender on the manifestations of celiac disease: evidence for greater malabsorption in men. Scand J Gastroenterol. 2005;40:183–187. doi: 10.1080/00365520510011498. [DOI] [PubMed] [Google Scholar]
- 5.Ferguson A, Arranz E, O’Mahony S. Clinical and pathological spectrum of coeliac disease--active, silent, latent, potential. Gut. 1993;34:150–151. doi: 10.1136/gut.34.2.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Malamut G, Afchain P, Verkarre V, Lecomte T, Amiot A, Damotte D, Bouhnik Y, Colombel JF, Delchier JC, Allez M, et al. Presentation and long-term follow-up of refractory celiac disease: comparison of type I with type II. Gastroenterology. 2009;136:81–90. doi: 10.1053/j.gastro.2008.09.069. [DOI] [PubMed] [Google Scholar]
- 7.Biagi F, Corazza GR. Defining gluten refractory enteropathy. Eur J Gastroenterol Hepatol. 2001;13:561–565. doi: 10.1097/00042737-200105000-00016. [DOI] [PubMed] [Google Scholar]
- 8.Fasano A, Catassi C. Current approaches to diagnosis and treatment of celiac disease: an evolving spectrum. Gastroenterology. 2001;120:636–651. doi: 10.1053/gast.2001.22123. [DOI] [PubMed] [Google Scholar]
- 9.Catassi C, Yachha SK. The global village of celiac disease. In: Fasano A, Troncone R, Branski D, editors. Frontiers in celiac disease. Basel: Switzerland Karger; 2008. pp. 23–31. [Google Scholar]
- 10.Greco L. Epidemiology of coeliac disease. Proceedings of the Seventh International Symposyum on coeliac disease. Tampere: Finland; 1996. [Google Scholar]
- 11.Koskinen LL, Einarsdottir E, Korponay-Szabo IR, Kurppa K, Kaukinen K, Sistonen P, Pocsai Z, Széles G, Adány R, Mäki M, et al. Fine mapping of the CELIAC2 locus on chromosome 5q31-q33 in the Finnish and Hungarian populations. Tissue Antigens. 2009;74:408–416. doi: 10.1111/j.1399-0039.2009.01359.x. [DOI] [PubMed] [Google Scholar]
- 12.Holopainen P, Naluai AT, Moodie S, Percopo S, Coto I, Clot F, Ascher H, Sollid L, Ciclitira P, Greco L, et al. Candidate gene region 2q33 in European families with coeliac disease. Tissue Antigens. 2004;63:212–222. doi: 10.1111/j.1399-0039.2004.00189.x. [DOI] [PubMed] [Google Scholar]
- 13.Van Belzen MJ, Meijer JW, Sandkuijl LA, Bardoel AF, Mulder CJ, Pearson PL, Houwen RH, Wijmenga C. A major non-HLA locus in celiac disease maps to chromosome 19. Gastroenterology. 2003;125:1032–1041. doi: 10.1016/s0016-5085(03)01205-8. [DOI] [PubMed] [Google Scholar]
- 14.Wapenaar MC, Monsuur AJ, van Bodegraven AA, Weersma RK, Bevova MR, Linskens RK, Howdle P, Holmes G, Mulder CJ, Dijkstra G, et al. Associations with tight junction genes PARD3 and MAGI2 in Dutch patients point to a common barrier defect for coeliac disease and ulcerative colitis. Gut. 2008;57:463–467. doi: 10.1136/gut.2007.133132. [DOI] [PubMed] [Google Scholar]
- 15.López-Vázquez A, Fuentes D, Rodrigo L, González S, Moreno M, Fernández E, Martínez-Borra J, López-Larrea C. MHC class I region plays a role in the development of diverse clinical forms of celiac disease in a Saharawi population. Am J Gastroenterol. 2004;99:662–667. doi: 10.1111/j.1572-0241.2004.04134.x. [DOI] [PubMed] [Google Scholar]
- 16.Jabbar AA. HLA and disease associations in Iraq. Dis Markers. 1993;11:161–170. doi: 10.1155/1993/192019. [DOI] [PubMed] [Google Scholar]
- 17.Dawood FH, Jabbar AA, Al-Mudaris AF, Al-Hasani MH. Association of HLA antigens with coeliac disease among Iraqi children. Tissue Antigens. 1981;18:35–39. doi: 10.1111/j.1399-0039.1981.tb01358.x. [DOI] [PubMed] [Google Scholar]
- 18.Erkan T, Kutlu T, Yilmaz E, Cullu F, Tümay GT. Human leukocyte antigens in Turkish pediatric celiac patients. Turk J Pediatr. 1999;41:181–188. [PubMed] [Google Scholar]
- 19.Martín-Pagola A, Pérez-Nanclares G, Ortiz L, Vitoria JC, Hualde I, Zaballa R, Preciado E, Castaño L, Bilbao JR. MICA response to gliadin in intestinal mucosa from celiac patients. Immunogenetics. 2004;56:549–554. doi: 10.1007/s00251-004-0724-8. [DOI] [PubMed] [Google Scholar]
- 20.Karell K, Louka AS, Moodie SJ, Ascher H, Clot F, Greco L, Ciclitira PJ, Sollid LM, Partanen J. HLA types in celiac disease patients not carrying the DQA1*05-DQB1*02 (DQ2) heterodimer: results from the European Genetics Cluster on Celiac Disease. Hum Immunol. 2003;64:469–477. doi: 10.1016/s0198-8859(03)00027-2. [DOI] [PubMed] [Google Scholar]
- 21.Megiorni F, Mora B, Bonamico M, Barbato M, Nenna R, Maiella G, Lulli P, Mazzilli MC. HLA-DQ and risk gradient for celiac disease. Hum Immunol. 2009;70:55–59. doi: 10.1016/j.humimm.2008.10.018. [DOI] [PubMed] [Google Scholar]
- 22.Sollid LM. Coeliac disease: dissecting a complex inflammatory disorder. Nat Rev Immunol. 2002;2:647–655. doi: 10.1038/nri885. [DOI] [PubMed] [Google Scholar]
- 23.Cummins AG, Roberts-Thomson IC. Prevalence of celiac disease in the Asia-Pacific region. J Gastroenterol Hepatol. 2009;24:1347–1351. doi: 10.1111/j.1440-1746.2009.05932.x. [DOI] [PubMed] [Google Scholar]
- 24.Middleton D. In worldwide populations, 2010. Available from: http// www.allelefrequencies.net.
- 25.Layrisse Z, Guedez Y, Domínguez E, Paz N, Montagnani S, Matos M, Herrera F, Ogando V, Balbas O, Rodríguez-Larralde A. Extended HLA haplotypes in a Carib Amerindian population: the Yucpa of the Perija Range. Hum Immunol. 2001;62:992–1000. doi: 10.1016/s0198-8859(01)00297-x. [DOI] [PubMed] [Google Scholar]
- 26.Rostom A, Murray JA, Kagnoff MF. American Gastroenterological Association (AGA) Institute technical review on the diagnosis and management of celiac disease. Gastroenterology. 2006;131:1981–2002. doi: 10.1053/j.gastro.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 27.Green PH, Cellier C. Celiac disease. N Engl J Med. 2007;357:1731–1743. doi: 10.1056/NEJMra071600. [DOI] [PubMed] [Google Scholar]
- 28.Fasano A, Berti I, Gerarduzzi T, Not T, Colletti RB, Drago S, Elitsur Y, Green PH, Guandalini S, Hill ID, et al. Prevalence of celiac disease in at-risk and not-at-risk groups in the United States: a large multicenter study. Arch Intern Med. 2003;163:286–292. doi: 10.1001/archinte.163.3.286. [DOI] [PubMed] [Google Scholar]
- 29.Thomas HJ, Ahmad T, Rajaguru C, Barnardo M, Warren BF, Jewell DP. Contribution of histological, serological, and genetic factors to the clinical heterogeneity of adult-onset coeliac disease. Scand J Gastroenterol. 2009;44:1076–1083. doi: 10.1080/00365520903100473. [DOI] [PubMed] [Google Scholar]
- 30.Hin H, Bird G, Fisher P, Mahy N, Jewell D. Coeliac disease in primary care: case finding study. BMJ. 1999;318:164–167. doi: 10.1136/bmj.318.7177.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cataldo F, Montalto G. Celiac disease in the developing countries: a new and challenging public health problem. World J Gastroenterol. 2007;13:2153–2159. doi: 10.3748/wjg.v13.i15.2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.NIH Consensus Development Conference on Celiac Disease. NIH Consensus and State of the Science Statements. 2004;21:1–23. [PubMed] [Google Scholar]
- 33.Gandolfi L, Pratesi R, Cordoba JC, Tauil PL, Gasparin M, Catassi C. Prevalence of celiac disease among blood donors in Brazil. Am J Gastroenterol. 2000;95:689–692. doi: 10.1111/j.1572-0241.2000.01847.x. [DOI] [PubMed] [Google Scholar]
- 34.Pratesi R, Gandolfi L, Garcia SG, Modelli IC, Lopes de Almeida P, Bocca AL, Catassi C. Prevalence of coeliac disease: unexplained age-related variation in the same population. Scand J Gastroenterol. 2003;38:747–750. doi: 10.1080/00365520310003255. [DOI] [PubMed] [Google Scholar]
- 35.Gomez JC, Selvaggio GS, Viola M, Pizarro B, la Motta G, de Barrio S, Castelletto R, Echeverría R, Sugai E, Vazquez H, et al. Prevalence of celiac disease in Argentina: screening of an adult population in the La Plata area. Am J Gastroenterol. 2001;96:2700–2704. doi: 10.1111/j.1572-0241.2001.04124.x. [DOI] [PubMed] [Google Scholar]
- 36.Cataldo F, Pitarresi N, Accomando S, Greco L. Epidemiological and clinical features in immigrant children with coeliac disease: an Italian multicentre study. Dig Liver Dis. 2004;36:722–729. doi: 10.1016/j.dld.2004.03.021. [DOI] [PubMed] [Google Scholar]
- 37.Vanciková Z, Chlumecký V, Sokol D, Horáková D, Hamsíková E, Fucíková T, Janatková I, Ulcová-Gallová Z, Stĕpán J, Límanová Z, et al. The serologic screening for celiac disease in the general population (blood donors) and in some high-risk groups of adults (patients with autoimmune diseases, osteoporosis and infertility) in the Czech republic. Folia Microbiol (Praha) 2002;47:753–758. doi: 10.1007/BF02818684. [DOI] [PubMed] [Google Scholar]
- 38.Volta U, Bellentani S, Bianchi FB, Brandi G, De Franceschi L, Miglioli L, Granito A, Balli F, Tiribelli C. High prevalence of celiac disease in Italian general population. Dig Dis Sci. 2001;46:1500–1505. doi: 10.1023/a:1010648122797. [DOI] [PubMed] [Google Scholar]
- 39.Rostami K, Kerckhaert J, Tiemessen R, von Blomberg BM, Meijer JW, Mulder CJ. Sensitivity of antiendomysium and antigliadin antibodies in untreated celiac disease: disappointing in clinical practice. Am J Gastroenterol. 1999;94:888–894. doi: 10.1111/j.1572-0241.1999.983_f.x. [DOI] [PubMed] [Google Scholar]
- 40.Mäki M, Mustalahti K, Kokkonen J, Kulmala P, Haapalahti M, Karttunen T, Ilonen J, Laurila K, Dahlbom I, Hansson T, et al. Prevalence of Celiac disease among children in Finland. N Engl J Med. 2003;348:2517–2524. doi: 10.1056/NEJMoa021687. [DOI] [PubMed] [Google Scholar]
- 41.Riestra S, Fernández E, Rodrigo L, Garcia S, Ocio G. Prevalence of Coeliac disease in the general population of northern Spain. Strategies of serologic screening. Scand J Gastroenterol. 2000;35:398–402. doi: 10.1080/003655200750023967. [DOI] [PubMed] [Google Scholar]
- 42.Rutz R, Ritzler E, Fierz W, Herzog D. Prevalence of asymptomatic celiac disease in adolescents of eastern Switzerland. Swiss Med Wkly. 2002;132:43–47. doi: 10.4414/smw.2002.09793. [DOI] [PubMed] [Google Scholar]
- 43.Catassi C, Fabiani E, Gasparin M, Troncone R. Quantitative antigliadin antibody measurement in clinical practice: an Italian multicentre study. SIGEP Working Group on Quantitative AGA Standardization. Ital J Gastroenterol Hepatol. 1999;31:366–370. [PubMed] [Google Scholar]
- 44.Ashabani A, Errabtea H, Shapan A, Tuckova L, Tlaskalova-Hogenova H. Serologic markers of untreated celiac disease in Libyan children: antigliadin, antitransglutaminase, antiendomysial, and anticalreticulin antibodies. J Pediatr Gastroenterol Nutr. 2001;33:276–282. doi: 10.1097/00005176-200109000-00009. [DOI] [PubMed] [Google Scholar]
- 45.Mankaï A, Landolsi H, Chahed A, Gueddah L, Limem M, Ben Abdessalem M, Yacoub-Jemni S, Ghannem H, Jeddi M, Ghedira I. Celiac disease in Tunisia: serological screening in healthy blood donors. Pathol Biol (Paris) 2006;54:10–13. doi: 10.1016/j.patbio.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 46.Catassi C, Abu-Zakey M, Kriszad D, Fasano A. Celiac disease among school-children in Egypt: results of a pilot study. Belfast: 11th International Symposium on Celiac Disease; 2004. [Google Scholar]
- 47.Boudraa G, Hachelaf W, Benbouabdellah M, Belkadi M, Benmansour FZ, Touhami M. Prevalence of coeliac disease in diabetic children and their first- degree relatives in west Algeria: screening with serological markers. Acta Paediatr Suppl. 1996;412:58–60. doi: 10.1111/j.1651-2227.1996.tb14254.x. [DOI] [PubMed] [Google Scholar]
- 48.Bouguerra F, Babron MC, Eliaou JF, Debbabi A, Clot J, Khaldi F, Greco L, Clerget-Darpoux F. Synergistic effect of two HLA heterodimers in the susceptibility to celiac disease in Tunisia. Genet Epidemiol. 1997;14:413–422. doi: 10.1002/(SICI)1098-2272(1997)14:4<413::AID-GEPI6>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 49.Lionetti P, Favilli T, Chiaravalloti G, Ughi C, Maggiore G. Coeliac disease in Saharawi children in Algerian refugee camps. Lancet. 1999;353:1189–1190. doi: 10.1016/S0140-6736(05)74414-7. [DOI] [PubMed] [Google Scholar]
- 50.Xu M, Jin YL, Fu J, Huang H, Chen SZ, Qu P, Tian HM, Liu ZY, Zhang W. The abnormal expression of retinoic acid receptor-beta, p 53 and Ki67 protein in normal, premalignant and malignant esophageal tissues. World J Gastroenterol. 2002;8:200–202. doi: 10.3748/wjg.v8.i2.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rätsch IM, Catassi C. Coeliac disease: a potentially treatable health problem of Saharawi refugee children. Bull World Health Organ. 2001;79:541–545. [PMC free article] [PubMed] [Google Scholar]
- 52.Meloni G, Dore A, Fanciulli G, Tanda F, Bottazzo GF. Subclinical coeliac disease in schoolchildren from northern Sardinia. Lancet. 1999;353:37. doi: 10.1016/S0140-6736(05)74871-6. [DOI] [PubMed] [Google Scholar]
- 53.Cook HB, Burt MJ, Collett JA, Whitehead MR, Frampton CM, Chapman BA. Adult coeliac disease: prevalence and clinical significance. J Gastroenterol Hepatol. 2000;15:1032–1036. doi: 10.1046/j.1440-1746.2000.02290.x. [DOI] [PubMed] [Google Scholar]
- 54.Hovell CJ, Collett JA, Vautier G, Cheng AJ, Sutanto E, Mallon DF, Olynyk JK, Cullen DJ. High prevalence of coeliac disease in a population-based study from Western Australia: a case for screening? Med J Aust. 2001;175:247–250. doi: 10.5694/j.1326-5377.2001.tb143555.x. [DOI] [PubMed] [Google Scholar]
- 55.Freeman HJ. Biopsy-defined adult celiac disease in Asian-Canadians. Can J Gastroenterol. 2003;17:433–436. doi: 10.1155/2003/789139. [DOI] [PubMed] [Google Scholar]
- 56.Kaur G, Sarkar N, Bhatnagar S, Kumar S, Rapthap CC, Bhan MK, Mehra NK. Pediatric celiac disease in India is associated with multiple DR3-DQ2 haplotypes. Hum Immunol. 2002;63:677–682. doi: 10.1016/s0198-8859(02)00413-5. [DOI] [PubMed] [Google Scholar]
- 57.Agrawal S, Srivastava SK, Borkar M, Chaudhuri TK. Genetic affinities of north and northeastern populations of India: inference from HLA-based study. Tissue Antigens. 2008;72:120–130. doi: 10.1111/j.1399-0039.2008.01083.x. [DOI] [PubMed] [Google Scholar]
- 58.Shanmugalakshmi S, Balakrishnan K, Manoharan K, Pitchappan RM. HLA-DRB1*, -DQB1* in Piramalai Kallars and Yadhavas, two Dravidian-speaking castes of Tamil Nadu, South India. Tissue Antigens. 2003;61:451–464. doi: 10.1034/j.1399-0039.2003.00061.x. [DOI] [PubMed] [Google Scholar]
- 59.Sher KS, Fraser RC, Wicks AC, Mayberry JF. High risk of coeliac disease in Punjabis. Epidemiological study in the south Asian and European populations of Leicestershire. Digestion. 1993;54:178–182. doi: 10.1159/000201035. [DOI] [PubMed] [Google Scholar]
- 60.Sood A, Midha V, Sood N, Avasthi G, Sehgal A. Prevalence of celiac disease among school children in Punjab, North India. J Gastroenterol Hepatol. 2006;21:1622–1625. doi: 10.1111/j.1440-1746.2006.04281.x. [DOI] [PubMed] [Google Scholar]
- 61.Sood A, Midha V, Sood N, Kaushal V, Puri H. Increasing incidence of celiac disease in India. Am J Gastroenterol. 2001;96:2804–2805. doi: 10.1111/j.1572-0241.2001.04150.x. [DOI] [PubMed] [Google Scholar]
- 62.Simoons FJ. Celiac disease as geographic problem. In: Walcher DN, Kretcher N, editors. Food, Nutrition and Evolution. New York: Masson; 1981. pp. 179–199. [Google Scholar]
- 63.Srivastava A, Yachha SK, Mathias A, Parveen F, Poddar U, Agrawal S. Prevalence, human leukocyte antigen typing and strategy for screening among Asian first-degree relatives of children with celiac disease. J Gastroenterol Hepatol. 2010;25:319–324. doi: 10.1111/j.1440-1746.2009.06044.x. [DOI] [PubMed] [Google Scholar]
- 64.Yachha SK, Poddar U. Celiac disease in India. Indian J Gastroenterol. 2007;26:230–237. [PubMed] [Google Scholar]
- 65.Accomando S, Cataldo F. The global village of celiac disease. Dig Liver Dis. 2004;36:492–498. doi: 10.1016/j.dld.2004.01.026. [DOI] [PubMed] [Google Scholar]
- 66.Shahbazkhani B, Malekzadeh R, Sotoudeh M, Moghadam KF, Farhadi M, Ansari R, Elahyfar A, Rostami K. High prevalence of coeliac disease in apparently healthy Iranian blood donors. Eur J Gastroenterol Hepatol. 2003;15:475–478. doi: 10.1097/01.meg.0000059118.41030.96. [DOI] [PubMed] [Google Scholar]
- 67.Shamir R, Lerner A, Shinar E, Lahat N, Sobel E, Bar-or R, Kerner H, Eliakim R. The use of a single serological marker underestimates the prevalence of celiac disease in Israel: a study of blood donors. Am J Gastroenterol. 2002;97:2589–2594. doi: 10.1111/j.1572-0241.2002.06028.x. [DOI] [PubMed] [Google Scholar]
- 68.Challar MH, Jouma M, Sitzmann FC, Seferian V, Shahin E. Prevalence of asymptomatic celiac disease in a Syrian population sample. JABMS. 2004;6:155–160. [Google Scholar]
- 69.Tatar G, Elsurer R, Simsek H, Balaban YH, Hascelik G, Ozcebe OI, Buyukasik Y, Sokmensuer C. Screening of tissue transglutaminase antibody in healthy blood donors for celiac disease screening in the Turkish population. Dig Dis Sci. 2004;49:1479–1484. doi: 10.1023/b:ddas.0000042250.59327.91. [DOI] [PubMed] [Google Scholar]
- 70.Gursoy S, Guven K, Simsek T, Yurci A, Torun E, Koc N, Patiroglu TE, Ozbakir O, Yucesoy M. The prevalence of unrecognized adult celiac disease in Central Anatolia. J Clin Gastroenterol. 2005;39:508–511. doi: 10.1097/01.mcg.0000165664.87153.e1. [DOI] [PubMed] [Google Scholar]
- 71.Imanzadeh F, Sayyari AA, Yaghoobi M, Akbari MR, Shafagh H, Farsar AR. Celiac disease in children with diarrhea is more frequent than previously suspected. J Pediatr Gastroenterol Nutr. 2005;40:309–311. doi: 10.1097/01.mpg.0000154012.10420.08. [DOI] [PubMed] [Google Scholar]
- 72.Ertekin V, Selimoğlu MA, Kardaş F, Aktaş E. Prevalence of celiac disease in Turkish children. J Clin Gastroenterol. 2005;39:689–691. doi: 10.1097/01.mcg.0000174026.26838.56. [DOI] [PubMed] [Google Scholar]
- 73.Freeman HJ. Risk factors in familial forms of celiac disease. World J Gastroenterol. 2010;16:1828–1831. doi: 10.3748/wjg.v16.i15.1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Book L, Zone JJ, Neuhausen SL. Prevalence of celiac disease among relatives of sib pairs with celiac disease in U.S. families. Am J Gastroenterol. 2003;98:377–381. doi: 10.1111/j.1572-0241.2003.07238.x. [DOI] [PubMed] [Google Scholar]
- 75.Farré C, Humbert P, Vilar P, Varea V, Aldeguer X, Carnicer J, Carballo M, Gassull MA. Serological markers and HLA-DQ2 haplotype among first-degree relatives of celiac patients. Catalonian Coeliac Disease Study Group. Dig Dis Sci. 1999;44:2344–2349. doi: 10.1023/a:1026685527228. [DOI] [PubMed] [Google Scholar]
- 76.Rubio-Tapia A, Van Dyke CT, Lahr BD, Zinsmeister AR, El-Youssef M, Moore SB, Bowman M, Burgart LJ, Melton LJ, Murray JA. Predictors of family risk for celiac disease: a population-based study. Clin Gastroenterol Hepatol. 2008;6:983–987. doi: 10.1016/j.cgh.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Larsson K, Carlsson A, Cederwall E, Jönsson B, Neiderud J, Jonsson B, Lernmark A, Ivarsson SA. Annual screening detects celiac disease in children with type 1 diabetes. Pediatr Diabetes. 2008;9:354–359. doi: 10.1111/j.1399-5448.2008.00367.x. [DOI] [PubMed] [Google Scholar]
- 78.Sollid LM. Molecular basis of celiac disease. Annu Rev Immunol. 2000;18:53–81. doi: 10.1146/annurev.immunol.18.1.53. [DOI] [PubMed] [Google Scholar]
- 79.Kupper C. Dietary guidelines and implementation for celiac disease. Gastroenterology. 2005;128:S121–S127. doi: 10.1053/j.gastro.2005.02.024. [DOI] [PubMed] [Google Scholar]
- 80.Di Sabatino A, Corazza GR. Coeliac disease. Lancet. 2009;373:1480–1493. doi: 10.1016/S0140-6736(09)60254-3. [DOI] [PubMed] [Google Scholar]
- 81.Shan L, Molberg Ø, Parrot I, Hausch F, Filiz F, Gray GM, Sollid LM, Khosla C. Structural basis for gluten intolerance in celiac sprue. Science. 2002;297:2275–2279. doi: 10.1126/science.1074129. [DOI] [PubMed] [Google Scholar]
- 82.Arentz-Hansen H, Körner R, Molberg O, Quarsten H, Vader W, Kooy YM, Lundin KE, Koning F, Roepstorff P, Sollid LM, et al. The intestinal T cell response to alpha-gliadin in adult celiac disease is focused on a single deamidated glutamine targeted by tissue transglutaminase. J Exp Med. 2000;191:603–612. doi: 10.1084/jem.191.4.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Meresse B, Ripoche J, Heyman M, Cerf-Bensussan N. Celiac disease: from oral tolerance to intestinal inflammation, autoimmunity and lymphomagenesis. Mucosal Immunol. 2009;2:8–23. doi: 10.1038/mi.2008.75. [DOI] [PubMed] [Google Scholar]
- 84.Corrao G, Corazza GR, Bagnardi V, Brusco G, Ciacci C, Cottone M, Sategna Guidetti C, Usai P, Cesari P, Pelli MA, et al. Mortality in patients with coeliac disease and their relatives: a cohort study. Lancet. 2001;358:356–361. doi: 10.1016/s0140-6736(01)05554-4. [DOI] [PubMed] [Google Scholar]
- 85.Mäki M, Collin P. Coeliac disease. Lancet. 1997;349:1755–1759. doi: 10.1016/S0140-6736(96)70237-4. [DOI] [PubMed] [Google Scholar]
- 86.Stepniak D, Koning F. Celiac disease--sandwiched between innate and adaptive immunity. Hum Immunol. 2006;67:460–468. doi: 10.1016/j.humimm.2006.03.011. [DOI] [PubMed] [Google Scholar]
- 87.Lähdeaho ML, Lehtinen M, Rissa HR, Hyöty H, Reunala T, Mäki M. Antipeptide antibodies to adenovirus E1b protein indicate enhanced risk of celiac disease and dermatitis herpetiformis. Int Arch Allergy Immunol. 1993;101:272–276. doi: 10.1159/000236457. [DOI] [PubMed] [Google Scholar]
- 88.Kagnoff MF, Paterson YJ, Kumar PJ, Kasarda DD, Carbone FR, Unsworth DJ, Austin RK. Evidence for the role of a human intestinal adenovirus in the pathogenesis of coeliac disease. Gut. 1987;28:995–1001. doi: 10.1136/gut.28.8.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kagnoff MF, Austin RK, Hubert JJ, Bernardin JE, Kasarda DD. Possible role for a human adenovirus in the pathogenesis of celiac disease. J Exp Med. 1984;160:1544–1557. doi: 10.1084/jem.160.5.1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sjöberg K, Lindgren S, Eriksson S. Frequent occurrence of non-specific gliadin antibodies in chronic liver disease. Endomysial but not gliadin antibodies predict coeliac disease in patients with chronic liver disease. Scand J Gastroenterol. 1997;32:1162–1167. doi: 10.3109/00365529709002997. [DOI] [PubMed] [Google Scholar]
- 91.Ruggeri C, La Masa AT, Rudi S, Squadrito G, Di Pasquale G, Maimone S, Caccamo G, Pellegrino S, Raimondo G, Magazzù G. Celiac disease and non-organ-specific autoantibodies in patients with chronic hepatitis C virus infection. Dig Dis Sci. 2008;53:2151–2155. doi: 10.1007/s10620-007-0146-1. [DOI] [PubMed] [Google Scholar]
- 92.Verdu EF, Mauro M, Bourgeois J, Armstrong D. Clinical onset of celiac disease after an episode of Campylobacter jejuni enteritis. Can J Gastroenterol. 2007;21:453–455. doi: 10.1155/2007/169591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Carroccio A, Cavataio F, Montalto G, Paparo F, Troncone R, Iacono G. Treatment of giardiasis reverses “active” coeliac disease to “latent” coeliac disease. Eur J Gastroenterol Hepatol. 2001;13:1101–1105. doi: 10.1097/00042737-200109000-00018. [DOI] [PubMed] [Google Scholar]
- 94.Stene LC, Honeyman MC, Hoffenberg EJ, Haas JE, Sokol RJ, Emery L, Taki I, Norris JM, Erlich HA, Eisenbarth GS, et al. Rotavirus infection frequency and risk of celiac disease autoimmunity in early childhood: a longitudinal study. Am J Gastroenterol. 2006;101:2333–2340. doi: 10.1111/j.1572-0241.2006.00741.x. [DOI] [PubMed] [Google Scholar]
- 95.Carlsson AK, Lindberg BA, Bredberg AC, Hyöty H, Ivarsson SA. Enterovirus infection during pregnancy is not a risk factor for celiac disease in the offspring. J Pediatr Gastroenterol Nutr. 2002;35:649–652. doi: 10.1097/00005176-200211000-00011. [DOI] [PubMed] [Google Scholar]
- 96.Ivarsson A, Persson LA, Nyström L, Ascher H, Cavell B, Danielsson L, Dannaeus A, Lindberg T, Lindquist B, Stenhammar L, et al. Epidemic of coeliac disease in Swedish children. Acta Paediatr. 2000;89:165–171. doi: 10.1080/080352500750028771. [DOI] [PubMed] [Google Scholar]
- 97.Farrell RJ, Kelly CP. Celiac sprue. N Engl J Med. 2002;346:180–188. doi: 10.1056/NEJMra010852. [DOI] [PubMed] [Google Scholar]
- 98.Snook JA, Dwyer L, Lee-Elliott C, Khan S, Wheeler DW, Nicholas DS. Adult coeliac disease and cigarette smoking. Gut. 1996;39:60–62. doi: 10.1136/gut.39.1.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Qiao SW, Bergseng E, Molberg Ø, Xia J, Fleckenstein B, Khosla C, Sollid LM. Antigen presentation to celiac lesion-derived T cells of a 33-mer gliadin peptide naturally formed by gastrointestinal digestion. J Immunol. 2004;173:1757–1762. doi: 10.4049/jimmunol.173.3.1757. [DOI] [PubMed] [Google Scholar]
- 100.Sollid LM, Thorsby E. HLA susceptibility genes in celiac disease: genetic mapping and role in pathogenesis. Gastroenterology. 1993;105:910–922. doi: 10.1016/0016-5085(93)90912-v. [DOI] [PubMed] [Google Scholar]
- 101.Maiuri L, Ciacci C, Ricciardelli I, Vacca L, Raia V, Auricchio S, Picard J, Osman M, Quaratino S, Londei M. Association between innate response to gliadin and activation of pathogenic T cells in coeliac disease. Lancet. 2003;362:30–37. doi: 10.1016/S0140-6736(03)13803-2. [DOI] [PubMed] [Google Scholar]
- 102.Gianfrani C, Auricchio S, Troncone R. Adaptive and innate immune responses in celiac disease. Immunol Lett. 2005;99:141–145. doi: 10.1016/j.imlet.2005.02.017. [DOI] [PubMed] [Google Scholar]
- 103.Maiuri L, Troncone R, Mayer M, Coletta S, Picarelli A, De Vincenzi M, Pavone V, Auricchio S. In vitro activities of A-gliadin-related synthetic peptides: damaging effect on the atrophic coeliac mucosa and activation of mucosal immune response in the treated coeliac mucosa. Scand J Gastroenterol. 1996;31:247–253. doi: 10.3109/00365529609004874. [DOI] [PubMed] [Google Scholar]
- 104.Ciclitira PJ, Ellis HJ. In vivo gluten ingestion in coeliac disease. Dig Dis. 1998;16:337–340. doi: 10.1159/000016887. [DOI] [PubMed] [Google Scholar]
- 105.Norris JM, Barriga K, Hoffenberg EJ, Taki I, Miao D, Haas JE, Emery LM, Sokol RJ, Erlich HA, Eisenbarth GS, et al. Risk of celiac disease autoimmunity and timing of gluten introduction in the diet of infants at increased risk of disease. JAMA. 2005;293:2343–2351. doi: 10.1001/jama.293.19.2343. [DOI] [PubMed] [Google Scholar]
- 106.Ivarsson A. The Swedish epidemic of coeliac disease explored using an epidemiological approach--some lessons to be learnt. Best Pract Res Clin Gastroenterol. 2005;19:425–440. doi: 10.1016/j.bpg.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 107.Feighery C. Fortnightly review: coeliac disease. BMJ. 1999;319:236–239. doi: 10.1136/bmj.319.7204.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ciclitira PJ, King AL, Fraser JS. AGA technical review on Celiac Sprue. American Gastroenterological Association. Gastroenterology. 2001;120:1526–1540. doi: 10.1053/gast.2001.24056. [DOI] [PubMed] [Google Scholar]
- 109.Freeman HJ. Clinical spectrum of biopsy-defined celiac disease in the elderly. Can J Gastroenterol. 1995;9:42–46. [Google Scholar]
- 110.Swinson CM, Levi AJ. Is coeliac disease underdiagnosed? Br Med J. 1980;281:1258–1260. doi: 10.1136/bmj.281.6250.1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Logan RF, Tucker G, Rifkind EA, Heading RC, Ferguson A. Changes in clinical features of coeliac disease in adults in Edinburgh and the Lothians 1960-79. Br Med J (Clin Res Ed) 1983;286:95–97. doi: 10.1136/bmj.286.6359.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Paré P, Douville P, Caron D, Lagacé R. Adult celiac sprue: changes in the pattern of clinical recognition. J Clin Gastroenterol. 1988;10:395–400. [PubMed] [Google Scholar]
- 113.Nisticò L, Fagnani C, Coto I, Percopo S, Cotichini R, Limongelli MG, Paparo F, D’Alfonso S, Giordano M, Sferlazzas C, et al. Concordance, disease progression, and heritability of coeliac disease in Italian twins. Gut. 2006;55:803–808. doi: 10.1136/gut.2005.083964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Mazzarella G, Maglio M, Paparo F, Nardone G, Stefanile R, Greco L, van de Wal Y, Kooy Y, Koning F, Auricchio S, et al. An immunodominant DQ8 restricted gliadin peptide activates small intestinal immune response in in vitro cultured mucosa from HLA-DQ8 positive but not HLA-DQ8 negative coeliac patients. Gut. 2003;52:57–62. doi: 10.1136/gut.52.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Al-Toma A, Goerres MS, Meijer JW, Peña AS, Crusius JB, Mulder CJ. Human leukocyte antigen-DQ2 homozygosity and the development of refractory celiac disease and enteropathy-associated T-cell lymphoma. Clin Gastroenterol Hepatol. 2006;4:315–319. doi: 10.1016/j.cgh.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 116.Heap GA, van Heel DA. Genetics and pathogenesis of coeliac disease. Semin Immunol. 2009;21:346–354. doi: 10.1016/j.smim.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 117.van Heel DA, Hunt K, Greco L, Wijmenga C. Genetics in coeliac disease. Best Pract Res Clin Gastroenterol. 2005;19:323–339. doi: 10.1016/j.bpg.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 118.Hunt KA, McGovern DP, Kumar PJ, Ghosh S, Travis SP, Walters JR, Jewell DP, Playford RJ, van Heel DA. A common CTLA4 haplotype associated with coeliac disease. Eur J Hum Genet. 2005;13:440–444. doi: 10.1038/sj.ejhg.5201357. [DOI] [PubMed] [Google Scholar]
- 119.Holmes GK. Coeliac disease and Type 1 diabetes mellitus - the case for screening. Diabet Med. 2001;18:169–177. doi: 10.1046/j.1464-5491.2001.00498.x. [DOI] [PubMed] [Google Scholar]
- 120.van Heel DA, Franke L, Hunt KA, Gwilliam R, Zhernakova A, Inouye M, Wapenaar MC, Barnardo MC, Bethel G, Holmes GK, et al. A genome-wide association study for celiac disease identifies risk variants in the region harboring IL2 and IL21. Nat Genet. 2007;39:827–829. doi: 10.1038/ng2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zhernakova A, Alizadeh BZ, Bevova M, van Leeuwen MA, Coenen MJ, Franke B, Franke L, Posthumus MD, van Heel DA, van der Steege G, et al. Novel association in chromosome 4q27 region with rheumatoid arthritis and confirmation of type 1 diabetes point to a general risk locus for autoimmune diseases. Am J Hum Genet. 2007;81:1284–1288. doi: 10.1086/522037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Todd JA, Walker NM, Cooper JD, Smyth DJ, Downes K, Plagnol V, Bailey R, Nejentsev S, Field SF, Payne F, et al. Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat Genet. 2007;39:857–864. doi: 10.1038/ng2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Liu Y, Helms C, Liao W, Zaba LC, Duan S, Gardner J, Wise C, Miner A, Malloy MJ, Pullinger CR, et al. A genome-wide association study of psoriasis and psoriatic arthritis identifies new disease loci. PLoS Genet. 2008;4:e1000041. doi: 10.1371/journal.pgen.1000041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Folk JE, Finlayson JS. The epsilon-(gamma-glutamyl)lysine crosslink and the catalytic role of transglutaminases. Adv Protein Chem. 1977;31:1–133. doi: 10.1016/s0065-3233(08)60217-x. [DOI] [PubMed] [Google Scholar]
- 125.Molberg O, Mcadam SN, Körner R, Quarsten H, Kristiansen C, Madsen L, Fugger L, Scott H, Norén O, Roepstorff P, et al. Tissue transglutaminase selectively modifies gliadin peptides that are recognized by gut-derived T cells in celiac disease. Nat Med. 1998;4:713–717. doi: 10.1038/nm0698-713. [DOI] [PubMed] [Google Scholar]
- 126.Caputo I, Barone MV, Martucciello S, Lepretti M, Esposito C. Tissue transglutaminase in celiac disease: role of autoantibodies. Amino Acids. 2009;36:693–699. doi: 10.1007/s00726-008-0120-z. [DOI] [PubMed] [Google Scholar]
- 127.Di Sabatino A, Ciccocioppo R, Cupelli F, Cinque B, Millimaggi D, Clarkson MM, Paulli M, Cifone MG, Corazza GR. Epithelium derived interleukin 15 regulates intraepithelial lymphocyte Th1 cytokine production, cytotoxicity, and survival in coeliac disease. Gut. 2006;55:469–477. doi: 10.1136/gut.2005.068684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Bernardo D, Garrote JA, Allegretti Y, León A, Gómez E, Bermejo-Martin JF, Calvo C, Riestra S, Fernández-Salazar L, Blanco-Quirós A, et al. Higher constitutive IL15R alpha expression and lower IL-15 response threshold in coeliac disease patients. Clin Exp Immunol. 2008;154:64–73. doi: 10.1111/j.1365-2249.2008.03743.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Clemente MG, De Virgiliis S, Kang JS, Macatagney R, Musu MP, Di Pierro MR, Drago S, Congia M, Fasano A. Early effects of gliadin on enterocyte intracellular signalling involved in intestinal barrier function. Gut. 2003;52:218–223. doi: 10.1136/gut.52.2.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Matysiak-Budnik T, Heyman M, Megraud F. Gastric permeability and Helicobacter pylori. Gastroenterologia Polska. 2003;10:305–315. doi: 10.1016/s0399-8320(04)94954-8. [DOI] [PubMed] [Google Scholar]
- 131.Hüe S, Mention JJ, Monteiro RC, Zhang S, Cellier C, Schmitz J, Verkarre V, Fodil N, Bahram S, Cerf-Bensussan N, et al. A direct role for NKG2D/MICA interaction in villous atrophy during celiac disease. Immunity. 2004;21:367–377. doi: 10.1016/j.immuni.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 132.Rossi M, Maurano F, Luongo D. Immunomodulatory strategies for celiac disease. Int Rev Immunol. 2005;24:479–499. doi: 10.1080/08830180500371082. [DOI] [PubMed] [Google Scholar]
- 133.Nilsen EM, Lundin KE, Krajci P, Scott H, Sollid LM, Brandtzaeg P. Gluten specific, HLA-DQ restricted T cells from coeliac mucosa produce cytokines with Th1 or Th0 profile dominated by interferon gamma. Gut. 1995;37:766–776. doi: 10.1136/gut.37.6.766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Rossi M, Maurano F, Luongo D. Immunomodulatory strategies for celiac disease. Int Rev Immunol. 2005;24:479–499. doi: 10.1080/08830180500371082. [DOI] [PubMed] [Google Scholar]
- 135.Matysiak-Budnik T, Moura IC, Arcos-Fajardo M, Lebreton C, Ménard S, Candalh C, Ben-Khalifa K, Dugave C, Tamouza H, van Niel G, et al. Secretory IgA mediates retrotranscytosis of intact gliadin peptides via the transferrin receptor in celiac disease. J Exp Med. 2008;205:143–154. doi: 10.1084/jem.20071204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Heyman M, Menard S. Pathways of gliadin transport in celiac disease. Ann N Y Acad Sci. 2009;1165:274–278. doi: 10.1111/j.1749-6632.2009.04032.x. [DOI] [PubMed] [Google Scholar]
- 137.Schumann M, Richter JF, Wedell I, Moos V, Zimmermann-Kordmann M, Schneider T, Daum S, Zeitz M, Fromm M, Schulzke JD. Mechanisms of epithelial translocation of the alpha(2)-gliadin-33mer in coeliac sprue. Gut. 2008;57:747–754. doi: 10.1136/gut.2007.136366. [DOI] [PubMed] [Google Scholar]
- 138.Fasano A, Baudry B, Pumplin DW, Wasserman SS, Tall BD, Ketley JM, Kaper JB. Vibrio cholerae produces a second enterotoxin, which affects intestinal tight junctions. Proc Natl Acad Sci USA. 1991;88:5242–5246. doi: 10.1073/pnas.88.12.5242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Fasano A, Fiorentini C, Donelli G, Uzzau S, Kaper JB, Margaretten K, Ding X, Guandalini S, Comstock L, Goldblum SE. Zonula occludens toxin modulates tight junctions through protein kinase C-dependent actin reorganization, in vitro. J Clin Invest. 1995;96:710–720. doi: 10.1172/JCI118114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Fasano A, Uzzau S, Fiore C, Margaretten K. The enterotoxic effect of zonula occludens toxin on rabbit small intestine involves the paracellular pathway. Gastroenterology. 1997;112:839–846. doi: 10.1053/gast.1997.v112.pm9041245. [DOI] [PubMed] [Google Scholar]
- 141.Baudry B, Fasano A, Ketley J, Kaper JB. Cloning of a gene (zot) encoding a new toxin produced by Vibrio cholerae. Infect Immun. 1992;60:428–434. doi: 10.1128/iai.60.2.428-434.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wang W, Uzzau S, Goldblum SE, Fasano A. Human zonulin, a potential modulator of intestinal tight junctions. J Cell Sci. 2000;113 Pt 24:4435–4440. doi: 10.1242/jcs.113.24.4435. [DOI] [PubMed] [Google Scholar]
- 143.Lammers KM, Lu R, Brownley J, Lu B, Gerard C, Thomas K, Rallabhandi P, Shea-Donohue T, Tamiz A, Alkan S, et al. Gliadin induces an increase in intestinal permeability and zonulin release by binding to the chemokine receptor CXCR3. Gastroenterology. 2008;135:194–204.e3. doi: 10.1053/j.gastro.2008.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Fasano A. Innovative strategies for the oral delivery of drugs and peptides. Trends Biotechnol. 1998;16:152–157. doi: 10.1016/s0167-7799(97)01170-0. [DOI] [PubMed] [Google Scholar]
- 145.Fasano A, Nataro JP. Intestinal epithelial tight junctions as targets for enteric bacteria-derived toxins. Adv Drug Deliv Rev. 2004;56:795–807. doi: 10.1016/j.addr.2003.10.045. [DOI] [PubMed] [Google Scholar]
- 146.Fina D, Sarra M, Caruso R, Del Vecchio Blanco G, Pallone F, MacDonald TT, Monteleone G. Interleukin 21 contributes to the mucosal T helper cell type 1 response in coeliac disease. Gut. 2008;57:887–892. doi: 10.1136/gut.2007.129882. [DOI] [PubMed] [Google Scholar]
- 147.Rescigno M, Di Sabatino A. Dendritic cells in intestinal homeostasis and disease. J Clin Invest. 2009;119:2441–2450. doi: 10.1172/JCI39134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Granzotto M, dal Bo S, Quaglia S, Tommasini A, Piscianz E, Valencic E, Ferrara F, Martelossi S, Ventura A, Not T. Regulatory T-cell function is impaired in celiac disease. Dig Dis Sci. 2009;54:1513–1519. doi: 10.1007/s10620-008-0501-x. [DOI] [PubMed] [Google Scholar]
- 149.Meeuwisse GW. Diagnostic criteria in CD. Acta Paediatr Scand. 1970;59:461–463. [Google Scholar]
- 150.Revised criteria for diagnosis of coeliac disease. Report of Working Group of European Society of Paediatric Gastroenterology and Nutrition. Arch Dis Child. 1990;65:909–911. doi: 10.1136/adc.65.8.909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Grefte JM, Bouman JG, Grond J, Jansen W, Kleibeuker JH. Slow and incomplete histological and functional recovery in adult gluten sensitive enteropathy. J Clin Pathol. 1988;41:886–891. doi: 10.1136/jcp.41.8.886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Rostom A, Dube C, Cranney A, Saloojee N, Sy R, Garritty C, Sampson M, Zhang L, Yazdi F, Mamaladze V, et al. Celiac Disease: Evidence Report/Technology Assessment. Rockville, MD: Agency for Healthcare Research and Quality; 2004. p. 104. [PMC free article] [PubMed] [Google Scholar]
- 153.Rashtak S, Murray JA. Tailored testing for celiac disease. Ann Intern Med. 2007;147:339–341. doi: 10.7326/0003-4819-147-5-200709040-00009. [DOI] [PubMed] [Google Scholar]
- 154.Wolters VM, Wijmenga C. Genetic background of celiac disease and its clinical implications. Am J Gastroenterol. 2008;103:190–195. doi: 10.1111/j.1572-0241.2007.01471.x. [DOI] [PubMed] [Google Scholar]
- 155.Lewis NR, Scott BB. Systematic review: the use of serology to exclude or diagnose coeliac disease (a comparison of the endomysial and tissue transglutaminase antibody tests) Aliment Pharmacol Ther. 2006;24:47–54. doi: 10.1111/j.1365-2036.2006.02967.x. [DOI] [PubMed] [Google Scholar]
- 156.Freeman HJ. Strongly positive tissue transglutaminase antibody assays without celiac disease. Can J Gastroenterol. 2004;18:25–28. doi: 10.1155/2004/912053. [DOI] [PubMed] [Google Scholar]
- 157.Bizzaro N, Villalta D, Tonutti E, Doria A, Tampoia M, Bassetti D, Tozzoli R. IgA and IgG tissue transglutaminase antibody prevalence and clinical significance in connective tissue diseases, inflammatory bowel disease, and primary biliary cirrhosis. Dig Dis Sci. 2003;48:2360–2365. doi: 10.1023/b:ddas.0000007875.72256.e8. [DOI] [PubMed] [Google Scholar]
- 158.Leffler DA, Dennis M, Edwards George JB, Jamma S, Magge S, Cook EF, Schuppan D, Kelly CP. A simple validated gluten-free diet adherence survey for adults with celiac disease. Clin Gastroenterol Hepatol. 2009;7:530–536, 536.e1-2. doi: 10.1016/j.cgh.2008.12.032. [DOI] [PubMed] [Google Scholar]
- 159.Rubio-Tapia A, Kelly DG, Lahr BD, Dogan A, Wu TT, Murray JA. Clinical staging and survival in refractory celiac disease: a single center experience. Gastroenterology. 2009;136:99–107; quiz 352-353. doi: 10.1053/j.gastro.2008.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Oxentenko AS, Grisolano SW, Murray JA, Burgart LJ, Dierkhising RA, Alexander JA. The insensitivity of endoscopic markers in celiac disease. Am J Gastroenterol. 2002;97:933–938. doi: 10.1111/j.1572-0241.2002.05612.x. [DOI] [PubMed] [Google Scholar]
- 161.Cellier C, Delabesse E, Helmer C, Patey N, Matuchansky C, Jabri B, Macintyre E, Cerf-Bensussan N, Brousse N. Refractory sprue, coeliac disease, and enteropathy-associated T-cell lymphoma. French Coeliac Disease Study Group. Lancet. 2000;356:203–208. doi: 10.1016/s0140-6736(00)02481-8. [DOI] [PubMed] [Google Scholar]
- 162.Verbeek WH, Goerres MS, von Blomberg BM, Oudejans JJ, Scholten PE, Hadithi M, Al-Toma A, Schreurs MW, Mulder CJ. Flow cytometric determination of aberrant intra-epithelial lymphocytes predicts T-cell lymphoma development more accurately than T-cell clonality analysis in Refractory Celiac Disease. Clin Immunol. 2008;126:48–56. doi: 10.1016/j.clim.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 163.Al-Toma A, Verbeek WH, Hadithi M, von Blomberg BM, Mulder CJ. Survival in refractory coeliac disease and enteropathy-associated T-cell lymphoma: retrospective evaluation of single-centre experience. Gut. 2007;56:1373–1378. doi: 10.1136/gut.2006.114512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Catassi C, Bearzi I, Holmes GK. Association of celiac disease and intestinal lymphomas and other cancers. Gastroenterology. 2005;128:S79–S86. doi: 10.1053/j.gastro.2005.02.027. [DOI] [PubMed] [Google Scholar]
- 165.Gao Y, Kristinsson SY, Goldin LR, Björkholm M, Caporaso NE, Landgren O. Increased risk for non-Hodgkin lymphoma in individuals with celiac disease and a potential familial association. Gastroenterology. 2009;136:91–98. doi: 10.1053/j.gastro.2008.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Chand N, Mihas AA. Celiac disease: current concepts in diagnosis and treatment. J Clin Gastroenterol. 2006;40:3–14. doi: 10.1097/01.mcg.0000190644.01661.2b. [DOI] [PubMed] [Google Scholar]
- 167.Schuppan D. Current concepts of celiac disease pathogenesis. Gastroenterology. 2000;119:234–242. doi: 10.1053/gast.2000.8521. [DOI] [PubMed] [Google Scholar]
- 168.Collin P, Kaukinen K, Vogelsang H, Korponay-Szabó I, Sommer R, Schreier E, Volta U, Granito A, Veronesi L, Mascart F, et al. Antiendomysial and antihuman recombinant tissue transglutaminase antibodies in the diagnosis of coeliac disease: a biopsy-proven European multicentre study. Eur J Gastroenterol Hepatol. 2005;17:85–91. doi: 10.1097/00042737-200501000-00017. [DOI] [PubMed] [Google Scholar]
- 169.Csizmadia CG, Mearin ML, Oren A, Kromhout A, Crusius JB, von Blomberg BM, Peña AS, Wiggers MN, Vandenbroucke JP. Accuracy and cost-effectiveness of a new strategy to screen for celiac disease in children with Down syndrome. J Pediatr. 2000;137:756–761. doi: 10.1067/mpd.2000.110421. [DOI] [PubMed] [Google Scholar]
- 170.Kaukinen K, Partanen J, Mäki M, Collin P. HLA-DQ typing in the diagnosis of celiac disease. Am J Gastroenterol. 2002;97:695–699. doi: 10.1111/j.1572-0241.2002.05471.x. [DOI] [PubMed] [Google Scholar]
- 171.Hadithi M, von Blomberg BM, Crusius JB, Bloemena E, Kostense PJ, Meijer JW, Mulder CJ, Stehouwer CD, Peña AS. Accuracy of serologic tests and HLA-DQ typing for diagnosing celiac disease. Ann Intern Med. 2007;147:294–302. doi: 10.7326/0003-4819-147-5-200709040-00003. [DOI] [PubMed] [Google Scholar]
- 172.Björck S, Brundin C, Lörinc E, Lynch KF, Agardh D. Screening detects a high proportion of celiac disease in young HLA-genotyped children. J Pediatr Gastroenterol Nutr. 2010;50:49–53. doi: 10.1097/MPG.0b013e3181b477a6. [DOI] [PubMed] [Google Scholar]
- 173.American Gastroenterological Association medical position statement: Celiac Sprue. Gastroenterology. 2001;120:1522–1525. doi: 10.1053/gast.2001.24055. [DOI] [PubMed] [Google Scholar]
- 174.Kaukinen K, Collin P, Laurila K, Kaartinen T, Partanen J, Mäki M. Resurrection of gliadin antibodies in coeliac disease. Deamidated gliadin peptide antibody test provides additional diagnostic benefit. Scand J Gastroenterol. 2007;42:1428–1433. doi: 10.1080/00365520701452217. [DOI] [PubMed] [Google Scholar]
- 175.Volta U, Granito A, Parisi C, Fabbri A, Fiorini E, Piscaglia M, Tovoli F, Grasso V, Muratori P, Pappas G, et al. Deamidated gliadin peptide antibodies as a routine test for celiac disease: a prospective analysis. J Clin Gastroenterol. 2010;44:186–190. doi: 10.1097/MCG.0b013e3181c378f6. [DOI] [PubMed] [Google Scholar]
- 176.Hill ID. What are the sensitivity and specificity of serologic tests for celiac disease? Do sensitivity and specificity vary in different populations? Gastroenterology. 2005;128:S25–S32. doi: 10.1053/j.gastro.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 177.Leeds JS, Hopper AD, Sanders DS. Coeliac disease. Br Med Bull. 2008;88:157–170. doi: 10.1093/bmb/ldn044. [DOI] [PubMed] [Google Scholar]
- 178.Marsh MN. Gluten, major histocompatibility complex, and the small intestine. A molecular and immunobiologic approach to the spectrum of gluten sensitivity (‘celiac sprue’) Gastroenterology. 1992;102:330–354. [PubMed] [Google Scholar]
- 179.Rostami K, Mulder CJ, Werre JM, van Beukelen FR, Kerchhaert J, Crusius JB, Peña AS, Willekens FL, Meijer JW. High prevalence of celiac disease in apparently healthy blood donors suggests a high prevalence of undiagnosed celiac disease in the Dutch population. Scand J Gastroenterol. 1999;34:276–279. doi: 10.1080/00365529950173681. [DOI] [PubMed] [Google Scholar]
- 180.Oberhuber G, Granditsch G, Vogelsang H. The histopathology of coeliac disease: time for a standardized report scheme for pathologists. Eur J Gastroenterol Hepatol. 1999;11:1185–1194. doi: 10.1097/00042737-199910000-00019. [DOI] [PubMed] [Google Scholar]
- 181.Mohamed BM, Feighery C, Coates C, O’Shea U, Delaney D, O’Briain S, Kelly J, Abuzakouk M. The absence of a mucosal lesion on standard histological examination does not exclude diagnosis of celiac disease. Dig Dis Sci. 2008;53:52–61. doi: 10.1007/s10620-007-9821-5. [DOI] [PubMed] [Google Scholar]
- 182.Auricchio R, Granata V, Borrelli M, Troncone R. Italian paediatricians’ approach to coeliac disease diagnosis. J Pediatr Gastroenterol Nutr. 2009;49:374–376. doi: 10.1097/MPG.0b013e3181940e18. [DOI] [PubMed] [Google Scholar]
- 183.Feighery C, Weir DG, Whelan A, Willoughby R, Youngprapakorn S, Lynch S, O’Moráin C, McEneany P, O’Farrelly C. Diagnosis of gluten-sensitive enteropathy: is exclusive reliance on histology appropriate? Eur J Gastroenterol Hepatol. 1998;10:919–925. [PubMed] [Google Scholar]
- 184.Hopper AD, Hadjivassiliou M, Hurlstone DP, Lobo AJ, McAlindon ME, Egner W, Wild G, Sanders DS. What is the role of serologic testing in celiac disease? A prospective, biopsy-confirmed study with economic analysis. Clin Gastroenterol Hepatol. 2008;6:314–320. doi: 10.1016/j.cgh.2007.12.008. [DOI] [PubMed] [Google Scholar]
- 185.Corazza GR, Villanacci V, Zambelli C, Milione M, Luinetti O, Vindigni C, Chioda C, Albarello L, Bartolini D, Donato F. Comparison of the interobserver reproducibility with different histologic criteria used in celiac disease. Clin Gastroenterol Hepatol. 2007;5:838–843. doi: 10.1016/j.cgh.2007.03.019. [DOI] [PubMed] [Google Scholar]
- 186.Corazza GR, Villanacci V. Coeliac disease. J Clin Pathol. 2005;58:573–574. doi: 10.1136/jcp.2004.023978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Hayat M, Cairns A, Dixon MF, O’Mahony S. Quantitation of intraepithelial lymphocytes in human duodenum: what is normal? J Clin Pathol. 2002;55:393–394. doi: 10.1136/jcp.55.5.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Cummins AG, Alexander BG, Chung A, Teo E, Woenig JA, Field JB, Thompson FM, Roberts-Thomson IC. Morphometric evaluation of duodenal biopsies in celiac disease. Am J Gastroenterol. 2011;106:145–150. doi: 10.1038/ajg.2010.313. [DOI] [PubMed] [Google Scholar]
- 189.Cellier C, Patey N, Mauvieux L, Jabri B, Delabesse E, Cervoni JP, Burtin ML, Guy-Grand D, Bouhnik Y, Modigliani R, et al. Abnormal intestinal intraepithelial lymphocytes in refractory sprue. Gastroenterology. 1998;114:471–481. doi: 10.1016/s0016-5085(98)70530-x. [DOI] [PubMed] [Google Scholar]
- 190.Patey-Mariaud De Serre N, Cellier C, Jabri B, Delabesse E, Verkarre V, Roche B, Lavergne A, Brière J, Mauvieux L, Leborgne M, et al. Distinction between coeliac disease and refractory sprue: a simple immunohistochemical method. Histopathology. 2000;37:70–77. doi: 10.1046/j.1365-2559.2000.00926.x. [DOI] [PubMed] [Google Scholar]
- 191.Carbonnel F, Grollet-Bioul L, Brouet JC, Teilhac MF, Cosnes J, Angonin R, Deschaseaux M, Châtelet FP, Gendre JP, Sigaux F. Are complicated forms of celiac disease cryptic T-cell lymphomas? Blood. 1998;92:3879–3886. [PubMed] [Google Scholar]
- 192.Daum S, Hummel M, Weiss D, Peters M, Wiedenmann B, Schäper F, Stein H, Riecken EO, Foss H. Refractory sprue syndrome with clonal intraepithelial lymphocytes evolving into overt enteropathy-type intestinal T-cell lymphoma. Digestion. 2000;62:60–65. doi: 10.1159/000007779. [DOI] [PubMed] [Google Scholar]
- 193.Collin P, Wahab PJ, Murray JA. Intraepithelial lymphocytes and coeliac disease. Best Pract Res Clin Gastroenterol. 2005;19:341–350. doi: 10.1016/j.bpg.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 194.Daum S, Weiss D, Hummel M, Ullrich R, Heise W, Stein H, Riecken EO, Foss HD. Frequency of clonal intraepithelial T lymphocyte proliferations in enteropathy-type intestinal T cell lymphoma, coeliac disease, and refractory sprue. Gut. 2001;49:804–812. doi: 10.1136/gut.49.6.804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Catassi C. The world map of celiac disease. Acta Gastroenterol Latinoam. 2005;35:37–55. [PubMed] [Google Scholar]
- 196.Lohi S, Mustalahti K, Kaukinen K, Laurila K, Collin P, Rissanen H, Lohi O, Bravi E, Gasparin M, Reunanen A, et al. Increasing prevalence of coeliac disease over time. Aliment Pharmacol Ther. 2007;26:1217–1225. doi: 10.1111/j.1365-2036.2007.03502.x. [DOI] [PubMed] [Google Scholar]
- 197.Haines ML, Anderson RP, Gibson PR. Systematic review: The evidence base for long-term management of coeliac disease. Aliment Pharmacol Ther. 2008;28:1042–1066. doi: 10.1111/j.1365-2036.2008.03820.x. [DOI] [PubMed] [Google Scholar]
- 198.Collin P, Mäki M, Kaukinen K. It is the compliance, not milligrams of gluten, that is essential in the treatment of celiac disease. Nutr Rev. 2004;62:490; author reply 491. doi: 10.1111/j.1753-4887.2004.tb00022.x. [DOI] [PubMed] [Google Scholar]
- 199.Catassi C, Fabiani E, Iacono G, D’Agate C, Francavilla R, Biagi F, Volta U, Accomando S, Picarelli A, De Vitis I, et al. A prospective, double-blind, placebo-controlled trial to establish a safe gluten threshold for patients with celiac disease. Am J Clin Nutr. 2007;85:160–166. doi: 10.1093/ajcn/85.1.160. [DOI] [PubMed] [Google Scholar]
- 200.Lohiniemi S, Mäki M, Kaukinen K, Laippala P, Collin P. Gastrointestinal symptoms rating scale in coeliac disease patients on wheat starch-based gluten-free diets. Scand J Gastroenterol. 2000;35:947–949. doi: 10.1080/003655200750023002. [DOI] [PubMed] [Google Scholar]
- 201.Donnelly SC, Ellis HJ, Ciclitira PJ. Pharmacotherapy and management strategies for coeliac disease. Expert Opin Pharmacother. 2011;12:1731–1744. doi: 10.1517/14656566.2011.592140. [DOI] [PubMed] [Google Scholar]
- 202.Matysiak-Budnik T, Candalh C, Cellier C, Dugave C, Namane A, Vidal-Martinez T, Cerf-Bensussan N, Heyman M. Limited efficiency of prolyl-endopeptidase in the detoxification of gliadin peptides in celiac disease. Gastroenterology. 2005;129:786–796. doi: 10.1053/j.gastro.2005.06.016. [DOI] [PubMed] [Google Scholar]
- 203.Pyle GG, Paaso B, Anderson BE, Allen DD, Marti T, Li Q, Siegel M, Khosla C, Gray GM. Effect of pretreatment of food gluten with prolyl endopeptidase on gluten-induced malabsorption in celiac sprue. Clin Gastroenterol Hepatol. 2005;3:687–694. doi: 10.1016/s1542-3565(05)00366-6. [DOI] [PubMed] [Google Scholar]
- 204.Tack GJ, van de Water JM, Kooy-Winkelaar EM, van Bergen J, Meijer GA, von Blomberg BM, Schreurs MW, Bruins MJ, Edens L, Mulder CJ, et al. Can prolyl endoprotease enzyme treatment mitigate the toxic effect of gluten in coeliac patients? Gastroenterology. 2010;138:S–54. [Google Scholar]
- 205.Tye-Din JA, Anderson RP, Ffrench RA, Brown GJ, Hodsman P, Siegel M, Botwick W, Shreeniwas R. The effects of ALV003 pre-digestion of gluten on immune response and symptoms in celiac disease in vivo. Clin Immunol. 2010;134:289–295. doi: 10.1016/j.clim.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 206.Lhdeaho ML, Mki M, Kaukinen K, Laurila K, Marcantonio A, Adelman D. OP050B: ALV003, A novel glutenase, attenuates gluten-induced small intestinal mucosal injury in celiac disease patients: A randomized controlled phase IIa clinical trial. Stockholm: 19th United European Gastroenterology Week; 2011. [Google Scholar]
- 207.De Angelis M, Rizzello CG, Fasano A, Clemente MG, De Simone C, Silano M, De Vincenzi M, Losito I, Gobbetti M. VSL#3 probiotic preparation has the capacity to hydrolyze gliadin polypeptides responsible for Celiac Sprue. Biochim Biophys Acta. 2006;1762:80–93. doi: 10.1016/j.bbadis.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 208.Rizzello CG, De Angelis M, Di Cagno R, Camarca A, Silano M, Losito I, De Vincenzi M, De Bari MD, Palmisano F, Maurano F, et al. Highly efficient gluten degradation by lactobacilli and fungal proteases during food processing: new perspectives for celiac disease. Appl Environ Microbiol. 2007;73:4499–4507. doi: 10.1128/AEM.00260-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Di Cagno R, De Angelis M, Auricchio S, Greco L, Clarke C, De Vincenzi M, Giovannini C, D’Archivio M, Landolfo F, Parrilli G, et al. Sourdough bread made from wheat and nontoxic flours and started with selected lactobacilli is tolerated in celiac sprue patients. Appl Environ Microbiol. 2004;70:1088–1096. doi: 10.1128/AEM.70.2.1088-1096.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Di Cagno R, De Angelis M, Lavermicocca P, De Vincenzi M, Giovannini C, Faccia M, Gobbetti M. Proteolysis by sourdough lactic acid bacteria: effects on wheat flour protein fractions and gliadin peptides involved in human cereal intolerance. Appl Environ Microbiol. 2002;68:623–633. doi: 10.1128/AEM.68.2.623-633.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Greco L, Gobbetti M, Auricchio R, Di Mase R, Landolfo F, Paparo F, Di Cagno R, De Angelis M, Rizzello CG, Cassone A, et al. Safety for patients with celiac disease of baked goods made of wheat flour hydrolyzed during food processing. Clin Gastroenterol Hepatol. 2011;9:24–29. doi: 10.1016/j.cgh.2010.09.025. [DOI] [PubMed] [Google Scholar]
- 212.Drago S, El Asmar R, Di Pierro M, Grazia Clemente M, Tripathi A, Sapone A, Thakar M, Iacono G, Carroccio A, D’Agate C, et al. Gliadin, zonulin and gut permeability: Effects on celiac and non-celiac intestinal mucosa and intestinal cell lines. Scand J Gastroenterol. 2006;41:408–419. doi: 10.1080/00365520500235334. [DOI] [PubMed] [Google Scholar]
- 213.Paterson BM, Lammers KM, Arrieta MC, Fasano A, Meddings JB. The safety, tolerance, pharmacokinetic and pharmacodynamic effects of single doses of AT-1001 in coeliac disease subjects: a proof of concept study. Aliment Pharmacol Ther. 2007;26:757–766. doi: 10.1111/j.1365-2036.2007.03413.x. [DOI] [PubMed] [Google Scholar]
- 214.Kelly CP, Green PH, Murray JA. Intestinal permeability of larazotide acetate in celiac disease: results of a phase IIB 6-week gluten-challenge clinical trial (abstr) Gastroenterology. 2009;136:A–474. [Google Scholar]
- 215.Liang L, Pinier M, Leroux JC, Subirade M. Interaction of alpha-gliadin with poly(HEMA-co-SS): structural characterization and biological implication. Biopolymers. 2009;91:169–178. doi: 10.1002/bip.21109. [DOI] [PubMed] [Google Scholar]
- 216.Liang L, Pinier M, Leroux JC, Subirade M. Interaction of alpha-gliadin with polyanions: design considerations for sequestrants used in supportive treatment of celiac disease. Biopolymers. 2010;93:418–428. doi: 10.1002/bip.21352. [DOI] [PubMed] [Google Scholar]
- 217.Pinier M, Verdu EF, Nasser-Eddine M, David CS, Vézina A, Rivard N, Leroux JC. Polymeric binders suppress gliadin-induced toxicity in the intestinal epithelium. Gastroenterology. 2009;136:288–298. doi: 10.1053/j.gastro.2008.09.016. [DOI] [PubMed] [Google Scholar]
- 218.Pinier M, Fuhrmann G, Verdu EF, Leroux JC. Prevention measures and exploratory pharmacological treatments of celiac disease. Am J Gastroenterol. 2010;105:2551–2561; quiz 2562. doi: 10.1038/ajg.2010.372. [DOI] [PubMed] [Google Scholar]
- 219.Reilly RM, Domingo R, Sandhu J. Oral delivery of antibodies. Future pharmacokinetic trends. Clin Pharmacokinet. 1997;32:313–323. doi: 10.2165/00003088-199732040-00004. [DOI] [PubMed] [Google Scholar]
- 220.Carlander D, Kollberg H, Wejåker PE, Larsson A. Peroral immunotherapy with yolk antibodies for the prevention and treatment of enteric infections. Immunol Res. 2000;21:1–6. doi: 10.1385/IR:21:1:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Gujral N, Löbenberg R, Suresh M, Sunwoo H. In-vitro and in-vivo binding activity of chicken egg yolk immunoglobulin Y (IgY) against gliadin in food matrix. J Agric Food Chem. 2012;60:3166–3172. doi: 10.1021/jf205319s. [DOI] [PubMed] [Google Scholar]
- 222.Griffin M, Casadio R, Bergamini CM. Transglutaminases: nature’s biological glues. Biochem J. 2002;368:377–396. doi: 10.1042/BJ20021234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Siegel M, Khosla C. Transglutaminase 2 inhibitors and their therapeutic role in disease states. Pharmacol Ther. 2007;115:232–245. doi: 10.1016/j.pharmthera.2007.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Watts RE, Siegel M, Khosla C. Structure-activity relationship analysis of the selective inhibition of transglutaminase 2 by dihydroisoxazoles. J Med Chem. 2006;49:7493–7501. doi: 10.1021/jm060839a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Siegel M, Xia J, Khosla C. Structure-based design of alpha-amido aldehyde containing gluten peptide analogues as modulators of HLA-DQ2 and transglutaminase 2. Bioorg Med Chem. 2007;15:6253–6261. doi: 10.1016/j.bmc.2007.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Pardin C, Roy I, Lubell WD, Keillor JW. Reversible and competitive cinnamoyl triazole inhibitors of tissue transglutaminase. Chem Biol Drug Des. 2008;72:189–196. doi: 10.1111/j.1747-0285.2008.00696.x. [DOI] [PubMed] [Google Scholar]
- 227.Schuppan D, Junker Y, Barisani D. Celiac disease: from pathogenesis to novel therapies. Gastroenterology. 2009;137:1912–1933. doi: 10.1053/j.gastro.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 228.Vader W, Kooy Y, Van Veelen P, De Ru A, Harris D, Benckhuijsen W, Peña S, Mearin L, Drijfhout JW, Koning F. The gluten response in children with celiac disease is directed toward multiple gliadin and glutenin peptides. Gastroenterology. 2002;122:1729–1737. doi: 10.1053/gast.2002.33606. [DOI] [PubMed] [Google Scholar]
- 229.Arentz-Hansen H, McAdam SN, Molberg Ø, Fleckenstein B, Lundin KE, Jørgensen TJ, Jung G, Roepstorff P, Sollid LM. Celiac lesion T cells recognize epitopes that cluster in regions of gliadins rich in proline residues. Gastroenterology. 2002;123:803–809. doi: 10.1053/gast.2002.35381. [DOI] [PubMed] [Google Scholar]
- 230.Keech CL, Dromey J, Tye-Din JA. Immune tolerance induced by peptide immunotherapy in an HLA Dq2-dependent mouse model of gluten immunity. Gastroenterology. 2009;136:A–57. [Google Scholar]
- 231.Huibregtse IL, Marietta EV, Rashtak S. Induction of antigen-specific tolerance by oral administration of Lactococcus lactis delivered immunodominant DQ8-restricted gliadin peptide in sensitized nonobese diabetic Ab Dq8 transgenic mice. J Immunol. 2009;183:2390–2396. doi: 10.4049/jimmunol.0802891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Senger S, Luongo D, Maurano F, Mazzeo MF, Siciliano RA, Gianfrani C, David C, Troncone R, Auricchio S, Rossi M. Intranasal administration of a recombinant alpha-gliadin down-regulates the immune response to wheat gliadin in DQ8 transgenic mice. Immunol Lett. 2003;88:127–134. doi: 10.1016/s0165-2478(03)00069-5. [DOI] [PubMed] [Google Scholar]
- 233.Weinstock JV, Elliott DE. Helminths and the IBD hygiene hypothesis. Inflamm Bowel Dis. 2009;15:128–133. doi: 10.1002/ibd.20633. [DOI] [PubMed] [Google Scholar]
- 234.Daveson AJ, Jones DM, Gaze S, McSorley H, Clouston A, Pascoe A, Cooke S, Speare R, Macdonald GA, Anderson R, et al. Effect of hookworm infection on wheat challenge in celiac disease--a randomised double-blinded placebo controlled trial. PLoS One. 2011;6:e17366. doi: 10.1371/journal.pone.0017366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Falcioni F, Ito K, Vidovic D, Belunis C, Campbell R, Berthel SJ, Bolin DR, Gillespie PB, Huby N, Olson GL, et al. Peptidomimetic compounds that inhibit antigen presentation by autoimmune disease-associated class II major histocompatibility molecules. Nat Biotechnol. 1999;17:562–567. doi: 10.1038/9865. [DOI] [PubMed] [Google Scholar]
- 236.Anderson RP, van Heel DA, Tye-Din JA, Jewell DP, Hill AV. Antagonists and non-toxic variants of the dominant wheat gliadin T cell epitope in coeliac disease. Gut. 2006;55:485–491. doi: 10.1136/gut.2005.064550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Silano M, Vincentini O, Iapello A, Mancini E, De Vincenzi M. Antagonist peptides of the gliadin T-cell stimulatory sequences: a therapeutic strategy for celiac disease. J Clin Gastroenterol. 2008;42 Suppl 3 Pt 2:S191–S192. doi: 10.1097/MCG.0b013e31817df76a. [DOI] [PubMed] [Google Scholar]
- 238.Anderson RP, van Heel DA, Tye-Din JA, Jewell DP, Hill AV. Antagonists and non-toxic variants of the dominant wheat gliadin T cell epitope in coeliac disease. Gut. 2006;55:485–491. doi: 10.1136/gut.2005.064550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Baslund B, Tvede N, Danneskiold-Samsoe B, Larsson P, Panayi G, Petersen J, Petersen LJ, Beurskens FJ, Schuurman J, van de Winkel JG, et al. Targeting interleukin-15 in patients with rheumatoid arthritis: a proof-of-concept study. Arthritis Rheum. 2005;52:2686–2692. doi: 10.1002/art.21249. [DOI] [PubMed] [Google Scholar]
- 240.Mention JJ, Ben Ahmed M, Bègue B, Barbe U, Verkarre V, Asnafi V, Colombel JF, Cugnenc PH, Ruemmele FM, McIntyre E, et al. Interleukin 15: a key to disrupted intraepithelial lymphocyte homeostasis and lymphomagenesis in celiac disease. Gastroenterology. 2003;125:730–745. doi: 10.1016/s0016-5085(03)01047-3. [DOI] [PubMed] [Google Scholar]
- 241.Salvati VM, Mazzarella G, Gianfrani C, Levings MK, Stefanile R, De Giulio B, Iaquinto G, Giardullo N, Auricchio S, Roncarolo MG, et al. Recombinant human interleukin 10 suppresses gliadin dependent T cell activation in ex vivo cultured coeliac intestinal mucosa. Gut. 2005;54:46–53. doi: 10.1136/gut.2003.023150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Kjellev S, Haase C, Lundsgaard D, Ursø B, Tornehave D, Markholst H. Inhibition of NKG2D receptor function by antibody therapy attenuates transfer-induced colitis in SCID mice. Eur J Immunol. 2007;37:1397–1406. doi: 10.1002/eji.200636473. [DOI] [PubMed] [Google Scholar]
- 243.Zhao J, de Vera J, Narushima S, Beck EX, Palencia S, Shinkawa P, Kim KA, Liu Y, Levy MD, Berg DJ, et al. R-spondin1, a novel intestinotrophic mitogen, ameliorates experimental colitis in mice. Gastroenterology. 2007;132:1331–1343. doi: 10.1053/j.gastro.2007.02.001. [DOI] [PubMed] [Google Scholar]